

Abstract
Increased resting metabolic rate and skeletal muscle wasting are hallmarks of the pathophysiological stress response to severe burn trauma. However, whether these two responses occur independently in burn patients or are in fact related remains unclear. In light of recent evidence demonstrating that increased proteolysis in skeletal muscle of burned patients is accompanied by mitochondrial hypermetabolism, oxidative stress, and protein damage; in this article, we discuss the evidence for a role for the mitochondrion in skeletal muscle wasting following severe burn trauma. In particular, we focus on the role of mitochondrial superoxide production in oxidative stress and subsequent proteolysis, and discuss the role of the mitochondrion as a signaling organelle resulting in protein catabolism in other cellular compartments following severe burn trauma.
Skeletal muscle wasting, an undesired loss of skeletal muscle mass, is a hallmark of the long-term pathophysiological stress response to severe burn trauma, which significantly contributes to the long-term morbidity of burn survivors.1 Patients with a ≥30% TBSA burn can lose up to 25% of their body mass in the first month post injury.2 In the acute period post burn, muscle wasting is associated with delayed wound healing, while in the long-term, muscle wasting is associated with reduced muscle strength and function.1 From a physiological perspective, muscle wasting in burn patients results from an increase in the rate at which muscle protein is broken down. In fact, the synthetic rate of skeletal muscle proteins is also elevated in burn survivors, which may be consequence of enhanced proteolysis increasing intracellular availability of amino acids.3–5 However, the magnitude of this increase in muscle anabolism is lesser than that of increased protein breakdown. This results in a net loss of protein from skeletal muscle of burn patients, even when aggressive nutritional support is provided.6
While our understanding of the pathophysiological stress response to severe burn trauma has significantly improved in the past few decades,1 the mechanisms underlying chronic skeletal muscle wasting in response to burn trauma remain poorly understood, particularly at the cellular and molecular levels. Several clinical studies have been undertaken to help improve outcomes by attenuating muscle wasting in burn patients.7–21 However, these studies have typically focused on augmenting muscle protein synthesis to better match elevated protein breakdown after burn. In our view, there is still a need to better understand the cellular mechanisms underlying burn-induced skeletal muscle proteolysis to devise new strategies that mitigate this deleterious response and hasten the recovery of burn survivors.
Hypermetabolism (increased whole body oxygen consumption) is another hallmark of the stress response to severe burn injury.1 The destruction of the body’s skin barrier and subsequent adrenergic stimulation accompanying a large burn necessitates a hypermetabolic response, which principally serves to fuel wound healing, fight infection, and maintain core temperature.1 Whether this hypermetabolic response is linked to muscle wasting in burn patients is unclear. Hart et al22 have previously reported a significant correlation between the degree of hypermetabolism and magnitude of amino acid loss from the leg in severely burned patients. However, because there is a clear link between the degree of injury (ie, TBSA burned) and both hypermetabolism and muscle catabolism, it remains difficult to discern whether hypermetabolism directly contributes to muscle wasting post burn.
Skeletal muscle is a significant contributor to resting metabolic rate in both healthy individuals23 and severely burned patients.24 Indeed, in patients with massive burns, skeletal muscle oxygen consumption rates can double.25 The mitochondrion is the cellular organelle responsible for the vast majority (~90%) of cellular oxygen consumption.23 Given the central role of mitochondria in oxidative phosphorylation and thermogenesis, it is perhaps not surprising that burn patients muscle mitochondria have been estimated to consume approximately 50% more oxygen than those of an average healthy individual,26,27 at least in the first few months post injury. While hypermetabolism, altered mitochondrial function, and muscle wasting are well-documented facets of the stress response to severe burn trauma, few studies have sought to determine the roles of hypermetabolism, and in particular mitochondrial hypermetabolism, in burn-induced skeletal muscle wasting.
Following severe burn trauma, chronically elevated respiration rates predispose mitochondria to stress in tissues such as skeletal muscle, which can increase protein oxidation and contribute to activation of mitochondrial unfolded protein stress response (mtUPR). Indeed, due to their central role in oxidative phosphorylation, mitochondria are the principal sites of cellular reactive oxygen species (ROS) generation.28 Increased electron transfer to support oxidative phosphorylation is accompanied by an obligatory leak of electrons from the electron transport chain at sites other than the heme domains of cytochrome C oxidase, ultimately resulting in increased formation of superoxide anions (O2•−). Greater O2•− production can contribute to the inhibition of mitochondrial aconitase (mtAcn), because mtAcn is sensitive to the presence of ROS and in particular, O2•−.29,30 Inactivation of mtAcn has been shown to enhance hydrogen peroxide (H2O2) production and iron (Fe2+) accumulation, which drives the Fenton reaction.30,31 Specifically, through the Fenton reaction, mtAcn is a major source of hydroxyl radical ions (•OH), a particularly damaging ROS implicated in protein oxidation.31,32 Therefore, persistent mitochondrial hypermetabolism in response to burn trauma increases the production of “byproducts” of electron transfer, namely O2•− and •OH. The continuous generation of O2•−and •OH endanger both the mitochondrial proteins and other structures within the cell.2,33 Indeed, as we will discuss in detail below, this mitochondrial stress response can also be relayed to other cellular compartments, therefore contributing to increased protein damage in other parts of the cell (Figure 1).
