

Abstract
The oncogene ETS-related gene (ERG) encodes a transcription factor with roles in the regulation of haematopoiesis, angiogenesis, vasculogenesis, inflammation, migration and invasion. The ERG oncogene is activated in >50% of prostate cancer cases, generally through a gene fusion with the androgen-responsive promoter of transmembrane protease serine 2. Phosphatase and tensin homologue (PTEN) is an important tumour suppressor gene that is often inactivated in cancer. ERG overexpression combined with PTEN inactivation or loss is often associated with aggressive prostate cancer. The present study aimed to determine whether or not ERG regulates PTEN transcription directly. ERG was demonstrated to bind to the PTEN promoter and repress its transcription. ERG overexpression reduced endogenous PTEN expression, whereas ERG knockdown increased PTEN expression. The ability of ERG to repress PTEN may contribute to a more cancer-permissive environment.
Keywords: ERG, TMPRSS2 fusion, prostate cancer, PTEN
Introduction
Prostate cancer is the most common form of cancer in men. It accounts for 10% of malignant tumours worldwide and 13% of male cancer deaths in the UK. It is most frequently seen in older men, with 80% of cases being diagnosed in the over 65 years (1). It is estimated that approximately 70% of men will develop some form of prostate cancer but the majority of cases will not be clinically relevant (2). Prostate cancer is not necessarily lethal; it is a multifocal and heterogeneous cancer with a wide range of outcomes. Its heterogeneity provides a real challenge for accurate prognosis and making appropriate treatment decisions (3). The difficulty lies in distinguishing between indolent and aggressive forms of the disease. The current non-invasive diagnostic test of choice is the PSA serum test. However, raised PSA levels can be due to factors other than prostate cancer such as benign prostatic hyperplasia and inflammation (4,5).
Gleason grading on histopathological examination is currently the gold standard prognostic test; however it cannot always give a correct prognosis. Inconsistencies arise due to differences in sampling procedures, and morphologically similar cancers can behave very differently. Approximately 70–80% of Gleason 6 and 20% of Gleason 7 tumours may be non-aggressive and not require intervention for 15 years or more (6); conversely, an undetected, aggressive cancer may become lethal within 2–12 years (7). Aggressive prostate cancer can be treated successfully if caught in early, organ-confined stages. Therefore it is imperative to develop a method able to discern indolent cases not requiring immediate intervention from the aggressive ones which do. Treatment can have a detrimental effect on the patients' quality of life by causing urinary and sexual dysfunction (8). Furthermore, approximately 30% of patients suffer from disease recurrence with metastases subsequent to radical prostatectomy (9).
Prostatic intraepithelial neoplasia (PIN) lesions are benign alterations thought to appear approximately 10 years before the development of prostatic carcinoma. PIN is often unidentified as it does not produce high levels of PSA and can only be detected by biopsy (3). A marker which could easily detect PIN would be extremely useful in both diagnostic and prognostic testing. Much attention has recently focused on a gene fusion, TMPRSS2:ETS-related gene (ERG), that is frequently found in aggressive prostate cancer. The TMPRRSS2 portion of the fusion contains an androgen-responsive promoter which drives the aberrant expression of the oncogenic transcription factor ERG (10). ERG is involved in homeostasis, survival, differentiation, angiogenesis and vasculogenesis (11).
Phosphatase and tensin homologue (PTEN) is one of the most studied tumour suppressor genes that influences a wide range of cellular processes including survival, proliferation, adhesion, migration, metabolism and differentiation. Loss of functional PTEN protein accelerates cancer by allowing the PI3K/AKT pathway to be constitutively switched on, promoting epithelial-mesenchymal transition (EMT) and metastasis (3,12). PTEN is lost or mutated in 50–80% of primary prostate cancer (but not in all cases). Its loss is involved in tumour initiation, is associated with highly aggressive and metastatic cancer, predicts poor clinical outcome (13), and is linked with progression to androgen-independence and biochemical recurrence (14). Tumours in which the loss of PTEN protein is observed at biopsy are more likely to have higher Gleason scores (15). Thus both ERG and PTEN are especially important players in prostate cancer; but the extent to which they might interact is not yet clear (16,17). Given the presence several potential ERG binding sites in the PTEN promoter, we sought to determine whether or not the transcription factor ERG might regulate PTEN expression directly.
