

Abstract
Objective
This study aimed to explore the effects of overexpression of sirtuin 6 (SIRT6) on the tumorigenicity of hepatocellular carcinoma (HCC) cells
Methods
Stable SIRT6-overexpressed HCC cell lines were established by transfecting SIRT6 plasmid. Soft agar assay and tumor xenograft assay in nude mice were applied. Flow cytometry was employed to detect cell cycle distribution. Western blotting analysis was used to detect the expression of proteins.
Results
Overexpression of SIRT6 attenuated HepG2 and HCCLM3 cells proliferation, colony formation in vitro and tumor formation in nude mice, and resulted in the G1 phase cell cycle arrest. Overexpression of SIRT6 reduced the expression of cyclin D1 and p-ERK proteins in both HepG2 and HCCLM3 cells.
Conclusion
Overexpression of SIRT6 attenuates the tumorigenicity of HCC cells.
Keywords: SIRT6, HCC cell, tumorigenicity, cell cycle
INTRODUCTION
Sirtuin 6 (SIRT6) is a member of the mammalian sirtuins family, which functions as a mono-ADP-ribosyltransferase and NAD+-dependent deacylase of both acetyl groups and long-chain fatty acyl groups [1]. SIRT6 is involved in multiple cellular processes including transcription [2], genome instability [3], telomere integrity [4, 5], DNA repair [6, 7], inflammation [8], metabolism and ageing [3, 9, 10], and cancer [11–14]. In the year of 2012, SIRT6 was identified as a novel tumor suppressor by regulating aerobic glycolysis in cancer cells [11].
Recently, a number of studies have identified SIRT6 as a key regulator of hepatocellular carcinoma (HCC), but the functional roles of SIRT6 in liver cancer are inconsistent. Min et al. reported that liver cancer initiation was controlled by AP-1 through SIRT6-dependent inhibition of survivin [15]. Marquardt et al. reported that loss of SIRT6 induced epigenetic changes which might be relevant to HCC development. Downregulation of SIRT6 and genes dysregulated by loss of SIRT6 possessed oncogenic effects in hepatocarcinogenesis [12]. Zhang et al. reported that overexpression of SIRT6 suppressed HCC cell proliferation and induced apoptosis [16]. Additional two studies [17, 18] suggested that SIRT6 acted as an oncogene in HCC development. Their results showed that SIRT6 was upregulated in a subset of HCC tissues and SIRT6 knockdown by shRNA suppressed the growth of HCC cells, induced apoptosis, and inhibited tumor growth of HCC cells in vivo. Therefore, the conflicts on the roles of SIRT6 in HCC promoted us to explore the functional roles of SIRT6 in the development of HCC and its underlying mechanisms. In this study, two stable SIRT6-overexpressed HCC cell lines were established to investigate the effects of overexpression of SIRT6 on HCC cell proliferation, cell cycle distribution, tumorigenicity in vitro and in vivo, and its related molecular mechanisms.
RESULTS
Identification of SIRT6 overexpression
The overexpression of SIRT6 was identified by western blotting analysis. The expression of SIRT6 was only detected in HepG2-SIRT6 and HCCLM3-SIRT6 cells by using primary anti-Flag antibody, but not in HepG2-3.1 and HCCLM3-3.1 cells (Figure 1A and 1B), which suggests that stable SIRT6-overexpressed HCC cell lines were successfully established.
Figure 1. The identification of SIRT6-overexpressed HCC cells and the effects of SIRT6 overexpression on HCC cell proliferation.
