

Abstract
Stromal cells like synovial fibroblasts gained great interest over the years, since it has become clear that they strongly influence their environment and neighbouring cells. The current review describes the role of synovial fibroblasts as cells of the innate immune system and expands on their involvement in inflammation and cartilage destruction in rheumatoid arthritis (RA). Furthermore, epigenetic changes in RA synovial fibroblasts and studies that focused on the identification of different subsets of synovial fibroblasts are discussed.
Keywords: rheumatoidarthritis, fibroblasts, inflammation
Key messages.
Synovial fibroblasts are carry a variety of innate immune receptors and are able to present antigen.
In rheumatoid arthritis (RA), synovial fibroblasts promote joint destruction via their attachment to cartilage and thus are key cells in RA pathogenesis.
RA synovial fibroblasts are stably activated and show alterations in their epigenetic landscape.
Synovial fibroblasts of different joints and of the lining and sublining layer differ in their phenotype and thus represent distinct subtypes of the synovial fibroblast population.
In most autoimmune diseases, including rheumatic diseases, research focused on the analysis of immune cells and their role in initiation and perpetuation of disease. Since a couple of years, however, it has been acknowledged that immune cells and local stroma have a significant role in disease pathogenesis. Stromal cells reveal an unexpected complexity in phenotype and function in different organs and in different diseases.
The stroma builds the structural framework of an organ or tissue. Studies analysing stromal cells in different settings revealed interactions between stromal cells and immune cells, which substantially contribute to shaping the immune response. Furthermore, stromal cells were recognised to have immunological functions and to be able to recognise pathogens and elicit an immune response. Intestinal, dermal, gingival and synovial fibroblasts are only some examples of stromal cells that have been shown to express innate immune receptors, in particular toll-like receptors (TLR). They react with the production of cytokines and chemokines to pathogen sensing and are able to process and to present antigens via major histocompatibility complex (MHC) II receptors.1–5 Therefore, these cells are part of the innate immune system and play an important role as local pathogen and damage sensors and activators of the immune system.
Synovial fibroblasts are the main stromal cells of the joint synovium. They are found in the synovial sublining and lining layer, which, in a healthy joint, is one to two cell layers thick and is interspersed with tissue-resident macrophages. Synovial fibroblasts produce the extracellular matrix (ECM) components of the synovial fluid and thus are important for cartilage integrity and lubrication of the joint. In rheumatoid arthritis (RA), the lining layer thickens and immune cells infiltrate the sublining layer of the synovium (figure 1). Synovial fibroblasts proliferate, become activated and invade and destroy the adjacent cartilage.
Figure 1.
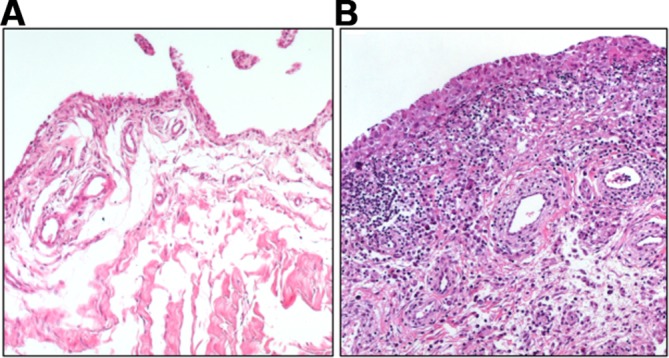
Osteoarthritis (A) and rheumatoid arthritis (RA) synovium (B). Synovial fibroblasts are found in the lining as well as in the sublining layer of the synovium. Thickening of the lining layer, lymphocytic infiltration and increased formation of blood vessels can be seen in RA synovium.
The analysis of synovial fibroblast in RA reaches back to the late 60s and early 70s with first studies done in particular by HG Fassbender and CW Castor and colleagues. Already in 1972, Castor and Buckingham noted that ‘extracts of Gram-negative bacteria, applied to fibroblast cultures, markedly increased hyaluronic acid production, glucose utilisation, and lactate output’,6 anticipating studies on TLR signalling and metabolic changes in synovial fibroblasts that were published decades later. Since then, numerous studies have characterised many different aspects of synovial fibroblast biology. Usually, cultured synovial fibroblasts from patients undergoing joint replacement surgery or synovial biopsies were used for these studies. Thereby, the synovium is enzymatically digested or fragmented, and adherent cells are used for analysis after three to four passages.7 These cultures are highly homogeneous for the expression of fibroblasts markers such as CD90 or proly-4-hydroxylase. However, it should be kept in mind that long-term culture of synovial fibroblasts can have an influence on their phenotype and that some characteristics of synovial fibroblasts, for example, the proliferative potential change over several passages.8 9 Furthermore, it has not yet been clarified how homogeneous this cultured cell population is, considering that different subtypes of synovial fibroblasts have been described as mentioned in more detail below.
Due to the vast amount of studies conducted with synovial fibroblasts in various rheumatic diseases, not all aspects of synovial fibroblast function in health and disease can be covered by the current review article. Instead, it focuses on the role of synovial fibroblasts as innate immune cells, their interaction with other immune cells and how this influences the pathogenesis of RA. Furthermore, epigenetic changes and their potential consequences in RA synovial fibroblasts are mentioned, and studies that subcategorise synovial fibroblasts into different subsets based on functional and embryonic differences are discussed.
Synovial fibroblasts as innate immune cells
By the expression of various pattern recognition receptors, synovial fibroblasts are well equipped to sense invading pathogens as well as joint damage. Synovial fibroblasts express TLR1–7, but very low or not-detectable basal transcript levels of TLR8–10.2 10
TLR3 and TLR7 bind double-stranded and single-stranded RNA, respectively. In addition, the cytosolic pattern recognition receptors (PRR) RIG-I and MDA5, which both bind double-stranded RNA are expressed and active in synovial fibroblasts.10 Necrotic cells in the synovial fluid were suggested to be a possible source of endogenous ligands leading to the activation of TLR3-mediated pathways in synovial fibroblasts in RA.11 In a large scale comparison of cytokine profiles between RA synovial fluid and stimulated synovial fibroblasts, the cytokine profiles obtained by stimulation of synovial fibroblasts with tumour necrosis factor (TNF) α, interleukin (IL) 1 or the TLR3 ligand Poly(I:C) reached the highest similarity with RA synovial fluids among 10 different stimuli used.12 The seven cytokines most strongly induced by these stimuli in synovial fibroblasts were also among the 12 cytokines with the highest levels in RA synovial fluids, corroborating the strong contribution of synovial fibroblasts to the inflammatory environment in RA joints. Unfortunately, other TLR ligands were not included in the stimulation experiments in this study.
TLR2, which forms heterodimers with TLR1 or TLR6, and TLR4 are both sensors of bacterial lipoproteins. A variety of endogenous ligands have been described to bind and activate TLR2 and TLR4, and many of them have been found at increased levels in RA joints.13–17 Some like heat-shock proteins, low-molecular-weight hyaluronan or high-mobility group protein 1 were initially described as direct ligands, but highly purified preparations were later not confirmed to stimulate TLR activation.18–20 Instead, it was suggested that these molecules facilitate binding of TLR ligands and thus amplify the cellular response to TLR activation. However, it is not clear in how far recombinant or highly purified preparations resemble their counterparts in vivo. Post-translational modifications and the formation of immune complexes can influence binding and activity of TLR ligands in vivo. Fibrinogen, which was described as endogenous ligand of TLR4,21 was shown to be more potent to activate macrophages and synovial fibroblasts in its citrullinated form.22 23 Also free histones that can activate TLR2 and TLR424 were found to be more potent stimulators if citrullinated and even more if incorporated in an immune complex.25 Interestingly, activation of monocytes by synovial fluids was significantly better blocked with an anti-TLR4 antibody if the patients had anti-citrullinated protein antibodies (ACPA) directed against citrullinated fibrinogen and citrullinated histone 2A, pointing toward a strong contribution of TLR4-mediated disease pathways in these patients.26 Thus, in the RA joint environment, the high levels of protein citrullination and the formation of immune complexes comprising citrullinated proteins and ACPA might further promote the binding of endogenous ligands to TLRs and cellular activation.
