

Abstract
Objective
The objectives of the current experiments were to develop and characterize primary rat nasal epithelial (RNE) cultures and evaluate their usefulness as a model of sinonasal transepithelial transport and CFTR function.
Study Design
Laboratory in vitro and animal studies.
Methods
CFTR+/+ and CFTR−/− rat nasal septal epithelia (RNSE) were cultured on semipermeable supports at an air-liquid interface (ALI) to confluence and full differentiation. Monolayers were mounted in Ussing chambers for pharmacologic manipulation of ion transport and compared to similar filters containing murine (MNSE) and human (HSNE) epithelia. Histology and scanning electron microscopy (SEM) were completed. Real-time PCR (RT-PCR) of CFTR+/+ RNSE, MNSE, and HSNE was performed to evaluate relative CFTR gene expression.
Results
Forskolin-stimulated anion transport (ΔIsc in μA/cm2) was significantly greater in epithelia derived from CFTR+/+ when compared to CFTR−/− animals (100.9+/−3.7 vs. 10.5+/−0.9, p<0.0001). Amiloride-sensitive ISC was equivalent (−42.3 ± 2.8 vs. −46.1 ± 2.3 p=0.524). No inhibition of CFTR-mediated Cl− secretion was exhibited in CFTR−/− epithelia with the addition of the specific CFTR inhibitor, CFTRInh-172. However, calcium-activated Cl− secretion (UTP) was significantly increased in CFTR−/− RNSE (CFTR−/− −106.8 ± 1.6 vs. CFTR+/+ −32.2 ± 3.1;p<0.0001). All responses were larger in RNSE when compared to CFTR+/+ and CFTR−/− (or F508del/F508del) murine and human cells (p<0.0001). SEM demonstrated 80–90% ciliation in all RNSE cultures. There was no evidence of infection in CFTR−/− rats at 4 months. CFTR expression was similar among species.
Conclusion
The successful development of the CFTR−/− rat enables improved evaluation of CF sinus disease based on characteristic abnormalities of ion transport.
Keywords: cystic fibrosis, CFTR, ion transport, sinusitis, rat nasal culture, mucociliary clearance, chronic sinusitis, chronic rhinosinusitis, Ussing chamber, electrophysiology
INTRODUCTION
Cystic fibrosis (CF) is the most common lethal genetic disease among Caucasians, affecting 1 in 2,500–3,500 newborns annually and has a prevalence of 30,000 in the United States alone.1,2 The fatal disorder leads to widespread multi-organ pathology due to the characteristic triad of chronic stasis of inspissated mucus, microbe trapping, and persistent inflammation.3 This results in respiratory complications, gastrointestinal obstruction, exocrine pancreatic dysfunction, biliary blockage, and absence of the vas deferens in males.4 CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene on the long arm of chromosome 7, which encodes a cell surface anion channel that permits chloride (Cl−) and bicarbonate (HCO3) transportation.5 Of the CFTR mutations, F508del accounts for two-thirds and results in a three-nucleotide deletion that leads to the absence of phenylalanine at position 508.2 Consequently, this leads to production of a misfolded protein that is unable to be transported to the cell surface.1 As such, there is a resultant increase in sodium and water absorption that cause exocrine secretions to become viscous and vulnerable to infection and inflammation.6
The majority of patients with CF develop chronic rhinosinusitis (CRS) due to mucous-filled, infected sinuses, and swelling of the intranasal and sinus mucosa.7–14 Symptoms of CRS can be incapacitating and characteristically include facial pain and/or pressure, headache, nasal obstruction, congestion, and nasal discharge.15–18 Furthermore, sinus anatomy is frequently abnormal in CF patients, which additionally contributes to CF sinus disease. In fact, many present with evidence of pansinus hypoplasia or aplasia on radiographic imaging.19 While CRS in CF patients is symptomatically managed with antibiotics, topical irrigations, and surgery, no definitive cure or prevention strategies currently exist. Consequently, this often translates to a reduced quality of life (QOL) in these patients. Moreover, the study of CF-related sinus disease and its pathogenic role in the unified airway are increasingly important as CF patients continue to experience premature mortality at 33.4 years20 due to pulmonary infection, the leading cause of death in this population.21
Since researchers first linked CF to the loss of CFTR in 198922,23, a number of animal models have been studied in an attempt to replicate human pathogenesis of the disease. Historically, transgenic CF murine models have been useful for pharmaceutical testing and gene modification studies.24 While mice develop CF-related intestinal disease, they fail to reproduce many of the additional CF-related manifestations exhibited by most patients.21 Most notably, mice do not develop upper airway disease and their small size precludes thorough examination of CF-related sinus pathology. While both CF porcine25 and ferret26 models display robust respiratory phenotypes that bear a strong resemblance to that observed in humans, they are associated with a number of limitations that hinder their widespread use. In addition to high cost, for example, larger models have prolonged gestational periods, time to sexual maturation, and require specialized care.21 Not surprisingly, ethical concerns regarding studies in the neonatal period preclude the research of CF pathogenesis in human models.
The recent development of the CFTR knockout (KO) rat model (Rattus norvegicus; SD-CFTRtm1sage), generated by Sigma Advanced Genetic Engineering Labs in collaboration with researchers at the University of Alabama at Birmingham (UAB), affords several advantages in comparison to previously studied animal models.21 Unlike porcine models, rats have a brief gestational period (21–23 days) followed by swift sexual maturation (8 weeks), which facilitates rapid colony propagation, breeding studies, and longitudinal maturation studies.21 Furthermore, rats develop a considerable amount of submucosal glands throughout their airway27, which is similar to humans and is thought to represent significant underlying pathology in CF.21 Relative to their murine counterparts, CF rat models are appreciably larger, which enables the sampling of larger tissue specimens and ameliorates the mechanistic exploratory limitations of smaller models.28 Lastly, rats have been widely used for both pharmacology and toxicology studies due to their favorable pharmacokinetic and biodistribution profiles.29,30 As such, use of rats would eliminate the need for additional species by allowing safety and efficacy studies of prospective therapeutic interventions to be performed in a single species, which is useful for drug development.21
In the present study, we aim to develop rat nasal septal epithelial (RNSE) cultures and evaluate their utility as a model for investigating sinonasal transepithelial transport and CFTR function in order to better understand the pathophysiology of CF-related sinus disease.