Materials and methods
Cell culture
The PC3 and LNCaP prostate cancer cell lines, and the PNT2 normal prostatic epithelium cell lines were maintained in RPMI-1640 and the DU145 and VCaP prostate cancer cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco-Invitrogen, Paisley, UK). All media were supplemented with 2 mM L-glutamine + 10% (v/v) Donor Bovine Serum (Sigma-Aldrich, Poole, UK). The PC3 and VCaP cell lines were obtained from the Health Protection Agency (HPA)-European Collection of Cell Cultures (ECACC, Salisbury, UK). The DU145, LNCaP and PNT2 cell lines were kindly provided by Professor Jeff Holly's group (Department of Clinical Science at North Bristol, University of Bristol, Bristol, UK).
RNA extraction and cDNA synthesis
RNA was extracted using the Isolate RNA mini kit (BioLine, London, UK) according to the manufacturer's protocols. RNA was quantified using a Nanodrop 1000 (Thermo Fisher Scientific, Inc., Waltham, MA, USA). A total of 500 ng of RNA was reverse transcribed using the cDNA Synthesis kit (BioLine) spiked with 0.2 µg of Arabidopsis thaliana RuBisCO RNA exogenous control.
Quantitative polymerase chain reaction (qPCR)
qPCR reactions were set up using BioLine's SensiFAST SYBR Hi-ROX kit consisting of master mix, primers (0.25 nM each), cDNA (6.25 ng) and run on an ABI (Applied Biosystems; Thermo Fisher Scientific, Inc.) 7300 qPCR thermal cycler for 95°C for 10 min, followed by 95°C for 15 sec and 60°C for 1 min for 40 cycles. Primers were designed using FastPCR software (PrimerDigital Ltd., Helsinki, Finland). Data was analysed using the relative standard curve method. For each experiment a standard curve was generated for both the gene of interest and the RuBisCO control. Reverse transcribed control cDNA (including the RuBisCO spike) was serially diluted over seven points and assigned arbitrary values. These values were then converted to log base 10 and plotted against the Ct data points for the target gene or RuBisCO to generate a line equation y=mx+c. To find the relative log values (x) the following equation was used: x=Ct-c/m. The antilog was taken to reach the original relative values. To calculate the relative abundance, target gene values were normalised to their corresponding RuBisco values.
Protein extraction and quantitation
Standard RIPA cell lysis buffer (20 mM Tris-HCl pH 7.4, with 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 5 mM EDTA) plus protease inhibitors (Pierce A32953; Pierce; Thermo Fisher Scientific, Inc.) was added to the cells and left to incubate on ice for 15 min. Wells were then scraped and the lysate homogenised by aspiration using a needle and syringe. Cells were pelleted at 14,000 rpm for 10 min at 4°C in a microcentrifuge. Clarified supernatant was then transferred to a fresh tube and frozen at −80°C. Protein quantitation was performed using the Pierce BCA (bicinchoninic acid) assay and a Nanodrop 1000 (Thermo Fisher Scientific, Inc.).
Western blotting
SDS-PAGE was performed according to standard procedures (10% acrylamide gels). Immunodetection was undertaken with 30 µg of protein lysate using the chemiluminescent Luminata Forte kit (Merck Millipore, Watford, UK). The ERG primary antibody (rabbit polyclonal, ERG-1/2/3 (C-20) antibody sc-353; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) was used at a dilution of 1:500. The PTEN antibody (mouse monoclonal 26H9; Cell Signaling Technology, Inc., Danvers, MA, USA) was used at a dilution 1:1,000. The secondary horse-radish peroxidise (HRP)-conjugated anti-rabbit IgG antibody was used at a 1:6,000 dilution. For a loading control a GAPDH primary antibody [rabbit polyclonal, GAPDH (sc-25778; Santa Cruz Biotechnology, Inc.) was used at a 1:6,000 dilution. Immunoblots were developed and imaged using an Amersham Imager 600 (GE Healthcare, Buckinghamshire, UK) or LI-COR Odyssey Fc gel-doc system. Densitometric analysis of the blots was performed using ImageJ software.