Interestingly, high content of citrullinated peptides was found in exosome preparations from synovial fluids of patients with RA, osteoarthritis (OA) and reactive arthritis, suggesting that extracellular vesicles might be of importance in the distribution and presentation of citrullinated peptides to various cell populations.27 Even though the amount of exosomes was similar between the patient’s groups, the content of proteins differed in that α2-macroglobulin, IgG1 γ-chain and fibronectin were only present in RA exosomes.27 Plasma-derived extracellular vesicles from RA patients but not from healthy individuals were shown to activate TLR4 on human embryonic kidney cells, on human monocytic cell lines and on murine bone-marrow derived monocytes.28 The authors could show that oxidised lipids in the membrane of RA exosomes were responsible for the activation of TLR4 and that the subsequent response resulted in the upregulation of genes that are important in resolving rather than promoting inflammation and substantially differed from a lipopolysaccharide-induced response. Synovial fibroblasts were found to produce proinflammatory mediators on incubation with extracellular vesicles.29–31 Even though it remains to be analysed whether extracellular vesicles from RA synovial fluid can elicit a specific TLR4 response in synovial fibroblasts, these studies support the hypothesis that extracellular vesicles with RA-specific content and/or modifications might activate synovial fibroblasts via TLR4 and thus modulate inflammation and joint destruction.
There are not many published studies investigating the expression and function of TLR9 and other DNA sensors such as AIM2 in synovial fibroblasts, and thus, it is not yet clarified in how much synovial fibroblasts can contribute to an immunoresponse induced by DNA sensing.32 After 24-hour stimulation with unmethylated CpG-rich DNA, the ligand of TLR9, no increase in protein levels of IL 6 or IL-8 could be measured,33 and only a small increase in transcript levels of IL-6 and IL-8 was measured after 4 hours of stimulation.34 An interesting study published in 2017 could show that neutrophil extracellular traps (NETs) can be internalised by synovial fibroblasts via a pathway involving the receptor for advanced glycation end-products and TLR9.35 Internalisation of NETs led to upregulation of MHC class II by synovial fibroblasts via IL-17B and presentation of citrullinated peptides to antigen-specific T cells. Injection of synovial fibroblasts that had internalised NETs into humanised HLA-DRB*04:01 transgenic mice led to formation of auto-antibodies against citrullinated histones H3 and H4 and citrullinated myeloperoxidase, which are all constituents of NETs,35 proving the effectiveness of antigen presentation by synovial fibroblasts. Earlier studies also showed that synovial fibroblasts in vivo and in vitro,9 mainly after stimulation with interferon γ (IFNγ),36 37 express MHC class II and that they can present antigens to T cells.38 It would be interesting to see whether also TLR stimulation can influence the expression and function of MHC class II in synovial fibroblasts as it was described in other cell types.
Once activated, TLRs were shown to induce the production of cytokines, chemokines and matrix metalloproteinases (MMPs) in synovial fibroblasts.2 12 39 High levels of the B cell survival factors B cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) produced by synovial fibroblasts after TLR3 stimulation promote antibody production and class switching of B cells.40 Interestingly, stimulation of one PRR also influences the expression of others. Stimulation of TLR3 in synovial fibroblasts leads to a robust upregulation of TLR3 itself, TLR2 and the cytoplasmic PRRs NOD1 and NOD2, which both sense bacterial products.2 41 42 Activation of NOD1 in turn synergises with TLR2 and TLR4 pathways.42 Therefore, activation of one PRR increases the reactivity of synovial fibroblasts to various endogenous and exogenous PRR ligands, which in an environment with high levels of endogenous ligands such as the RA synovium might lead to a constant build-up of the proinflammatory response. Furthermore, in contrast to macrophages, the production of proinflammatory cytokines is not dampened after repeated stimulation of TLRs in synovial fibroblasts.43 This lack of tolerisation might further contribute to sustaining the inflammatory response in RA synovium.
For the sake of completeness, it should be mentioned that several other stimuli, for example, a variety of cytokines, growth factors44 and adipokines,45–47 were found to activate synovial fibroblasts and promote their proinflammatory and joint destructive behaviour. However, it would go beyond the scope of this review to discuss all of them.
Interactions of synovial fibroblasts with T cells
Based on findings that various types of stromal cells such as bone marrow-derived mesenchymal stem cells and dermal and synovial fibroblasts inhibit T cell proliferation, it has been proposed that stromal cells in general possess immunosuppressive properties.48 49 For the suppression of T cell proliferation by synovial fibroblasts, soluble factors are needed that are only induced in synovial fibroblasts via direct cell-cell contact between synovial fibroblasts and T cells.49 50 A recent study could show that the inhibition of T cell proliferation is at least partly mediated via depletion of tryptophan by synovial fibroblasts.50 This study also showed that OA synovial fibroblasts are more efficient in suppressing T cell proliferation than RA synovial fibroblasts.
Whereas some studies showed that co-culture of T cells with synovial fibroblast reduced the production of IFNγ and IL-10, but not tumour necrosis factor (TNF) or IL-17A by T cells,49 50 other studies showed increased production of IFNγ, TNF and IL-17 by T cells co-cultured with synovial fibroblasts.51 52 This effect was mediated via membrane-bound IL-1551 and the CD40 pathway.52 The discrepancies between these studies probably stem from differences in isolation, culturing and stimulation techniques. It is hard to estimate which in vitro system most closely reflects the situation in vivo.
Stromal cells, including synovial fibroblasts, maintain T cells in a resting G0/G1 state and extend their survival.49 53 Thus, synovial fibroblasts keep T cells in the tissue in a non-proliferative state and protect them from undergoing apoptosis. In an inflammatory state, this might have profound effects on the persistence of inflammation and might actively counteract resolution of inflammation. Indeed, in the synovium of RA patients, differentiated T cells are protected from apoptotic cell death.54 Together, these data point to a modulation of the T cell phenotype and function by synovial fibroblasts, rather than general immunosuppressive properties. Whether this interaction between stroma and invading T cells is promoting or counteracting disease development remains, however, elusive up to now.
Various different direct adhesive and signalling interactions have been described between synovial fibroblasts and T cells as summarised in figure 2. These interactions first promote infiltration and retention of T cells in the synovium, for example, very late antigen-4 (VLA-4) and vascular cell adhesion protein 1 (VCAM-1) interactions, and second activate intracellular signalling pathways in both T cells and synovial fibroblasts. The connection between lymphocyte function-associated antigen 1 (LFA-1/CD11a) on T cells and intercellular adhesion molecule 1 (ICAM-1/CD54) on synovial fibroblasts is of particular interest because of its ability to enhance antigen presentation and T cell activation.55 LFA-1/ICAM-1 binding lowers the threshold for presented antigens to induce T cell activation, thereby allowing T cell activation by antigens that without this co-stimulation would not elicit a T cell response.56 LFA-1 binding by ICAM-1 also amplifies signalling pathways activated by T cell receptor binding.56 Interestingly, presentation of superantigens in conjunction with ICAM-1 strongly inhibits the production of IL-10 by T cells and thus counteracts self-limitation of the immune response.57 Whether this ICAM-1-dependent modulation of the T cell cytokine profile occurs in RA synovium is, however, not known. Similar to ICAM-1/LFA-1, binding of LFA-3 to CD2 and of activated leukocyte cell adhesion molecule (ALCAM/CD166) to CD6 optimises antigen-induced T cell responses.58 59
Figure 2.

