

Abstract
t-Darpp is the truncated form of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa (Darpp-32) and has been demonstrated to confer resistance to trastuzumab, a Her2-targeted anticancer agent, via sustained signaling through the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Akt pathway and activation of protein kinase A (PKA). The mechanism of t-Darpp-mediated PKA activation is poorly understood. In the PKA holoenzyme, when the catalytic subunits are bound to regulatory subunits RI or RII, kinase activity is inhibited. We investigated PKA activity and holoenzyme composition in cell lines overexpressing t-Darpp (SK.tDp) or a T39A phosphorylation mutant (SK.tDpT39A), as well as an empty vector control cell line (SK.empty). We also evaluated protein-protein interactions between t-Darpp and PKA catalytic (PKAc) or regulatory subunits RI and RII in those cell lines. SK.tDp cells had elevated PKA activity and showed diminished association of RI with PKAc, whereas SK.tDpT39A cells did not have these properties. Moreover, wild type t-Darpp associates with RI. Concurrent expression of Darpp-32 reversed t-Darrp’s effects on PKA holoenzyme state, consistent with earlier observations that Darpp-32 reverses t-Darpp’s activation of PKA. Together, t-Darpp phosphorylation at T39 seems to be crucial for t-Darpp—mediated PKA activation and this activation appears to occur through an association with RI and sequestering of RI away from PKAc. The t-Darpp-RI interaction could be a druggable target to reduce PKA activity in drug-resistant cancer.
Keywords: breast cancer, protein kinase A, regulatory subunit RI, t-Darpp
1. Introduction
The Her2 (erbB2/neu) oncogene is overexpressed in 20–25% of invasive breast cancers and its expression levels correlate with prognosis, making this EGFR family member an ideal target for stratified breast cancer therapy [1,2]. Trastuzumab is a humanized monoclonal antibody that inhibits Her2-mediated anti-apoptotic and pro-survival signaling through the PI3K/Akt axis [3]. Its clinical application has dramatically improved breast cancer outcomes, but response rates to trastuzumab monotherapy are only 35% and clinical benefit is estimated at 48% in patients with Her2 overexpression [4]. Combining trastuzumab with cytotoxic chemotherapy is associated with a longer time to disease progression, a higher rate and longer duration of response, longer survival and reduced risk of death, but this has only been observed during a follow-up of 30 months. Together, the data indicate that there is intrinsic or acquired resistance to trastuzumab in the majority of patients [5,6].
The precise mechanisms of resistance are not completely proven, but sustained PI3K/Akt signaling despite Her2 blockage seems to be crucial [7]. This sustained signaling can be accomplished by different cellular mechanisms, including inhibition of receptor-antibody interaction [8]; gain-of-function mutations in the catalytic subunit of PI3K [9] or down-regulation or loss-of-function mutations in phosphatases such as PTEN [10]; or signaling through alternative receptor tyrosine kinases [11]. PKA activation can also mediate trastuzumab resistance [12,13] and several genes involved in PKA regulation are differentially expressed in cells selected for trastuzumab resistance [12]. One of them is PPP1R1B, which encodes Darpp-32 and also an amino-truncated isoform called t-Darpp [12,14] (Fig. 1). t-Darpp is overexpressed in several cancers, including breast cancer [14,15], and it confers trastuzumab resistance through activation of PI3K/Akt signaling [16–18]. Cells that overexpress t-Darpp also have elevated PKA activity, thus potentially linking the resistance phenotype associated with PKA to the resistance phenotype mediated by t-Darpp [16]. High levels of Darpp-32 reverse t-Darpp’s effects on trastuzumab resistance and PKA activity [16].
Fig. 1.
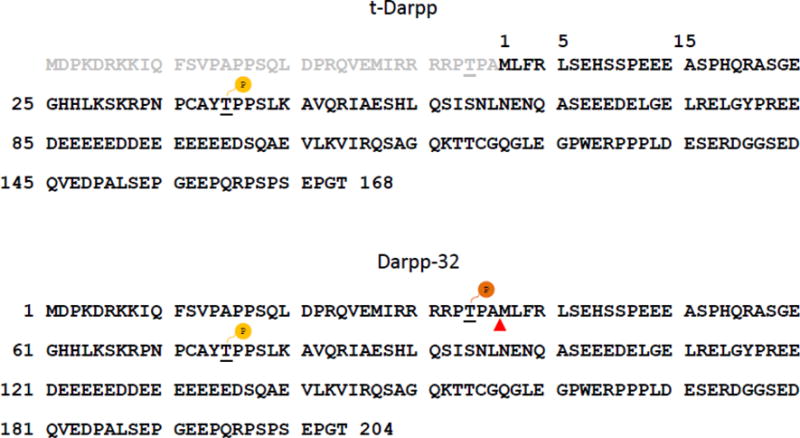
Amino acid sequences of human Darpp-32 protein (lower panel) and its truncated form t-Darpp (upper panel). Full-length Darpp-32 is a bifunctional phosphoprotein whose role is determined by its phosphorylation state. Darpp-32 phosphorylated at threonine 34 (T34) acts as an inhibitor of protein phosphatase 1 (PP1). When it is phosphorylated at threonine 75 (T75), Darpp-32 is converted into an inhibitor of protein kinase A (PKA). t-Darpp is lacking the amino-terminal 36 amino acids of full-length Darpp-32, resulting in the absence of the T34 phosphorylation site. The T75 residue is renumbered as threonine 39 (T39) in the t-Darpp protein. Phosphorylation at T39 is crucial for t-Darpp’s effects on trastuzumab resistance of breast cancer cells [17].
The molecular mechanism of t-Darpp-mediated effects on PKA activity is not known, but it most likely functions through direct protein-protein interactions, as does Darpp-32 [19]. Both proteins are regulated by phosphorylation. Phosphorylation at the T75 site in Darpp-32 converts it into a PKA inhibitor [20], whereas phosphorylation at T39, the analogous site in t-Darpp (see Fig. 1), is required for trastuzumab resistance and for activation of the PI3K/Akt pathway [17,21]. The role for T39 phosphorylation in t-Darpp-mediated PKA activation has not been previously reported.
PKA enzymatic activity is primarily controlled by regulatory subunits (RI or RII) that form a holoenzyme complex with the catalytic subunit (PKAc) and inhibit its activity until they are released from the complex by cAMP binding [22]. In this report, we sought to determine if the T39 phosphorylation site is involved in t-Darpp’s activating effect on PKA signaling and whether that effect, and Darpp-32’s inhibitory effect on PKA [16,20], could be mediated by direct interactions with one or more of the PKA holoenzyme subunits. Our findings demonstrate a physical mechanism by which t-Darpp overexpression stimulates PKA activity in cancer cells.