Two-step chromatin immunoprecipitation
Putative ETS transcription factor binding sites within the PTEN promoter were determined by searching for GGAA or TTCC sequences within the promoter's nucleotide sequence (NCBI Accession no. AF067844.1). Primers for each gene were designed using the FastPCR programme (PrimerDigital Ltd.). Chromatin immunoprecipitation was performed on extracts derived from the ERG expressing VCaP cell line; 6×106 cells were seeded into a 100 mm tissue culture dish and left to adhere for 72 h. Chromatin immunoprecipitation was then carried out using the Champion ChIP Assay kit (SABiosciences; Qiagen, Frederick, MD, USA) following the manufacturer's recommended protocol but with an additional protein-protein cross-linking step before formaldehyde fixation. Cells were fixed in 2 mM disuccinimidyl glutarate (DSG) with 1 mM MgCl2 in PBS at room temperature for 45 min. Cells were then washed in PBS and fixed in 1% formaldehyde + 1 mM MgCl2 in PBS (pH 8) and incubated for 15 min at room temperature. Sonication was performed using a MSE Soniprep 150 set at 7 amplitude microns with 4 cycles of 15 sec on, 30 sec off. 4 µg of ERG antibody (ERG-1/2/3 (C-20): sc-353 for ChIP; Santa Cruz Biotechnology, Inc.), RNA Pol II antibody (positive control; Sigma-Aldrich) and mouse IgG (negative control; Sigma-Aldrich) were used in the immunoprecipitations. DNA was extracted from the immunoprecipitate using the GenElute Mammalian Genomic DNA Purification kit (Sigma-Aldrich) and amplified by SYBR-Green qPCR. Primers corresponding to the target promoters were as follows (all 5′ to 3′): PTEN forward tcaacggctatgtgttcacg, and reverse gtcttagcacaaagagcaacctgc (163 bp amplicon); IGFBP2 forward tgctgctactgggcgcgagt, and reverse acaagtgccctcgcccatgaccag (329 bp). Data was analysed using the % input method. Firstly, input Ct was adjusted to 100% (Ct input-6.64). Results from immunoprecipitated samples were analysed using the following calculation: 100*2^ [adjusted input-Ct (IP)]. Fold difference was calculated against the negative control (mouse IgG).
Knockdown of ERG through splice-switching oligonucleotides
Splice-switching oligonucleotides (Vivo-Morpholinos) were designed and provided by Gene Tools, LLC (Philomath, OR, USA). A standard morpholino control (5′-CCTCTTACCTCAGTTACAATTTATA-3′) and two Vivo-Morpholinos targeting the 3′ and 5′ splice sites of exon 4 of ERG pre-mRNA were used (E43′ and E45′, respectively, sequences available on request). A total of 700,000 VCaP (ATCC CRL-2876) cells were seeded into compartments of a 6-well plate and cultured at 37°C for 72 h. Media (DMEM with 10% FBS) was then replaced with fresh media containing 0.006% endoporter delivery agent (Gene Tools, LLC) and one of the following: 6 µM standard morpholino control; 6 µM E45′ Vivo-Morpholinos; 6 µM E43′ Vivo-Morpholinos. After 72 h of Morpholino treatment cells were lysed in RIPA buffer (20 mM Tris-HCl pH 7.4, with 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% deoxycholate, 5 mM EDTA) plus protease inhibitors (Pierce A32953; Pierce; Thermo Fisher Scientific, Inc.).