Overview of key interactions between T cells and synovial fibroblasts. ALCAM, activated leukocyte cell adhesion molecule; CD40L, CD40 ligand; ICAM-1, intercellular adhesion molecule 1; LFA, lymphocyte function-associated antigen; MHC II, major histocompatibility complex II; TNFR1/2, tumour necrosis factor receptor 1/2; VCAM-1, vascular cell adhesion protein 1; VLA-4, very late antigen-4.
Also, synovial fibroblast functions are modulated by T cells, and synovial fibroblasts produce higher levels of cytokines such as IL-6 and IL-8 in co-culture with T cells.51 52 60 61 Blockade of TNF could inhibit this response of synovial fibroblasts, even though no soluble TNF was found in the co-cultures.61 The authors therefore concluded that membrane-bound TNF on T cells must be responsible for this effect.
In summary, there is clear evidence that synovial fibroblasts can present antigens to T cells with concurrent activation of an immune response and that synovial fibroblasts themselves are activated by this cell-cell contact. The effect on the phenotype and function of such cell-cell contacts on T cells is, however, not clarified in detail, and studies are complicated by the complexity of the in vivo situation in the RA synovium, which is hard to mimic in an artificial in vitro environment.
The role of synovial fibroblasts in the formation of ectopic lymphoid structures and B cell survival
Based on the histology of the synovium, patients with RA can be grouped into three subtypes. The fibroid pathotype is characterised by little infiltration of immune cells; in the myeloid pathotype, immune cells invade diffusely and monocytes/macrophages are the predominant immune cell type, and in the lymphoid pathotype, ectopic lymphoid structures (ELS) that are formed by B and T cell aggregates are found.62 63 Whether and how ELS contribute to disease pathogenesis has not been clarified in detail, but affinity maturation of B cells in ELS and local production of auto-antibodies in ELS could be shown in RA synovium.64 65 Based on their cytokine expression profile, synovial fibroblasts are believed to promote ELS formation and function. Synovial fibroblasts of patients with RA and psoriatic arthritis were shown to express higher levels of CCL19 and CCL21 than synovial fibroblasts from patients with OA.66 Both factors are being considered important for the development and maintenance of ELS (for review, see Bombardieri et al67). Similarly, receptor activator of NF-κB ligand (RANKL)68 and IL-769 are expressed at higher levels by synovial fibroblasts from RA patients compared with OA and are crucial factors in ELS development. Synovial fibroblasts furthermore promote retention and survival of plasma cells that matured in ELS or infiltrated through the circulation in the synovium by the production of CXCL12, IL-6, BAFF and APRIL.40 70 71 It is, however, still unclear to which extent synovial fibroblasts are actually involved in the appearance of ELS and why they only develop in a subgroup of RA patients.
Interactions of synovial fibroblasts with monocytes and macrophages
There is an intimate relationship between synovial fibroblasts and tissue resident synovial macrophages (type A synoviocytes) in the synovial tissue. The origin of these tissue resident macrophages in the synovium is not known to date. Apart from the long-lived, self-renewing tissue macrophages, monocytes are recruited from the circulation and mature into monocyte-derived macrophages (MDM), in particular during inflammation.72 MDM show remarkable plasticity, and in K/BxN serum transfer-induced arthritis in mice, they were shown to switch from a proinflammatory phenotype promoting arthritis to an anti-inflammatory phenotype necessary to resolve arthritis at a later stage of disease.73 The real extent of variability of MDM in inflamed synovium can probably only be estimated by applying singe cell based technologies in the future.
Surprisingly, little data are available addressing the role of synovial fibroblasts in macrophage differentiation and phenotypes, even though in other organs, it has been shown that the local stroma is crucial for the formation of tissue-specialised phenotypes of macrophages.74 Synovial fibroblasts can support the survival of monocytes in two-dimensional and three-dimensional co-cultures.75 76 Co-culture experiments with synovial fibroblasts and MDM showed that synovial fibroblasts suppressed the TNF-induced IFN response of MDM.77 This effect was found to be based on yet unidentified, soluble factors produced by synovial fibroblasts after stimulation with TNF. Strikingly, co-culture of synovial fibroblasts regulated almost one-third of TNF-induced genes in MDM, and there was substantial overlap of synovial fibroblast-regulated genes with genes that were found to be specific for synovial macrophages isolated from RA patients.77 These data strongly support a role of synovial fibroblasts in modulating the phenotype of macrophages in the RA synovium, and more studies focusing on this issue would be of high interest.
How do synovial fibroblasts invade cartilage
One of the first described and most characteristic features of RA synovial fibroblasts is their capability of invading and destroying cartilage. There is evidence from mouse models that cartilage structure has to be impaired before synovial fibroblasts can attach to cartilage. In TNF transgenic (hTNFtg) mice, loss of cartilage proteoglycan is a prerequisite for the attachment of and thus cartilage damage by synovial fibroblasts.78 Furthermore, collagenase or IL-1 injection into mouse joints attracts intravenously injected human RA synovial fibroblasts to the joint and promotes attachment and degradation.79 80 These data support the interesting hypothesis that synovial fibroblasts that leave the inflamed synovium can migrate through the body and spread RA to additional joints. Which factors are responsible for this attraction and whether the same mechanisms are operative in human RA joints are, however, not known to date.
Integrins and syndecans are adhesion molecules that have been implicated in the attachment of synovial fibroblasts to cartilage and ECM proteins. In hTNFtg mice, attachment of synovial fibroblasts to cartilage was substantially diminished when α2β1 integrin or syndecan 4 expression was knocked out.78 81 The functions of syndecan 4 are, however, manifold, and apart from promoting binding to ECM proteins, it binds a variety of growth factors (eg, basic fibroblast growth factor) induces cytoskeletal rearrangement and activates intracellular signalling pathways (for review, see Afratis et al82). Therefore, also other properties than ECM binding could be responsible for the positive effect of syndecan 4 knockout in the hTNFtg arthritis model. In RA synovium, the expression of the integrin subunits α5, αv and β1 was specifically increased at the site of synovial invasion into cartilage.83 Cultured RA synovial fibroblasts adhered significantly more to collagen type IV, fibronectin, laminin and tenascin than normal synovial fibroblasts and expressed increased levels of α5β1 integrins.84 Interestingly, syndecan 4 functions as co-receptor of α5β1 integrins in the formation of focal adhesions and cell migration as shown in a melanoma cell line.85 They also might cooperate in the RA synovium to promote cartilage destruction.
α5β1 integrins bind to fibronectin, and levels of fibronectin are increased in RA synovium and synovial fluids.86 Shiozawa et al found fibronectin at the surface of RA, but not OA or healthy cartilage, and staining increased inversely to proteoglycan content.87 It was suggested that cartilage impairment in RA exposes cartilage fibronectin or stimulates fibronectin deposition, which might promote binding by synovial fibroblasts.88 However, additional factors are necessary to promote attachment of synovial fibroblasts to cartilage because the content of fibronectin is also greatly increased in OA cartilage,89 but cartilage damage in OA patients does not lead to attachment and invasion of synovial fibroblasts.
Synovial fibroblasts damage cartilage by the production of MMPs, a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTs) and cathepsins. In particular, MMP1, 3, 9, 13, 14 and 15; ADAMTS4 and 5; and cathepsins B, L and K are highly expressed by synovial fibroblasts in RA joints (for detailed review, see Rengel et al90). The production of these proteinases is induced by the proinflammatory environment, for example, TNF and IL-1 in the inflamed joint. Also, epigenetic changes have been found that maintain the expression of MMPs at a high level in RA synovial fibroblasts.91 Even though many attempts have been made to influence cartilage destruction by pharmacological targeting of MMPs, none of the trials up to now was successful, partly because of lack of effect and partly because of serious side effects. An overview of factors that are implicated in the attachment of RA synovial fibroblasts to cartilage and cartilage destruction is given in figure 3.
