

ABSTRACT
New chemotherapeutic agents with novel mechanisms of action are urgently required to combat the challenge imposed by the emergence of drug-resistant mycobacteria. In this study, a phenotypic whole-cell screen identified 5-nitro-1,10-phenanthroline (5NP) as a lead compound. 5NP-resistant isolates harbored mutations that were mapped to fbiB and were also resistant to the bicyclic nitroimidazole PA-824. Mechanistic studies confirmed that 5NP is activated in an F420-dependent manner, resulting in the formation of 1,10-phenanthroline and 1,10-phenanthrolin-5-amine as major metabolites in bacteria. Interestingly, 5NP also killed naturally resistant intracellular bacteria by inducing autophagy in macrophages. Structure-activity relationship studies revealed the essentiality of the nitro group for in vitro activity, and an analog, 3-methyl-6-nitro-1,10-phenanthroline, that had improved in vitro activity and in vivo efficacy in mice compared with that of 5NP was designed. These findings demonstrate that, in addition to a direct mechanism of action against Mycobacterium tuberculosis, 5NP also modulates the host machinery to kill intracellular pathogens.
KEYWORDS: 5-nitro-1,10-phenanthroline; autophagy; F420 dependence; Mycobacterium tuberculosis; phenotypic screening
INTRODUCTION
Tuberculosis (TB) continues to be one of the major causes of unnatural morbidity and mortality affecting the world today (1). The outcome of TB treatment is hampered due to the ability of Mycobacterium tuberculosis to switch to a phenotypically drug-tolerant dormant state (2). This situation is further aggravated due to the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) M. tuberculosis strains (3, 4). Approximately, 5% of TB cases across the globe are estimated to be MDR-TB, which includes 5% of newly diagnosed TB cases and 20.5% of previously treated TB cases. Recently, a few reports have also suggested the emergence of totally drug-resistant (TDR) M. tuberculosis strains, infection with which results in a very limited chance of successful treatment (5, 6). The current WHO-recommended regimens for the treatment of TB caused by drug-susceptible and -resistant bacteria last 6 months and 18 to 24 months, respectively. These lengthy durations of treatment are probably required due to the inherently low bacterial growth rates and the poor penetration of drugs into TB lesions. In order to tackle this situation, there is an urgent need to develop a regimen that (i) includes drugs with a novel mechanism of action, (ii) targets bacteria in different metabolic states, (iii) is able to shorten the duration of treatment, and (iv) is effective against MDR-TB and XDR-TB.
Phenotypic or target-based screening has led to the identification of various scaffolds that possess activity against drug-resistant M. tuberculosis (7). Target-based platforms rely on protein essentiality in vivo; however, the translation of in vitro potency into whole-cell activity has been a major challenge. Unlike target-based screens, cell-based screens result in the identification of scaffolds that are able to penetrate the cell membrane and inhibit their intracellular target. Phenotypic screening in association with the use of a next-generation sequencing platform has led to the identification of various prodrugs and scaffolds with novel mechanisms of action. FDA has approved treatment with bedaquiline (TMC-207) and delamanid (OPC-67683) for individuals with MDR-TB (8–10). TMC-207 inhibits the ATP synthase enzyme and is active against both replicating and nonreplicating bacteria (11, 12). Bicyclic nitroimidazoles (OPC-67683 and PA-824) are prodrugs activated by Rv3547 (the deazaflavin-dependent nitroreductase [Ddn]) in an F420-dependent manner, resulting in the formation of the des-nitro metabolite and the generation of a reactive nitrogen intermediate (13–15). Phenotypic screens have also led to the identification of structurally diverse compounds that target either MmpL3, a lipoprotein involved in the transport of mycobacterial trehalose monomycolate (TMM) into the M. tuberculosis cell wall; DprE1, an enzyme involved in the synthesis of cell wall arabinans; or QcrB, a component of the cytochrome bc1 complex of the respiratory chain (7, 16, 17). These screens have also resulted in the identification of newer scaffolds targeting already validated drug targets, such as InhA or KasA (18, 19). Additionally, various repurposed drugs are being evaluated in various clinical trials in an effort to decrease the duration of chemotherapy for individuals with MDR-TB (20–24).
In this study, we performed phenotypic whole-cell screening using M. bovis BCG as a surrogate host and identified 5-nitro-1,10-phenanthroline (5NP) to be a lead compound. We demonstrate that, similar to PA-824 and 5-nitrothiophenes, 5NP is activated in an F420-dependent manner, but a clear difference in the nitroreductase mediating 5NP activation compared to PA-824 and 5-nitrothiophene exists. Unlike PA-824, 5NP is able to induce autophagy in macrophages, a phenomenon reversed in the presence of 3-methyladenine (3MA). We report that the nitro group is essential for both in vitro activity and the induction of autophagy in macrophages. The structural analog with a methyl substitution at the 3′ position possessed better in vitro activity than the other analogs and showed in vivo potency. These observations demonstrate that 5NP exhibits a dual mechanism of action against M. tuberculosis by inhibiting mycolic acid biosynthesis and augmenting host antimicrobial pathways.
RESULTS
Phenotypic whole-cell-based screening to identify M. tuberculosis growth inhibitors.