Over-expression of ERG
A pCMV-SPORT6 plasmid containing the full length cDNA clone of ERG variant 1 (accession no: BC040168) in DH10B TonA cells was purchased from Open Biosystems (Thermo Fisher Scientific, Inc., Ashford, UK). Cells were revived overnight in 5 ml of lysogeny broth with 100 µg/ml ampicillin (Sigma-Aldrich). Plasmids were then purified using the PureYield Plasmid Miniprep Sytem (Promega Corp., Madison, WI, USA) according to the manufacturer's instructions. The day before transfection 1.0×106 cells per well were seeded into a 6-well plate. After 24 h the cells were starved for two h in Optimem. Cells were then transfected with 2 µg pCMV-SPORT6-ERG DNA with transfection reagent (Fugene HD; Promega Corp.) added at a ratio of 1:3 (DNA: reagent). Cells were incubated for four h and then refreshed in complete media. Cells were transfected for up to 72 h, after which cells were subjected to either RNA or protein extraction.
Construction of transcription reporter plasmids
The promoter constructs for IGF1, IGFBP2 and PTEN were made as follows. DNA containing the upstream promoter sequences of IGF1, IGFBP2 and PTEN was amplified from human cheek cell genomic DNA by PCR using KOD Hot Start DNA polymerase (Novagen; Merck Biosciences, Bad Soden, Germany) supplemented with 5% (v/v) DMSO final volume. Primers were designed to introduce KpnI restriction sites at the 5′ end of each amplified fragment. The PCR product was cut with KpnI/NcoI, and ligated into KpnI/NcoI digested luciferase-expressing pGL3 plasmid (Promega Corp.). Plasmids were transformed into E. coli (Topo cloning kit; Invitrogen; Thermo Fisher Scientific, Inc.), amplified and purified. The promoter sequences were verified by restriction analysis and sequencing (DNA Sequencing and Services; University of Dundee, Dundee, Scotland).
Dual-luciferase transcription assay
DU145 and VCaP cells were transfected using the method previously described (overexpression of ERG). Promoter constructs were transfected at 400 ng per well in a 12-well plate along with 40 ng of pRLTk Renilla-expressing plasmid as an internal control or 400 ng promoter + 40 ng Renilla + 0–1000 ng pCMVSport6-ERG. Untransfected cells were used as a control for background fluorescence. The dual-luciferase assay kit (Promega Corp.) was used according to the manufacturer's instructions and luminescence measured using a Centro XS LB 960 Microplate Luminometer and Microwin2000 software (BERTHOLD TECHNOLOGIES GmbH & Co. KG, Bad Wildbad, Germany). Read-outs for untreated cells were subtracted from query sample readings to remove background noise. Query samples were normalised against Renilla outputs, normalised query results were compared to control samples (promoter construct only). Results were calculated as fold differences.
siRNA-mediated ERG knockdown combined with DLR assay
At 72 h before transfection VCaP (ATCC CRL-2876) cells were seeded at 1.0×106 cells per well in a 6-well plate. On the day of transfection, cells were starved in reduced-serum medium (Optimem; Invitrogen; Thermo Fisher Scientific, Inc.) for 2 h, followed by transfection with 100 nM ERG-targeting siRNA (Qiagen) or non-targeting siRNA (allstars negative control, Qiagen). Cells were incubated for four h before removal of the transfection media and replacement with complete media. Cells were transfected for up to 120 h, then subjected to dual-luciferase assays, as described above.
Results and Discussion
ERG is a member of the ETS family of transcription factors that share a DNA-binding domain termed the ETS-binding domain (EBD). The EBD is an 85 amino-acid domain that forms a winged helix-turn-helix motif that binds to DNA sequences that contain a core GGAA/T sequence (18). ERG has been shown to bind to this core sequence (19).
The cell line VCaP, established from a vertebral metastasis, retains PTEN expression and is positive for the TMPRSS2:ERG fusion (20). We first confirmed that ERG is expressed exclusively in VCaP cells and that PTEN is expressed in VCaP, DU145 and LNCaP cells (Fig. 1). We examined the PTEN promoter and identified several clusters of GGAA sequences ranging from 1,300 bp upstream to 500 downstream of the transcription start site (Fig. 2C). We performed a ChIP (chromatin immunoprecipitation) assay using extracts from VCaP cells and observed that ERG interacts with the PTEN promoter (Fig. 2). As a positive control an antibody against RNA polymerase II co-precipitated with the PTEN promoter (as PTEN is expressed in VCaP cells). We also looked at whether the ERG antibody could co-precipitate with another promoter that contains putative ETS binding sites, and examined IGFBP2. Only the PTEN promoter co-precipitated with ERG.