Figure 3.
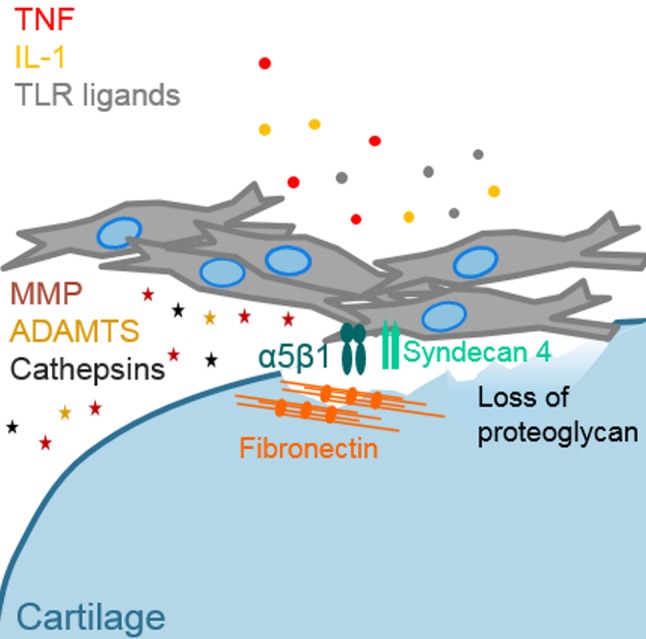
Attachment of rheumatoid arthritis (RA) synovial fibroblasts to cartilage and cartilage destruction. Impairment of cartilage structure and loss of proteoglycan were described as initiating events in the attachment of synovial fibroblasts to cartilage. Increased expression of α5β1 and its co-receptor syndecan 4 in RA synovial fibroblasts promotes binding of fibronectin, which can be deposited or exposed in damaged cartilage. High levels of proinflammatory factors such as tumour necrosis factor (TNF), interleukin (IL) 1 and toll-like receptor (TLR) ligands stimulate synovial fibroblasts to produce matrix-degrading molecules such as matrix metalloproteinases (MMPs), a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) and cathepsins.
Interaction of synovial fibroblasts with endothelial cells and osteoclasts
Blood vessel growth is increased in RA synovium, and synovial fibroblasts have been shown to produce a variety of factors that promote angiogenesis such as vascular endothelial growth factor and CXCL12 (for detailed review, see Elshabrawy et al92). Nevertheless, oxygen concentrations are strongly reduced in RA synovium.93 This hypoxic environment has severe consequences for the function of synovial fibroblasts by promoting mutations of mitochondrial DNA leading to mitochondrial dysfunction, changes in metabolic activity and increased migratory and invasive properties.94 In addition, hypoxia was recently shown to induce the release of extracellular RNA from synovial fibroblasts, which might influence the attachment of synovial fibroblasts to cartilage and subsequent invasion.95 The increase in the amount of blood vessels supports the recruitment of leukocytes in the inflamed synovium. Interactions between synovial, but not dermal fibroblasts with endothelial cells, were shown to increase the adhesion and migration of lymphocytes and neutrophils.96 97 Interestingly, under inflammatory conditions (in vitro stimulation with TNF and IFNγ), synovial fibroblasts from non-inflamed and resolving arthritis inhibited lymphocyte recruitment, while RA synovial fibroblasts did not.98 This was also true for synovial fibroblasts isolated from RA patients before they fulfilled RA classification criteria, showing that this change in fibroblast behaviour occurs at a very early stage of disease.98
Bone erosion is another classical feature of RA that is influenced by synovial fibroblasts. Production of proinflammatory cytokines and RANKL by synovial fibroblasts directly promotes osteoclastogenesis (for detailed review, see Wehmeyer et al99). Myostatin, a member of the transforming growth factor-β family, was shown to be strongly expressed by synovial fibroblasts in RA synovium and to enhance RANKL-mediated osteoclastogenesis.100
Epigenetic imprinting of synovial fibroblasts
Epigenetic mechanisms can induce stable changes in gene expression without altering the DNA sequence. They are essential during development and define tissue- and cell-specific gene expression. Later in life, the epigenetic landscape can still be modulated by environmental influences, which is a possibility for cells to adapt to an altered environment. However, shifts in the epigenetic landscape can also lead to the stable imprinting of pathological changes in gene expression. In RA synovial fibroblasts, a stably activated, aggressive phenotype has been described more than 20 years ago.101 102 Since then, a variety of epigenetic modifications, mostly DNA methylation, but also histone modifications and changes in non-coding RNA have been described to be altered in RA synovial fibroblasts compared with OA or healthy synovial fibroblasts and have been made responsible for the development of this disease-specific phenotype. The findings of the various studies analysing epigenetic changes in RA synovial fibroblasts will only briefly discussed here, since several review articles were recently published that focused in more detail on this particular topic.103–105
Studies analysing changes in DNA methylation in RA synovial fibroblasts could show that specific genes and pathways connected to the pathogenesis of RA are affected by these changes, which suggests that these alterations do not occur arbitrarily but are introduced by directed processes.106–108 Which processes might cause the changes in DNA methylation in RA synovial fibroblasts is largely unknown. Besides hypomethylation and hypermethylation connected to coding regions and gene expression in RA, a globally hypomethylated state, which presumably stems from a loss of DNA methylation in sequences harbouring retrotransposons, were found.109 Global hypomethylation often occurs in high proliferative cells and is connected to an increased recycling of polyamines, competing with the DNA methylating enzyme for the same methyl donor.110 How the subsequent expression of retrotransposable elements such as long interspersed element-1 (LINE-1) contributes to the aggressive phenotype of synovial fibroblasts is, however, not known.
The study of histone modifications is technically much more challenging than the analysis of DNA methylation. Accordingly, few studies analysing histone modifications were conducted in synovial fibroblasts. As yet, mainly histone acetylation and its modifying enzymes have been studied.111 Histone deacetylases (HDACs) have emerged as therapeutic targets in RA with HDAC inhibitors showing promising anti-inflammatory properties in a variety of cell types and animal models. Probably, this effect is based on modulation of histone acetylation as well as of changes in acetylation of non-histone proteins, such as transcription factors.
Apart from DNA methylation and histone modifications, non-coding RNAs, in particular microRNAs, are altered in RA synovial fibroblasts.112 113 MicroRNAs are small non-coding RNA molecules that can inhibit translation of proteins by binding of complementary messenger RNA (mRNA). The identification of the targets and pathways that are altered by differentially expressed microRNAs in RA synovial fibroblasts is, however, challenging, since one microRNA can bind several hundred different target mRNAs and one mRNA can be targeted by several microRNAs.
In summary, major advances have been made in the identification of epigenetic modifications that are altered in RA synovial fibroblasts. Epigenetic research now needs to go a step further and decipher how and at which stage of the disease these changes are introduced and what functional consequences they really have.
Subtypes of synovial fibroblasts
In earlier days, it was assumed that all fibroblasts throughout the body are similar and their function is limited to providing the extracellular framework for the more specialised tissue and immune cells. Nowadays, it is well known that fibroblasts from different anatomical sites are distinct and fulfil specialised functions; for example, synovial fibroblasts interact differently with local cells and immune cells than dermal fibroblasts and produce different molecules on stimulation.2 96
Newer data support the notion that the population of synovial fibroblasts is also heterogeneous and that several distinct subtypes of synovial fibroblasts exist. Synovial fibroblasts isolated from different joints show distinct phenotypes.114 115 They differ in their gene expression patterns, epigenetic marks and functions, which could explain why some joints are more susceptible to develop certain kinds of arthritis than others are.114 In particular, Hox genes that are expressed in a strictly controlled, time- and location-dependent manner during limb development are differentially expressed in synovial fibroblasts from different joints and maintain the same pattern of expression that was established during embryogenesis.114 Not a lot is known about the function of Hox genes in adult cells, but their continuous expression in a joint-specific manner together with some preliminary experiments114 suggests that they have a role in joint physiology and probably also in joint pathology.