Figure 1.

Schematic overview of the mitochondrial stress response to burn trauma in skeletal muscle. ETC, electron transport chain; O2•−, Superoxide ion; •OH, hydroxyl ion; PRDX3, thioredoxin-dependent peroxide reductase; GPRX, glutathione peroxidase; HSP, heat shock protein; TOM, translocase of the outer membrane; TIM, translocase of the inner membrane; CHOP, C/enhancer-binding protein homologous protein; C/EBPβ, CCAAT/enhancer-binding protein β.
Although mitochondria are normally capable of eliminating ROS and damaged proteins, the magnitude and persistence of the hypermetabolic stress to severe burns may overwhelm the mitochondrial quality control system.28,34 In the present review, we discuss recent data regarding the response of skeletal muscle mitochondria to hypermetabolism in burn patients, and the role of mitochondrial stress in skeletal muscle catabolism. Moreover, we propose a hypothesis whereby mitochondrial hypermetabolism and subsequent oxidative stress contributes to muscle wasting in patients with severe burns. Finally, we will briefly discuss the utility of strategies aimed at mitigating muscle wasting in response to severe burn trauma by blunting oxidative stress.
BURN TRAUMA INDUCES MITOCHONDRIAL STRESS IN SKELETAL MUSCLE
Increased O2•− and •OH production in skeletal muscle of burn patients predisposes mitochondrial proteins to proteotoxic stress.2 Accumulation of damaged and/or misfolded proteins results in mitochondrial dysfunction, as seen in skeletal muscle of burn patients,26,27 which may in turn potentiate further ROS production. A mtUPR is activated when proteotoxic stress exceeds protein-folding capacity of chaperones. A nuclear response is then initiated to import new replacement proteins to re-establish homeostasis within the mitochondrial protein-folding environment.2,35,36
In response to mitochondria proteotoxic stress, several regulatory transcription factors such as C/enhancer-binding protein (EBP) homologous protein (CHOP) and CCAAT/EBP β (C/EBPβ) are activated.37,38 CHOP is induced early during mitochondrial stress, because it contributes to the transcription of heat shock protein (HSP60) to prevent aggregation of oxidatively damaged and unfolded proteins.37,39 Along with the upregulation of CHOP and C/EBPβ, elevated c-jun n-terminal kinase 2 (JNK2) and activator protein-1 (AP-1) has been reported in response to mitochondrial stress in vitro.37,38 The ultimate goal of the activation of these transcription factors is to increase the production of nuclear-encoded mitochondrial chaperones, such as HSP60 and HSP10. Increased production of these chaperones facilitates the assembly of new proteins in the mitochondria to meet increased protein demands in the face of greater protein misfolding and degradation.2,37,39
Interestingly, we have recently shown increased mRNA expression and protein concentrations of a number of mediators of the mtUPR response in skeletal muscle of severely burned patients.2 Specifically, we reported the increased transcription of JNK2, Lon peptidase 1 (LONP1), c-JUN, CHOP, C/EBPβ, C/EBPα, HSP60, ATP-dependent Clp protease (CLpP), JUNB, and Activating transcription factor 6 (ATF6) mRNA, and elevated relative protein expression of LONP1, CHOP, and HSP90 in muscle of burn patients. This mtUPR was accompanied by a concurrent increase in whole body hypermetabolism, skeletal muscle mitochondrial hypermetabolism, oxidative stress, and marked skeletal muscle catabolism.2 Collectively these data suggest that mtUPR is activated in skeletal muscle of burn patients in response to hypermetabolism and increased oxidative stress, and likely functions to prevent mitochondrial protein aggregation, dysfunction, and ultimately, mitophagy. However, it is important to note that while HSP60, CLpP, and LONP1 are specific to the mitochondrion, the majority of the transcription factors induced in skeletal muscle following burn trauma are not. Thus, while these data support the notion that a mitochondrial stress response is mounted in skeletal muscle of severely burned individuals, they also suggest a more global stress response that likely effects multiple cellular organelles.
MITOCHONDRIAL STRESS: TRANSMISSION TO THE CELL CYTOSOL
Cellular ROS are predominantly produced within the mitochondrion.28 A potentially deleterious outcome of prolonged mitochondrial hypermetabolism following burn trauma is the generation and transduction of mitochondrial ROS into the cell cytosol; a concept supported by the recent study by Maharjan et al,40 who demonstrated that the transmission of mitochondrial stress to the cell cytosol is feasible. These researchers further showed that treating cells with mitochondrial-targeted antioxidants and antioxidant enzymes attenuated cellular damage and protected cellular integrity.40 Furthermore, Kirstein et al41 demonstrated the transduction of redox state and peroxide between tissue (neurons and muscle) and between organelles (endoplasmic reticulum [ER] and cytosol) in vitro and in vivo. Collectively, these studies suggest that mitochondrial ROS production can result in oxidative damage in other cellular compartments/organelles.