M. bovis BCG was used as a surrogate host for phenotypic screening of a small-molecule library due to its high degree of relatedness to M. tuberculosis and a lack of pathogenicity. This small-molecule library consisting of 2,300 compounds was procured from the National Cancer Institute Developmental Therapeutic Program (NCI-DTP; https://dtp.cancer.gov/). For preliminary screening, all compounds were evaluated for their ability to inhibit the growth of M. bovis BCG at a single concentration of 50 μM. We observed that out of the 2,300 compounds, 52 compounds possessed antimycobacterial activity at the tested concentration. These primary hits were further rescreened for activity against either M. bovis BCG or isoniazid (INH)-sensitive or isoniazid-resistant M. tuberculosis strains in a dose-response assay determining their MICs. We observed that 35 scaffolds displayed similar activity against both isoniazid-sensitive and isoniazid-resistant M. tuberculosis strains, suggesting a mechanism of action unlike that of isoniazid (Table 1; Fig. 1A). As shown in Table 1, most of these compounds displayed comparable in vitro activity against both M. bovis BCG and M. tuberculosis.
TABLE 1.
Activity of small molecules against M. bovis BCG and INH-susceptible and INH-resistant M. tuberculosis H37Rv and cytotoxicity against THP-1 macrophages
| Serial no. | NSC no. | MIC99 (μM) |
CC50 (μM) against THP-1 macrophages | ||
|---|---|---|---|---|---|
| M. bovis BCG | M. tuberculosis H37Rv | M. tuberculosis H37Rv INHr | |||
| 1 | NSC 18415 | 3.125 | 6.25 | 3.125 | >25 |
| 2 | NSC 122131 | 1.25 | 2.5 | NDa | ND |
| 3 | NSC 4263 | 1.56 | 1.56 | 3.125 | >25 |
| 4 | NSC 26371 | 1.56 | 25 | 25 | >25 |
| 5 | NSC 94945 | 0.78 | 1.56 | 1.56 | >25 |
| 6 | NSC 149286 | 1.56 | 1.56 | 3.125 | >25 |
| 7 | NSC 69187 | 1.56 | 3.125 | 3.125 | 5 |
| 8 | NSC 125531 | 1.56 | 3.125 | 1.56 | >25 |
| 9 | NSC 139021 | 1.56 | 3.125 | 3.125 | >25 |
| 10 | NSC 228155 | 0.78 | 1.56 | 1.56 | >25 |
| 11 | NSC 47147 | 6.25 | 3.125 | 6.25 | 5 |
| 12 | NSC 83318 | 0.39 | 12.5 | 12.5 | >25 |
| 13 | NSC 105827 | 0.78 | 1.56 | 3.125 | 5 |
| 14 | NSC 60339 | 1.56 | 3.125 | 3.125 | >25 |
| 15 | NSC 60340 | 0.39 | 3.125 | 3.125 | >25 |
| 16 | NSC 67436 | 6.25 | 25 | 25 | >25 |
| 17 | NSC 3852 | 6.25 | 12.5 | 12.5 | 5 |
| 18 | NSC 285166 | 3.125 | 12.5 | 12.5 | >25 |
| 19 | NSC 148958 | 6.25 | 50 | 25 | >25 |
| 20 | NSC 4280 | 6.25 | 6.25 | 3.125 | >25 |
| 21 | NSC 163501 | 0.78 | 0.39 | ND | >25 |
| 22 | NSC 659501 | 3.125 | 3.125 | 3.125 | >25 |
| 23 | NSC 697726 | 0.39 | 1.56 | 3.125 | 5 |
| 24 | NSC 328587 | 3.125 | 1.56 | 3.125 | 5 |
| 25 | NSC 82116 | 6.25 | 12.5 | 12.5 | >25 |
| 26 | NSC 693632 | 12.5 | 12.5 | 25 | >25 |
| 27 | NSC 687849 | 0.78 | 1.56 | 1.56 | >25 |
| 28 | NSC 273829 | 1.56 | 6.25 | 6.25 | >25 |
| 29 | NSC 183359 | 1.56 | 0.78 | ND | 5 |
| 30 | NSC 311153 | 1.56 | 1.56 | ND | >25 |
| 31 | NSC 641245 | 1.56 | 6.25 | ND | >25 |
| 32 | NSC 145366 | 1.56 | 1.56 | ND | 5 |
| 33 | NSC 690634 | 0.39 | 1.56 | 1.56 | 5 |
| 34 | NSC 218439 | 0.39 | 0.39 | 0.39 | >25 |
| 35 | NSC 10010 | 1.56–3.125 | 6.25 | 3.125 | 5 |
| 36 | INH | 0.39–0.78 | 0.39–0.78 | >50 | >25 |
ND, not done.
FIG 1.

(A) Chemical structures of a few of the primary hits (therapeutic index [Ti] > 21) identified in this study. (B) Antimycobacterial activity of identified hits in liquid cultures. For in vitro killing experiments, M. bovis BCG was grown until the OD600 was 0.2 and subsequently exposed to the drugs at 8× MIC. After 7 days of incubation, bacterial counts were enumerated by plating 100 μl of 10-fold serial dilutions on MB 7H11 plates at 37°C for 3 to 4 weeks. The data presented in this panel are means ± SEs for duplicate samples and are representative of those from three independent experiments. Significant differences were observed for the indicated groups and were determined by a paired (two-tailed) t test. *, P < 0.05; **, P < 0.01. (C and D) Antimycobacterial activity of the identified hits in macrophages. THP-1 macrophages were seeded at a density of 2 × 105 and infected with either M. bovis BCG (C) or M. tuberculosis (D) at an MOI of 1:10. At 24 h postinfection, the macrophages were washed and overlaid with RPMI medium containing the various drugs (8× MIC) for 4 days. For bacterial enumeration, macrophages were lysed in 1 ml of 1× PBS–0.1% Triton X-100, and 100 ml of 10-fold serial dilutions was plated on MB 7H11 plates at 37°C for 3 to 4 weeks. The data shown in this panel are means ± SEs for triplicate wells and are representative of those from three independent experiments. Significant differences were observed for the indicated groups and were determined by a paired (two-tailed) t test. *, P < 0.05; **, P < 0.01. UT, untreated.