Figure 1.

Expression of ERG in representative prostate cancer cell lines. (A) ERG transcript levels were normalised to a spiked exogenous plant mRNA (RuBisCO). The TMPRSS2:ERG positive VCaP cell line expressed high levels of ERG. In contrast ERG mRNA was barely detected in PC3, DU145 and LNCaP cells. (B) PTEN transcript levels were similarly normalised to plant RuBisCO. PTEN mRNA was detected in all cell lines except the PTEN-/−PC3 cells. (C) ERG protein was also detected by western blotting in VCaP cells, but not in PC3 or PNT2 (normal prostate epithelium) cell lines. 20 µg total cell extract was loaded into each well. ERG, ETS-related gene; PTEN, phosphatase and tensin homologue.
Figure 2.

Chromatin immunoprecipitation of PTEN and IGFBP2 promoters with an anti-ERG antibody in the VCaP cell line. The signal was normalised to a control mouse IgG. (A) For the PTEN promoter, the RNA Pol II signal was 15.38 (±0.32; P<0.0001) relative to the control, whereas the ERG signal was 7.53 (±0.72; P<0.001). There was no significant binding of ERG to the promoter of IGFBP2 (B). (C) Putative ERG binding sites in the promoter region of human PTEN promoter. GGAA (black diamonds; +strand) and the reverse complement TTCC are shown (grey diamonds; -strand). ERG, ETS-related gene; PTEN, phosphatase and tensin homologue
Having obtained evidence that ERG interacts with the PTEN promoter, we transfected DU145 prostate cancer cells (ERG negative and PTEN positive) with a plasmid that expresses full length ERG. After 24–48 h we observed a significant increase in ERG expression; this was confirmed by qPCR and western blotting (Fig. 3). At 48 h post-transfection we observed a significant reduction in PTEN expression both at the RNA and protein level in cells transfected with the ERG expressing plasmid compared to the control. Next we determined whether the knockdown of ERG resulted in an upregulation of PTEN. To knockdown ERG in VCaP cells we used a splice switching oligonucleotide (SSO) approach. Morpholino SSOs were generated against both the 5′ and 3′ splice sites of ERG's exon 4. Transfection of the SSOs results in exon 4 skipping (data not shown). Exon 4 skipping creates a premature stop codon which leads to nonsense mediated decay and a resulting drop in ERG protein. Reduction of ERG protein achieved with the exon 4 SSOs resulted in a clear increase of PTEN protein.
Figure 3.

Effect of ERG overexpression in DU145 and knockdown in VCaP cells on PTEN expression. (A) Expression of ERG in DU145 cells was measured by qPCR, relative to 18S rRNA, 24 and 48 h post-transfection. There was no detectable expression in the control (empty vector) transfection compared to cells transfected with ERG-expressing plasmid (ERG O/E). (B) ERG overexpression caused a significant decrease in PTEN expression. (C) A reduction of PTEN expression as a result of ERG overexpression was also confirmed by western blotting. Three replicate experiments are shown; increased ERG protein levels correspond in each case to reduced PTEN levels. (D) ERG expression was knocked down in VCaP cells using splice-switching morpholino oligonucleotides (SSOs, provided by Gene Tools, LLC) directed against the 5′ and 3′ splice sites of exon 4. ERG knockdown resulted in increased endogenous PTEN expression. A control SSO did not affect ERG or PTEN expression. A total of 6 µM of morpholino was transfected in each experiment. (C and D) A total of 20 µg of protein were loaded into each track. ***P<0.001 vs. control. ERG, ETS-related gene; PTEN, phosphatase and tensin homologue.