2. Material and methods
2.1 Cell models
The SK-Br-3 human breast cancer cell line was obtained from the American Type Culture Collection (Rockville, MD). Stably transfected SK-Br-3 cells expressing pcDNA3 empty vector (SK.empty), t-Darpp (SK.tDp) or both t-Darpp and Darpp-32 (SK.dDp) were described previously [16]. SK-Br-3 cells transfected with a phosphorylation mutant of t-Darpp in which T39 was mutated to an alanine (SK.tDpT39A) were also described [21]. Stably transfected clones were maintained in McCoy’s 5A media with 10% v/v heat-inactivated fetal bovine serum, 1% penicillin/streptomycin (Gibco by Life Technologies), 1% L-glutamine (Gibco by Life Technologies) and 500 μg/ml G418 (Omega Scientific) for sustained selection pressure.
The expression of Darpp-32, t-Darpp, their phosphorylated forms, and each PKA subunit in four cell clones was detected by Western hybridization with antibodies specific to total Darpp (Darpp-32 plus t-Darpp, Cell Signaling #2306), phospho-Darpp Thr75 (Cell Signaling #2301), PKA-RIα (Cell Signaling #5675), PKA-RIβ (ThermoFisher Scientific #PA5-13798), PKA-RIIα (BD Biosciences #612242), PKA-RIIβ (BD Biosciences #610625), and PKAc (BD Biosciences #610980). α-tubulin (Sigma-Aldrich #T5168) or β-actin (Sigma-Aldrich #A4700) was used as loading control. Protein expression levels were quantified as relative intensity normalized to loading control values using ImageJ densitometry software.
2.2 PKA activity assays
Two different assays were used to assess PKA activity. The first was a CREB DNA binding assay using the CREB (Phospho-Serine133) Transcription Factor Assay Kit (Cayman Chemical #10009846, Ann Arbor, MI). In brief, nuclear extracts from 1.5 × 107 cells were collected using the Nuclear Extraction Kit (Cayman Chemical #10009846). Nuclear extracts were incubated in a 96-well plate coated with a specific CRE consensus sequence. The activated CREB-CRE complex was detected by the addition of a phospho-Ser133 CREB antibody and a secondary horseradish peroxidase-conjugated antibody. Absorbance measurements at 655 nm (to monitor the signal increase) were obtained on a Molecular Devices microplate reader at 5 min intervals after addition of the Transcription Factor Developing Solution at room temperature. At a reading of 0.4–0.5, the Stop Solution was added, leading to a yellow color change. The final absorbance was measured at 450 nm on the microplate reader. Assays were performed in triplicate and repeated with extracts from three independent experiments.
The second assay was a PKA substrate phosphorylation assay using the FRET-based AKAR4 reporter, kindly provided by J. Zhang (University of California, San Diego). AKAR4 consists of an enhanced CFP domain and YFP domain separated by a PKA substrate domain. When PKA phosphorylates the substrate domain, a conformation change in the reporter brings the CFP and YFP domains together, leading to a FRET signal [23]. Cells were transiently transfected with 4 μg of the cDNA encoding the AKAR4 reporter using Lipofectamine 2000 (Life Technologies) in appropriate media without antibiotics. After 48 h, cells were washed with PBS (Mediatech Inc., cat. no. 21-030-CV) and fixed using 4% v/v paraformaldehyde. The CFP and YFP channels were excited at 458 nm and 514 nm, respectively. The FRET channel was excited using CFP (458 nm) and captured using the emission range of YFP (535-590 nm). Images were obtained using a Zeiss LSM510 Meta with AIM4.2 LSM imaging software. Relative FRET levels were calculated using the device-implemented nFRET macro 4.2 and normalized to the level present in SK.empty cells.
2.3 Proximity ligation assay
A Duolink proximity ligation assay (Sigma-Aldrich) was used to assess PKA holoenzyme composition and protein-protein interactions in live cells. In brief, 5,000 cells/well (triplicate wells for each experiment condition) were seeded in black, clear-bottom 96-well plates (Corning #3340) and incubated for 24 h at 37°C. After washing with PBS, fixing with 4% paraformaldehyde, permeabilizing with 0.2% v/v Tween-20 in PBS, washing again with 0.05% v/v Tween-20 in PBS (all at room temperature) and blocking (solution contained in the kit) for 1 h at 37°C, cells were exposed to the primary antibodies (diluted 1:1000 in antibody dilution solution contained in the kit) at 4°C overnight. The following antibodies were used: mouse anti-RI generated by using mouse RIβ as the immunogen and that recognizes both RIα and RIβ subunits (BD Bioscience #610165), mouse anti-RIIα (BD Bioscience #612242), rabbit anti-PKAc (Cell Signaling #4782), mouse anti-PKAc (BD Bioscience #610980), rabbit anti-Darpp-32 that recognizes Darpp-32 only (Cell Signaling #2302), and rabbit anti-Darpp that recognizes both t-Darpp and Darpp-32 (Cell Signaling #2306). After primary antibody incubations, cells were washed with wash buffer A (contained in the kit) and exposed to secondary antibodies (PLUS and MINUS probes targeting mouse and rabbit antibodies, respectively, conjugated with oligonucleotides) at room temperature. After 1 h exposure to antibodies, cells were re-washed with wash buffer A and incubated for 1 h at room temperature with the ligation solution (diluted 1:40 in dilution solution) that contained a DNA ligase and oligonucleotides that hybridized to the probes to generate a closed circle if the two probes were in close proximity (<40 nm). Cells were washed with wash buffer A and exposed to the amplification solution (polymerase diluted 1:80 in dilution solution) for 100 min at 37°C to begin rolling circle amplification. During this step, fluorescently labeled oligonucleotides were incorporated into the amplification product. Finally, cells were washed with wash buffer B and mounted with mounting media containing DAPI for nuclear counterstaining. Fluorescence (DsRed filter and DAPI filter) was recorded with a Zeiss Z1 Observer microscope and original, non-compressed gray scale pictures (40× magnification) were taken and analyzed with ImageJ. The corrected integrated cell fluorescence (CICF) of a cell was calculated according to the following formula: CICF = integrated cell fluorescence of the cell − (area of the cell × mean fluorescence of the background reading). The CICF value for each cell was normalized to the average CICF from SK.empty cells in the entire experiment and then the normalized CICF values in each well of the experiment were averaged. The average normalized CICF values from three independent wells were averaged to give the final CICF values (±S.D.) represented in the figures. To prevent bias from outliers (very low fluorescence or extremely high fluorescence values from artifact staining), the highest and lowest CICF values in each well were excluded from the CICF calculations. The number of cells analyzed of each cell line was 40–50 in each experiment and each experiment was repeated a minimum of one time.