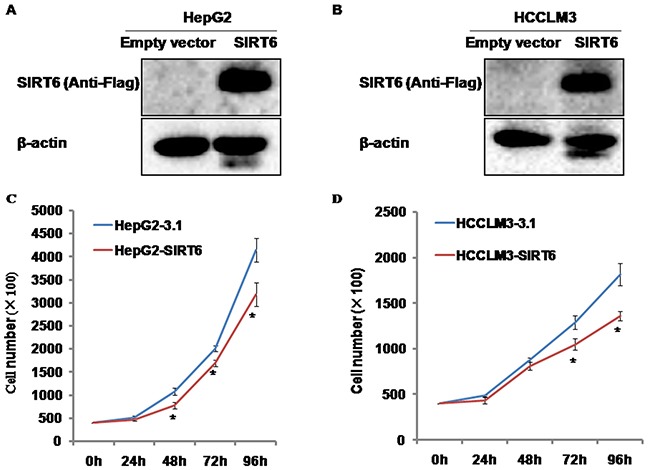
(A and B) SIRT6 expression was only detected in HepG2-SIRT6 cells and HCCLM3-SIRT6 cells by anti-Flag, which suggests that stable SIRT6-overexpressed cell lines were successfully established. (C and D) Overexpression of SIRT6 decreased the proliferation of HepG2 cells from 48 hours and HCCLM3 cells from 72 hours. *P<0.05.
Effects of SIRT6 overexpression on HCC cell proliferation
To test the effects of SIRT6 overexpression on HCC cell proliferation, we generated stable SIRT6-overexpressed HepG2 and HCCLM3 cell lines. The number of cells was counted at different time points indicated. SIRT6 overexpression significantly slowed down the growth of HepG2 cells from 48 hours and HCCLM3 cells from 72 hours compared with its controls, respectively (Figure 1C and 1D).
Effects of SIRT6 overexpression on colony formation
Soft agar assay was employed to test the capability of colony formation of SIRT6-overexpressed HCC cells in vitro. We observed that overexpression of SIRT6 dramatically decreased the formation of cell colonies compared with its controls in both HepG2 and HCCLM3 cell lines (Figure 2A and 2B).
Figure 2. Effects of SIRT6 overexpression on colony formation.
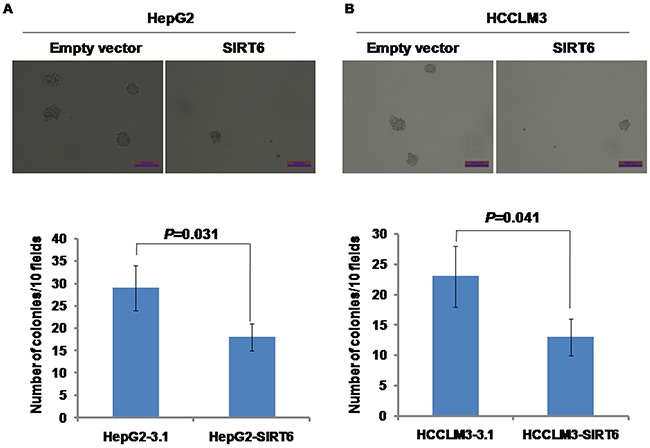
Overexpression of SIRT6 significantly reduced the rate of colony formation in both HepG2 and HCCLM3 cells. The scale of bar is 1000μm.
Effects of SIRT6 overexpression on tumor growth in nude mice
To test the effects of SIRT6 overexpression on the capability of tumor growth in vivo, BALB/c nude mice xenograft models were established by subcutaneous injection with HepG2-SIRT6, HepG2-3.1, HCCLM3-SIRT6 and HCCLM3-3.1 cells. The tumor weight was significantly decreased in both HepG2-SIRT6 (0.2555 ± 0.1295 g) and HCCLM3-SIRT6 (0.1814 ± 0.0934 g) groups compared with HepG2-3.1 (0.5849 ± 0.2699 g) and HCCLM3-3.1 (0.4898 ± 0.1455 g) groups, respectively (Figure 3A and 3B), which suggests that overexpression of SIRT6 significantly reduced tumor formation and tumor growth of HepG2 and HCCLM3 cells in vivo.
Figure 3. Effects of SIRT6 overexpression on tumor growth in nude mice.