MATERIALS AND METHODS
Acquisition of Rat Models
Institutional Animal Care and Use Committee (IACUC) approval was obtained prior to the initiation of the study. Male CFTR+/+ and CFTR−/− rats were bred in the CF Animal Core facility at the University of Alabama at Birmingham (UAB) and generated by SAGE Labs, Inc. using zinc-finger endonuclease based gene disruption on a Sprague-Dawley rat background as previously described.21 CFTR−/− rats have a 16 base-pair deletion of exon 3, which leads to loss of CFTR expression.21 Special diets and administration of laxatives were administered to CFTR−/− rat models to allow longitudinal evaluation of CF-related pathology for a period of months rather than weeks post-weaning. This curtailed prior issues regarding reduced body weight and decreased survival in CF rats, which prohibited such longitudinal studies.
Scanning Electron Microscopy (SEM)
SEM was performed on rat septal ALI monolayers after two weeks in concordance with our previously described study of porcine NSE cultures.31 Filters were rinsed with phosphate buffered saline (PBS) and fixed with Karnovsky’s fixative (2.5% glutaraldehyde, 2% paraformaldehyde in 0.2M cacodylate buffer; pH 7.4). Ethanol (up to 100%) was used to progressively dehydrate monolayers and was followed by treatment with HMDS. Monolayers were then air-dried in a fume hood overnight. Specimens were subsequently fixed to SEM stubs and mucosal surface was sputter-coated with gold palladium to a depth of 12 nm as previously described.31 An AMR-1400 (Philips Electronics, Austin, TX) scanning electron microscope set at an accelerating voltage of 20 kV was used to examine mucosal surfaces of ALI monolayers. Photomicrographs of ALI monolayers were taken at various angles and then analyzed for percentage of ciliated epithelium.
Histology of Rat Nasal Tissue
CF rats were euthanized and sinonasal cavities assessed for evidence of inflammation and infection on histology. Rat nasal septum samples were fixed in 10% formalin and blocks were subsequently decalcified, grossly sectioned, embedded in paraffin, thinly sectioned, mounted on glass sides, and stained with alcian blue periodic acid Schiff (AB-PAS) for identification of mucosubstances. Three random windows of tissue were analyzed for submucosal glands per mm basement membrane using an EVO FLC microscope at 20× magnification. ImageJ was used to analyze images.
Tissue Culture
With IACUC approval, the nasal septa of wild type (CFTR+/+) and knockout (CFTR−/−) rats were collected for this study using our previously described murine dissociation method and technique.10,32–40 Harvested tissue was subsequently transferred to 50-mL aliquots of dissociation media consisting of minimal essential medium (MEM; Invitrogen), penicillin (60 IU/mL), streptomycin (60μg/mL), 1.4 mg/mL Pronase (Roche Applied Science, Indianapolis, IN, USA), 0.1μg/mL DNase (Roche Applied Science) and then incubated in a 5% CO2 chamber at 37°C for 60 minutes previously described.31 Termination of enzymatic dissociation was achieved using 5 ml of sterile 5% fetal bovine serum (FBS; Sigma Aldrich). To facilitate epithelial cell dissociation, each tube was inverted 12 times. Tissue was subsequently removed and the remaining cell suspension was centrifuged before being re-suspended in cultured media. Cells were incubated at 37°C for 2 hours in 100 mm Primaria™ culture dishes (BD Biosciences) to remove any potential contamination by fibroblasts. Cell suspensions were then collected, centrifuged, and resuspended. A hemocytometer was used to calculate cell yield.
Once cells were collected, they were subsequently suspended with growth medium (BEGM BulletKit (CC-3171 & CC-4175) from LONZA, Cat#: CC-3170) and seeded to a density of 2×105/cm2 on semipermeable support membranes with 24-well transwell filters (Costar® Transwell® clear 24-well plate inserts, 0.4μm pore; Corning Life Sciences, Lowell, MA, USA). After 48 hours, both basal and apical culture media were replaced with differentiation media consisting of Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (DMEM/F-12, Life Technologies), 10% Nuserum (BD Biosciences), 2% fetal clone II (HyClone™), 2.5μg/mL insulin (Sigma Aldrich), 0.25% bovine brain extract (Lonza), 20nM hydrocortisone (Sigma Aldrich), 500nM triodothyronine (Sigma Aldrich), 2.5μg/mL transferrin (Life Technologies), 250nM ethanolamine (Sigma Aldrich), 1.5μM epinephrine (Sigma Aldrich), 250nM phosphoetheanolamine (Sigma Aldrich), and 10nM all trans-retinoic acid (Sigma Aldrich). After another 24 hours, apical differentiation media and non-adherent cellular debris were removed and discarded. Basal differentiation media was discarded and replaced every 48 hours.
Primary sinonasal epithelia from F508del/F508del humans and CFTR−/− mice were additionally utilized for this study and cultured at an air-liquid interface according to previously established protocols.32–34
Bioelectric Measurements
Solutions and chemicals
All chemicals were obtained from Sigma Aldrich. The contents of the bath solution consisted of (in mM): 120 NaCl, 25 NaHCO3, 3.3 KH2PO4, 0.8 K2HPO4, 1.2 MgCl2, 1.2 CaCl2, and 10 glucose, with a pH of 7.3–7.4 in a bubbled mixture of 95% O2:5% CO2 at 37°C. All studies were conducted in low Cl− (6 mM) mucosal baths. Pharmacologic manipulations were prepared with the following: amiloride (100 μM), forskolin (20μM), CFTRINH-172 (10μM), and UTP (150 μM) as previously described.31 Amiloride was administered to inhibit epithelial sodium channels (ENaC) in order to assure that any changes in short-circuit current (ISC) are independent of effects upon ENaC activity. Forskolin is an indirect CFTR activator that acts via a cAMP-dependent pathway while CFTRInh-172 is a highly specific inhibitor of CFTR that blocks CFTR dependent chloride current. Treatment with UTP results in stimulation of calcium-activated chloride channels (i.e. TMEM16A21), increased cytosolic Ca2+, and promotes calcium-mediated ciliary beat frequency (CBF).