2.4 Co-immunoprecipitation and Western analysis
Cell lysates from SK.empty, SK.tDp, SK.tDpT39A and SK.dDp cells were prepared in lysis buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, and 1% Nonidet P-40. Protein concentrations were determined using the Pierce BCA protein assay kit (Thermo Scientific). For immunoprecipitation, each cell lysate (500 μg total protein) was pre-cleared by incubating with 20 μl protein G-agarose beads (Cell Signaling) at 4°C for 1 h. The pre-cleared lysates were incubated with a mouse monoclonal anti-RIα/β antibody (BD Biosciences #610165) or a rabbit monoclonal anti-Darpp antibody (Cell Signaling #2306), overnight with gentle shaking at 4°C. A 20 μl aliquot of protein G-agarose beads was added and incubated at 4°C for 4 h. The beads were pelleted and washed three times with wash buffer (50 mM Tris, 0.1% Nonidet P-40, pH 8.0) and proteins were eluted in Laemmli sample buffer (125 mM Tris-HCl (pH 6.8), 5 % SDS, 20 % glycerol, 0,2 % bromphenol blue) containing 1% (v/v) ß-mercaptoethanol by boiling for 5 min. The proteins were separated by 10% SDS-PAGE, transferred to a nitrocellulose membrane (GE Healthcare Life Sciences), and analyzed by Western hybridization with anti-Darpp antibody (#2306) or anti-RIα/β antibody (#610165), accordingly. As a negative control, the identical pre-cleared cell lysates were incubated with beads without the precipitation antibody. To confirm the specificity of co-immunoprecipitation, membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Scientific) and re-probed for RIα using rabbit monoclonal antibody (Cell Signaling #5675) or t-Darpp + Darpp-32 using rabbit anti-Darpp antibody (#2306).
The same co-immunoprecipitation procedure was used to detect possible co-complexes of PKAc with t-Darpp/Darpp-32 and protein phosphatase-1 (PP1). Rabbit polyclonal antibody recognizing both t-Darpp and Darpp-32 and mouse monoclonal anti-PKAc antibody (BD Biosciences #610980) were used for the immunoprecipitations; the same rabbit anti-Darpp and mouse anti-PKAc antibodies as well as rabbit polyclonal anti-PP1α antibody (Santa Cruz Biotechnology #sc-443) were used for the Western hybridizations. The rabbit polyclonal anti-Darpp antibody was generated against recombinant His-tagged human t-Darpp [24] and affinity-purified on a t-Darpp-conjugated column (ThermoFisher Pierce Custom Services). The antibody concentration was 0.8 mg/ml.
2.5 Statistical analysis
Data were analyzed using GraphPad Prism Version 6.01 (GraphPad Software, San Diego, USA). Differences in CREB-CRE complex formation, nFRET levels or CICF values were evaluated by one-way ANOVA corrected for multiple comparisons using Tukey’s post-hoc test.
3. Results
3.1. Comparison of protein expression in four stably transfected SK-Br-3 cell clones
Fig. 2 shows the results of Western analysis to quantify relevant protein expression levels in the cell lines used in the current work. There was no detectable Darpp-32 or t-Darpp expression in SK.empty cells, whereas SK.tDp and SK.tDpT39A cells respectively overexpress exogenous wild type t-Darpp or the T39A mutant, respectively, at similar levels. SK.dDp cells co-express high levels of Darpp-32 and t-Darpp. t-Darpp was phosphorylated at residue T39 to the same extent that Darpp-32 was phosphorylated at the corresponding residue T75 (Fig. 2, left panel). RIα, RIIα, and PKAc were at virtually the same levels in all cell clones; RIβ was elevated and RIIβ was lower in SK.dDp cells, compared with other three cell lines (Fig. 2, middle and right panels).
Fig. 2.
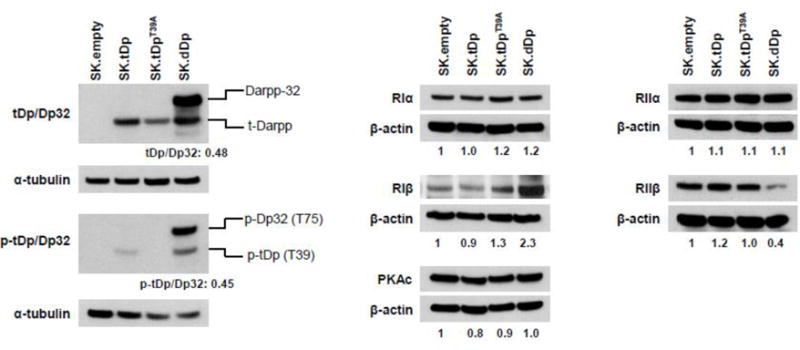
Protein expression levels in relevant cell lines. Levels of t-Darpp and Darpp-32 (tDp/Dp32); phospho-t-Darpp and phospho-Darpp-32 (p-tDp/Dp32) and PKA subunits, as labeled, in the indicated cell lines were analyzed by Western hybridization with specific antibodies. α-tubulin or β-actin was used as loading control. All quantifications were performed using ImageJ software. PKA subunit levels were normalized to the corresponding protein level in SK.empty cell after correcting for loading differences; normalized values are shown below the actin image for each cell line. Values in the left panel indicate the band density ratio of t-Darpp to Darpp-32 and phospho-tDp (T39) to phospho-Dp32 (T75) in SK.dDp cells.
3.2. T39 phosphorylation is required for t-Darpp to enhance PKA activity
We have previously reported that cells overexpressing t-Darpp have elevated CREB DNA binding activity (as determined by electrophoretic mobility shift assay), an indirect measure of PKA activity [16]. To confirm this observation and determine if T39 phosphorylation on t-Darpp was required for the phenotype, we used two different assays for PKA activity. In a plate-based CREB DNA binding assay, nuclear extracts from SK.tDp cells (which overexpress t-Darpp) had three-fold higher CREB DNA binding activity than SK.empty cells (which carry an empty expression vector) (p ≤0.05). SK.tDpT39A cells (which overexpress the T39A mutant of t-Darpp) had CREB DNA binding activity similar to SK.empty cells (Fig. 3A). To validate this result, we used a PKA substrate phosphorylation assay with a FRET-based AKAR4 reporter [23]. Forty-eight hours after transfection of the AKAR4 reporter, >2-fold higher FRET levels were recorded in SK.tDp cells, relative to SK.empty cells, but not in SK.tDpT39A (Fig. 3B). H89 (PKA inhibitor) and forskolin (PKA activator) were used as controls to confirm functionality of the assay (Supplemental Fig. 1).
Fig. 3.