In the skeletal muscle of patients with severe burns, we have recently reported profound oxidative stress, induction of cytoprotective transcription factors, antioxidants, and cytosolic proteasomes (i.e., the 26S proteasome).2 Interestingly, we also reported higher mRNA level of nuclear factor erythroid 2-related factor 2/nuclear factor (erythroid derived 2)-like 2 (NFE2L2) and NFE2L1 transcription factors.2 NFE2L2 is known as a transcription factor responsible for protection of cells from oxidative stress.2,42,43 Activation of NFE2L2 protects cells from mitochondrial ROS by up regulating nuclear respiratory factor1 (NrF1) and peroxisome proliferator-activated receptor γ co-activator 1α.43 Cell in the cytoplasm, NFE2L2 dissociates from kelch-like ECH-associated protein 1 (KEAP1) in the presence of oxidative stress to activate antioxidant defense transcription factors and ubiquitin proteasome pathways, which facilitates the neutralization of excess oxidants and the degradation of denatured proteins, respectively.44–46 Elevated mRNA levels of both NFE2L2 and KEAP1 in skeletal muscle of patients with burns provide evidence that oxidative stress is transmitted from the mitochondrion to the cell cytosol.28
The degradation of damaged proteins is performed by the proteasomes in the cell cytosol, a process which can be induced by oxidative stress.47 In agreement with Majetschak et al,48 who reported higher levels of 20S proteasomes in the plasma of patients with severe burns, we recently demonstrated the upregulation of proteasomes in skeletal muscle of severely burned individuals, which was accompanied by the accumulation of damaged proteins, indicating greater protein oxidation and degradation in skeletal muscle of burn patients.2 Collectively, these data are consistent with the hypothesis that hypermetabolism-induced mitochondrial ROS can be transmitted to the cytosol, resulting in protein damage and breakdown.
The above observation may have important physiological implications. Specifically, skeletal muscle wasting in response to severe burns has long been thought to function to provide substrate (amino acids) for other important processes in the body (i.e., the acute phase response and wound healing), suggesting that the process of skeletal muscle protein wasting post burn is not futile or inefficient, and likely serves an important function in the healing process. Thus, attempting to block this response may in fact be facile if adequate counter measures to support the bodies’ enhanced demand for amino acids are not provided. However, if a portion of muscle proteolysis following burn trauma results from the oxidative damage of cytosolic proteins, then this component of the proteolytic response to burns may indeed be wasteful, and therefore could represent a potential target for further therapeutic manipulation. Crucially, this would not carry the same constraints as strategies targeting proteolysis per se.
INCREASED MITOCHONDRIAL PROTEIN TURNOVER IN SKELETAL MUSCLE OF BURN SURVIVORS
The integrity of mitochondrial proteins is of obvious importance. Mitochondria exposed to unabated ROS will accumulate damaged proteins, ultimately impairing their function. Thus, mitochondria must protect their proteins from the potentially harmful effects of ROS. Mitochondria use various means to ensure the quality of their protein components.49,50 However, proteasomes responsible for the degradation of oxidative damaged and misfolded proteins are present only in the cell cytosol.51 Subsequently, mitochondria are equipped with specialized proteolytic systems responsible for the degradation of damaged mitochondrial proteins before they aggregate and cause dysfunction.50
The mitochondrial AAA protease family of proteases, LONP1, YME1 Like 1 ATPase (YME1L1), and CLpP, play critical roles in mitochondrial protein turnover.5,10 LONP1 facilitates the degradation of oxidized and misfolded proteins within the mitochondrial matrix, while YME1L1 is localized in the mitochondrial inner membrane, where it carries out a similar function to LONP1.49,51,52 CLpP also resides within the mitochondrial matrix.52 CLpP is also responsible for the degradation of misfolded proteins and participates in the activation of mtUPR within the mitochondrial matrix.53
LONP1 and CLpP are responsible for selective degradation of mitochondrial matrix proteins, preventing the aggregation of damaged proteins.54,55 Bezawork-Geleta et al55 tracked the fate of ornithine transcarbamylase (OTC-Δ), a mitochondria UPRmt-specific protein, in mammalian cells to determine the role of CLpP and LONP1 in the degradation of unfolded mitochondrial proteins.39 Their data demonstrated that LONP1 plays a more important role in degrading OTC-Δ, suggesting that LONP1 may be a more important player in mitochondrial protein degradation.55 Furthermore, in addition to degrading unfolded proteins, LONP1 has been shown to degrade oxidatively damaged mitochondrial proteins such as aconitase.29,32 However, we should note that while CLpP may not play as an important role in degrading OTC-Δ, it may be critical for the removal of other damaged mitochondrial proteins.56,57 Indeed, Zhao et al39 demonstrated that CLpP protein is upregulated following accumulation of unfolded protein in mammalian cells.