Next, we determined the cellular cytotoxicity of these hits against THP-1 macrophages and observed that 24 of these primary hits were noncytotoxic even at a 25 μM concentration (Table 1). A few of these noncytotoxic scaffolds were further evaluated for their in vitro mode of activity (bactericidal or bacteriostatic) and ability to inhibit the growth of intracellular M. bovis BCG (Fig. 1A). In our in vitro growth inhibition experiments, we observed that exposure of M. bovis BCG to either NSC 125531, NSC 94945, NSC 60339, or NSC 228155 resulted in up to 60.0- to 100.0-fold reductions in the bacterial counts in comparison to those for the untreated cultures (P < 0.05) (Fig. 1B). As shown in Fig. 1B, an approximately 8.0-fold reduction in viable bacterial counts was observed for NSC 4263-exposed M. bovis BCG (P < 0.05) (Fig. 1B). The exposure of M. bovis BCG to either NSC 26371, NSC 163501, or NSC 149286 resulted in a bacteriostatic effect in liquid cultures (Fig. 1B). In THP-1 macrophages, we did not observe any intracellular killing of M. bovis BCG after 4 days of exposure to either NSC 228155, NSC 60339, or NSC 26371 (Fig. 1C). In concordance with the results obtained in vitro, a bacteriostatic effect was observed in macrophages exposed to NSC 163501 (Fig. 1C). As shown in Fig. 1C, exposure to either NSC 125531, NSC 94945, or NSC 4263 resulted in approximately 20.0-fold, 5.0-fold, and 8.0-fold reductions in intracellular bacterial counts, respectively, in comparison to the counts for the untreated wells (P < 0.01) (Fig. 1C). These lead compounds, NSC 125531, NSC 94945, and NSC 4263, identified in our screen were also able to inhibit the growth of intracellular M. tuberculosis in THP-1 macrophages (P < 0.01) (Fig. 1D). These three scaffolds displayed MICs in the range of 0.78 to 1.56 μM against slowly growing mycobacteria. These three scaffolds, NSC 125531, NSC 9495, and NSC 4263, lacked activity against Escherichia coli and displayed MIC values of >50 μM, 50 μM, and 25 μM, respectively, against M. smegmatis.
We next determined the activity of the lead compounds against both drug-sensitive and drug-resistant clinical strains (25) (Tables 2 and 3). These drug-resistant clinical strains were as sensitive as the drug-susceptible strains to these scaffolds, indicating a lack of cross-resistance to known anti-TB drugs (Table 2). Additionally, a number of scaffolds have recently been shown to target M. tuberculosis by inhibiting MmpL3; therefore, we also tested these lead compounds for cross-resistance to MmpL3 inhibitors using M. tuberculosis strains resistant to SQ109 and DA8. As shown in Table 2, these strains were found to be susceptible to the identified lead compounds, ruling out the possibility that MmpL3 is their putative target. These data suggest a novel mechanism of action for these scaffolds and underscore the developmental potential of these compounds for use against drug-resistant TB.
TABLE 2.
MICs for various strains in vitro
| Strain | Drug resistancea | MIC (μM) |
||
|---|---|---|---|---|
| NSC 94945 | NSC 4263 | NSC 125531 | ||
| H37Rv | None | 0.61 | 1.25 | 2.5 |
| 210 | None | 0.61 | 1.25 | 5 |
| 31b (RpoBS531T KatGS315T EmbM306V) | INH, Rif, Emb, Kan, SM, Cap | 1.25 | 1.25 | 5 |
| 36b (RpoBH526D KatGS315T EmbM306V) | INH, Rif, Emb | 0.3 | 0.3 | 2.5 |
| 116b (RpoBL511P KatGS315T EmbM306VD354A) | INH, Emb, PAS | 1.25 | 1.25 | 5 |
| 692 | None | 0.61 | 1.25 | 5 |
| 8.1 (MmpL3L567P) | DA8, SQ109 | 0.61 | 1.25 | 2.5 |
| 8.3 (MmpL3D40R) | DA8, SQ109 | 0.61 | 1.25 | 2.5 |
INH, isoniazid; Rif, rifampin; Emb, ethambutol; Kan, kanamycin; SM, streptomycin; Cap, capreomycin; PAS, para-aminosalicylic acid.
The genotype known from target gene sequencing is shown.
TABLE 3.