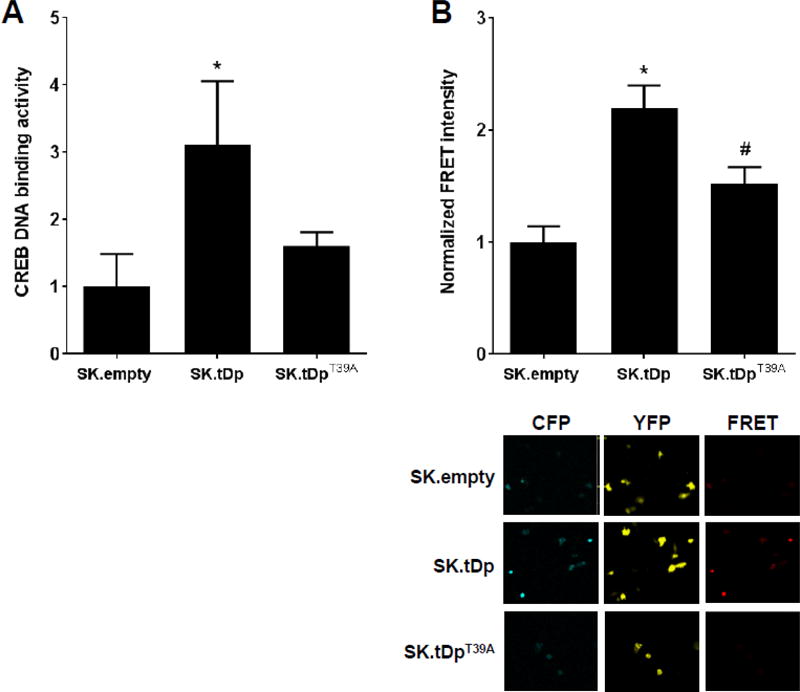
t-Darpp overexpression enhances PKA activity. (A) An ELISA-based CREB DNA binding activity assay was performed with SK.empty, SK.tDp and SK.tDpT39A cells to assess the relative PKA activity in each cell line. Shown are the means (± S.D.) of three independent experiments, with normalization to the activity in SK.empty cells. Statistical significance was evaluated by one-way ANOVA corrected for multiple comparisons using Tukey’s post-hoc test. *p ≤0.05 compared to SK.empty. (B) Top, nFRET quantification of SK.empty, SK.tDp and SK.tDpT39A cells transiently transfected with AKAR4 for 48 h, normalized to the average nFRET quantification in untreated SK.empty cells. Shown are the averages (± S.D.) from ≥30 cells imaged per cell line. Bottom, Representative confocal images of SK.empty, SK.tDp, and SK.tDpT39A cells transiently transfected with AKAR4. The CFP and YFP channels were excited at 458 nm and 514 nm, respectively. The FRET channel was excited using CFP (458 nm) and captured using the emission range of YFP (535–590 nm).
3.3. t-Darpp overexpression disrupts PKA holoenzyme composition
Since PKA activity is primarily controlled by its holoenzyme composition or state, we used the Duolink proximity ligation assay to assess spatial proximity of PKAc to its regulatory subunits, RI or RII, in SK.empty, SK.tDp, and SK.tDpT39A cells. For the RI interaction, we used an antibody that detects both the α and β subunits of RI. We detected bright red fluorescence (indicating proximity within 40 nm) in SK.empty and SK.tDpT39A cells but not in SK.tDp cells (Fig. 4A). CICF, a measure of the fluorescence signal intensity, was diminished by 76% in SK.tDp cells compared to SK.empty cells (p ≤0.001), indicating PKAc-RIα/β dissociation upon overexpression of t-Darpp. CICF in SK.tDpT39A cells was also lower than in SK.empty cells (p ≤0.05), but was still significantly higher (p ≤0.05) than in SK.tDp cells (Fig. 4B).
Fig. 4.
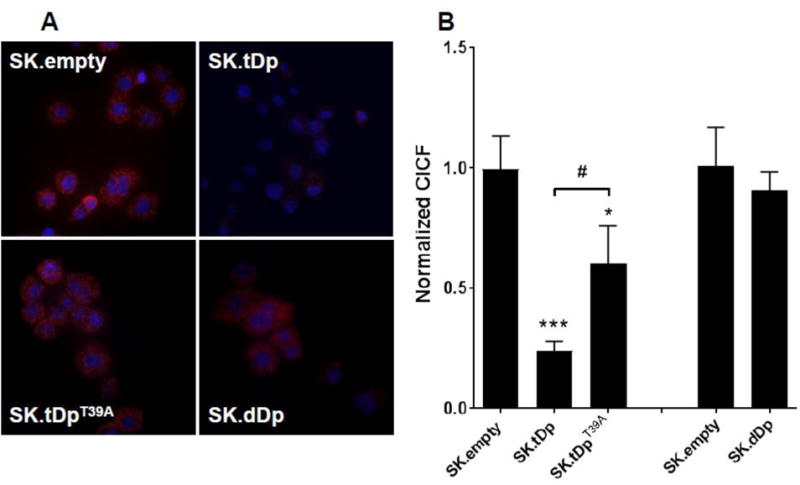
t-Darpp overexpression leads to dissociation of RI from PKAc. (A) Using the proximity ligation assay, the association of PKAc with RIα/β in SK.empty, SK.tDp, SK.tDpT39A and SK.dDp cells was visualized (red fluorescence). DAPI was used to counterstain cell nuclei (blue fluorescence). (B) Cellular fluorescence was quantified using ImageJ and CICF (integrated cell fluorescence corrected for nonspecific background staining) was calculated. Shown are the average normalized CICF values (±S.D.) from three independent wells in a single experiment. Statistical significances were evaluated using ANOVA corrected for multiple comparisons using Tukey’s post-hoc test. *p ≤0.05, ***p≤0.001 compared to SK.empty; #p ≤0.05 compared to SK.tDp. Each cell line was analyzed in 2–3 independent experiments with similar results.
We also explored potential alterations in PKAc-RII interactions in these cell lines, using an antibody that detects the α subunit of RII. The PKAc-RIIα interaction was slightly increased in SK.tDp cells (36% higher; p ≤0.05) and reduced in SK.tDpT39A cells (67% lower; p ≤0.05), relative to SK.empty cells (Supplemental Fig. 2A). The data suggest that t-Darpp overexpression causes an alteration in the PKA holoenzyme composition, predominantly a decrease in PKAc-RIα/β and a small increase in PKAc-RIIα. The t-Darpp T39A mutant does not appear to decrease PKAc-RIα/β significantly but it may decrease PKAc-RIIα.
Darpp-32 is known to reverse t-Darpp’s effect on PKA-mediated CREB DNA binding activity [16]. Thus, we evaluated the PKAc-RIα/β interaction in SK.dDp cells that overexpress both t-Darpp and Darpp-32. We found that SK.dDp cells had significantly higher levels of PKAc-RIα/β interaction than SK.tDp cells and SK.tDpT39A cells (p ≤0.05), essentially the same as the interaction seen in SK.empty cells (Fig. 4A, 4B). This suggests that Darpp-32 overexpression reverses t-Darpp’s effect on the PKAc-RIα/β holoenzyme composition.
3.4. t-Darpp interacts directly with RI regulatory subunit
To investigate further the molecular reasons for enhanced PKA activity and altered PKA holoenzyme composition in SK.tDp cells, we used the proximity ligation assay to evaluate t-Darpp protein-protein interaction with PKA subunits RI, RII or PKAc itself. t-Darpp interaction with α-tubulin was used as a negative control. Whereas the t-Darpp interaction with RIIα, PKAc or α-tubulin did not result in CICF values exceeding background (data not shown), high CICF values were detected for the interaction between t-Darpp and RIα/β in SK.tDp cells (14-fold higher CICF in SK.tDp than in SK.empty; p ≤0.05) (Fig. 5A). The t-Darpp-RIα/β interaction was also detected in SK-Br-3 cells expressing a HA-tagged variant of t-Darpp and using an antibody against HA for the primary antibody to detect t-Darpp; this analysis also showed the negligible interaction between t-Darpp and PKAc or RIIα (Supplemental Fig. 2B). We also saw an interaction between t-Darpp and RIα/β in a single experiment with several breast cancer cell lines that express t-Darpp to varying degrees, including two lines selected for trastuzumab resistance that over-express t-Darpp relative to their parental controls (Supplemental Fig. 3).