In addition to LONP1 and CLpP, YME1L1, which is localized within the mitochondrial intermembrane space, plays an important role in mitochondrial protein turnover. Protein quality control in this compartment of the mitochondrion is of paramount importance, particularly when considering that it is the site of electron transfer and oxidative phosphorylation. Accordingly, the phospholipid membranes and the transmembrane protein complexes that reside within them are particularly susceptible to oxidative damage. The role of YME1L1 in protein turnover in the mitochondrial intermembrane space is well documented.58,59 A recent report by Rainbolt et al58 demonstrated that oxidative stress reduces YME1L1 levels and function, which sensitizes cells to oxidative stress. Furthermore, the loss of YME1L1 in mouse embryonic fibroblast cells results in elevated mitochondrial fragmentation and leads to significant increased mitochondrial fusion,59 suggesting a critical role for YME1L1 in mitochondrial proteostasis and function.
Recently, we reported that the expression of YME1L1, LONP1, and CLpP were upregulated in skeletal muscle of patients with severe burns.2 Furthermore, we found that LONP1 protein abundance was about 60% higher in burn patients skeletal muscle compared with healthy controls.2 Interestingly, we report here that LONP1 mRNA levels are positively correlated with increased leak of mitochondrial respiration in skeletal muscle of burn patients (Figure 2), supporting an association between hypermetabolism and mitochondrial protein turnover. We hypothesize that the significant increase in transcription of these mitochondrial proteases reflects an adaptive response to prevent aggregation of damaged proteins within the mitochondrion. Collectively, these data suggest that persistent mitochondrial hypermetabolism seen in patients with severe burns results in oxidative stress, induction of mitochondrial proteases, and increased mitochondrial protein breakdown in skeletal muscle.
Figure 2.
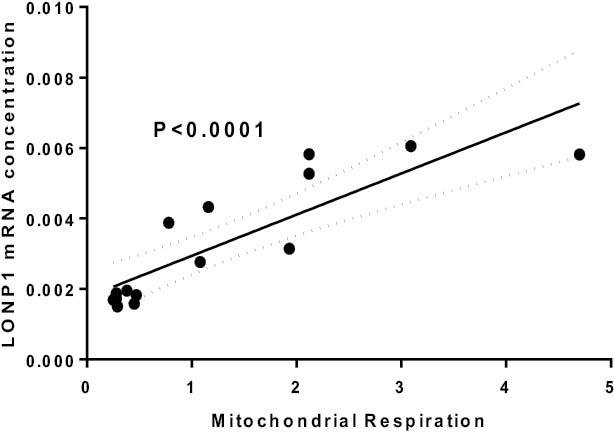
The relationship between Lon peptidase 1 (LONP1) transcription and mitochondrial hypermetabolism in skeletal muscle of patients with severe burns. Greater mitochondrial hypermetabolism in response to burns is associated with greater transcription of the mitochondrial peptidase LONP1. Raw data were originally published in reference 1.
DOES MITOCHONDRIAL PROTEIN DEGRADATION IMPACT CYTOSOL PROTEIN TURNOVER IN SKELETAL MUSCLE OF BURN PATIENTS?
The increased mitochondrial protein turnover in skeletal muscle of burn patients may have important implications for protein homeostasis in the rest of the cell. Since ~98% of mitochondrial proteins are encoded in the nucleus and thus synthesized outside of the mitochondrion,35,49,50,61–63 mitochondrial and cytosolic protein turnover are closely linked, where increased mitochondrial protein degradation must be met by an increase in synthesis and transport of new proteins into mitochondria. Indeed, the vast majority of proteins that constitute the electron transport chain are imported from the cell cytosol.64 Thus, the maintenance of the mitochondrial protein pool is heavily dependent on the nucleus and protein availability within the cell cytosol.35,65 Therefore, it is logical to suppose that increased stress-induced mitochondrial protein degradation in skeletal muscle of burn patients likely contributes to skeletal muscle wasting by diverting resources away from the synthesis of myofibrillar proteins.