MICs for various resistant strains in vitro
| Strain | MIC (μM) |
|||
|---|---|---|---|---|
| PA-824 | NSC 4263 | Levofloxacin | INH | |
| NSC 4263-resistant mutant strains | ||||
| M. bovis BCG (wild type) | 0.78 | 1.56 | 0.78 | 0.78 |
| 4263 (resistant mutant 1, FbiB−) | >50 | 12.5 | 0.78 | 0.78 |
| 4263 (resistant mutant 2, FbiB−) | >50 | 12.5 | 0.78 | 0.78 |
| 4263 (resistant mutant 3, FbiB−) | >50 | 12.5 | 0.78 | 0.78 |
| 4263 (resistant mutant 4, FbiB−) | >50 | 12.5 | 0.78 | 0.78 |
| 4263 (resistant mutant 5, FbiB−) | >50 | 12.5 | 0.78 | 0.78 |
| 4263R::pVV16-FbiB 1 | 1.56 | 1.56 | 0.78 | 0.78 |
| 4263R::pVV16-FbiB 2 | 1.56 | 1.56 | 0.78 | 0.78 |
| PA-824-resistant mutant strains | ||||
| H37Rv (wild type) | 0.78 | 1.56 | 0.78 | 0.78 |
| T3-MT (FGD− F420+) | >50 | 25 | 0.78 | 0.78 |
| 50-5A1 (FGD FbiC−) | >50 | 25 | 0.78 | 0.78 |
| 50-14A1 (FGD+ Ddn−) | >50 | 1.56 | 0.78 | 0.78 |
| 50-7A2 (FGD+ FbiAB−) | >50 | 25 | 0.78 | 0.78 |
| 50-14A1-3547 (50-14A1::Rv3547) | 0.78 | 1.56 | 0.78 | 0.78 |
Generation and characterization of drug-specific spontaneous resistant mutants.
In order to gain insight into their mechanism of action and identify the cellular targets for the lead compounds, we attempted to raise spontaneous drug-resistant mutants. We successfully raised such mutants resistant to NSC 4263, but repeated efforts to raise such mutants resistant to NSC 94945 and NSC 125531 were futile, indicating that M. bovis BCG had an unusually high barrier to genetic resistance to those compounds, that the compounds had a nonspecific mechanism of action, or that the mutations in the targets were not well tolerated. For M. bovis BCG raised to be resistant to NSC 4263 (5-nitro-1,10-phenanthroline [5NP]), the MIC of the parent compound was increased >8.0-fold in comparison to that for the wild-type strain (Table 3). The in vitro frequency of 5NP resistance in M. bovis BCG was approximately 10−6, which was 10-fold higher than the frequency of resistance to PA-824 and isoniazid. These resistant strains consistently displayed resistance to 5NP but remained susceptible to standard antitubercular drugs (Table 3; see also Table S1 in the supplemental material). For target identification, genomic DNA was isolated from both the wild-type strain and the 5NP-resistant strain and subjected to whole-genome sequencing. Detailed analysis of the sequencing data revealed an inactivating frameshift mutation in the fbiB gene, which encodes the F420 coenzyme l-glutamate ligase, an essential enzyme involved in F420 biosynthesis. Sanger sequencing of five additional independent isolates also revealed mutations in fbiB, confirming the role of FbiB in resistance to this compound. The complementation of resistant strains with a functional FbiB expressed from an episomal plasmid under the control of a constitutive hsp60 promoter restored their sensitivity to 5NP without affecting their susceptibilities to standard anti-TB drugs, such as isoniazid and levofloxacin (Table 3).
F420 (8-hydroxy-5-deazaflavin), a two-electron transfer cofactor, has been implicated in redox balance, methane biosynthesis, and activation of bicyclic nitroimidazoles (26–28). Mycobacterial strains deficient in F420 biosynthesis are also resistant to other nitro group-containing drugs, such as 5-nitrothiophenes and bicyclic nitroimidazoles (13, 14, 29). Therefore, we next investigated whether our resistant strains were also resistant to killing by PA-824, a bicyclic nitroimidazole, and observed that they were resistant to killing by PA-824 (Table 3). PA-824-resistant mutants were characterized by mutations in either (i) FGD-1, an enzyme involved in the regeneration of reduced F420; (ii) FbiA, FbiB, or FbiC, each of which is an enzyme involved in F420 biosynthesis; or (iii) Ddn, a nitroreductase involved in PA-824 activation (13). As shown in Table 3, we observed that the loss of functional FGD-1 (T3 mutant) or F420 biosynthetic enzymes (the FbiC 5A1 mutant or the FbiB 7A2 mutant) also resulted in cross-resistance to 5NP. However, an FGD+ F420+ PA-824-resistant strain harboring a mutation in Ddn was as sensitive as the wild-type strain to 5NP. These results suggest the presence of a discrete F420-dependent nitroreductase that might activate 5NP in mycobacteria.
In vitro metabolite profiling of 5NP by LC-MS.
It is well-known that activation of bicyclic nitroimidazoles, such as PA-824 and OPC-67683, by Ddn results in the formation of the des-nitro metabolite in bacteria (14, 15). Based on our observation that the killing activity of 5NP is F420 dependent, we hypothesized that 5NP is a prodrug and its activation results in the formation of the des-nitro product in bacteria. In support of our hypothesis, we observed two major peaks with m/z corresponding to 181.09 and 195.10 in M. bovis BCG exposed to 5NP (Fig. 2A). We next performed a detailed analytical characterization of a synthetic des-nitro metabolite and confirmed that the peak with m/z 181.09 was indeed that for 1,10-phenanthroline. As shown in Fig. 2B, we did not observe any endogenous peaks corresponding to m/z 181.09 or m/z 195.10 in samples prepared from untreated M. bovis BCG.
FIG 2.