Short circuit current and conductance measurements
Transwell inserts were mounted in Ussing chambers for ion transport monitoring and pharmacologic blockade as previously described.31 Cell monolayers were evaluated under short-circuit conditions after compensation of fluid resistance with automatic VCC 600 voltage clamps (Physiologic Instruments, San Diego, CA, USA). Inserts were then fixed in the bath solution bubbled with 95% O2:5% CO2 at 37°C. Short-circuit measurements were taken at 1 sample/second with the assumption that positive deflections represent a net movement of the anion from the serosal to the mucosal surface.
CFTR Gene Expression
Total CFTR RNA was isolated from CFTR+/+ rat, human (HSNE), and murine nasal septal epithelium (MNSE) with RNeasy mini kit (Qiagen, Valencia, Ca) per manufacturer’s instructions, and quantitative real-time polymerase chain reaction (RT-PCR) was performed as previously described.11
Statistical Analysis
Statistical analysis was performed using two-tailed, unpaired t-tests for all Ussing chamber studies of CFTR+/+ and CFTR−/− RNSE cultures and tissue. When comparing Ussing chamber studies and CFTR expression among the different species, statistical analysis was performed using one-way ANOVA. A post-hoc Tukey’s Honest Significant Difference (HSD) test was conducted in instances where group means exhibited significant difference. For all calculations, a p-value < 0.05 was considered statistically significant. Values are reported as mean ± standard error of the mean.
RESULTS
Ciliary differentiation visualized with SEM
Primary RNSE cultured ALI monolayers demonstrated confluency and complete differentiation by day 14 with a resistance of >350 Ω/cm2. At 14 days, scanning electron micrographs of RNSE cultured ALI monolayers exhibited 80 to 90% ciliation throughout the apical monolayer surface (figure 1).32
Figure 1.
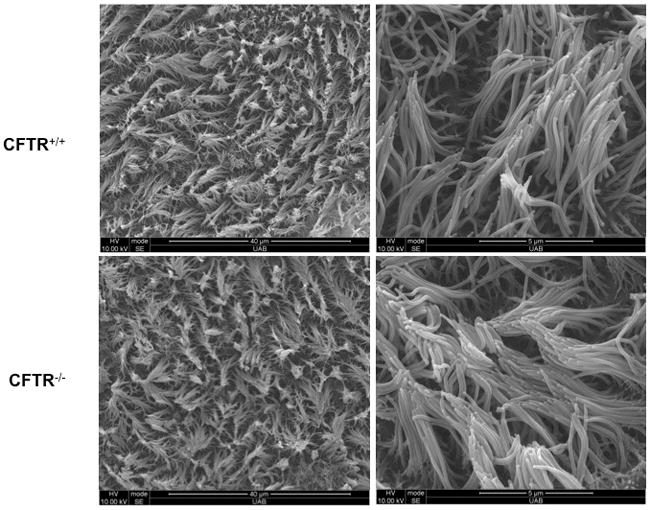
CFTR+/+ and CFTR −/− rat nasal epithelia at high (25×) and low (5×) magnification are well differentiated when cultured at an air-liquid interface. Polarized epithelia are highly differentiated with widespread ciliogenesis. Robust ciliary beating with 80 to 90% ciliation was identified in all cultures.
CF sinus epithelia demonstrated normal histology
AB-PAS staining of 5 CFTR+/+ and 4 CFTR−/− rat maxillary sinonasal epithelia were examined for submucosal gland density, measured as submucosal gland per mm basement membrane (figure 2). CFTR+/+ rats exhibited an average of 11 ± 0.9 submucosal glands per mm while CFTR−/− exhibited 12.8 ± 1.0 submucosal glands per mm (p=0.25). Although the average number of submucosal glands was increased in the CFTR−/− rats, both groups exhibited normal submucosal gland morphology with no evidence of distention or plugging.
Figure 2.
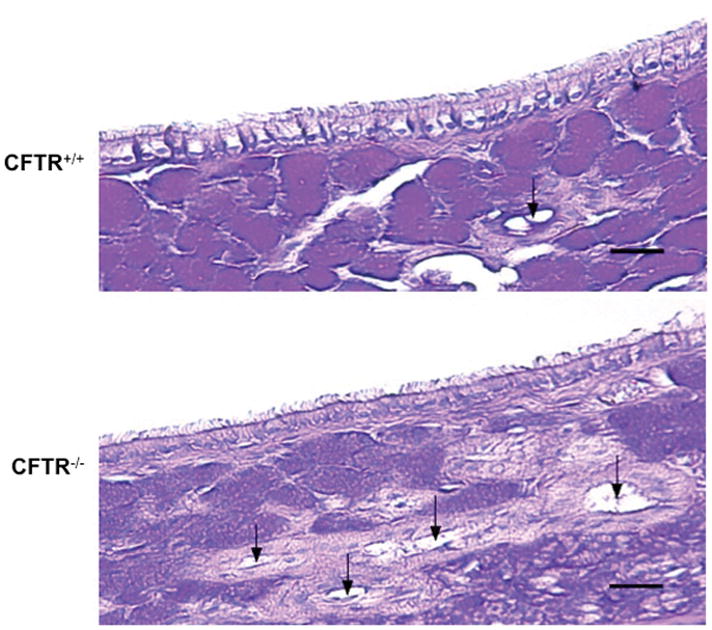
AB-PAS staining of CFTR+/+ and CFTR−/− rat maxillary sinonasal epithelia was examined for submucosal gland density at 25× magnification and measured as submucosal gland per mm basement membrane. CFTR−/− rats demonstrate higher density of glands, which areindicated by the black arrows.