Transport of mitochondrial proteins depends on loosely assembled nuclear-encoded proteins from the cell cytosol.62,66 The specialized translocase complex on the outer mitochondrial membrane that interacts with newly synthesized preproteins is known as the translocase of the outer membrane (TOM) complex.66 After translocation of the preproteins through the TOM complex, they proceed into the mitochondrial matrix by interacting with and moving through the inner mitochondrial membrane via the translocase of inner membrane (TIM) complex.62,66,67 Besides these translocase protein complexes, translocation of the preproteins through the inner membrane space also depends on the presence of HSP70, co-chaperone Mge1, ATP hydrolysis, and an appropriate mitochondrial membrane potential.66 In the mitochondrial matrix, processing peptidases selectively remove presequence amino acids and the proteins are folded into mature peptides with or without the help of chaperones.62,66
In line with increased expression of mitochondrial proteases and increased mitochondrial protein degradation, we found that mitochondrial membrane translocase proteins (TOM40, TIM23, and TIM17), responsible for protein transport into the mitochondria, were also upregulated in skeletal muscle of burn patients.2 We suggest that increased protein expression of TOM and TIM translocases supports greater protein transport from the cell cytosol into mitochondria to replace the damaged proteins that are removed by mitochondrial proteases. However, this increased demand for new proteins by mitochondria poses a threat to proteostasis in the cell cytosol. Furthermore, efflux of ROS from the mitochondria of burn patients also results in damage to cytosol proteins, leading to their degradation by the proteasomes (Figure 1). Indeed, recent studies have demonstrated increased protein damage and activation of proteosomes in skeletal muscle of burn patients.2,48 Thus, oxidative stress in response to severe burns likely results in protein degradation in both the mitochondrion and cytosol of skeletal muscle. Furthermore, the reliance of the mitochondrion on the cell cytosol for the bulk of its newly synthesized protein may constitute a double hit on cytosolic amino acids stores.
Most data on the impact of severe burn trauma on skeletal muscle protein turnover have assayed amino acid flux into and out of bound proteins within a mixed muscle homogenate. The prevailing opinion is that the net efflux of amino acids from these bound proteins largely originates from the contractile myofibrilar protein pool. Because myofibrilar protein represents the bulk of the total protein pool within skeletal muscle, this is most likely true. However, the mitochondrial protein pool represents ~5% of the protein pool in skeletal muscle,68 and may have a greater turnover rate than that of contractile proteins.69 Thus, increased damage to the mitochondrial protein pool may significantly influence intracellular amino acid utilization in skeletal muscle of burn patients. While skeletal muscle contractile proteins likely represent an amino acid reservoir used to buffer circulating amino acid levels in patients recovering from burns,6 our new data suggest that increased mitochondrial protein turnover likely places additional demands on the skeletal muscle protein pool, contributing to muscle protein wasting following severe burn trauma.
THERAPEUTIC STRATEGIES TO BLUNT MITOCHONDRIAL STRESS IN BURN-INDUCED HYPERMETABOLISM WASTING
Modulating oxidative stress may be a plausible approach to blunt hypermetabolism and skeletal muscle protein losses following severe burn trauma. Interestingly, a growing body of evidence suggests that uncoupled mitochondrial respiration may be an important component of the hypermetabolic stress response to severe burns.26,27,70–74 Increased uncoupled respiration is mediated by mitochondrial uncoupling proteins. Of interest, UCP2 and UCP3, both of which are expressed in skeletal muscle, have been postulated to play a role in reducing mitochondrial ROS production.75 This suggests that greater uncoupled mitochondrial respiration in tissues such as skeletal muscle may be a protective response aimed at reducing ROS production. Either way, modulating ROS production and/or providing exogenous antioxidants compounds to mop up excess ROS may hold value in terms of reducing hypermetabolism and attenuating oxidative stress and protein damage in skeletal muscle (and other tissues following major burn trauma).76,77
A hurdle to this approach is the need to develop antioxidants that can penetrate the mitochondrial matrix to help directly mop up the excess mitochondrial ROS. Indeed, the outer mitochondrial membrane is made up of a phospholipid bilayer with a membrane potential of approximately of ~150 mV.78,79 As a result, antioxidant molecules need to be lipid soluble and positively charged to enter into the mitochondrial membrane space or matrix.79 Interestingly, despite the obvious role of mitochondria in oxidative stress following burn trauma, data on mitochondrial-targeted antioxidants in burn patients are scarce, perhaps owing to the difficultly in providing antioxidant therapy that will reach the mitochondrion. A report by Carter et al80 showed that mitochondrial-targeted antioxidant peptide SS-31 ameliorated burn-induced insulin resistance. In addition, Righi et al34 demonstrated that SS-31 blunted hypermetabolism and improved mitochondrial redox status and coupling post burn. These studies suggest that this mitochondrial-specific antioxidant has the potential of restoring mitochondrial function post burn. Human trials on SS-31 and other similar compounds are eagerly awaited.
As mentioned previously, we recently demonstrated that nuclear factor erythroid 2-related factor 2 mRNA was elevated following burn trauma but interestingly, its relative protein abundances were in fact lower in burn patients compared with healthy adults.2 In response to accumulation of oxidized proteins and/or stress, transcription factors such as the NFE2L2 are activated,42 resulting in the transcription of a battery of cytoprotective genes. The low expression of this NRF2 protein in skeletal muscle of burn patients suggests that therapeutic administration of this protein might help ameliorate the effect of oxidative stress in burn patients. An interesting study by Nelson et al60 showed that administration of protandim, a synergetic activator of NRF2, to healthy human subjects increased superoxide dismutase protein 100-fold.81 The therapeutic advantage of NRF2 is that it has the capacity to induce several cytoprotective genes including membrane transporters.42 This might provide a potential therapeutic drug to help protect both the mitochondrial and cytoplasmic proteins in burn patients but remains to be tested.