(A and B) Metabolic analysis of M. bovis BCG treated with 5NP. For metabolic analysis, a mid-log-phase culture of M. bovis BCG was exposed to 25 μM 5NP. After exposure for 8 h, metabolites were extracted by lysing the cells with acetonitrile and subjected to LC-MS analysis. The data shown were extracted from ion chromatograms for 5NP-treated (A) or untreated (B) M. bovis BCG. In panels A and B, the y axis shows ion intensity. (C) Proposed mechanism for activation of 5NP. The first step for 5NP activation involves initial hydride transfer from H2F420 to the 4′ position of phenanthroline, resulting in the formation of nitronic acid. This nitronic acid intermediate can subsequently undergo either protonation or reduction, thereby leading to the formation of metabolite 1 or metabolite 2, respectively.
Based on these observations, we propose the following mechanism for 5NP activation in mycobacteria (Fig. 2C). The first step of activation involves hydride transfer from H2F420 to the 4 position of 5NP, resulting in the formation of a nitronic acid intermediate. This is followed by the protonation of nitronic acid, resulting in the formation of the des-nitro metabolite (metabolite 1; m/z 181.09 and 182.10 [isotope]) and the release of nitrous acid (Fig. 2C). Alternatively, the reduction of the nitronic acid intermediate leads to the formation of a hydroxylamine intermediate, which upon further reduction results in the formation of 1,10-phenanthrolin-5-amine (metabolite 2; m/z 195.10 and 196.11 [13]). We observed metabolite peaks corresponding to metabolite 1 and metabolite 2 in our liquid chromatography-mass spectrometry (LC-MS) experiments (Fig. 2A). In our MIC determination assays, synthetically derived 1,10-phenanthroline (compound 2) and 1,10-phenanthrolin-5-amine (compound 3) displayed an MIC value of 12.5 μM against M. tuberculosis (Table 4; see also the text in the supplemental material). These findings suggest that the antimycobacterial component of 5NP-mediated M. tuberculosis killing occurred during the process of its activation.
TABLE 4.
MIC99s of 1,10-phenanthroline derivatives against M. tuberculosis H37Rv, M. bovis BCG, and 5NP-resistant M. bovis BCG

aStrains 2, 5, and 8 were bought commercially (Sigma-Aldrich).
5NP inhibition of M. tuberculosis growth is independent of chelation of metal ions.
The compounds in the 1,10-phenanthroline series act as powerful chelating agents by virtue of their ability to form thermodynamically stable metal ion complexes. These phenanthroline-metal complexes have been demonstrated to inhibit the growth of drug-resistant Pseudomonas aeruginosa and Clostridium difficile strains (30, 31) and also inhibit biofilm formation by P. aeruginosa and Enterococcus faecalis (31, 32). The 1,10-phenanthroline compounds also possess fungistatic activity by inhibiting the secretory metallopeptidase of Phialophora verrucosa (33). Therefore, we next investigated whether 5NP inhibits M. tuberculosis growth by chelating intracellular metal ions. To determine this, we measured the MIC of 5NP against M. bovis BCG both in the presence and in the absence of various metal ions (Table S2). As shown in Table S2, the addition of metal ions, such as MgCl2, ZnCl2, CaCl2, or Fe2(SO4)3, in liquid medium did not alter the MIC values of 5NP against M. bovis BCG. As shown in Table S2, we observed that addition of 200 μg/ml and 1 μg/ml ZnCl2 in liquid medium completely inhibited the growth of M. bovis BCG. These results suggest that metal chelation is not the plausible mechanism by which 5NP inhibits mycobacterial growth.
5NP inhibits the mycolic acid biosynthesis of M. tuberculosis.
Bicyclic nitroimidazoles are known to inhibit mycolic acid biosynthesis; therefore, we next investigated the effect of 5NP exposure on M. tuberculosis mycolic acid biosynthesis. The analysis of extracted polar lipids revealed that exposure of M. tuberculosis to 5NP results in inhibition of trehalose monomycolate (TMM) and trehalose dimycolate (TDM) biosynthesis, a phenomenon similar to that observed in the case of INH-exposed bacteria (Fig. S1). TMMs and TDMs were identified by exposing M. tuberculosis to DA5, which inhibits the MmpL3-dependent transport of TMM across the membrane, thereby resulting in the accumulation of TMM and a reduction of TDM levels (Fig. S1) (34). We did not observe inhibition of the biosynthesis of TDM and TMM in M. tuberculosis bacteria exposed to either NSC 125531 or NSC 94945 (Fig. S1). In order to confirm the inhibition of mycolic acid biosynthesis in 5NP-treated samples, we analyzed total mycolates by preparing mycolic acid methyl esters (MAMEs) and fatty acid methyl esters (FAMEs). Consistent with our observation of reduced levels of TDMs and TMMs in 5NP-treated bacteria, we noticed a dose-dependent reduction in the amounts of MAMEs and FAMEs (Fig. 3A and B). A similar reduction in the levels of MAMEs was also noticed with treatment with INH, a known inhibitor of fatty acid synthase II (FAS-II). This observed reduction in the amount of FAMEs indicates that the FAS-I pathway might be inhibited in 5NP-treated M. tuberculosis. As reported earlier (54), we also observed the inhibition of MAMEs and FAMEs in PA-824-treated M. tuberculosis.
FIG 3.