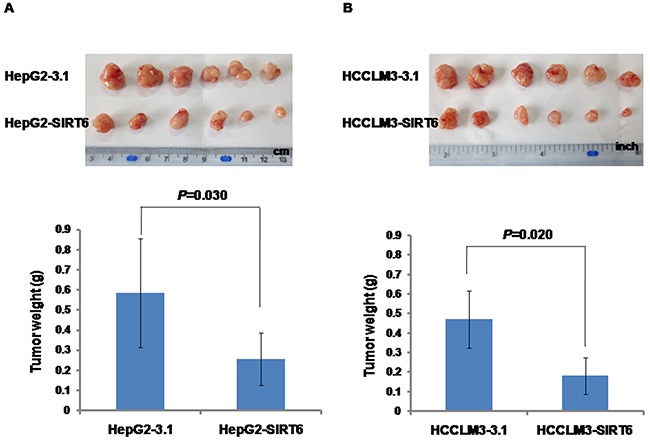
Significantly decreased tumor weight was observed in the group injected with SIRT6-overexpressed HCC cells compared with the group injected with control cells.
Effects of SIRT6 overexpression on cell cycle
Above, we observed that overexpression of SIRT6 decreased HCC cell proliferation. To further investigate whether or not the reduction of growth speed of SIRT6-overexpressed HCC cells is resulted from cell cycle arrest, the distribution of cell cycle was detected by flow cytometry. We observed the significantly increased proportion of cells stably overexpressing SIRT6 in the G1 phase population (60.5 ± 2.9% for HepG2-SIRT6 and 57.5 ± 1.7% for HCCLM3-SIRT6) compared with the control cells (49.9 ± 2.6% for HepG2-3.1 and 50.0 ± 2.3% for HCCLM3-3.1), respectively (Figure 4A and 4B). We observed the significantly decreased proportion of cells stably overexpressing SIRT6 in the S phase population (34.9 ± 1.9% for HepG2-SIRT6 and 36.1 ± 0.9% for HCCLM3-SIRT6) compared with the control cells (44.2 ± 1.0% for HepG2-3.1 and 42.2 ± 0.4% for HCCLM3-3.1), respectively (Figure 4A and 4B). Our results suggested that overexpression of SIRT6 might decrease HCC cell proliferation by arresting the cell cycle in the G1 phase.
Figure 4. Effects of SIRT6 overexpression on cell cycle distribution.
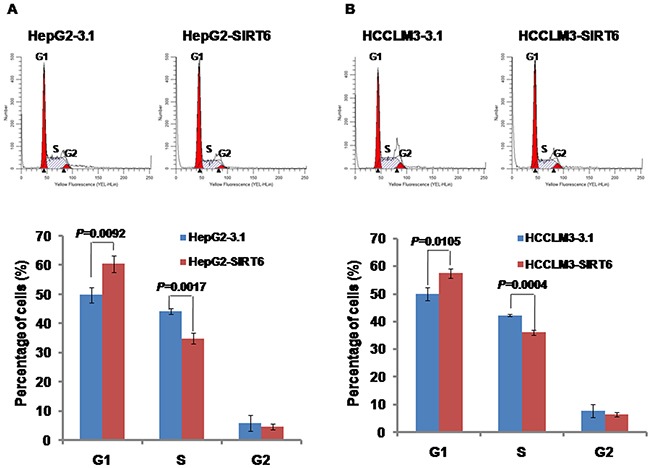
The detection of cell cycle was performed in a Muse Cell Analyzer. Our data showed that overexpression of SIRT6 increased the cell percentage in the G1 phase and decreased the cell percentage in the S phase in both HCC cell lines.
Overexpression of SIRT6 repressed the expression of cyclin D1
To explore the molecular mechanisms of the cell cycle arrest while HCC cells stably overexpressing SIRT6, AQ:the expression of a series of cell cycle-related proteins was detected by western blotting analysis. Our data showed that the level of cyclin D1 protein expression was significantly decreased in HepG2-SIRT6 cells and HCCLM3-SIRT6 cells compared with its controls, respectively (Figure 5). There was no significant difference in the expression of the Rb, p-Rb, p21, cyclin A, cyclin B1 and CDK4 proteins between SIRT6-overexpressed HCC cells and its control cells (Figure 5).
Figure 5. Effects of SIRT6 overexpression on the expression of cell cycle-related proteins.