SUMMARY
Because burn-induced skeletal muscle wasting contributes significantly to morbidity, it is imperative to understand the molecular mechanisms underlying this response to devise novel treatment solutions. Recent data suggest a link between hypermetabolism and skeletal muscle wasting following burn trauma. Specifically, hypermetabolism at the level of the mitochondrion results in oxidative stress. Mitochondrial hypermetabolism may place a significant burden on the skeletal muscle protein pool of burn survivors by i) causing damage to and degradation of proteins in the mitochondrion and thus increasing the protein demands of the mitochondria to replace nuclear-encoded proteins critical to mitochondrial function; and ii) the transmission of oxidative stress to the cell cytosol and resultant protein damage and proteolysis. Of potential clinical importance, unlike muscle wasting to support processes such as wound healing, oxidative damage induced turnover of protein confers little benefit to the patient (other than the need to remove and replace damaged proteins). Thus, this mechanism of skeletal muscle protein damage secondary to mitochondrial hypermetabolism represents a new target for therapeutic interventions aimed at blunting two of the greatest problems facing burn survivors: hypermetabolism and muscle wasting.
REFERENCES
- 1. Porter C, Tompkins RG, Finnerty CC, Sidossis LS, Suman OE, Herndon DN. The metabolic stress response to burn trauma: current understanding and therapies. Lancet 2016;388:1417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ogunbileje JO, Porter C, Herndon DN, et al. . Hypermetabolism and hypercatabolism of skeletal muscle accompany mitochondrial stress following severe burn trauma. Am J Physiol Endocrinol Metab 2016;311:E436–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Biolo G, Fleming RY, Maggi SP, Nguyen TT, Herndon DN, Wolfe RR. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. J Clin Endocrinol Metab 2002;87:3378–84. [DOI] [PubMed] [Google Scholar]
- 4. Chao T, Herndon DN, Porter C, et al. . Skeletal muscle protein breakdown remains elevated in pediatric burn survivors up to one-year post-injury. Shock 2015;44:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hart DW, Wolf SE, Mlcak R, et al. . Persistence of muscle catabolism after severe burn. Surgery 2000;128:312–9. [DOI] [PubMed] [Google Scholar]
- 6. Porter C, Herndon DN, Sidossis LS, Børsheim E. The impact of severe burns on skeletal muscle mitochondrial function. Burns 2013;39:1039–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Diaz E, Herndon D, Porter C, Sidossis L, Suman O, Børsheim E. Effects of pharmacological interventions on muscle protein synthesis and breakdown in recovery from burns. Burns 2015;41:649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gore DC, Wolf SE, Sanford A, Herndon DN, Wolfe RR. Influence of metformin on glucose intolerance and muscle catabolism following severe burn injury. Ann Surg 2005;241:334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gore DC, Wolf SE, Sanford AP, Herndon DN, Wolfe RR. Extremity hyperinsulinemia stimulates muscle protein synthesis in severely injured patients. Am J Physiol Endocrinol Metab 2004;286:E529–34. [DOI] [PubMed] [Google Scholar]
- 10. Hart DW, Herndon DN, Klein G, et al. . Attenuation of posttraumatic muscle catabolism and osteopenia by long-term growth hormone therapy. Ann Surg 2001;233:827–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hart DW, Wolf SE, Chinkes DL, et al. . Effects of early excision and aggressive enteral feeding on hypermetabolism, catabolism, and sepsis after severe burn. J Trauma 2003;54:755–61; discussion 761–4. [DOI] [PubMed] [Google Scholar]
- 12. Hart DW, Wolf SE, Chinkes DL, Lal SO, Ramzy PI, Herndon DN. Beta-blockade and growth hormone after burn. Ann Surg 2002;236:450–6; discussion 456–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hart DW, Wolf SE, Ramzy PI, et al. . Anabolic effects of oxandrolone after severe burn. Ann Surg 2001;233:556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hart DW, Wolf SE, Zhang XJ, et al. . Efficacy of a high-carbohydrate diet in catabolic illness. Crit Care Med 2001;29:1318–24.. [DOI] [PubMed] [Google Scholar]
- 15. Herndon DN, Rodriguez NA, Diaz EC, et al. . Long-term propranolol use in severely burned pediatric patients: a randomized controlled study. Ann Surg 2012;256:402–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herndon DN, Hart DW, Wolf SE, Chinkes DL, Wolfe RR. Reversal of catabolism by beta-blockade after severe burns. N Engl J Med 2001;345:1223–9. [DOI] [PubMed] [Google Scholar]
- 17. Mlcak RP, Suman OE, Murphy K, Herndon DN. Effects of growth hormone on anthropometric measurements and cardiac function in children with thermal injury. Burns 2005;31:60–6. [DOI] [PubMed] [Google Scholar]
- 18. Pereira C, Murphy K, Jeschke M, Herndon DN. Post burn muscle wasting and the effects of treatments. Int J Biochem Cell Biol 2005;37:1948–61. [DOI] [PubMed] [Google Scholar]