Also within the synovium, heterogeneous populations of synovial fibroblasts are found. Synovial fibroblasts in the lining layer express the adhesion molecule VCAM-1, phosphodiesterase, cadherin-11 and gp38 (podoplanin), whereas synovial fibroblasts in the sublining layer express CD90 and CD248.116 117 This separation is even more pronounced in the hyperplastic lining layer of RA synovium and might play a role in the adhesive properties of RA synovium. Cadherin-11-deficient mice lack the typical organisation of the synovium into lining and sublining layer and develop ameliorated arthritis compared with wild-type mice as shown in the K/BxN serum transfer model.116 However, also CD248 knockout mice develop less severe collagen antibody-induced arthritis than wild-type mice, with less synovial hyperplasia and cartilage destruction, suggesting that also molecules expressed primarily in the sublining layer can influence synovial hyperplasia and invasion.118 These studies underline the importance of synovial fibroblasts in arthritis development, but further studies are needed to fully characterise differences in synovial fibroblasts within the synovium and their specific roles in arthritis development.
Conclusion and outlook
There is an increasing interest of the research community in analysing synovial fibroblasts and their role in arthritis development. From the many in vitro and in vivo studies, we learnt that synovial fibroblasts are critical in promoting destructive arthritis and produce a variety of factors that drive joint inflammation and joint destruction. They are innate immune cells that can sense pathogens and danger molecules in their environment and respond by the production of chemokines and cytokines. Furthermore, they are able to present antigen and elicit an adaptive immune response. In RA, they exhibit a stably altered phenotype and attach to and destroy cartilage. At the same time, they show profound changes in their epigenome, which might be responsible for this alteration in phenotype.
Despite this wealth of knowledge gained in the last years, many questions are still open that need to be answered to fully understand the role of synovial fibroblasts in the development of destructive arthritis. It is not clear at which stage of disease and how synovial fibroblasts become activated and phenotypically altered in RA. Furthermore, studies that connect epigenetic changes with functional changes in synovial fibroblasts are still rare. Cell adhesion pathways are predicted to be altered by changes in DNA methylation in RA synovial fibroblasts,107 but it is not really known which adhesion molecules are crucial for the attachment of synovial fibroblasts to cartilage, a process that is typical for RA synovium and a key step in disease development. Finally, the question why this process only happens in some joints, but not in others, remains unresolved. Further characterisation of the different subtypes of synovial fibroblasts might clarify the question which type of synovial fibroblasts drive destruction at which anatomical site.
The fascinating technical advances of recent years, such as single cell based transcriptome and proteome analysis, gene editing by the CRISPR/Cas9 system and high-resolution imaging techniques, will certainly drive scientific insights in the years to come. With the help of these techniques, it will be possible to get a clear picture of the biology of synovial fibroblasts in health and disease and their interactions with their neighbouring cells and tissues. Interdisciplinary integration of data and knowledge gained from genetics, immune and stroma cell analysis will be key to develop a comprehensive model of disease pathogenesis and preventive treatment strategies.
Footnotes
Contributors: CO planned and wrote the manuscript.
Competing interests: None declared.
Provenance and peer review: Commissioned; externally peer reviewed.
References
- 1.Owens BM, Steevels TA, Dudek M, et al. CD90(+) stromal cells are non-professional innate immune effectors of the human colonic mucosa. Front Immunol 2013;4:307 10.3389/fimmu.2013.00307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ospelt C, Brentano F, Rengel Y, et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum 2008;58:3684–92. 10.1002/art.24140 [DOI] [PubMed] [Google Scholar]
- 3.Umetsu DT, Katzen D, Jabara HH, et al. Antigen presentation by human dermal fibroblasts: activation of resting T lymphocytes. J Immunol 1986;136:440–5. [PubMed] [Google Scholar]
- 4.Wassenaar A, Snijders A, Abraham-Inpijn L, et al. Antigen-presenting properties of gingival fibroblasts in chronic adult periodontitis. Clin Exp Immunol 1997;110:277–84. 10.1111/j.1365-2249.1997.tb08328.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uehara A, Takada H. Functional TLRs and NODs in human gingival fibroblasts. J Dent Res 2007;86:249–54. 10.1177/154405910708600310 [DOI] [PubMed] [Google Scholar]
- 6.Buckingham RB, Castor CW. The effect of bacterial products on synovial fibroblast function: hypermetabolic changes induced by endotoxin. J Clin Invest 1972;51:1186–94. 10.1172/JCI106912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosengren S, Boyle DL, Firestein GS. Acquisition, culture, and phenotyping of synovial fibroblasts. Methods Mol Med 2007;135:365–75. [DOI] [PubMed] [Google Scholar]
- 8.Neumann E, Riepl B, Knedla A, et al. Cell culture and passaging alters gene expression pattern and proliferation rate in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther 2010;12:R83 10.1186/ar3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmermann T, Kunisch E, Pfeiffer R, et al. Isolation and characterization of rheumatoid arthritis synovial fibroblasts from primary culture—primary culture cells markedly differ from fourth-passage cells. Arthritis Res 2001;3:72–6. 10.1186/ar142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrión M, Juarranz Y, Pérez-García S, et al. RNA sensors in human osteoarthritis and rheumatoid arthritis synovial fibroblasts: immune regulation by vasoactive intestinal peptide. Arthritis Rheum 2011;63:1626–36. 10.1002/art.30294 [DOI] [PubMed] [Google Scholar]
- 11.Brentano F, Schorr O, Gay RE, et al. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum 2005;52:2656–65. 10.1002/art.21273 [DOI] [PubMed] [Google Scholar]
- 12.Jones DS, Jenney AP, Swantek JL, et al. Profiling drugs for rheumatoid arthritis that inhibit synovial fibroblast activation. Nat Chem Biol 2017;13:38–45. 10.1038/nchembio.2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamura Y, Watari M, Jerud ES, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 2001;276:10229–33. 10.1074/jbc.M100099200 [DOI] [PubMed] [Google Scholar]
- 14.Kokkola R, Sundberg E, Ulfgren AK, et al. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum 2002;46:2598–603. 10.1002/art.10540 [DOI] [PubMed] [Google Scholar]
- 15.Hasegawa M, Nakoshi Y, Muraki M, et al. Expression of large tenascin-C splice variants in synovial fluid of patients with rheumatoid arthritis. J Orthop Res 2007;25:563–8. 10.1002/jor.20366 [DOI] [PubMed] [Google Scholar]
- 16.Huang QQ, Sobkoviak R, Jockheck-Clark AR, et al. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol 2009;182:4965–73. 10.4049/jimmunol.0801563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connolly M, Rooney PR, McGarry T, et al. Acute serum amyloid A is an endogenous TLR2 ligand that mediates inflammatory and angiogenic mechanisms. Ann Rheum Dis 2016;75:1392–8. 10.1136/annrheumdis-2015-207655 [DOI] [PubMed] [Google Scholar]