Fig. 5.
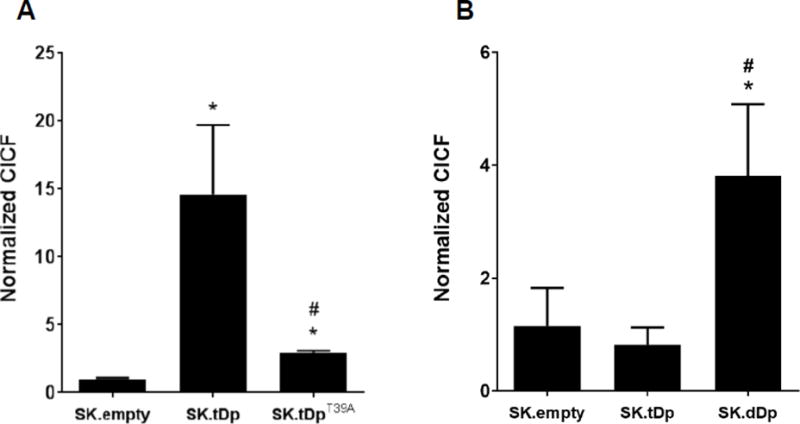
t-Darpp and Darpp-32 interact with RI. Using the proximity ligation assay, the association of t-Darpp with RIα/β in SK.empty, SK.tDp, SK.tDpT39A, and SK.dDp cells was visualized, cellular fluorescence was quantified using ImageJ, and CICF was calculated. (A) #2306 antibody that detects t-Darpp + Darpp-32 and #610165 antibody that detects RIα/β. (B) #2302 antibody that only detects Darpp-32 and #610165 antibody that detects RIα/β. Shown are the average normalized CICF values (±S.D.) from three independent wells in a single experiment. Statistical significances were evaluated using ANOVA corrected for multiple comparisons using Tukey’s post-hoc test. *p ≤0.05 compared to SK.empty; #p ≤0.05 compared to SK.tDp. Each cell line was analyzed in 1–2 independent experiments for each antibody combination.
In SK.tDpT39A cells, the t-Darpp-RIα/β interaction was significantly diminished compared to SK.tDp cells (p≤ 0.05), but was still slightly increased (3-fold; p ≤0.05) compared to SK.empty cells (Fig. 5A). In addition, a single proximity ligation experiment showed a concentration-dependent loss of t-Darpp-RIα/β interaction and concomitant increase in the PKAc-RIα/β interaction signal in the presence of small-molecule inhibitors of cdk1 and cdk5, the kinases responsible for T39 phosphorylation on t-Darpp [20,24] (Supplemental Fig. 4).
In a separate experiment, CICF values for the interaction between RIα/β and t-Darpp + Darpp-32 in SK.dDp cells (using an antibody that detects both t-Darpp and Darpp-32 and an antibody that detects RIα/β) were approximately twice the CICF values seen in SK.tDp cells (data not shown). This prompted us to evaluate the specific interaction between Darpp-32 and RIα/β using an antibody to the amino-terminal domain of Darpp-32 that is absent from t-Darpp. The proximity ligation assay revealed CICF values that were about 4-fold higher in SK.dDp cells than SK.empty and SK.tDp cells (Fig. 5B), suggesting that Darpp-32 can also bind the RIα/β regulatory subunit.
To evaluate the suspected interaction between t-Darpp and RIα/β and to determine the relevance of the T39A mutation using another experimental approach, we conducted co-immunoprecipitation assays followed by Western analysis using lysates from SK.empty, SK.tDp, SK.tDpT39A, and SK.dDp cells. Immunoprecipitation with an antibody against RIα/β pulled down t-Darpp from SK.tDp lysates and to a much lesser degree from SK.tDpT39A lysates (Fig. 6A). The reciprocal approach, immunoprecipitation with anti-Darpp antibody followed by Western detection of RIα/β, confirmed the interaction between t-Darpp and RIα/β (Fig. 6B). The RIα/β immunoprecipitation and reciprocal Darpp immunoprecipitation further substantiated the interaction between RIα/β and both t-Darpp and Darpp-32 when lysates from SK.dDp cells were analyzed, with Darpp-32 perhaps interacting with RIα/β to a greater extent even than t-Darpp (Fig. 6C).
Fig. 6.
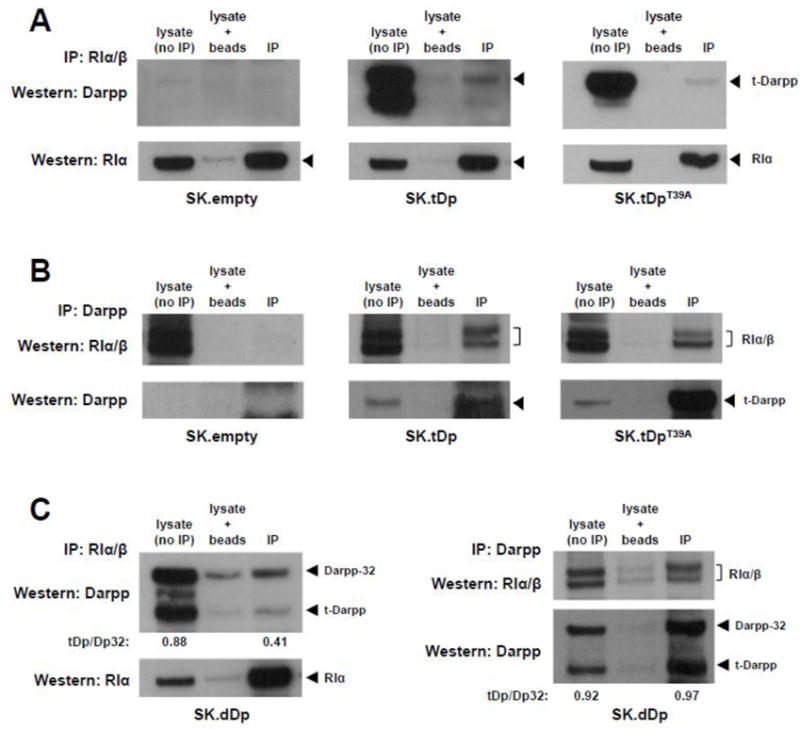
t-Darpp and Darpp-32 co-immunoprecipitate with RI. (A) Lysates from SK.empty, SK.tDp and SK.tDpT39A cells were subjected to precipitation with mouse anti-RIα/β antibody (BD Biosciences #610165) followed by Western analysis using rabbit anti-Darpp antibody (#2306, Cell Signaling). Western analysis of lysates (without prior immunoprecipitation), lysates incubated with protein G-agarose beads (no primary antibody), and immunoprecipitated lysates (IP) are shown. Stripping and re-probing for RIα (Cell Signaling antibody #5675) is shown in the bottom panels. (B) Lysates were subjected to precipitation with a rabbit anti-Darpp antibody (#2306) followed by Western analysis using mouse anti-RIα/β antibody (#610165). Stripping and re-probing for t-Darpp (#2306) is shown in the bottom panels. (C) Left, lysates from SK.dDp cells were subjected to precipitation with mouse anti-RIα/β antibody (#610165) followed by Western analysis using rabbit anti-Darpp antibody (#2306), with re-probing for RIα (#5675) in the bottom panel. Right, lysates were subjected to precipitation with rabbit anti-Darpp antibody (#2306) followed by Western analysis using mouse anti-RIα/β antibody (#610165), with re-probing for t-Darpp and Darpp-32 (#2306) in the bottom panel. The ratios of t-Darpp to Darpp-32 in lysate and IP elution lanes were quantified by measuring the bands densities using ImageJ software.