(A) Effect of 5NP, INH, and PA824 on mycolic acid biosynthesis in M. tuberculosis. Polar lipids were extracted from M. tuberculosis exposed to either 5NP (1×, 5×, 20×, or 50× MIC), INH (5× or 20× MIC), or PA824 (1×, 5×, 20×, or 50× MIC). Equal volumes of samples were loaded, and MAME and FAME levels were determined by developing normal-phase TLC plates using hexane-ethyl acetate (19:1 vol/vol; two runs). (B) The intensities of the bands corresponding to MAMEs (combined) and FAMEs from untreated and drug-exposed M. tuberculosis samples were quantified using ImageQuant (version 5.2) software. The values shown on the x axis represent the multiples of the MICs of the respective drugs. RFU, relative fluorescent units. (C) Chemical structures of the most active compounds identified in structure-activity relationship studies: 3-methyl-6-nitro-1,10-phenanthroline (compound 10) and 3-ethyl-6-nitro-1,10-phenanthroline (compound 11). (D and E) 3-Methyl-6-nitro-1,10-phenanthroline inhibits the growth of M. tuberculosis in mouse tissues. Female BALB/c mice were infected via the aerosol route and gavaged daily starting at 4 weeks postinfection. The bacterial loads in the lungs and spleens of the animals were enumerated at 2 weeks (D) and 4 weeks (E) posttreatment. For bacterial enumeration, mouse tissues were homogenized in normal saline, and 100 μl of a 10-fold serial dilution was plated on MB 7H11 plates at 37°C for 3 to 4 weeks. The data shown in this panel are means ± SEs for 5 animals. Significant differences were observed for the indicated groups and were determined by a paired (two-tailed) t test. *, P < 0.05; **, P < 0.01; ***, P < 0.0001. cpd, compound.
Structure-activity relationship studies of 5NP.
We initially synthesized the parent compound, 5NP (compound 1), and evaluated its activity against M. tuberculosis. The synthesized lead compound 5NP (compound 1) exhibited an MIC value of 0.78 μM against M. tuberculosis, which was similar to that observed in our phenotypic whole-cell screen. We next started a medicinal chemistry approach to design, synthesize, and evaluate the activity of 5NP structural analogs against M. tuberculosis (Table 4). The detailed synthesis and analytical characterization of these structural analogs are included in the text in the supplemental material. We observed that the removal of a nitro group (compound 2) reduced the activity of the parent compound against M. tuberculosis. The des-nitro derivative (compound 2) exhibited approximately 10.0-fold less activity than 5NP against M. tuberculosis (MIC, 12.5 μM). In order to further evaluate the role of the nitro group on the antimycobacterial activity of this series, we synthesized various compounds with modifications at the R4 position. As shown in Table 3, substitution of a nitro group with either electron-donating groups, such as amino (compound 3), methoxy (compound 4), methyl (compound 5), and carbamoyl (compound 6) groups, or electron-withdrawing groups, such as bromo (compound 7), chloro (compound 8), and cyano (compound 9) groups, was unfavorable, as these analogs possessed reduced activity against both M. bovis BCG and M. tuberculosis in comparison to that of the parent compound (5NP, compound 1; Table 4). These results demonstrate that the nitro group at the R4 position is important for the antimycobacterial activity of 5NP. We also observed that the 5NP-resistant M. bovis BCG strain was as sensitive as the parental strain to these structural analogs, indicating that their antimicrobial activity is independent of F420 levels in bacteria (Table 4).
Next, we synthesized the various compounds to determine the effect of a substitution at the R2 position on the antitubercular activity of 5NP (Table 4). We observed that the addition of electron-donating groups, such as methyl (compound 10) and ethyl (compound 11) groups, at the R2 position increased the potency of the parent compound by 4.0-fold (Table 4; Fig. 3C). Furthermore, replacement of the methyl group with other functional groups, such as vinyl (compound 12), phenyl (compound 13), substituted phenyl (compounds 14 to 19), thiophen-2-yl (compound 20), and 3-(6-fluoro pyridin-3-yl) (compound 21) groups, was unfavorable, as these substitutions reduced the antimycobacterial activity of compound 10 (Table 4). In vitro MIC determination experiments revealed that derivatives with a halogen substitution, such as a bromo (compound 22) or iodo (compound 23) substitution, were more potent than analogs with a cyano substitution (compound 24) (Table 4). Therefore, structure-activity relationship studies led to the identification of 3-methyl-6-nitro-1,10-phenanthroline (compound 10) and 3-ethyl-6-nitro-1,10-phenanthroline (compound 11) as the most active compounds, each of which displayed a MIC value of 0.195 μM against both M. bovis BCG and M. tuberculosis. As expected, in comparison to the MIC for the wild-type strain, most of these analogs displayed >4.0-fold increases in the MICs for the 5NP-resistant M. bovis BCG strain (Table 4).
In vivo activity of 3-methyl-6-nitro-1,10-phenanthroline in a murine model of tuberculosis.