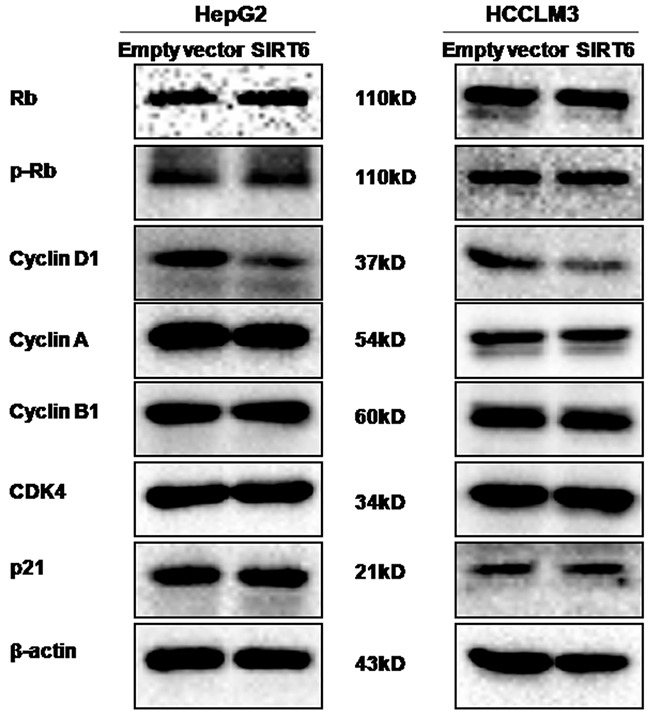
The western blotting assay was performed. Our data showed that overexpression of SIRT6 could reduce the expression of cyclin D1 protein in both HCC cell lines.
Overexpression of SIRT6 repressed p-ERK expression
Since several proteins including β-catenin, TGIF, STIM1, p-Akt, p-ERK, p53 and PTEN are involved in HCC tumorigenesis, these proteins were detected by western blotting analysis. Our results showed that SIRT6 overexpression markedly reduced the level of p-ERK protein expression in both HepG2 and HCCLM3 cells (Figure 6). We did not observe obvious alterations in the expression of β-catenin, TGIF, STIM1, p-Akt, p53 and PTEN proteins between SIRT6-overexpressed HCC cells and its control cells (Figure 6).
Figure 6. Effects of SIRT6 overexpression on the expression of several proteins.
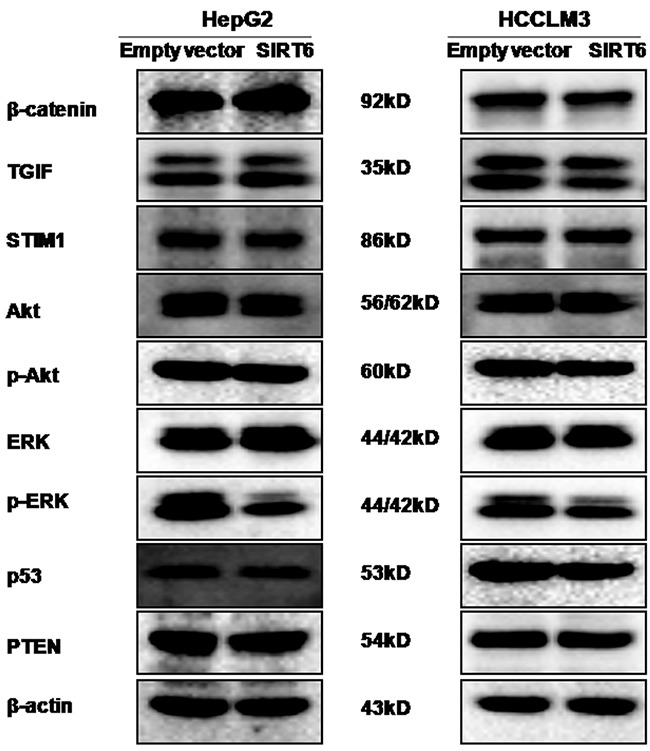
The western blotting assay was performed. Our data showed that overexpression of SIRT6 could reduce the level of p-ERK protein expression in both HCC cell lines.