Ion transport phenotype of CFTR+/+ and CFTR−/− rat nasal septal tissue
Ussing chamber analysis of nasal septal tissue was performed on both wild type (CFTR+/+, n=3) and knockout (CFTR−/−, n=3) rats after pharmacologic manipulation with amiloride, forskolin, CFTRInh-172, and UTP (figure 3). Amiloride-sensitive ion transport (ΔIsc in μA/cm2) was significantly more reduced in nasal septal tissue of CFTR−/− rats vs CFTR+/+ (−207.0 ± 28.3 [CFTR−/−] vs −56.1 ± 19.4 [CFTR+/+], p=0.0098). Forskolin-stimulated ISC was significantly greater in rat nasal septal tissue derived from the CFTR+/+ when compared to CFTR−/− (1143.1 ± 59.9 [CFTR+/+] vs 298.4 ± 119.0 [CFTR−/−], p=0.002). Addition of the CFTR-specific inhibitor, CFTRInh-172, led to inhibition of CFTR-mediated Cl− secretion in both CFTR+/+ and CFTR−/− rats. However, this was not statistically significant (−58.1 ± 41.8 [CFTR+/+] vs −21.4 ± 1.5 [CFTR−/−], p=0.22). Calcium-activated Cl− secretion with UTP was increased in both the CFTR+/+ and CFTR−/− rats; however, this was not significant (259.0 ± 68.3 [CFTR+/+] vs 638.0 ± 251.5 [CFTR−/−], p=0.714).
Figure 3.
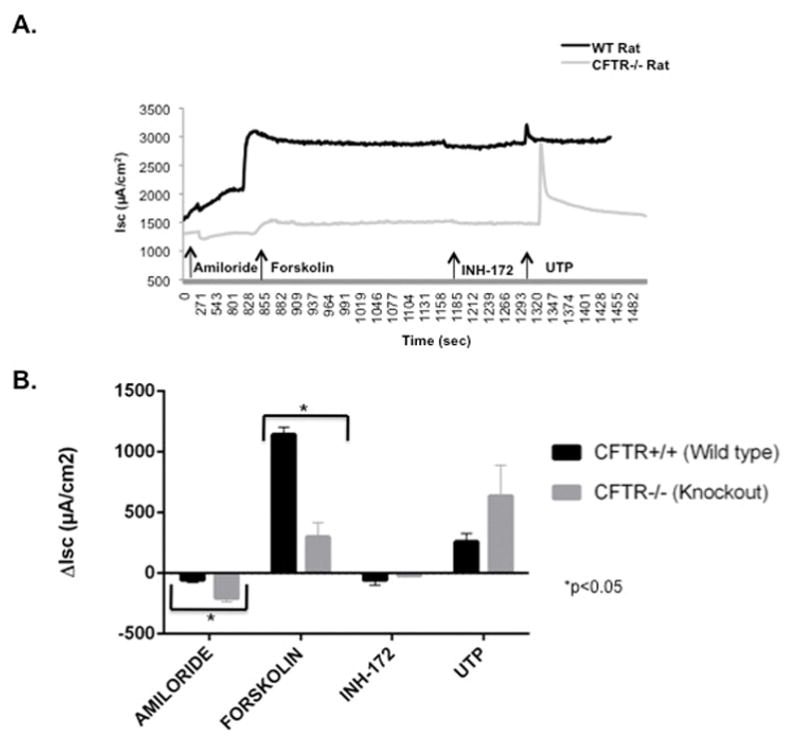
(A) Representative Ussing chamber current tracings and (B) summary of current measurements with SEM from CFTR+/+ and CFTR−/− rat nasal septal epithelial tissues following administration of amiloride, forskolin, CFTRInh-172, and UTP.
Ion transport phenotype of CFTR+/+ and CFTR−/− RNSE cultures (figure 4)
Figure 4.
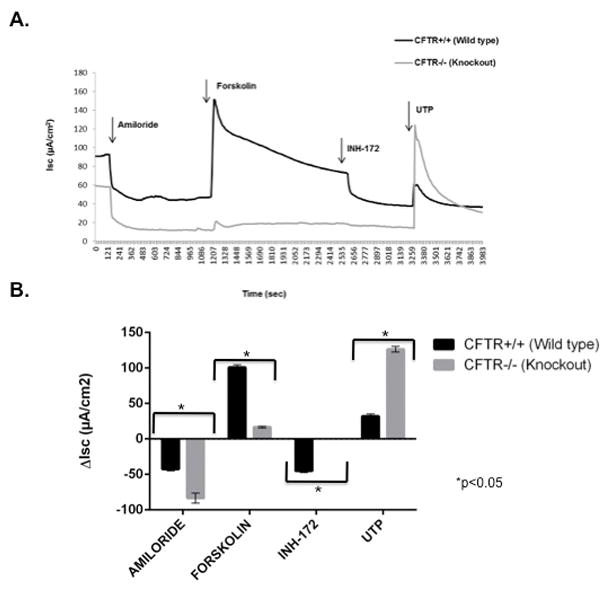
(A) Representative Ussing chamber current tracings and (B) summary of current measurements with SEM from CFTR+/+ and CFTR−/− rat nasal septal epithelial cultures following administration of amiloride, forskolin, CFTRInh-172, and UTP.