Taken together these results clearly suggested that ERG transcriptionally represses PTEN. To obtain further evidence to confirm this we fused the PTEN promoter to a luciferase reporter plasmid for use in a dual-luciferase:Renilla (DLR) transcription assay. We co-transfected increasing amounts of the ERG expressing plasmid (0-1,000 ng) with 1 µg of the PTEN promoter:luciferase construct in DU145 cells. We observed a progressive reduction in PTEN promoter activity with increasing amounts of co-transfected ERG expressing plasmid (Fig. 4A). To extend these findings, we also transfected the PTEN promoter:luciferase construct into VCaP cells (which express both ERG and PTEN) and then knocked-down ERG in VCaP cells using two independent siRNAs, observing in each case a significant increase in the activity of the PTEN promoter (Fig. 4B).
Figure 4.
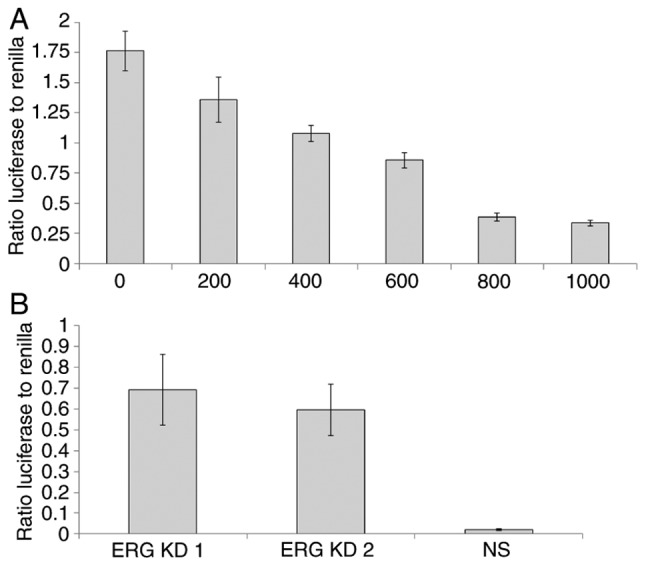
Dual-luciferase assay demonstrating transcriptional repression of PTEN by ERG. (A) ERG expressing plasmid (0-1,000 ng) was co-transfected into DU145 cells with a PTEN promoter fused to a luciferase reporter (400 ng; pGL3 vector) and a Renilla internal control (40 ng). (B) ERG was knocked down in VCaP cells using two independent siRNAs (ERG KD1 and KD2; NS, non-specific siRNA). The ratio of luciferase to Renilla signal is shown. N=3 repeats in each case. ERG, ETS-related gene; PTEN, phosphatase and tensin homologue.
The interest in ERG as a potential biomarker of prostate cancer has grown since the first report of its over-expression in clinical prostate cancer samples (10). The fusion with the TMPRSS2 promoter allows ERG expression to be driven by androgens. At the same time the literature indicates clearly that loss of expression of the PTEN tumour suppressor gene is also linked to the progression of prostate cancer. Mice that overexpress ERG and lack PTEN expression develop prostate tumours by the age of six months, further confirming the synergy between these two genetic alterations (21). PTEN deletions tend to occur after ERG activation-it has also been suggested that ERG might itself drive the development of PTEN aberrations (22). However it is also clear that the upregulation of ERG and the deletion of PTEN expression can occur independently and do not always occur in the same tumour (23).
In the current study we sought to look into the possibility that the transcription factor ERG affects the expression of PTEN directly. We present evidence that ERG represses PTEN transcription in DU145 and VCaP prostate cancer cells. By repressing the transcription of PTEN, ERG could help cancer development by activating the AKT/PI3K pathway, increasing angiogenesis, proliferation, invasion, motility and metastasis. This finding may have broader significance because ERG is not exclusively associated with prostate cancer. ERG is also implicated in leukaemia where it is linked to chemoresistance (24,25). The ability of ERG to repress PTEN transcription in leukaemia, or in other types of cancer, remains to be investigated.
ERG, like the vast majority of human genes, is alternatively spliced. We recently reported in advanced prostate cancer an increased inclusion rate of exons that encode the CAE domain in the middle of ERG (26). The CAE domain is thought to modulate ERG's transcriptional activities. Future experiments will compare the extent to which ERG splice isoforms can repress PTEN transcription and whether or not ERG's repression of PTEN plays a role in the development of aggressive prostate cancer.