DISCUSSION
In this study, we observed that overexpression of SIRT6 suppressed the growth of HepG2 and HCCLM3 cells in vitro. Our observation is consistent with Zhang et al.'s results. Their results indicated that adenovirus-mediated overexpression of SIRT6 significantly inhibited HepG2 cell growth, whereas, silencing SIRT6 by shRNA promoted HepG2 cell growth [16]. But Marquardt et al. reported that overexpression of SIRT6 did not lead to a change in cell proliferation [12]. On the contrary, Lee et al. reported that SIRT6 knockdown by shRNA suppressed the growth of HCC cells (Huh7, SNU475 and SNU449) [17]. Ran et al. reported that depletion of SIRT6 inhibited the growth of liver cancer cell lines (PLC/PRF/5, SMMC-7721, Huh-7 and SK-Hep-1) [18]. The discrepancy among these studies should be addressed further among different laboratories in the future studies.
To further explore the underlying mechanisms of overexpression of SIRT6 in decreasing HCC cell proliferation, cell cycle distribution was analyzed by flow cytometry. Our data showed that overexpression of SIRT6 arrested cell cycle in the G1 phase in HepG2 and HCCLM3 cells. Moreover, we observed that overexpression of SIRT6 significantly decreased the expression of cyclin D1 protein in HCC cells, accompanied with the G1 phase arrest. Studies showed that the increased expression of cyclin D1 in cancer cells resulted in an uncontrolled growth advantage [19]. Cyclin D1 is required for progression through the G1 phase of the cell cycle, which dimerizes with CDK4/6 to regulate the G1/S phase transition and entry in the S phase. Cai et al. reported that DLK1 knockdown delayed the cell cycle G1/S transition in HCC cells, along with the decreased expression of cyclin D1 [20]. Duan et al. reported that suppression of ARID2 expression accelerated G1/S transition associated with upregulation of cyclin D1 in HCC cells [21]. Lee et al. reported that VRK1 knockdown increased the number of G1 phase arrested cells by decreasing cyclin D1 expression in HCC cells [22]. Taken together, our observations suggested that overexpression of SIRT6 arrested cell cycle in the G1 phase via decreasing the expression of cyclin D1 in HCC cells. But the mechanism underlying the regulation of cyclin D1 by SIRT6 was not addressed in this paper, which should be explored in the future study.
Previous study reported that SIRT6 promoted cell death during liver cancer initiation in diethylnitrosamine (DEN)-induced mouse liver cancer model [15], which suggests that SIRT6 might be a tumor suppressor in HCC tumorigenesis. To address the potential roles of SIRT6 in tumorigenicity of HCC cells, we established stable SIRT6-overexpressed HCC cell lines. Our data showed that overexpression of SIRT6 reduced the colony formation of HCC cells in soft agar and tumor growth in nude mice, which supported the opinion of SIRT6 as a tumor suppressor in HCC to a certain extent. However, other two studies reported opposite results. Lee et al. reported that SIRT6 depletion reduced colony formation of HCC cells in soft agar and tumor growth in xenograft mouse model [17]. Feng et al. reported that upregulation of SIRT6 was required for TGF-β1/H2O2/HOCl to promote the tumorigenicity of HCC cells owing to that suppression of SIRT6 expression could abrogate the promoting effects of TGF-β1/H2O2/HOCl on the tumorigenicity of HCC cells [23].
The extracellular signal-regulated kinase (ERK) pathway is involved in crucial cellular processes, including proliferation, differentiation, angiogenesis and survival [24]. Overexpression or activation of ERK is believed to contribute to hepatocarcinogenesis. Studies have shown that an increase in the expression and activity of ERK was found in human HCC tissues [25–27] and in a rat model of experimental HCC [28]. ERK knockdown abolished liver tumor cell proliferation and DNA replication as well as the growth of xenografted tumors [29]. In this study, overexpression of SIRT6 decreased the level of p-ERK expression in two HCC cell lines. Consistent with our observations, one published paper reported that overexpression of SIRT6 significantly inhibited ERK1/2 phosphorylation in HCC cells [16]. Both of these studies suggested that the ERK signaling pathway might be involved in SIRT6 suppressing the proliferation and tumorigenicity of HCC cells.
In conclusion, our data suggested that overexpression of SIRT6 reduced HCC cell proliferation by arresting cell cycle in the G1 phase and attenuated the tumorigenicity. Therefore, this study extends our knowledge of liver tumorigenesis.