Ussing chamber analysis of NSE cultures was performed on both CFTR+/+ (n=12) and CFTR−/− (n=12) rats after pharmacologic manipulation with amiloride, forskolin, CFTRInh-172, and UTP. Similar to rat nasal septal tissue, amiloride-sensitive ISC was significantly enhanced in CFTR−/− RNSE cultures, indicating increased Na+ absorption in CFTR deficient states (−84.8 ± 6.9 vs −42.3 ± 2.8 [CFTR+/+] ; p<0.0001). Forskolin-stimulated anion transport was significantly greater in RNSE derived from CFTR+/+ when compared to CFTR−/− rats (100.9 ± 3.7 vs. 16.6 ± 1.2, p<0.0001). CFTR−/− epithelia demonstrated no change in baseline ISC with the addition of the CFTR-specific inhibitor, INH-172 (0 [CFTR−/−] vs. −45.1 ± 2.2 [CFTR+/+], p<0.0001). However, calcium-activated Cl− secretion with UTP was significantly increased in CFTR−/− RNSE (126.5 ± 3.9 vs. 32.2 ± 3.1 [CFTR+/+]; p<0.0001) and suggests that there is a compensatory increase of calcium-activated Cl− transport in CFTR deficient states, which is similar to that seen in mice.41
CFTR+/+ and CFTR−/− RNSE cultures have increased ion transport phenotype when compared to mice and humans
With the exception of UTP-stimulated ISC, both wild type (CFTR+/+) and knockout (CFTR−/−) nasal septal epithelial (NSE) cultures exhibited pharmacologic responses that were significantly larger than those observed in mice and humans (F508del/F508del).
Ussing chamber analysis was performed on NSE cultures derived from CFTR+/+ rats (n=12), mice (n=12), and humans (n=12) after pharmacologic manipultion with amiloride, forskolin, CFTRInh-172, and UTP (figure 5). Amiloride-sensitive ISC was significantly greater in RNSE (−42.3 ± 2.8 vs. −10.2 ± 0.9 [MNSE] vs. −8.1 ± 1.1 [HSNE], p<0.0001). Forskolin-stimulated ISC was also markedly larger in RNSE when compared to MNSE and HSNE (100.9 ± 3.7 vs. 27.9 ± 2.8 [MNSE] vs. 28.8 ± 1.2 [HSNE], p<0.0001). INH-172 blockade was also more robust in RNSE vs MNSE and HSNE (−45.1 ± 2.2 vs. −24.5 ± 2.3 [MNSE] vs. −29.5 ± 1.4 [HSNE], p<0.0001). Calcium-activated Cl− secretion with UTP-stimulated ISC was most robust in MNSE (42.8 ± 1.7 vs. 32.2 ± 3.1 [RNSE] vs. 23.6 ± 1.1 [HSNE], p<0.0001). Tukey post-hoc analysis indicated RNSE Isc was significantly different from MNSE and HSNE under all pharmacologic conditions (p<0.05).
Figure 5.
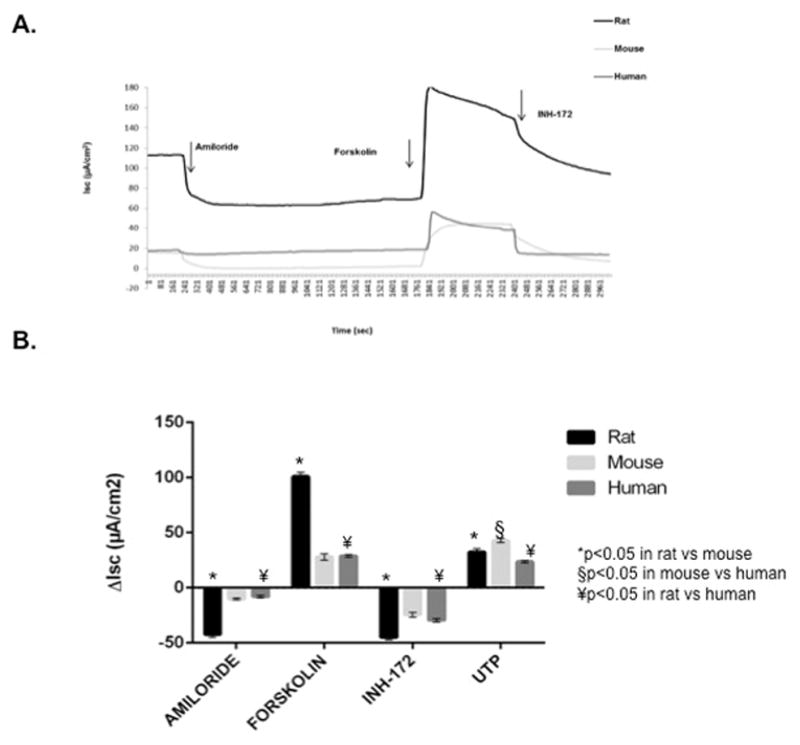
CFTR+/+ species comparison of ion transport phenotypes with (A) representative Ussing chamber current tracings and (B) summary of current measurements with SEM from CFTR+/+ rat, mouse, and human nasal septal epithelial cultures following administration of amiloride, forskolin, CFTRInh-172, and UTP.
Ussing chamber analysis was also completed on NSE cultures from CFTR−/− rats (n=12), mice (n=12), and humans (n=12) after pharmacologic manipulation with amiloride, forskolin, and UTP (figure 6). All responses were markedly greater in RNSE when compared to mice or humans. Amiloride-sensitive ISC (−83.4 ± 7.0 vs −21.1 ± 2.4 [MNSE] vs −2.4 ± 0.6 [HSNE], p<0.0001), forskolin-stimulated ISC (16.7 ± 1.2 vs 3.1 ± 1.0 [MNSE] vs 1.4 ± 0.3 [HSNE], p<0.001), and calcium-activated Cl− secretion with UTP (126.4 ± 4.1 vs 52.7 ± 3.9 [MNSE] vs 11.9 ± 2.4 [HSNE], p<0.0001) were all significantly more robust in RNSE cultures. Post-hoc Tukey’s HSD tests revealed amiloride, forskolin, and UTP-sensitive ISC were significantly different for all group comparisons (p<0.05) except for forskolin-stimulated ISC in MNSE vs HSNE.
Figure 6.