- 18.Warger T, Hilf N, Rechtsteiner G, et al. Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. J Biol Chem 2006;281:22545–53. 10.1074/jbc.M502900200 [DOI] [PubMed] [Google Scholar]
- 19.Hreggvidsdottir HS, Ostberg T, Wähämaa H, et al. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol 2009;86:655–62. 10.1189/jlb.0908548 [DOI] [PubMed] [Google Scholar]
- 20.Ebid R, Lichtnekert J, Anders H-J. Hyaluronan is not a ligand but a regulator of toll-like receptor signaling in mesangial cells: role of extracellular matrix in innate immunity. ISRN Nephrol 2014;2014:1–11. 10.1155/2014/714081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol 2001;167:2887–94. 10.4049/jimmunol.167.5.2887 [DOI] [PubMed] [Google Scholar]
- 22.Sokolove J, Zhao X, Chandra PE, et al. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum 2011;63:53–62. 10.1002/art.30081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez-Pernaute O, Filkova M, Gabucio A, et al. Citrullination enhances the pro-inflammatory response to fibrin in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis 2013;72:1400–6. 10.1136/annrheumdis-2012-201906 [DOI] [PubMed] [Google Scholar]
- 24.Xu J, Zhang X, Monestier M, et al. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol 2011;187:2626–31. 10.4049/jimmunol.1003930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sohn DH, Rhodes C, Onuma K, et al. Local joint inflammation and histone citrullination in a murine model of the transition from preclinical autoimmunity to inflammatory arthritis. Arthritis Rheumatol 2015;67:2877–87. 10.1002/art.39283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatterer E, Shang L, Simonet P, et al. A specific anti-citrullinated protein antibody profile identifies a group of rheumatoid arthritis patients with a toll-like receptor 4-mediated disease. Arthritis Res Ther 2016;18:224 10.1186/s13075-016-1128-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skriner K, Adolph K, Jungblut PR, et al. Association of citrullinated proteins with synovial exosomes. Arthritis Rheum 2006;54:3809–14. 10.1002/art.22276 [DOI] [PubMed] [Google Scholar]
- 28.Manček-Keber M, Frank-Bertoncelj M, Hafner-Bratkovič I, et al. Toll-like receptor 4 senses oxidative stress mediated by the oxidation of phospholipids in extracellular vesicles. Sci Signal 2015;8:ra60 10.1126/scisignal.2005860 [DOI] [PubMed] [Google Scholar]
- 29.Berckmans RJ, Nieuwland R, Kraan MC, et al. Synovial microparticles from arthritic patients modulate chemokine and cytokine release by synoviocytes. Arthritis Res Ther 2005;7:R536–44. 10.1186/ar1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messer L, Alsaleh G, Freyssinet JM, et al. Microparticle-induced release of B-lymphocyte regulators by rheumatoid synoviocytes. Arthritis Res Ther 2009;11:R40 10.1186/ar2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jüngel A, Distler O, Schulze-Horsel U, et al. Microparticles stimulate the synthesis of prostaglandin E(2) via induction of cyclooxygenase 2 and microsomal prostaglandin E synthase 1. Arthritis Rheum 2007;56:3564–74. 10.1002/art.22980 [DOI] [PubMed] [Google Scholar]
- 32.Paludan SR, Bowie AG. Immune sensing of DNA. Immunity 2013;38:870–80. 10.1016/j.immuni.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyburz D, Rethage J, Seibl R, et al. Bacterial peptidoglycans but not CpG oligodeoxynucleotides activate synovial fibroblasts by toll-like receptor signaling. Arthritis Rheum 2003;48:642–50. 10.1002/art.10848 [DOI] [PubMed] [Google Scholar]
- 34.Hu F, Mu R, Zhu J, et al. Hypoxia and hypoxia-inducible factor-1α provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Ann Rheum Dis 2014;73:928–36. 10.1136/annrheumdis-2012-202444 [DOI] [PubMed] [Google Scholar]
- 35.Carmona-Rivera C, Carlucci PM, Moore E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol 2017;2:eaag3358 10.1126/sciimmunol.aag3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boots AM, Wimmers-Bertens AJ, Rijnders AW. Antigen-presenting capacity of rheumatoid synovial fibroblasts. Immunology 1994;82:268–74. [PMC free article] [PubMed] [Google Scholar]
- 37.Burmester GR, Dimitriu-Bona A, Waters SJ, et al. Identification of three major synovial lining cell populations by monoclonal antibodies directed to Ia antigens and antigens associated with monocytes/macrophages and fibroblasts. Scand J Immunol 1983;17:69–82. 10.1111/j.1365-3083.1983.tb00767.x [DOI] [PubMed] [Google Scholar]
- 38.Tran CN, Davis MJ, Tesmer LA, et al. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum 2007;56:1497–506. 10.1002/art.22573 [DOI] [PubMed] [Google Scholar]
- 39.Pierer M, Rethage J, Seibl R, et al. Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands. J Immunol 2004;172:1256–65. 10.4049/jimmunol.172.2.1256 [DOI] [PubMed] [Google Scholar]
- 40.Bombardieri M, Kam NW, Brentano F, et al. A BAFF/APRIL-dependent TLR3-stimulated pathway enhances the capacity of rheumatoid synovial fibroblasts to induce AID expression and Ig class-switching in B cells. Ann Rheum Dis 2011;70:1857–65. 10.1136/ard.2011.150219 [DOI] [PubMed] [Google Scholar]
- 41.Ospelt C, Brentano F, Jüngel A, et al. Expression, regulation, and signaling of the pattern-recognition receptor nucleotide-binding oligomerization domain 2 in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2009;60:355–63. 10.1002/art.24226 [DOI] [PubMed] [Google Scholar]
- 42.Yokota K, Miyazaki T, Hemmatazad H, et al. The pattern-recognition receptor nucleotide-binding oligomerization domain—containing protein 1 promotes production of inflammatory mediators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2012;64:1329–37. 10.1002/art.34318 [DOI] [PubMed] [Google Scholar]
- 43.Klein K, Frank-Bertoncelj M, Karouzakis E, et al. The epigenetic architecture at gene promoters determines cell type-specific LPS tolerance. J Autoimmun 2017;83:122–33. 10.1016/j.jaut.2017.07.001 [DOI] [PubMed] [Google Scholar]
- 44.Lefèvre S, Schwarz M, Meier FMP, et al. Disease-specific effects of matrix and growth factors on adhesion and migration of rheumatoid synovial fibroblasts. J Immunol 2017;198:4588–95. 10.4049/jimmunol.1600989 [DOI] [PubMed] [Google Scholar]
- 45.Ehling A, Schäffler A, Herfarth H, et al. The potential of adiponectin in driving arthritis. J Immunol 2006;176:4468–78. 10.4049/jimmunol.176.7.4468 [DOI] [PubMed] [Google Scholar]
- 46.Boström EA, Svensson M, Andersson S, et al. Resistin and insulin/insulin-like growth factor signaling in rheumatoid arthritis. Arthritis Rheum 2011;63:2894–904. 10.1002/art.30527 [DOI] [PubMed] [Google Scholar]
- 47.Meier FM, Frommer KW, Peters MA, et al. Visfatin/pre-B-cell colony-enhancing factor (PBEF), a proinflammatory and cell motility-changing factor in rheumatoid arthritis. J Biol Chem 2012;287:28378–85. 10.1074/jbc.M111.312884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haniffa MA, Wang XN, Holtick U, et al. Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. J Immunol 2007;179:1595–604. 10.4049/jimmunol.179.3.1595 [DOI] [PubMed] [Google Scholar]
- 49.Jones S, Horwood N, Cope A, et al. The antiproliferative effect of mesenchymal stem cells is a fundamental property shared by all stromal cells. J Immunol 2007;179:2824–31. 10.4049/jimmunol.179.5.2824 [DOI] [PubMed] [Google Scholar]