MATERIALS AND METHODS
Cell culture
The cell lines of HepG2 and HCCLM3 were purchased from the Cell Resource Center, Peking Union Medical College (which is the headquarters of National Infrastructure of Cell Line Resource, NSTI). The two cell lines were cultured in DMEM medium containing 10% of fetal bovine serum (FBS), 2 mM of L-glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin at a condition of 37°C and 5% CO2.
Establishment of stable cell lines
The enhanced SIRT6 plasmid with flag tag was obtained from Addgene (#13817) and the empty vector (pcDNA3.1) was purchased from Invitrogen (V79020). The transfection was performed using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, USA). The transfected cells of HepG2 and HCCLM3 were selected with 500μg/ml of G418 (Gibco, USA). The stable cell lines were identified by western blotting and named as HepG2-SIRT6 and HepG2-3.1, and HCCLM3-SIRT6 and HCCLM3-3.1, respectively.
Detection of cell proliferation
The assay of cell proliferation was performed as previously described [30]. In brief, 4 × 104 cells were plated into each well of 12-well plates in triplicate. Cells were trypsinized and counted by using a CASY Cell Counter (Scharfe System, Reutlingen, Germany) at 24, 48, 72, and 96 hours after plating.
Soft agar assay
The assay of soft agar was performed as previously described [31]. 500 cells in 1 ml of DMEM full medium containing 0.35% low-melting point agarose were plated on top of the base layer (3 wells per group). Cells were maintained at a condition of 37°C and 5% CO2 for two weeks.
Tumor xenograft assay
This study was approved by the Ethics Committee of Henan Center for Disease Control and Prevention. All of the animal experiments were carried out according to the Guide for the Care and Use of Laboratory Animals. Male BALB/c nude mice aged four weeks were obtained from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Cells were trypsinized and washed with PBS. 5 × 106 cells in 150 μl of PBS were subcutaneously injected into the back neck of each mouse (six mice per group). The mice were monitored every day for tumor formation and were sacrificed at 18 days postinjection [31].
Western blotting assay
The western blotting analysis was performed as previously described [30]. In brief, cells were lysed by RIPA buffer containing protease inhibitors and phosphatase inhibitors (Pierce, USA). The protein concentration was measured by a BCA regent kit (Pierce, USA). The lysates were loaded into 10% of sodium dodecyl sulfate-polyacrylamide electrophoresis gels and then transferred to nitrocellulose (NC) membranes (PALL, USA). The membrane was blocked with 5% of bovine serum albumin (BSA) in Tris-buffered saline-Tween 20 and then incubated with a primary antibody at 4°C for overnight. The anti-Flag (F3165) was purchased from Sigma. The β-catenin (sc-7199), TGIF (sc-9084), p21 (sc-397), STIM1 (sc-68897), p53 (sc-6243), Akt (sc-8312), p-Akt (sc-33437), cyclin A (sc-751), cyclin B1 (sc-752), cyclin D1 (sc-718), CDK4 (sc-260), and β-actin (sc-8432) were purchased from Santa Cruz Biotechnology. The Rb (#9313S), phospho-Rb (#8516S), ERK1/2 (#4695S), p-ERK1/2 (#4370S), and PTEN (#9188S) were purchased from Cell Signaling Technology. The secondary antibodies (Peroxidase-Conjugated AffiniPure Goat Anti-Rabbit-IgG and Goat Anti-Mouse-IgG) were purchased from ZSGB-BIO (Beijing, China). The signals were detected in the ChemiDoc™ XRS+ Imaging System (Bio-Rad, USA) by a Bio-Rad Clarity™ western ECL substrate (Bio-Rad, USA).
Detection of cell cycle
Cells were plated in 60-mm dishes, and trypsinized while growing to 70-80% confluence. Detached cells were harvested, washed with PBS and fixed in 70% of ethanol at −20°C overnight. The detection of cell cycle was performed in a Muse Cell Analyzer (Millipore, Merck Group) by using Muse cell cycle assay kit (MCH100106, Millipore) according to the manufacturer's instructions.
Statistical analysis
The Student's t test was performed to estimate the statistical significance by using the SPSS 13.0 software (SPSS, Chicago, IL). Values were presented as mean ± standard deviation (SD). Difference was considered statistically significant when P < 0.05. All of the tests were two-sided.