3.5. t-Darpp/Darpp-32 and PP1 are not in the complex with PKAc
We also wanted to use co-immunoprecipitation to confirm the lack of a direct interaction between t-Darpp and PKAc. In this same analysis, we determined if PP1α, a known interaction partner of Darpp-32, was in the PKAc complex when Darpp-32 was present. Immunoprecipitation with anti-Darpp antibody followed by Western detection showed that PKAc was not significantly present in the pull-down complex with t-Darpp/Darpp-32 from SK.empty, SK.tDp, SK.tDpT39A, and SK.dDp cell lysates, although a low association was seen in SK.dDp lysates (Fig. 7A). We also detected a low PP1α association with Darpp in the co-immunoprecipitations performed on SK.empty and SK.dDp lysates (Fig. 7A). Both of these cell lines express Darpp-32, albeit at much different levels (see Fig. 2).
Fig. 7.

t-Darpp/Darpp-32 and PP1 are absent from the PKAc complex. (A) Lysates from SK.empty, SK.tDp, SK.tDpT39A, and SK.dDp cells were subjected to immunoprecipitation with polyclonal rabbit anti-Darpp antibody followed by Western analysis using mouse anti-PKAc antibody (BD Biosciences #610980) (upper panels) or rabbit anti-PP1α antibody (Santa Cruz Biotechnology #sc-443) (middle panels). Stripping and re-probing for t-Darpp/Darpp-32 is shown in the bottom panels. (B) Lysates were immunoprecipitated with mouse anti-PKAc antibody (#610980) followed by Western analysis using polyclonal rabbit anti-Darpp antibody or rabbit anti-PP1α antibody (sc-443). Stripping and reprobing for PKAc for each experiment is shown in the bottom panels.
The low level of PKAc that co-precipitated with Darpp from SK.dDp lysates prompted us to perform the reciprocal analysis — immunoprecipitation of PKAc followed by Western detection of associated t-Darpp, Darpp-32, and PP1α. t-Darpp/Darpp-32 and PP1α were absent from the PKAc immunoprecipitations from all four cell lysates (Fig. 7B).
4. Discussion
Sustained PI3K/Akt signaling is considered a hallmark of trastuzumab resistance and enhanced PKA activity is reported as one of the mechanisms contributing to trastuzumab resistance and possibly Akt activation [12,13,16,25]. Both pharmacological (e.g. forskolin treatment) and knock-down approaches to influence PKA holoenzyme composition are sufficient to alter PKA enzymatic activity, Akt phosphorylation state and trastuzumab resistance [12]. Since t-Darpp confers trastuzumab resistance and PKA activation [16–18], we wanted to know if t-Darpp could affect PKA holoenzyme composition as a possible mechanism of PKA activation.
Darpp-32 is an effector of PKA’s inhibitory effect on PP1 and it is a feedback inhibitor of PKA itself [19,20]. Darpp-32 inhibits PP1 via a protein-protein interaction that requires the N-terminal domain that is absent from t-Darpp, but the molecular mechanism by which Darpp-32 inhibits PKA is not known. We focused on the T39 phosphorylation site in t-Darpp because phosphorylation at this site is required for t-Darpp’s ability to increase trastuzumab resistance and PI3K/Akt signaling [17,21] and phosphorylation at the analogous T75 site in Darpp-32 converts Darpp-32 from a PP1 inhibitor into a PKA inhibitor [20].
Consistent with our previous report [16], t-Darpp overexpression enhanced PKA activity as determined by CREB DNA binding and PKA substrate phosphorylation assays (Fig. 3). Concurrently, t-Darpp overexpression led to a dissociation of PKAc from its RI regulatory subunit (Fig. 4) and it appears that this dissociation was mediated by direct binding of t-Darpp to RIα/β (Fig. 5, Fig. 6), which sequestered RIα/β away from PKAc. Such an event would be consistent with canonical activation of PKA, similar to what is seen when cAMP binds RIα/β and causes dissociation and activation of PKAc. A protein-protein interaction-based mechanism of PKA activation has already been described for p90 ribosomal kinase 1, which also binds RI subunits and subsequently enhances the enzymatic activity of the released PKAc [26]. Our new data suggests the same mode of action for t-Darpp.
Cells expressing the T39A mutant of t-Darpp had only modestly elevated PKA activity (Fig. 3) and exhibited only slightly diminished PKAc-RIα/β interaction compared to cells transfected with an empty vector (Fig. 4). Comparing the t-Darpp-RIα/β interaction in the different cell lines using proximity ligation assays, the signal from the wild-type t-Darpp interaction exceeded that from the T39A mutant interaction by 5-fold (Fig. 5A). Likewise, a very low level of the T39A mutant co-precipitated with RIα/β in SK.tDpT39A cells (Fig. 6A). This is consistent with proximity ligation assays showing a weak interaction of the T39A mutant with RIα/β in SK.tDpT39A cells (Fig. 5A) and a slightly diminished PKAc-RIα/β interaction in SK.tDpT39A cells, compared to the interaction observed in SK.empty cells (Fig. 4). Together, these data suggest an interaction between the T39A mutant protein and RIα/β, but such interaction might be too weak to translate into significant PKAc-RIα/β disruption (Fig. 4) and subsequent PKA activation (Fig. 3). All of these results suggest that T39 phosphorylation plays an important role in t-Darpp’s capacity to modulate PKA activity. This same site has already been shown to be critical for t-Darpp’s ability to activate EGFR signaling [21], to cause sustained Akt phosphorylation and to confer trastuzumab resistance [17], perhaps suggesting that these outcomes are downstream effects of t-Darpp-mediated PKA stimulation.
It should be noted that the diminished PKAc-RIα/β interaction seen in SK.tDp cells (Fig. 4) was accompanied by a slight increase in the interaction between PKAc and RIIα, as determined by proximity ligation assay (Supplemental Fig. 2A). However, this increased PKAc-RIIα interaction did not prevent enhanced PKA activity, which suggests that there might not be enough PKAc-RIIα holoenzyme in these cells to inhibit PKA activity effectively and the net impact of t-Darpp overexpression would still be PKA activation. This speculation is supported by our previous finding that RIIα expression is down-regulated in other trastuzumab-resistant breast cancer cells that have enhanced PKA activity [12], suggesting a need for low RIIα levels concomitant with t-Darpp binding to RIα/β to achieve PKA activation. It is also possible that sequestering RIα/β away from PKAc (by t-Darpp) has an effect on PKAc localization in a way that precludes significant RIIα binding to PKAc, thus promoting PKA activation [27]. Further analysis of PKA, RI and RII subcellular localization is required to determine if enhanced PKA activity is due to altered localization in SK.tDp cells.