The lead compound (compound 10) and the positive control, rifampin (Rif), were further evaluated for efficacy in a model of chronic infection. For animal experiments, female BALB/c mice were infected via the aerosol route with a low dose of ∼100 bacilli of M. tuberculosis H37Rv. Following 4 weeks of infection, the mice were treated by continuous gavage with either compound 10 at 25 or 100 mg/kg of body weight or Rif at 10 mg/kg daily for 4 weeks. We observed that treatment with 25 mg/kg of compound 10 did not result in a reduction of the bacterial loads in either the spleens or lungs of the animals at 14 days and 28 days posttreatment (Fig. 3D and E). As expected, daily administration of Rif at 10 mg/kg reduced the bacterial loads by 1.6 log10 counts in both the lungs and spleens of infected animals at 4 weeks posttreatment (P < 0.001) (Fig. 3E). As shown in Fig. 3E, we also noticed that treatment with 100 mg/kg of compound 10 resulted in approximately 9.0- and 60.0-fold reductions in lung and splenic bacterial counts, respectively (P < 0.01 and P < 0.001 in the case of lungs and spleens, respectively). In the case of animals treated with 10 mg/kg Rif or 100 mg/kg compound 10, the splenic bacterial counts were below the limit of detection. We also observed that treatment with 10 mg/kg of Rif or 100 mg/kg of compound 10 resulted in 9.0- and 5.0-fold reductions in lung bacillary counts, respectively, at 2 weeks posttreatment (P < 0.01 in the case of 10 mg/kg Rif and P < 0.05 in the case of 100 mg/kg compound 10) (Fig. 3D). To the best of our knowledge, this is the first study to report that this series of compounds possesses antimycobacterial activity in mice, a finding that underscores their potential for further development.
5NP induces autophagy in differentiated THP-1 macrophages.
Numerous host pathways, such as the generation of reactive oxygen species, phagosome lysosome fusion, and autophagy pathways, have been demonstrated to control the growth of intracellular pathogens (35, 36). Therefore, we next investigated whether 5NP is able to induce autophagy, thereby resulting in the killing of intracellular bacteria. We compared the ability of 5NP (compound 1), 3-methyl-6-nitro-1,10-phenanthroline (compound 10), 1,10-phenanthroline (compound 2), and PA-824 to induce autophagy in macrophages by quantifying the expression of Atg-3, Atg-7, and Beclin-1 and the conversion of LC3-I to LC3-II. As shown in Fig. 4A and B, exposure to compounds 1 and 10 resulted in the significant induction of Beclin-1, Atg-7, and Atg-3 expression at 24 h posttreatment (P < 0.05 and P < 0.01, respectively). Interestingly, we did not observe any significant increase in the levels of expression of these proteins in macrophages treated with either compound 2 or PA-824 (Fig. 4A and B). As expected, a dose-dependent increase in the levels of expression of Beclin-1 and Atg-3 was observed in macrophages treated with compound 1 (Fig. 4C). These observations implicate that the nitro group is essential for the induction of host autophagy pathways by 5NP.
FIG 4.

(A) Effects of the various drugs on induction of autophagy in THP-1 macrophages. THP-1 cells were differentiated with PMA and treated with 25 μM either compound 1, compound 10, compound 2, or PA-824 for 24 h. The clarified lysates from these drug-treated macrophages were prepared and subjected to immunoblot analysis as described in Materials and Methods. The data presented in this panel are representative of those from an experiment performed in triplicate. (B) Statistical analysis of replicate immunoblots shown in panel A showing the normalized fold intensity of proteins in macrophages exposed to 25 μM either compound 1, compound 10, compound 2, or PA-824. The values depicted in this panel are means ± SEs from three independent experiments. Statistically significant differences were observed for the indicated groups and were determined by a paired (two-tailed) t test. *, P < 0.05; **, P < 0.01. (C) Dose dependence of 5NP on expression of Beclin-1 and Atg-3 in THP-1 macrophages. THP-1 macrophages were differentiated and treated with various concentrations of 5NP (3.125 μM to 25 μM, as indicated above the gels) for 24 h. As described in Materials and Methods, expression of Beclin-1 and Atg-3 was measured by immunoblot analysis. The blots shown in this panel are representative of those from three independent experiments. (D) Pretreatment with 5NP or its structural analogs increases LC3 punctum formation in THP-1 macrophages. THP-1 macrophages were differentiated and treated with 25 μM either compound 1 or compound 10 for 24 h. As described in Materials and Methods, LC3 punctum formation was visualized by staining with anti-LC3 using confocal microscopy. Bars, 10 μm. (E) Detailed quantitative analysis of LC3 punctum formation in THP-1 macrophages treated with 25 μM either compound 1 or compound 10 for 24 h. The data shown in this panel are the mean number of LC3 puncta ± SE obtained from random fields of the panels shown in panel D. Significant differences were observed for the indicated groups and were determined by a paired (two-tailed) t test. **, P < 0.05.
During the process of autophagy, the cytosolic form of LC3-I undergoes complex C-terminal proteolytic and lipid modification to generate LC3-II, which is then translocated to autophagosomal membranes (37). In concordance with earlier observations, we noticed increased levels of conversion of LC3-I to LC3-II in THP-1 macrophages treated with either compound 1 or compound 10 in comparison to those in compound 2- or PA-824-treated macrophages (P < 0.05) (Fig. 4A and B). In concordance with that finding, higher levels of LC3 punctum formation were observed in THP-1 macrophages treated with either compound 1 or 10 than in macrophages treated with dimethyl sulfoxide (DMSO) (Fig. 4D). A detailed quantitative analysis of confocal microscopy images also revealed that the level of LC3 punctum formation was significantly increased in compound 1- or 10-treated macrophages compared with that in DMSO-treated macrophages (P < 0.05) (Fig. 4E).