- 50.Tykocinski LO, Lauffer AM, Bohnen A, et al. Synovial fibroblasts selectively suppress Th1 cell responses through IDO1-mediated tryptophan catabolism. J Immunol 2017;198:3109–17. 10.4049/jimmunol.1600600 [DOI] [PubMed] [Google Scholar]
- 51.Miranda-Carús ME, Balsa A, Benito-Miguel M, et al. IL-15 and the initiation of cell contact-dependent synovial fibroblast-T lymphocyte cross-talk in rheumatoid arthritis: effect of methotrexate. J Immunol 2004;173:1463–76. 10.4049/jimmunol.173.2.1463 [DOI] [PubMed] [Google Scholar]
- 52.Cho ML, Yoon CH, Hwang SY, et al. Effector function of type II collagen-stimulated T cells from rheumatoid arthritis patients: cross-talk between T cells and synovial fibroblasts. Arthritis Rheum 2004;50:776–84. 10.1002/art.20106 [DOI] [PubMed] [Google Scholar]
- 53.Scott S, Pandolfi F, Kurnick JT. Fibroblasts mediate T cell survival: a proposed mechanism for retention of primed T cells. J Exp Med 1990;172:1873–6. 10.1084/jem.172.6.1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salmon M, Scheel-Toellner D, Huissoon AP, et al. Inhibition of T cell apoptosis in the rheumatoid synovium. J Clin Invest 1997;99:439–46. 10.1172/JCI119178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krzesicki RF, Fleming WE, Winterrowd GE, et al. T lymphocyte adhesion to human synovial fibroblasts. role of cytokines and the interaction between intercellular adhesion molecule 1 and CD11a/CD18. Arthritis Rheum 1991;34:1245–53. 10.1002/art.1780341007 [DOI] [PubMed] [Google Scholar]
- 56.Brunmark A, O’Rourke AM. Augmentation of mature CD4+ T cell responses to isolated antigenic class II proteins by fibronectin and intercellular adhesion molecule-1. J Immunol 1997;159:1676–85. [PubMed] [Google Scholar]
- 57.Labuda T, Wendt J, Hedlund G, et al. ICAM-1 costimulation induces IL-2 but inhibits IL-10 production in superantigen-activated human CD4+ T cells. Immunology 1998;94:496–502. 10.1046/j.1365-2567.1998.00540.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moingeon P, Chang HC, Wallner BP, et al. CD2-mediated adhesion facilitates T lymphocyte antigen recognition function. Nature 1989;339:312–4. 10.1038/339312a0 [DOI] [PubMed] [Google Scholar]
- 59.Hassan NJ, Simmonds SJ, Clarkson NG, et al. CD6 regulates T-cell responses through activation-dependent recruitment of the positive regulator SLP-76. Mol Cell Biol 2006;26:6727–38. 10.1128/MCB.00688-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamamura Y, Gupta R, Morita Y, et al. Effector function of resting T cells: activation of synovial fibroblasts. J Immunol 2001;166:2270–5. 10.4049/jimmunol.166.4.2270 [DOI] [PubMed] [Google Scholar]
- 61.Tran CN, Lundy SK, White PT, et al. Molecular interactions between T cells and fibroblast-like synoviocytes: role of membrane tumor necrosis factor-alpha on cytokine-activated T cells. Am J Pathol 2007;171:1588–98. 10.2353/ajpath.2007.070004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dennis G, Holweg CT, Kummerfeld SK, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther 2014;16:R90 10.1186/ar4555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pitzalis C, Kelly S, Humby F. New learnings on the pathophysiology of RA from synovial biopsies. Curr Opin Rheumatol 2013;25:334–44. 10.1097/BOR.0b013e32835fd8eb [DOI] [PubMed] [Google Scholar]
- 64.Scheel T, Gursche A, Zacher J, et al. V-region gene analysis of locally defined synovial B and plasma cells reveals selected B cell expansion and accumulation of plasma cell clones in rheumatoid arthritis. Arthritis Rheum 2011;63:63–72. 10.1002/art.27767 [DOI] [PubMed] [Google Scholar]
- 65.Humby F, Bombardieri M, Manzo A, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med 2009;6:e1 10.1371/journal.pmed.0060001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pickens SR, Chamberlain ND, Volin MV, et al. Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum 2011;63:914–22. 10.1002/art.30232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol 2017;13:141–54. 10.1038/nrrheum.2016.217 [DOI] [PubMed] [Google Scholar]
- 68.Takayanagi H, Iizuka H, Juji T, et al. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum 2000;43:259–69. [DOI] [PubMed] [Google Scholar]
- 69.Harada S, Yamamura M, Okamoto H, et al. Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum 1999;42:1508–16. [DOI] [PubMed] [Google Scholar]
- 70.Burger JA, Zvaifler NJ, Tsukada N, et al. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest 2001;107:305–15. 10.1172/JCI11092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ohata J, Zvaifler NJ, Nishio M, et al. Fibroblast-like synoviocytes of mesenchymal origin express functional B cell-activating factor of the TNF family in response to proinflammatory cytokines. J Immunol 2005;174:864–70. 10.4049/jimmunol.174.2.864 [DOI] [PubMed] [Google Scholar]
- 72.Thurlings RM, Wijbrandts CA, Bennink RJ, et al. Monocyte scintigraphy in rheumatoid arthritis: the dynamics of monocyte migration in immune-mediated inflammatory disease. PLoS One 2009;4:e7865 10.1371/journal.pone.0007865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Misharin AV, Cuda CM, Saber R, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep 2014;9:591–604. 10.1016/j.celrep.2014.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liddiard K, Taylor PR. Understanding local macrophage phenotypes in disease: shape-shifting macrophages. Nat Med 2015;21:119–20. 10.1038/nm.3798 [DOI] [PubMed] [Google Scholar]
- 75.Darrieutort-Laffite C, Boutet MA, Chatelais M, et al. IL-1β and TNFα promote monocyte viability through the induction of GM-CSF expression by rheumatoid arthritis synovial fibroblasts. Mediators Inflamm 2014;2014:1–10. 10.1155/2014/241840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kiener HP, Watts GF, Cui Y, et al. Synovial fibroblasts self-direct multicellular lining architecture and synthetic function in three-dimensional organ culture. Arthritis Rheum 2010;62:742–52. 10.1002/art.27285 [DOI] [PubMed] [Google Scholar]
- 77.Donlin LT, Jayatilleke A, Giannopoulou EG, et al. Modulation of TNF-induced macrophage polarization by synovial fibroblasts. J Immunol 2014;193:2373–83. 10.4049/jimmunol.1400486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Korb-Pap A, Stratis A, Mühlenberg K, et al. Early structural changes in cartilage and bone are required for the attachment and invasion of inflamed synovial tissue during destructive inflammatory arthritis. Ann Rheum Dis 2012;71:1004–11. 10.1136/annrheumdis-2011-200386 [DOI] [PubMed] [Google Scholar]
- 79.Hillen J, Geyer C, Heitzmann M, et al. Structural cartilage damage attracts circulating rheumatoid arthritis synovial fibroblasts into affected joints. Arthritis Res Ther 2017;19:40 10.1186/s13075-017-1245-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lefèvre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med 2009;15:1414–20. 10.1038/nm.2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Peters MA, Wendholt D, Strietholt S, et al. The loss of α2β1 integrin suppresses joint inflammation and cartilage destruction in mouse models of rheumatoid arthritis. Arthritis Rheum 2012;64:1359–68. 10.1002/art.33487 [DOI] [PubMed] [Google Scholar]
- 82.Afratis NA, Nikitovic D, Multhaupt HA, et al. Syndecans—key regulators of cell signaling and biological functions. Febs J 2017;284:27–41. 10.1111/febs.13940 [DOI] [PubMed] [Google Scholar]
- 83.Lowin T, Straub RH, Neumann E, et al. Glucocorticoids increase alpha5 integrin expression and adhesion of synovial fibroblasts but inhibit ERK signaling, migration, and cartilage invasion. Arthritis Rheum 2009;60:3623–32. 10.1002/art.24985 [DOI] [PubMed] [Google Scholar]