Footnotes
Author contributions
Yadong Wang contributed to study design, implementation, experiments, data analysis, and manuscript writing. Yadong Wang, Haiyu Wang and Jiangmin Li contributed to animal model experiments and soft agar assay. Teng Pan and Yadong Wang contributed to western blotting analysis. Yadong Wang, Li Li and Teng Pan contributed to cell cycle analysis. Ding Zhang and Haiyan Yang contributed to data analysis.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts interest.
FUNDING
This work was supported by a grant from National Natural Science Foundation of China (No. U1404815). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Lerrer B, Gertler AA, Cohen HY. The complex role of SIRT6 in carcinogenesis. Carcinogenesis. 2015;37:108–118. doi: 10.1093/carcin/bgv167. [DOI] [PubMed] [Google Scholar]
- 2.Gertler AA, Cohen HY. SIRT6, a protein with many faces. Biogerontology. 2013;14:629–639. doi: 10.1007/s10522-013-9478-8. [DOI] [PubMed] [Google Scholar]
- 3.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 4.Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, Chang HY, Bohr VA, Ried T, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, Chua KF. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. 2009;8:2664–2666. doi: 10.4161/cc.8.16.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329:1348–1353. doi: 10.1126/science.1192049. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011;71:5296–5306. doi: 10.1158/0008-5472.CAN-11-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lombard DB, Schwer B, Alt FW, Mostoslavsky R. SIRT6 in DNA repair, metabolism and ageing. J Intern Med. 2008;263:128–141. doi: 10.1111/j.1365-2796.2007.01902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, Cosentino C, Greenson JK, MacDonald AI, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marquardt JU, Fischer K, Baus K, Kashyap A, Ma S, Krupp M, Linke M, Teufel A, Zechner U, Strand D, Thorgeirsson SS, Galle PR, Strand S. Sirtuin-6-dependent genetic and epigenetic alterations are associated with poor clinical outcome in hepatocellular carcinoma patients. Hepatology. 2013;58:1054–1064. doi: 10.1002/hep.26413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukuda T, Wada-Hiraike O, Oda K, Tanikawa M, Makii C, Inaba K, Miyasaka A, Miyamoto Y, Yano T, Maeda D, Sasaki T, Kawana K, Fukayama M, et al. Putative tumor suppression function of SIRT6 in endometrial cancer. FEBS Lett. 2015;589:2274–2281. doi: 10.1016/j.febslet.2015.06.043. [DOI] [PubMed] [Google Scholar]
- 14.Bai L, Lin G, Sun L, Liu Y, Huang X, Cao C, Guo Y, Xie C. Upregulation of SIRT6 predicts poor prognosis and promotes metastasis of non-small cell lung cancer via the ERK1/2/MMP9 pathway. Oncotarget. 2016;7:40377–40386. doi: 10.18632/oncotarget.9750. https://doi.org/10.18632/oncotarget.9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, Chen L, Scheuch H, Zheng H, Qin L, Zatloukal K, Hui L, Wagner EF. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol. 2012;14:1203–1211. doi: 10.1038/ncb2590. [DOI] [PubMed] [Google Scholar]
- 16.Zhang ZG, Qin CY. Sirt6 suppresses hepatocellular carcinoma cell growth via inhibiting the extracellular signalregulated kinase signaling pathway. Mol Med Rep. 2014;9:882–888. doi: 10.3892/mmr.2013.1879. [DOI] [PubMed] [Google Scholar]
- 17.Lee N, Ryu HG, Kwon JH, Kim DK, Kim SR, Wang HJ, Kim KT, Choi KY. SIRT6 depletion suppresses tumor growth by promoting cellular senescence induced by DNA damage in HCC. PLoS One. 2016;11:e0165835. doi: 10.1371/journal.pone.0165835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ran LK, Chen Y, Zhang ZZ, Tao NN, Ren JH, Zhou L, Tang H, Chen X, Chen K, Li WY, Huang AL, Chen J. SIRT6 overexpression potentiates apoptosis evasion in hepatocellular carcinoma via BCL2-associated X protein-dependent apoptotic pathway. Clin Cancer Res. 2016;22:3372–3382. doi: 10.1158/1078-0432.CCR-15-1638. [DOI] [PubMed] [Google Scholar]