We have previously reported that Darpp-32 reverses t-Darpp’s effects on drug resistance, Akt phosphorylation and PKA activity [16]. This antagonistic effect of Darpp-32 was also seen in the proximity ligation assays presented here, in which concurrent expression of Darpp-32 with t-Darpp (in SK.dDp cells) appeared to reverse t-Darpp’s effect on the PKAc-RIα/β interaction. SK.dDp cells exhibited four-fold higher PKAc-RIα/β interaction than SK.tDp cells and the level of this interaction was not significantly different from what was observed in SK.empty cells (Fig. 4B). Interestingly, we did detect association of Darpp-32 with RIα/β in SK.dDp cells when we used a Darpp-32-specific antibody in the proximity ligation assay (Fig. 5B) and Darpp-32-RIα/β association was detected in co-immunoprecipitation assays (Fig. 6C). The interaction might be relatively weak, given the high levels of Darpp-32 in SK.dDp cells (Fig. 2) and the relatively lower interaction with RIα/β in the proximity ligation assay (4-fold over background vs. the 14-fold interaction seen for t-Darpp-RIα/β in SK.tDp cells, Fig. 5). The co-immunoprecipitation data, on the other hand, suggest a stronger pull-down of Darpp-32 than t-Darpp in the RIα/β immunoprecipitation (Fig. 6C).
Taken together, the data on PKA holoenzyme state (PKAc-RIα/β interaction) and PKA activity, together with published reports of Darpp-32 as a PKA inhibitor [20] and our own studies of t-Darpp and Darpp-32 [16] support a model in which Darpp-32 and t-Darpp are antagonizing proteins whose interactions with RIα/β produce very different effects on PKA holoenzyme and PKA activity (see Fig. 8). The molecular basis for this antagonism is unclear, but one possibility is that the aminoterminal domain unique to Darpp-32 (amino acids 1–36) plays a role, either by directly preventing PKAc-RIα/β dissociation (perhaps even by “masking” the T39 site that is crucial for PKA activation and PKAc-RIα/β dissociation mediated by t-Darpp) or by an indirect effect on t-Darpp and/or RIα/β localization or activity. We also observed that RIβ is elevated and RIIβ is lower in cells expressing t-Darpp and Darpp-32 compared to our other three cell lines (see Fig. 2). The mechanism for this is unclear, but it is possible that the effect of the different subunit levels is to influence PKA holoenzyme composition and thus make PKA refractory to CREB activation. Indeed, RIβ is associated with mitochondrial membranes and RIIβ is found in the soluble fraction of cells [28], so perhaps the altered levels of these competing PKA holoenzyme components cause RIβ to sequester PKAc to the mitochondria, thus preventing access of PKAc to CREB and other soluble substrates. Regardless of the molecular mechanism, the net effect in the presence of Darpp-32 is sustained PKAc-RIα/β interaction and resulting PKA inhibition, a known role for Darpp-32 in the PKA signaling pathway [19].
Fig. 8.
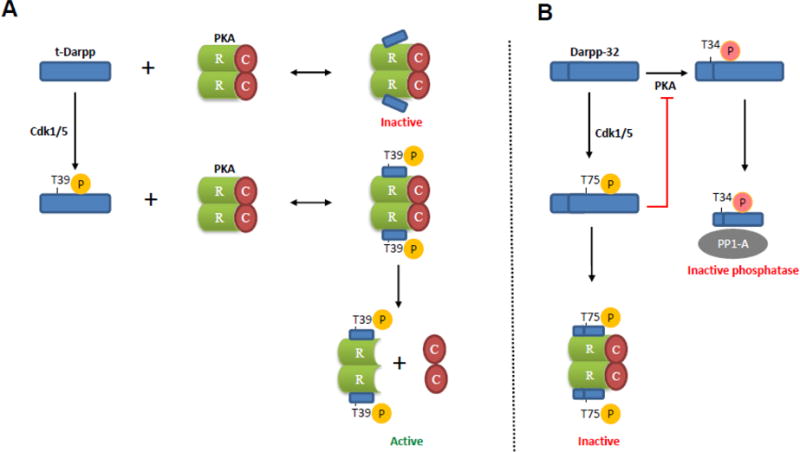
Model illustrating the effects of t-Darpp and Darpp-32 on PKA activity. (A) t-Darpp T39 site is phosphorylated by Cdk1/5 which promotes binding of t-Darpp to PKA regulatory subunit RI (R) and results in release and activation of PKA catalytic subunit (C). The interaction between non-phosphorylated t-Darpp (T39A mutant) and RI may be too weak to promote PKA catalytic subunit release, which would leave the PKA holoenzyme in the inactive state. (B) By contrast, Cdk1/5-mediated phosphorylation of Darpp-32 at T75 converts it into a PKA inhibitor [20]. This inhibition might be mediated by Darpp-32 interaction with PKA regulatory subunit RI, preventing dissociation of RI from the catalytic subunit of PKA. Darpp-32 phosphorylated at another site (T34), mediated by PKA, binds to protein phosphatase-1 (PP1) catalytic subunit, resulting in PP1 inhibition [19]. The inhibition of PP1 results in a net increase in phosphorylation of various downstream substrates.
Given the opposite effects of t-Darpp and Darpp-32 in this pathway and on the PKA holoenzyme, it seems reasonable to suggest that the ratio of t-Darpp and Darpp-32 proteins should influence the PKA signaling phenotype of cancer cells and eventually the growth/survival phenotype. Small molecule drugs that inhibit t-Darpp interaction with RIα/β or perturb the t-Darpp:Darpp-32 ratio (towards high Darpp-32 and low t-Darpp) could have a positive impact on tumor growth and drug resistance.
Conclusions
We present here a model explaining part of the trastuzumab-resistant phenotype of breast cancer cells that overexpress t-Darpp. Our model is that t-Darpp sequesters regulatory subunit RIα/β away from PKAc, leading to enhanced PKA activity and sustained PI3K/Akt signaling. t-Darpp phosphorylation at the T39 residue promotes its complex formation with RIα/β, thus enhancing its effect on PKA activity. The t-Darpp-RIα/β interaction could be a druggable target to reduce PKA activity and Akt signaling in drug-resistant cancer.
Supplementary Material
Highlights.
t-Darpp overexpression enhances protein kinase A (PKA) activity
t-Darpp overexpression leads to dissociation of the regulatory subunit (RI) from the catalytic subunit (PKAc) of PKA
Effect on PKA structure and function are likely mediated by binding of RI by t-Darpp
T39A mutation of t-Darpp modulates its effect on PKA structure and function
Darpp-32 overexpression reverses t-Darpp’s effects on PKA activity and structure
Acknowledgments
The authors wish to thank Cécile Donohue for technical assistance and Brian Armstrong and Loren Quintanar from the Light Microscopy Core for their technical and scientific assistance. The authors also acknowledge Patrycja Magdziarz for purification of His-tagged t-Darpp. Funding: This work was supported by grants from the National Institutes of Health (GM105898, CA33572).