In summary, the ability of ERG to repress the transcription of a critically important tumour suppressor such as PTEN further implicates ERG in carcinogenesis and underlines its clear potential as a diagnostic marker and therapeutic target.
Acknowledgements
We would like to thank Professor Jeff Holly for providing cell lines and reagents. This study was supported by a grant from the Bristol Urological Institute (no. BUI 256), the Rotary Club of Bristol, Funds for Women Graduates, and Prostate Cancer UK (no. RIA15-ST2-030).
References
- 1.Thompson JC, Wood J, Feuer D. Prostate cancer: Palliative care and pain relief. Br Med Bull. 2007;83:341–354. doi: 10.1093/bmb/ldm018. [DOI] [PubMed] [Google Scholar]
- 2.Taichman RS, Loberg RD, Mehra R, Pienta KJ. The evolving biology and treatment of prostate cancer. J Clin Invest. 2007;1179:2351–2361. doi: 10.1172/JCI31791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14:2410–2434. doi: 10.1101/gad.819500. [DOI] [PubMed] [Google Scholar]
- 4.Chu DC, Chuang CK, Liou YF, Tzou RD, Lee HC, Sun CF. The use of real-time quantitative PCR to detect circulating prostate-specific membrane antigen mRNA in patients with prostate carcinoma. Ann N Y Acad Sci. 2004;1022:157–162. doi: 10.1196/annals.1318.026. [DOI] [PubMed] [Google Scholar]
- 5.Parekh DJ, Ankerst DP, Troyer D, Srivastava S, Thompson IM. Biomarkers for prostate cancer detection. J Urol. 2007;178:2252–2259. doi: 10.1016/j.juro.2007.08.055. [DOI] [PubMed] [Google Scholar]
- 6.Kumar-Sinha C, Chinnaiyan AM. Molecular markers to identify patients at risk for recurrence after primary treatment for prostate cancer. Urology. 2003;62(Suppl 1):S19–S35. doi: 10.1016/j.urology.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Tryggvadóttir L, Vidarsdóttir L, Thorgeirsson T, Jonasson JG, Olafsdóttir EJ, Olafsdóttir GH, Rafnar T, Thorlacius S, Jonsson E, Eyfjord JE, Tulinius H. Prostate cancer progression and aurvival in BRCA2 mutation carriers. J Natl Cancer Inst. 2007;99:929–935. doi: 10.1093/jnci/djm005. [DOI] [PubMed] [Google Scholar]
- 8.Liatsikos EN, Assimakopoulos K, Stolzenburg JU. Quality of life after radical prostatectomy. Urol Int. 2008;80:226–230. doi: 10.1159/000127331. [DOI] [PubMed] [Google Scholar]
- 9.Gopalkrishnan RV, Kang DC, Fisher PB. Molecular markers and determinants of prostate cancer metastasis. J Cell Physiol. 2001;189:245–256. doi: 10.1002/jcp.10023. [DOI] [PubMed] [Google Scholar]
- 10.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 11.Birdsey GM, Dryden NH, Amsellem V, Gebhardt F, Sahnan K, Haskard DO, Dejana E, Mason JC, Randi AM. Transcription factor Erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood. 2008;111:3498–3506. doi: 10.1182/blood-2007-08-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Squire JA. TMPRSS2-ERG and PTEN loss in prostate cancer. Nat Genet. 2009;41:509–510. doi: 10.1038/ng0509-509. [DOI] [PubMed] [Google Scholar]
- 13.Mithal P, Allott E, Gerber L, Reid J, Welbourn W, Tikishvili E, Park J, Younus A, Sangale Z, Lanchbury JS, et al. PTEN loss in biopsy tissue predicts poor clinical outcomes in prostate cancer. Int J Urol. 2014;21:1209–1214. doi: 10.1111/iju.12571. [DOI] [PubMed] [Google Scholar]