- 19.Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- 20.Cai CM, Xiao X, Wu BH, Wei BF, Han ZG. Targeting endogenous DLK1 exerts antitumor effect on hepatocellular carcinoma through initiating cell differentiation. Oncotarget. 2016;7:71466–71476. doi: 10.18632/oncotarget.12214. https://doi.org/10.18632/oncotarget.12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan Y, Tian L, Gao Q, Liang L, Zhang W, Yang Y, Zheng Y, Pan E, Li S, Tang N. Chromatin remodeling gene ARID2 targets cyclin D1 and cyclin E1 to suppress hepatoma cell progression. Oncotarget. 2016;7:45863–45875. doi: 10.18632/oncotarget.10244. https://doi.org/10.18632/oncotarget.10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee N, Kwon JH, Kim YB, Kim SH, Park SJ, Xu W, Jung HY, Kim KT, Wang HJ, Choi KY. Vaccinia-related kinase 1 promotes hepatocellular carcinoma by controlling the levels of cell cycle regulators associated with G1/S transition. Oncotarget. 2015;6:30130–30148. doi: 10.18632/oncotarget.4967. https://doi.org/10.18632/oncotarget.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng XX, Luo J, Liu M, Yan W, Zhou ZZ, Xia YJ, Tu W, Li PY, Feng ZH, Tian DA. Sirtuin 6 promotes transforming growth factor-beta1/H2O2/HOCl-mediated enhancement of hepatocellular carcinoma cell tumorigenicity by suppressing cellular senescence. Cancer Sci. 2015;106:559–566. doi: 10.1111/cas.12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gollob JA, Wilhelm S, Carter C, Kelley SL. Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol. 2006;33:392–406. doi: 10.1053/j.seminoncol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt CM, McKillop IH, Cahill PA, Sitzmann JV. Increased MAPK expression and activity in primary human hepatocellular carcinoma. Biochem Biophys Res Commun. 1997;236:54–58. doi: 10.1006/bbrc.1997.6840. [DOI] [PubMed] [Google Scholar]
- 26.Tsuboi Y, Ichida T, Sugitani S, Genda T, Inayoshi J, Takamura M, Matsuda Y, Nomoto M, Aoyagi Y. Overexpression of extracellular signal-regulated protein kinase and its correlation with proliferation in human hepatocellular carcinoma. Liver Int. 2004;24:432–436. doi: 10.1111/j.1478-3231.2004.0940.x. [DOI] [PubMed] [Google Scholar]
- 27.Lee HC, Tian B, Sedivy JM, Wands JR, Kim M. Loss of Raf kinase inhibitor protein promotes cell proliferation and migration of human hepatoma cells. Gastroenterology. 2006;131:1208–1217. doi: 10.1053/j.gastro.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKillop IH, Schmidt CM, Cahill PA, Sitzmann JV. Altered expression of mitogen-activated protein kinases in a rat model of experimental hepatocellular carcinoma. Hepatology. 1997;26:1484–1491. doi: 10.1002/hep.510260615. [DOI] [PubMed] [Google Scholar]
- 29.Gailhouste L, Ezan F, Bessard A, Fremin C, Rageul J, Langouet S, Baffet G. RNAi-mediated MEK1 knock-down prevents ERK1/2 activation and abolishes human hepatocarcinoma growth in vitro and in vivo. Int J Cancer. 2010;126:1367–1377. doi: 10.1002/ijc.24950. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Pan T, Wang H, Li L, Li J, Zhang C, Yang H. Silencing of TGIF attenuates the tumorigenicity of A549 cells in vitro and in vivo. Tumour Biol. 2016;37:12725–12730. doi: 10.1007/s13277-016-5222-8. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Wang H, Gao H, Xu B, Zhai W, Li J, Zhang C. Elevated expression of TGIF is involved in lung carcinogenesis. Tumour Biol. 2015;36:9223–9231. doi: 10.1007/s13277-015-3615-8. [DOI] [PubMed] [Google Scholar]