Abbreviations
- CFP
cerulean fluorescent protein
- CICF
corrected integrated cell fluorescence
- CREB
cAMP response element binding protein
- CRE
cAMP response element
- Darpp-32
dopamine- and AMP-regulated phosphoprotein of 32 kDa
- DAPI
4′,6-diamidino-2-phenylindole
- EGFR
epidermal growth factor receptor
- FRET
fluorescence resonance energy transfer
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PKA
protein kinase A
- YFP
yellow fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflicts of interest.
References
- 1.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 2.Hung MC, Lau YK. Basic science of HER-2/neu: a review. Semin Oncol. 1999;26:51–9. [PubMed] [Google Scholar]
- 3.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62:4132–41. [PubMed] [Google Scholar]
- 4.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–26. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 5.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 6.Vici P, Pizzuti L, Natoli C, Moscetti L, Mentuccia L, Vaccaro A, Sergi D, Di Lauro L, Trenta P, Seminara P, Santini D, Iezzi L, Tinari N, Bertolini I, Sini V, Mottolese M, Giannarelli D, Giotta F, Maugeri-Saccà M, Barba M, Marchetti P, Michelotti A, Sperduti I, Gamucci T. Outcomes of HER2-positive early breast cancer patients in the pre-trastuzumab and trastuzumab eras: a real-world multicenter observational analysis. The RETROHER study. Breast Cancer Res Treat. 2014;147:599–607. doi: 10.1007/s10549-014-3133-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nahta R, Esteva FJ. Herceptin: mechanisms of action and resistance. Cancer Lett. 2006;232:123–38. doi: 10.1016/j.canlet.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 8.Price-Schiavi SA, Jepson S, Li P, Arango M, Rudland PS, Yee L, Carraway KL. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer. 2002;99:783–91. doi: 10.1002/ijc.10410. [DOI] [PubMed] [Google Scholar]
- 9.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 10.Fujita T, Doihara H, Kawasaki K, Takabatake D, Takahashi H, Washio K, Tsukuda K, Ogasawara Y, Shimizu N. PTEN activity could be a predictive marker of trastuzumab efficacy in the treatment of ErbB2-overexpressing breast cancer. Br J Cancer. 2006;94:247–52. doi: 10.1038/sj.bjc.6602926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–27. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Gu L, Lau SK, Loera S, Somlo G, Kane SE. Protein kinase A activation confers resistance to trastuzumab in human breast cancer cell lines. Clin Cancer Res. 2009;15:7196–206. doi: 10.1158/1078-0432.CCR-09-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moody SE, Schinzel AC, Singh S, Izzo F, Strickland MR, Luo L, Thomas SR, Boehm JS, Kim SY, Wang ZC, Hahn WC. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene. 2015;34:2061–2071. doi: 10.1038/onc.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El-Rifai W, Smith MF, Jr, Li G, Beckler A, Carl VS, Montgomery E, Knuutila S, Moskaluk CA, Frierson HF, Powell SM. Gastric cancers overexpress DARPP-32 and a novel isoform, t-DARPP. Cancer Res. 2002;62:4061–4. [PubMed] [Google Scholar]
- 15.Beckler A, Moskaluk CA, Zaika A, Hampton GM, Powell SM, Firerson HF, El-Rifai W. Overexpression of the 32-kilodalton dopamine and cyclic adenosine 3′,5′-monophosphate-regulated phosphoprotein in common adenocarcinomas. Cancer. 2003;98:1547–51. doi: 10.1002/cncr.11654. [DOI] [PubMed] [Google Scholar]
- 16.Gu L, Waliany S, Kane SE. Darpp-32 and its truncated variant t-Darpp have antagonistic effects on breast cancer cell growth and herceptin resistance. PLoS One. 2009;4:e6220. doi: 10.1371/journal.pone.0006220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamel S, Bouchard A, Ferrario C, Hassan S, Aguilar-Mahecha A, Buchanan M, Quenneville L, Miller W, Basik M. Both t-Darpp and DARPP-32 can cause resistance to trastuzumab in breast cancer cells and are frequently expressed in primary breast cancers. Breast Cancer Res Treat. 2010;120:47–57. doi: 10.1007/s10549-009-0364-7. [DOI] [PubMed] [Google Scholar]
- 18.Belkhiri A, Dar AA, Peng DF, Razvi MH, Rinehart C, Arteaga CL, El-Rifai W. Expression of t-DARPP mediates trastuzumab resistance in breast cancer cells. Clin Cancer Res. 2008;14:4564–71. doi: 10.1158/1078-0432.CCR-08-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–96. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- 20.Bibb, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC, Jr, Nairn AC, Greengard P. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–71. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- 21.Denny EC, Kane SE. t-Darpp Promotes Enhanced EGFR Activation and New Drug Synergies in Her2-Positive Breast Cancer Cells. PLoS One. 2015;10:e0132267. doi: 10.1371/journal.pone.0132267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Francis SH, Corbin JD. Structure and function of cyclic nucleotide-dependent protein kinases. Annu Rev Physiol. 1994;56:237–72. doi: 10.1146/annurev.ph.56.030194.001321. [DOI] [PubMed] [Google Scholar]
- 23.Depry C, Allen MD, Zhang J. Visualization of PKA activity in plasma membrane microdomains. Mol Biosyst. 2011;7:52–8. doi: 10.1039/c0mb00079e. [DOI] [PubMed] [Google Scholar]
- 24.Momand J, Magdziarz P, Feng Y, Jiang D, Parga E, Celis A, Denny E, Wang X, Phillips ML, Monterroso E, et al. t-Darpp is an elongated monomer that binds to calcium and is phosphorylated by cyclin-dependent kinases 1 and 5. FEBS Open Bio. 2017 doi: 10.1002/2211-5463.12269. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filippa N, Sable CL, Filloux C, Hemmings B, Van Obberghen E E. Mechanism of protein kinase B activation by cyclic AMP-dependent protein kinase. Mol Cell Biol. 1999;19:4989–5000. doi: 10.1128/mcb.19.7.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao X, Chaturvedi D, Patel TB. p90 ribosomal S6 kinase 1 (RSK1) and the catalytic subunit of protein kinase A (PKA) compete for binding the pseudosubstrate region of PKAR1alpha: role in the regulation of PKA and RSK1 activities. J Biol Chem. 2010;285:6970–9. doi: 10.1074/jbc.M109.083642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaturvedi D, Poppleton HM, Stringfield T, Barbier A, Patel TB. Subcellular localization and biological actions of activated RSK1 are determined by its interactions with subunits of cyclic AMP-dependent protein kinase. Mol Cell Biol. 2006;26:4586–600. doi: 10.1128/MCB.01422-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ilouz R, Bubis J, Wu JJ, Yim YY, Deal MS, Kornev AP, Ma Y, Blumenthal DK, Taylor SS. Localization and quaternary structure of the PKA RIβ holoenzyme. Proc Natl Acad Sci USA. 2012;109:12443–8. doi: 10.1073/pnas.1209538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.