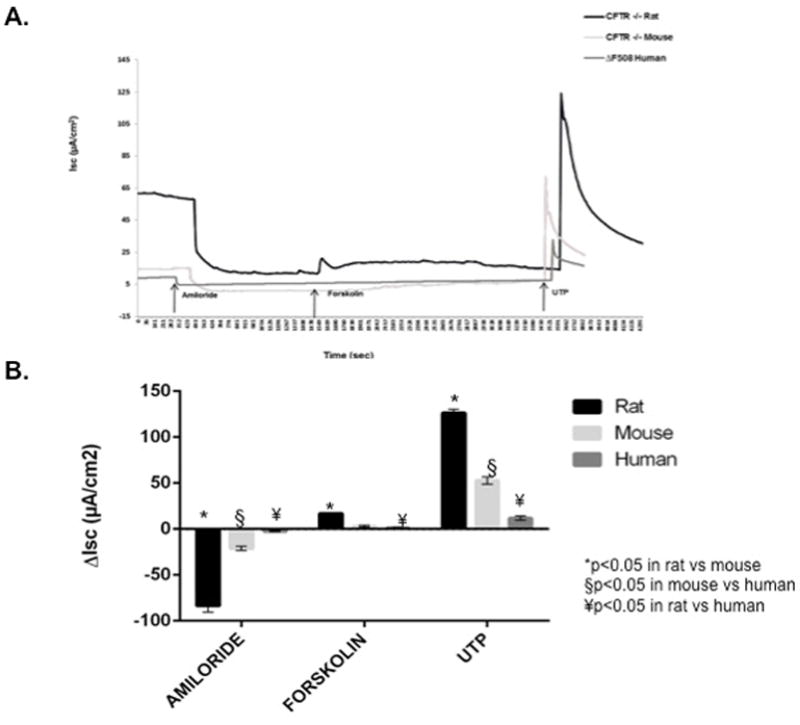
CFTR−/− species comparison of ion transport phenotypes with (A) representative Ussing chamber current tracings and (B) summary of current measurements with SEM from CFTR−/− rat, mouse, and human nasal septal epithelial cultures following administration of amiloride, forskolin, and UTP.
CFTR expression in wild type rats is comparable to that seen in mice and humans
PCR was performed to evaluate the relative amounts of CFTR mRNA expression in CFTR+/+ NSE cultures derived from CFTR+/+ rats (n=3), mice (n=3), and humans (n=3). CFTR expression was comparable between all 3 groups (97.0+/−3.0 [RNSE] vs 129.9+/−22.0 [MNSE] vs 91.1+/−30.0 [HSNE], p=0.376) (figure 7).
Figure 7.

CFTR+/+ species comparison demonstrating relative amount of CFTR expression in rat, mouse, and human.
DISCUSSION
The use of various animal models has been paramount in advancing our understanding of the pathophysiology of cystic fibrosis. Additionally, they facilitate the development of novel drug therapies to improve CF treatments. In the past, murine models have often dominated CF animal studies, as CFTR−/− mice are readily available for both in vivo and in vitro studies. While they do exhibit CF gastrointestinal pathology, mice fail to reproduce many of the characteristic traits observed in humans.23 As such, researchers have developed a number of animal models in an effort to more accurately imitate the wide range of multi-organ pathology seen in humans with CF.
Our current study aims to characterize RNSE cultures that were developed at an air-liquid interface (ALI) for the purpose of studying CF sinusitis. We found that the overall phenotype of ion transport in RNSE cell cultures is quite comparable to electrically robust, mucociliary epithelium of the sinonasal airways and exhibits Cl− -secretory and Na+-absorptive pathways. These findings are valuable for allowing future in vitro studies of CF airway physiology, particularly for the purpose of pharmacotherapeutic testing.
When comparing RNSE cultures and tissue, we found that cultures demonstrated a more vigorous ion transport phenotype as compared to tissue. Unlike CFTR−/− rat NSE tissue, RNSE cultures were found to have statistically significant CFTRInh-172 and UTP-stimulated ΔISC. However, both CFTR−/− cultures and tissue demonstrated a statistically significant reduction in forskolin-stimulated ΔISC. In contrast to previous CFTR−/− rat studies of tracheal short-circuit currents21, we found that forskolin-stimulated anion transport was significantly greater in both RNSE cultures and tissues derived from the WT when compared to KO rats. This is a relatively expected finding since forskolin enhances the activation of CFTR, which is almost negligible in the CFTR−/− rat. Whether the previous postulation by Tuggle et al. that CFTR in WT rats is constitutively active and thus less susceptible to forskolin-stimulated activation remains unclear.21 However, there appears to be an observable difference between tracheal and RNSE ISC measurements. Interestingly, we found that RNSE tissues were more sensitive to amiloride-stimulated ISC than RNSE cultures, which may indicate that tissues exhibit higher levels of Na+ transport at baseline. Nevertheless, we found that RNSE cultures are preferable over RNSE tissues for conducting ion transport studies with pharmacologic manipulation as changes in ISC appear to be more appreciable when directly comparing CFTR+/+ and CFTR−/− phenotypes.
Similar to what has been demonstrated in murine and prior rat CFTR−/− models, all RNSE preparations exhibited robust calcium-activated Cl− secretion as measured by UTP-stimulated ISC, which is also consistent with what is observed in humans.41,42 However, this is in stark contrast to what we found in our previous study of porcine NSE cultures, which demonstrated a markedly diminished calcium-activated Cl− secretion as measured by activation of UTP.31 Unlike the CFTR−/− porcine model31, rats do not appear to develop spontaneous sinus disease. In fact, the development of spontaneous sinus disease in CF pigs may be due, in part, to certain phenotypic differences when compared to CF rats. For example, pigs do not express abundant alternative Cl− channels as seen rats.43 Abundant calcium-activated Cl− secretion could provide alternative pathways that result in a normal CF rat sinus phenotype. This suggests that, similar to murine models, rats do not develop observable manifestations of CF-sinus disease in vivo. Thus, rats too are likely to be limited in their translational utility. Still, histopathologic evaluation of maxillary sinus tissue in WT and KO rats exhibited some observable differences, with CFTR−/− rats demonstrating a greater amount of submucosal glands per millimeter basement membrane. Although the mechanism for development of sinus disease is largely unknown, we previously attributed it to reduced alternative Cl− transport pathways in our study of porcine CFTR−/− models.31 We speculated that the absence or non-functionality of vigorous alternative pathways play a role in development of sinus disease in pigs. This is in contrast to what we observe in the current study, which demonstrates a strong UTP-mediated ISC in both rats and mice and suggests that alternative Cl− pathways are present, functional, and potentially serve to ameliorate Cl− ion imbalance in these models. While these conjectures require further evaluation, there are likely several factors involved in the development of CF sinonasal disease.