- 84.Rinaldi N, Schwarz-Eywill M, Weis D, et al. Increased expression of integrins on fibroblast-like synoviocytes from rheumatoid arthritis in vitro correlates with enhanced binding to extracellular matrix proteins. Ann Rheum Dis 1997;56:45–51. 10.1136/ard.56.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mostafavi-Pour Z, Askari JA, Parkinson SJ, et al. Integrin-specific signaling pathways controlling focal adhesion formation and cell migration. J Cell Biol 2003;161:155–67. 10.1083/jcb.200210176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vartio T, Vaheri A, Von Essen R, et al. Fibronectin in synovial fluid and tissue in rheumatoid arthritis. Eur J Clin Invest 1981;11:207–12. 10.1111/j.1365-2362.1981.tb01842.x [DOI] [PubMed] [Google Scholar]
- 87.Shiozawa K, Shiozawa S, Shimizu S, et al. Fibronectin on the surface of articular cartilage in rheumatoid arthritis. Arthritis Rheum 1984;27:615–22. 10.1002/art.1780270603 [DOI] [PubMed] [Google Scholar]
- 88.Shiozawa S, Yoshihara R, Kuroki Y, et al. Pathogenic importance of fibronectin in the superficial region of articular cartilage as a local factor for the induction of pannus extension on rheumatoid articular cartilage. Ann Rheum Dis 1992;51:869–73. 10.1136/ard.51.7.869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown RA, Jones KL. The synthesis and accumulation of fibronectin by human articular cartilage. J Rheumatol 1990;17:65–72. [PubMed] [Google Scholar]
- 90.Rengel Y, Ospelt C, Gay S. Proteinases in the joint: clinical relevance of proteinases in joint destruction. Arthritis Res Ther 2007;9:221 10.1186/ar2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Araki Y, Tsuzuki Wada T, Aizaki Y, et al. Histone methylation and STAT-3 differentially regulate interleukin-6-induced matrix metalloproteinase gene activation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol 2016;68:1111–23. 10.1002/art.39563 [DOI] [PubMed] [Google Scholar]
- 92.Elshabrawy HA, Chen Z, Volin MV, et al. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015;18:433–48. 10.1007/s10456-015-9477-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ng CT, Biniecka M, Kennedy A, et al. Synovial tissue hypoxia and inflammation in vivo. Ann Rheum Dis 2010;69:1389–95. 10.1136/ard.2009.119776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Biniecka M, Canavan M, McGarry T, et al. Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis 2016;75:2192–200. 10.1136/annrheumdis-2015-208476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zimmermann-Geller B, Köppert S, Fischer S, et al. Influence of extracellular RNAs, released by rheumatoid arthritis synovial fibroblasts, on their adhesive and invasive properties. J Immunol 2016;197:2589–97. 10.4049/jimmunol.1501580 [DOI] [PubMed] [Google Scholar]
- 96.McGettrick HM, Smith E, Filer A, et al. Fibroblasts from different sites may promote or inhibit recruitment of flowing lymphocytes by endothelial cells. Eur J Immunol 2009;39:113–25. 10.1002/eji.200838232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lally F, Smith E, Filer A, et al. A novel mechanism of neutrophil recruitment in a coculture model of the rheumatoid synovium. Arthritis Rheum 2005;52:3460–9. 10.1002/art.21394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Filer A, Ward LSC, Kemble S, et al. Identification of a transitional fibroblast function in very early rheumatoid arthritis. Ann Rheum Dis 2017:annrheumdis-2017-211286 10.1136/annrheumdis-2017-211286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wehmeyer C, Pap T, Buckley CD, et al. The role of stromal cells in inflammatory bone loss. Clin Exp Immunol 2017;189:1–11. 10.1111/cei.12979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dankbar B, Fennen M, Brunert D, et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med 2015;21:1085–90. 10.1038/nm.3917 [DOI] [PubMed] [Google Scholar]
- 101.Müller-Ladner U, Kriegsmann J, Franklin BN, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol 1996;149:1607–15. [PMC free article] [PubMed] [Google Scholar]
- 102.Müller-Ladner U, Kriegsmann J, Gay RE, et al. Oncogenes in rheumatoid arthritis. Rheum Dis Clin North Am 1995;21:675–90. [PubMed] [Google Scholar]
- 103.Doody KM, Bottini N, Firestein GS. Epigenetic alterations in rheumatoid arthritis fibroblast-like synoviocytes. Epigenomics 2017;9:479–92. 10.2217/epi-2016-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ospelt C, Gay S, Klein K. Epigenetics in the pathogenesis of RA. Semin Immunopathol 2017;39:409–19. 10.1007/s00281-017-0621-5 [DOI] [PubMed] [Google Scholar]
- 105.Cribbs A, Feldmann M, Oppermann U. Towards an understanding of the role of DNA methylation in rheumatoid arthritis: therapeutic and diagnostic implications. Ther Adv Musculoskelet Dis 2015;7:206–19. 10.1177/1759720X15598307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakano K, Whitaker JW, Boyle DL, et al. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis 2013;72:110–7. 10.1136/annrheumdis-2012-201526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Whitaker JW, Shoemaker R, Boyle DL, et al. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med 2013;5:40 10.1186/gm444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.de la Rica L, Urquiza JM, Gómez-Cabrero D, et al. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J Autoimmun 2013;41:6–16. 10.1016/j.jaut.2012.12.005 [DOI] [PubMed] [Google Scholar]
- 109.Karouzakis E, Gay RE, Michel BA, et al. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2009;60:3613–22. 10.1002/art.25018 [DOI] [PubMed] [Google Scholar]
- 110.Karouzakis E, Gay RE, Gay S, et al. Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2012;64:1809–17. 10.1002/art.34340 [DOI] [PubMed] [Google Scholar]
- 111.Angiolilli C, Baeten DL, Radstake TR, et al. The acetyl code in rheumatoid arthritis and other rheumatic diseases. Epigenomics 2017;9:447–61. 10.2217/epi-2016-0136 [DOI] [PubMed] [Google Scholar]
- 112.Churov AV, Oleinik EK, Knip M. MicroRNAs in rheumatoid arthritis: altered expression and diagnostic potential. Autoimmun Rev 2015;14:1029–37. 10.1016/j.autrev.2015.07.005 [DOI] [PubMed] [Google Scholar]
- 113.Kolarz B, Majdan M. Epigenetic aspects of rheumatoid arthritis: contribution of non-coding RNAs. Semin Arthritis Rheum 2017;46:724–31. 10.1016/j.semarthrit.2017.01.003 [DOI] [PubMed] [Google Scholar]
- 114.Frank-Bertoncelj M, Trenkmann M, Klein K, et al. Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions. Nat Commun 2017;8:14852 10.1038/ncomms14852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ai R, Hammaker D, Boyle DL, et al. Joint-specific DNA methylation and transcriptome signatures in rheumatoid arthritis identify distinct pathogenic processes. Nat Commun 2016;7:11849 10.1038/ncomms11849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lee DM, Kiener HP, Agarwal SK, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science 2007;315:1006–10. 10.1126/science.1137306 [DOI] [PubMed] [Google Scholar]
- 117.Croft AP, Naylor AJ, Marshall JL, et al. Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage. Arthritis Res Ther 2016;18:270 10.1186/s13075-016-1156-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maia M, de Vriese A, Janssens T, et al. CD248 and its cytoplasmic domain: a therapeutic target for arthritis. Arthritis Rheum 2010;62:3595–606. 10.1002/art.27701 [DOI] [PubMed] [Google Scholar]