- 19. Porter C, Cotter M, Diaz E, Jennings K, Herndon D, Børsheim E. Amino acid infusion fails to stimulate skeletal muscle protein synthesis up to 1 year after injury in children with severe burns. J Trauma Acute Care 2013;74: 1480–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakurai Y, Aarsland A, Herndon DN, et al. . Stimulation of muscle protein synthesis by long-term insulin infusion in severely burned patients. Ann Surg 1995;222:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wolf SE, Thomas SJ, Dasu MR, et al. . Improved net protein balance, lean mass, and gene expression changes with oxandrolone treatment in the severely burned. Ann Surg 2003;237:801–10; discussion 810–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hart DW, Wolf SE, Chinkes DL, et al. . Determinants of skeletal muscle catabolism after severe burn. Ann Surg 2000;232:455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 1997;77:731–58. [DOI] [PubMed] [Google Scholar]
- 24. Porter C, Chondronikola M, Sidossis LS. The therapeutic potential of brown adipocytes in humans. Front Endocrinol (Lausanne) 2015;6:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilmore DW, Aulick LH. Systemic responses to injury and the healing wound. JPEN J Parenter Enteral Nutr 1980;4:147–51. [DOI] [PubMed] [Google Scholar]
- 26. Porter C, Herndon DN, Børsheim E, et al. . Uncoupled skeletal muscle mitochondria contribute to hypermetabolism in severely burned adults. Am J Physiol Endocrinol Metab 2014;307:E462–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Porter C, Herndon DN, Børsheim E, et al. . Long-Term Skeletal Muscle Mitochondrial Dysfunction is Associated with Hypermetabolism in Severely Burned Children. J Burn Care Res 2016;37:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bota DA, Van Remmen H, Davies KJ. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett 2002;532:103–6. [DOI] [PubMed] [Google Scholar]
- 30. Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS One 2009;4:e7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vasquez-Vivar J, Kalyanaraman B, Kennedy MC. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J Biol Chem 2000;275:14064–9. [DOI] [PubMed] [Google Scholar]
- 32. Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol 2002;4:674–80. [DOI] [PubMed] [Google Scholar]
- 33. Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 2010;35:505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Righi V, Constantinou C, Mintzopoulos D, et al. . Mitochondria-targeted antioxidant promotes recovery of skeletal muscle mitochondrial function after burn trauma assessed by in vivo 31P nuclear magnetic resonance and electron paramagnetic resonance spectroscopy. FASEB J 2013;27:2521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mottis A, Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response in mammalian physiology. Mamm Genome 2014;25:424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta 2013;1833:410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haynes CM, Ron D. The mitochondrial UPR- protecting organelle protein homeostasis. J Cell Sci 2010;123(Pt 22):3849–55. [DOI] [PubMed] [Google Scholar]
- 38. Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol 2014;217(Pt 1):137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 2002;21:4411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maharjan S, Oku M, Tsuda M, Hoseki J, Sakai Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci Rep 2014;4:5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kirstein J, Morito D, Kakihana T, et al. . Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J 2015;34:2334–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Al-Sawaf O, Clarner T, Fragoulis A, et al. . Nrf2 in health and disease: current and future clinical implications. Clin Sci (Lond) 2015;129:989–99. [DOI] [PubMed] [Google Scholar]
- 43. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 2015;88(Pt B):179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jang J, Wang Y, Kim HS, Lalli MA, Kosik KS. Nrf2, a regulator of the proteasome, controls self-renewal and pluripotency in human embryonic stem cells. Stem Cells 2014;32:2616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kapeta S, Chondrogianni N, Gonos ES. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J Biol Chem 2010;285:8171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monaghan RM, Whitmarsh AJ. Mitochondrial proteins moonlighting in the nucleus. Trends Biochem Sci 2015;40:728–35. [DOI] [PubMed] [Google Scholar]
- 47. Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol 2003;285:C806–12. [DOI] [PubMed] [Google Scholar]
- 48. Majetschak M, Zedler S, Romero J, et al. . Circulating proteasomes after burn injury. J Burn Care Res 2010;31:243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koppen M, Langer T. Protein degradation within mitochondria: versatile activities of AAA proteases and other peptidases. Crit Rev Biochem Mol Biol 2007;42:221–42. [DOI] [PubMed] [Google Scholar]
- 50. Kotiadis VN, Duchen MR, Osellame LD. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim Biophys Acta 2014;1840:1254–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Major T, von Janowsky B, Ruppert T, Mogk A, Voos W. Proteomic analysis of mitochondrial protein turnover: identification of novel substrate proteins of the matrix protease pim1. Mol Cell Biol 2006;26:762–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Käser M, Langer T. Protein degradation in mitochondria. Semin Cell Dev Biol 2000;11:181–90. [DOI] [PubMed] [Google Scholar]