Overall, when compared to mice and humans, RNSE cultures demonstrate similar CFTR gene expression and nasal electrophysiological profiles but a significantly more robust ion transport phenotype. These findings suggest that RNSE cultures are more responsive and susceptible to ion transport modulators than their human and murine counterparts. This further supports our conclusions that RNSE cultures represent a good model for in vitro pharmacologic manipulation studies. The phenotype exhibited by RNSE in both CFTR+/+ and CFTR−/− rats suggests they may be a better model for in vitro ion transport studies when compared to mice.
CONCLUSIONS
While CFTR−/− rats do not appear to develop spontaneous sinus disease, our study characterizing NSE cultures in WT and KO rats demonstrated several findings to suggest that the rat is a useful model for in vitro ion transport studies. RNSE cultures could represent a physiologically applicable means for studying CF-related sinonasal disease and provide a useful model for pharmacologic studies in CF by providing a relevant comparison to in vivo rat nasal potential difference measurements. Furthermore, future studies attempting to induce sinus infection in the CF rat are planned.
Acknowledgments
Funding Support: This work was supported by National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (1 R01 HL133006-01) and National Institute of Diabetes and Digestive and Kidney Diseases (5P30DK072482-04, CF Research Center Pilot Award) to B.A.W. and National Institutes of Health (T32CA091078) to K.E.T.
Footnotes
Disclosures: All authors have read and approved the manuscript. Dr. Bradford A. Woodworth is a consultant for Cook Medical, Olympus, and Smith and Nephew. An oral presentation of this work was presented at the Triological Society Combined Sections Meeting on January 19, 2017 in New Orleans, Louisiana; manuscript submission number #185.
References
- 1.Grosse SD, Boyle CA, Botkin JR, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep. 2004;53:1–36. [PubMed] [Google Scholar]
- 2.Chang EH. New insights into the pathogenesis of cystic fibrosis sinusitis. Int Forum Allergy Rhinol. 2014;4:132–137. doi: 10.1002/alr.21252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavelle GM, White MM, Browne N, McElvaney NG, Reeves EP. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. Biomed Res Int. 2016;2016:5258727. doi: 10.1155/2016/5258727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derichs N. Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev. 2013;22:58–65. doi: 10.1183/09059180.00008412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knowles M, Gatzy J, Boucher R. Relative ion permeability of normal and cystic fibrosis nasal epithelium. J Clin Invest. 1983;71:1410–1417. doi: 10.1172/JCI110894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doull IJ. Recent advances in cystic fibrosis. Arch Dis Child. 2001;85:62–66. doi: 10.1136/adc.85.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaaban MR, Kejner A, Rowe SM, Woodworth BA. Cystic fibrosis chronic rhinosinusitis: a comprehensive review. Am J Rhinol Allergy. 2013;27:387–395. doi: 10.2500/ajra.2013.27.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Illing EA, Woodworth BA. Management of the upper airway in cystic fibrosis. Curr Opin Pulm Med. 2014;20:623–631. doi: 10.1097/MCP.0000000000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodworth BA, Ahn C, Flume PA, Schlosser RJ. The delta F508 mutation in cystic fibrosis and impact on sinus development. Am J Rhinol. 2007;21:122–127. doi: 10.2500/ajr.2007.21.2905. [DOI] [PubMed] [Google Scholar]
- 10.Woodworth BA. Resveratrol ameliorates abnormalities of fluid and electrolyte secretion in a hypoxia-Induced model of acquired CFTR deficiency. Laryngoscope. 2015;125(Suppl 7):S1–S13. doi: 10.1002/lary.25335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, Blount AC, McNicholas CM, et al. Resveratrol enhances airway surface liquid depth in sinonasal epithelium by increasing cystic fibrosis transmembrane conductance regulator open probability. PloS one. 2013;8:e81589. doi: 10.1371/journal.pone.0081589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azbell C, Zhang S, Skinner D, Fortenberry J, Sorscher EJ, Woodworth BA. Hesperidin stimulates cystic fibrosis transmembrane conductance regulator-mediated chloride secretion and ciliary beat frequency in sinonasal epithelium. Otolaryngol Head Neck Surg. 2010;143:397–404. doi: 10.1016/j.otohns.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Illing EA, Cho do Y, Riley KO, Woodworth BA. Draf III mucosal graft technique: long-term results. Int Forum Allergy Rhinol. 2016;6:514–517. doi: 10.1002/alr.21708. [DOI] [PubMed] [Google Scholar]
- 14.Conger BT, Jr, Riley K, Woodworth BA. The Draf III mucosal grafting technique: a prospective study. Otolaryngol Head Neck Surg. 2012;146:664–668. doi: 10.1177/0194599811432423. [DOI] [PubMed] [Google Scholar]
- 15.Cepero R, Smith RJ, Catlin FI, Bressler KL, Furuta GT, Shandera KC. Cystic fibrosis--an otolaryngologic perspective. Otolaryngol Head Neck Surg. 1987;97:356–360. doi: 10.1177/019459988709700403. [DOI] [PubMed] [Google Scholar]
- 16.Cuyler JP, Monaghan AJ. Cystic fibrosis and sinusitis. J Otolaryngol. 1989;18:173–175. [PubMed] [Google Scholar]
- 17.Duplechain JK, White JA, Miller RH. Pediatric sinusitis. The role of endoscopic sinus surgery in cystic fibrosis and other forms of sinonasal disease. Archives of otolaryngology--head & neck surgery. 1991;117:422–426. doi: 10.1001/archotol.1991.01870160076013. [DOI] [PubMed] [Google Scholar]
- 18.Ramsey B, Richardson MA. Impact of sinusitis in cystic fibrosis. The Journal of allergy and clinical immunology. 1992;90:547–552. doi: 10.1016/0091-6749(92)90183-3. [DOI] [PubMed] [Google Scholar]
- 19.Eggesbo HB, Sovik S, Dolvik S, Eiklid K, Kolmannskog F. CT characterization of developmental variations of the paranasal sinuses in cystic fibrosis. Acta Radiol. 2001;42:482–493. doi: 10.1080/028418501127347214. [DOI] [PubMed] [Google Scholar]
- 20.2016. CFNTCfshccc-f-sAA.