- 14.Chaux A, Peskoe SB, Gonzalez-Roibon N, Schultz L, Albadine R, Hicks J, De Marzo AM, Platz EA, Netto GJ. Loss of PTEN expression is associated with increased risk of recurrence after prostatectomy for clinically localized prostate cancer. Mod Pathol. 2012;25:1543–1549. doi: 10.1038/modpathol.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lotan TL, Carvalho FL, Peskoe SB, Hicks JL, Good J, Fedor HL, Humphreys E, Han M, Platz EA, Squire JA, et al. PTEN loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy. Mod Pathol. 2015;28:128–137. doi: 10.1038/modpathol.2014.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chetram MA, Odero-Marah V, Hinton CV. Loss of PTEN permits CXCR4-mediated tumorigenesis through ERK1/2 in prostate cancer cells. Mol Cancer Res. 2011;9:90–102. doi: 10.1158/1541-7786.MCR-10-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singareddy R, Semaan L, Conley-Lacomb MK, St John J, Powell K, Iyer M, Smith D, Heilbrun LK, Shi D, Sakr W, et al. Transcriptional regulation of CXCR4 in prostate cancer: Significance of TMPRSS2-ERG fusions. Mol Cancer Res. 2013;11:1349–1361. doi: 10.1158/1541-7786.MCR-12-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shore P, Whitmarsh AJ, Bhaskaran R, Davis RJ, Waltho JP, Sharrocks AD. Determinants of DNA-binding specificity of ETS-domain transcription factors. Mol Cell Biol. 1996;16:3338–3349. doi: 10.1128/MCB.16.7.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nhili R, Peixoto P, Depauw S, Flajollet S, Dezitter X, Munde MM, Ismail MA, Kumar A, Farahat AA, Stephens CE, et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013;41:125–138. doi: 10.1093/nar/gks971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He J, Sun X, Shi T, Schepmoes AA, Fillmore TL, Petyuk VA, Xie F, Zhao R, Gritsenko MA, Yang F, et al. Antibody-independent targeted quantification of TMPRSS2-ERG fusion protein products in prostate cancer. Mol Oncol. 2014;8:1169–1180. doi: 10.1016/j.molonc.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srivastava A, Price DK, Figg WD. Prostate tumor development and androgen receptor function alterations in a new mouse model with ERG overexpression and PTEN inactivation. Cancer Biol Ther. 2014;15:1293–1295. doi: 10.4161/cbt.29694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krohn A, Freudenthaler F, Harasimowicz S, Kluth M, Fuchs S, Burkhardt L, Stahl PC, Tsourlakis M, Bauer M, Tennstedt P, et al. Heterogeneity and chronology of PTEN deletion and ERG fusion in prostate cancer. Mod Pathol. 2014;27:1612–1620. doi: 10.1038/modpathol.2014.70. [DOI] [PubMed] [Google Scholar]
- 23.Heselmeyer-Haddad KM, Garcia Berroa LY, Bradley A, Hernandez L, Hu Y, Habermann JK, Dumke C, Thorns C, Perner S, Pestova E, et al. Single-cell genetic analysis reveals insights into clonal development of prostate cancers and indicates loss of PTEN as a marker of poor prognosis. Am J Pathol. 2014;184:2671–2686. doi: 10.1016/j.ajpath.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bock J, Mochmann LH, Schlee C, Farhadi-Sartangi N, Göllner S, Müller-Tidow C, Baldus CD. ERG transcriptional networks in primary acute leukemia cells implicate a role for ERG in deregulated kinase signaling. PLoS One. 2013;8:e52872. doi: 10.1371/journal.pone.0052872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mochmann LH, Neumann M, von der Heide EK, Nowak V, Kühl AA, Ortiz-Tanchez J, Bock J, Hofmann WK, Baldus CD. ERG induces a mesenchymal-like state associated with chemoresistance in leukemia cells. Oncotarget. 2014;5:351–362. doi: 10.18632/oncotarget.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagen RM, Adamo P, Karamat S, Oxley J, Aning JJ, Gillatt D, Persad R, Ladomery MR, Rhodes A. Quantitative analysis of ERG expression and its splice isoforms in formalin-fixed, paraffin-embedded prostate cancer samples: Association with seminal vesicle invasion and biochemical recurrence. Am J Clin Pathol. 2014;142:533–540. doi: 10.1309/AJCPH88QHXARISUP. [DOI] [PubMed] [Google Scholar]