- 53. Held NM, Houtkooper RH. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays 2015;37:867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci 2011;36:254–61. [DOI] [PubMed] [Google Scholar]
- 55. Bezawork-Geleta A, Brodie EJ, Dougan DA, Truscott KN. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci Rep 2015;5:17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Baker TA, Sauer RT. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta 2012;1823:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hoskins JR, Singh SK, Maurizi MR, Wickner S. Protein binding and unfolding by the chaperone ClpA and degradation by the protease ClpAP. Proc Natl Acad Sci U S A 2000;97:8892–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rainbolt TK, Saunders JM, Wiseman RL. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep 2015;16:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ruan Y, Li H, Zhang K, Jian F, Tang J, Song Z. Loss of Yme1L perturbates mitochondrial dynamics. Cell Death Dis 2013;4:e896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nelson SK, Bose SK, Grunwald GK, Myhill P, McCord JM. The induction of human superoxide dismutase and catalase in vivo: a fundamentally new approach to antioxidant therapy. Free Radic Biol Med 2006;40:341–7. [DOI] [PubMed] [Google Scholar]
- 61. Chan XC, Black CM, Lin AJ, Ping P, Lau E. Mitochondrial protein turnover: methods to measure turnover rates on a large scale. J Mol Cell Cardiol 2015;78:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kutik S, Guiard B, Meyer HE, Wiedemann N, Pfanner N. Cooperation of translocase complexes in mitochondrial protein import. J Cell Biol 2007;179:585–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vögtle FN, Meisinger C. Sensing mitochondrial homeostasis: the protein import machinery takes control. Dev Cell 2012;23:234–6. [DOI] [PubMed] [Google Scholar]
- 64. Poyton RO, McEwen JE. Crosstalk between nuclear and mitochondrial genomes. Annu Rev Biochem 1996;65:563–607. [DOI] [PubMed] [Google Scholar]
- 65. Baker MJ, Frazier AE, Gulbis JM, Ryan MT. Mitochondrial protein-import machinery: correlating structure with function. Trends Cell Biol 2007;17:456–64. [DOI] [PubMed] [Google Scholar]
- 66. Pfanner N, Meijer M. Mitochondrial biogenesis: the Tom and Tim machine. Curr Biol 1997;7: R100–3. [DOI] [PubMed] [Google Scholar]
- 67. Stojanovski D, Guiard B, Kozjak-Pavlovic V, Pfanner N, Meisinger C. Alternative function for the mitochondrial SAM complex in biogenesis of alpha-helical TOM proteins. J Cell Biol 2007;179:881–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hoppeler H, Lüthi P, Claassen H, Weibel ER, Howald H [Ultrastructure of normal human skeletal muscle; a morphometric analysis in controls and men trained in long-distance running.] Hoppe Seylers Z Physiol Chem 1973;354:229–30. [PubMed] [Google Scholar]
- 69. Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A 1996;93:15364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Patsouris D, Qi P, Abdullahi A, et al. . Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Rep 2015;13:1538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Porter C, Herndon DN, Bhattarai N, et al. . Differential acute and chronic effects of burn trauma on murine skeletal muscle bioenergetics. Burns 2016;42:112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Porter C, Herndon D, Bhattarai N, et al. . Severe burn injury induces thermogenically functional mitochondria in murine white adipose tissue. Shock 2015;44:258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sidossis LS, Porter C, Saraf MK, et al. . Browning of Subcutaneous White Adipose Tissue in Humans after Severe Adrenergic Stress. Cell Metab 2015;22:219–27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tzika AA, Mintzopoulos D, Mindrinos M, Zhang J, Rahme LG, Tompkins RG. Microarray analysis suggests that burn injury results in mitochondrial dysfunction in human skeletal muscle. Int J Mol Med 2009;24:387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hagen T, Vidal-Puig A. Mitochondrial uncoupling proteins in human physiology and disease. Minerva Med 2002;93:41–57. [PubMed] [Google Scholar]
- 76. Al-Jawad FH, Sahib AS, Al-Kaisy AA. Role of antioxidants in the treatment of burn lesions. Ann Burns Fire Disasters 2008;21:186–91. [PMC free article] [PubMed] [Google Scholar]
- 77. Horton JW, White DJ, Maass DL, Hybki DP, Haudek S, Giroir B. Antioxidant vitamin therapy alters burn trauma-mediated cardiac NF-kappaB activation and cardiomyocyte cytokine secretion. J Trauma 2001;50:397–406; discussion 407–8. [DOI] [PubMed] [Google Scholar]
- 78. Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004;53(Suppl 1):S110–8. [DOI] [PubMed] [Google Scholar]
- 79. Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 2011;50:98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Carter EA, Bonab AA, Goverman J, et al. . Evaluation of the antioxidant peptide SS31 for treatment of burn-induced insulin resistance. Int J Mol Med 2011;28:589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med 2011;32:234–46. [DOI] [PubMed] [Google Scholar]