- 21.Tuggle KL, Birket SE, Cui X, et al. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PloS one. 2014;9:e91253. doi: 10.1371/journal.pone.0091253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 23.Guilbault C, Saeed Z, Downey GP, Radzioch D. Cystic fibrosis mouse models. American journal of respiratory cell and molecular biology. 2007;36:1–7. doi: 10.1165/rcmb.2006-0184TR. [DOI] [PubMed] [Google Scholar]
- 24.Carvalho-Oliveira I, Scholte BJ, Penque D. What have we learned from mouse models for cystic fibrosis? Expert review of molecular diagnostics. 2007;7:407–417. doi: 10.1586/14737159.7.4.407. [DOI] [PubMed] [Google Scholar]
- 25.Rogers CS, Hao Y, Rokhlina T, et al. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. 2008;118:1571–1577. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun X, Sui H, Fisher JT, et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. 2010;120:3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smolich JJ, Stratford BF, Maloney JE, Ritchie BC. New features in the development of the submucosal gland of the respiratory tract. Journal of anatomy. 1978;127:223–238. [PMC free article] [PubMed] [Google Scholar]
- 28.Mashimo T, Serikawa T. Rat resources in biomedical research. Current pharmaceutical biotechnology. 2009;10:214–220. doi: 10.2174/138920109787315105. [DOI] [PubMed] [Google Scholar]
- 29.Aitman TJ, Critser JK, Cuppen E, et al. Progress and prospects in rat genetics: a community view. Nature genetics. 2008;40:516–522. doi: 10.1038/ng.147. [DOI] [PubMed] [Google Scholar]
- 30.Jacob HJ. Functional genomics and rat models. Genome research. 1999;9:1013–1016. doi: 10.1101/gr.9.11.1013. [DOI] [PubMed] [Google Scholar]
- 31.Dean N, Ranganath NK, Jones B, et al. Porcine nasal epithelial cultures for studies of cystic fibrosis sinusitis. Int Forum Allergy Rhinol. 2014;4:565–570. doi: 10.1002/alr.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodworth BA, Antunes MB, Bhargave G, Palmer JN, Cohen NA. Murine tracheal and nasal septal epithelium for air-liquid interface cultures: a comparative study. Am J Rhinol. 2007;21:533–537. doi: 10.2500/ajr.2007.21.3068. [DOI] [PubMed] [Google Scholar]
- 33.Antunes MB, Woodworth BA, Bhargave G, et al. Murine nasal septa for respiratory epithelial air-liquid interface cultures. Biotechniques. 2007;43:195–196. doi: 10.2144/000112531. 198, 200 passim. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, Fortenberry JA, Cohen NA, Sorscher EJ, Woodworth BA. Comparison of vectorial ion transport in primary murine airway and human sinonasal air-liquid interface cultures, models for studies of cystic fibrosis, and other airway diseases. Am J Rhinol Allergy. 2009;23:149–152. doi: 10.2500/ajra.2009.23.3285. [DOI] [PubMed] [Google Scholar]
- 35.Cho DY, Skinner D, Zhang S, et al. Cystic fibrosis transmembrane conductance regulator activation by the solvent ethanol: implications for topical drug delivery. Int Forum Allergy Rhinol. 2016;6:178–184. doi: 10.1002/alr.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Illing EA, Cho DY, Zhang S, et al. Chlorogenic Acid Activates CFTR-Mediated Cl- Secretion in Mice and Humans: Therapeutic Implications for Chronic Rhinosinusitis. Otolaryngol Head Neck Surg. 2015;153:291–297. doi: 10.1177/0194599815586720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang S, Skinner D, Hicks SB, et al. Sinupret activates CFTR and TMEM16A-dependent transepithelial chloride transport and improves indicators of mucociliary clearance. PloS one. 2014;9:e104090. doi: 10.1371/journal.pone.0104090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lazrak A, Jurkuvenaite A, Ness EC, et al. Inter-alpha-inhibitor blocks epithelial sodium channel activation and decreases nasal potential differences in DeltaF508 mice. American journal of respiratory cell and molecular biology. 2014;50:953–962. doi: 10.1165/rcmb.2013-0215OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S, Smith N, Schuster D, et al. Quercetin increases cystic fibrosis transmembrane conductance regulator-mediated chloride transport and ciliary beat frequency: therapeutic implications for chronic rhinosinusitis. Am J Rhinol Allergy. 2011;25:307–312. doi: 10.2500/ajra.2011.25.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blount A, Zhang S, Chestnut M, et al. Transepithelial ion transport is suppressed in hypoxic sinonasal epithelium. Laryngoscope. 2011;121:1929–1934. doi: 10.1002/lary.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clarke LL, Boucher RC. Chloride secretory response to extracellular ATP in human normal and cystic fibrosis nasal epithelia. Am J Physiol. 1992;263:C348–356. doi: 10.1152/ajpcell.1992.263.2.C348. [DOI] [PubMed] [Google Scholar]
- 42.Clarke LL, Grubb BR, Yankaskas JR, Cotton CU, McKenzie A, Boucher RC. Relationship of a non-cystic fibrosis transmembrane conductance regulator-mediated chloride conductance to organ-level disease in Cftr(−/−) mice. Proc Natl Acad Sci U S A. 1994;91:479–483. doi: 10.1073/pnas.91.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang EH, Pezzulo AA, Meyerholz DK, et al. Sinus hypoplasia precedes sinus infection in a porcine model of cystic fibrosis. Laryngoscope. 2012;122:1898–1905. doi: 10.1002/lary.23392. [DOI] [PMC free article] [PubMed] [Google Scholar]