We next combined 3-methyladenine (3MA), an inhibitor of autophagosome formation, with compound 1, to quantify the levels of expression of various autophagic markers in THP-1 macrophages. As shown in Fig. 5A and B, addition of 3MA abrogated the ability of compound 1 to induce expression of the Beclin-1, Atg-3, or Atg-7 protein or the conversion of LC3-I to LC3-II in THP-1 macrophages. As expected, pretreatment with 3MA reduced the level of LC3 punctum formation in THP-1 macrophages treated with compound 1 (P < 0.05) (Fig. S2). Taken together, these observations demonstrate that 5NP has the property to induce autophagy in THP-1 macrophages. Further, we also noticed significantly higher levels of LC3 punctum formation in THP-1 macrophages treated with other nitro group-containing 5NP analogs (compound 18, 22, or 24) than in macrophages treated with non-nitro group-containing derivatives (compound 2, 3, 4, 5, 7, or 8) (P < 0.001, in the case of compounds 18 and 22; P < 0.01, in the case of compounds 1 and 24; and P < 0.05, in the case of compounds 3, 4, 7, and 8) (Fig. S3; data not shown). We also investigated the ability of NSC 94945 and NSC 125531 to induce autophagy in macrophages. As shown in Fig. S4A and B, exposure to NSC 125531 also resulted in the upregulation of expression of Beclin-1, Atg-3, and Atg-7 (P < 0.05) or the conversion of LC3-I to LC3-II (P < 0.01) in THP-1 macrophages.
FIG 5.
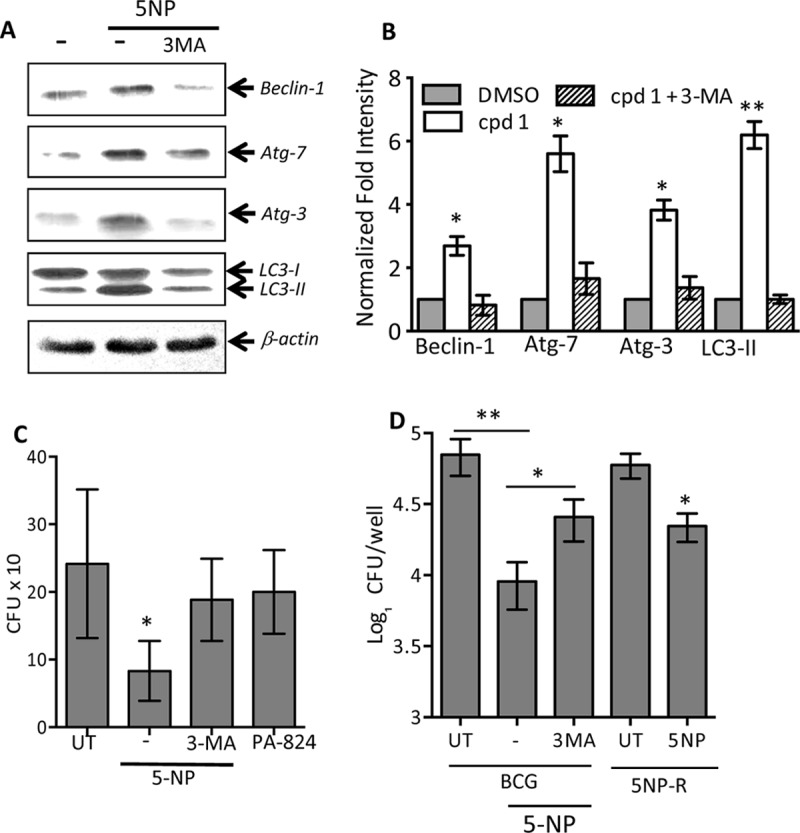
(A) Effect of 5NP on induction of autophagy in the presence or the absence of 3MA. THP-1 macrophages were differentiated and treated with 25 μM 5NP either in the presence or in the absence of 10 mM 3MA. At 24 h posttreatment, the expression of various proteins was quantified by immunoblot analysis. The data shown in this panel are representative of those from three independent experiments. (B) Statistical analysis of immunoblot replicates showing the normalized fold intensity of proteins in the different combinations tested, as shown in panel A. The values depicted are means ± SEs from three independent experiments. Statistically significant differences were observed for the indicated groups and were determined by paired (two-tailed) t test. *, P < 0.05; **, P < 0.01. (C) Effect of 5NP and PA-824 on the intracellular growth of M. smegmatis. THP-1 macrophages were seeded at a density of 5 × 105, differentiated, and infected with M. smegmatis at an MOI of 1:1. Following 3 h of infection, extracellular bacteria were removed and the macrophages were overlaid with RPMI medium containing 25 μM drugs. For bacterial enumeration, macrophages were lysed in 1 ml of PBS–0.1% Triton X-100, and 100 μl of 10-fold serial dilutions was plated on MB 7H11 plates at 37°C for 2 days. The data shown in this panel are means ± SEs from three independent experiments. Statistically significant differences were observed for the indicated groups and were determined by a paired (two-tailed) t test. *, P < 0.05. (D) Effect of 5NP on intracellular growth of M. bovis BCG wild-type and 5NP-resistant strains. THP-1 macrophages were seeded at a density of 2 × 105 and infected with either M. bovis BCG or an M. bovis BCG-resistant strain at an MOI of 1:10. At 24 h postinfection, macrophages were washed and overlaid with RPMI medium containing 25 μM drugs. Bacterial enumeration was performed as described in the legend to Fig. 1C. Statistically significant differences were observed for the indicated groups and were determined by a paired (two-tailed) t test. *, P < 0.05; **, P < 0.01. 5NP-R, 5NP resistant.
Host-directed therapies (HDTs) have recently emerged as a promising intervention strategy to tackle the problem of drug-resistant TB. Several reports have demonstrated that small molecules modulating host antimicrobial pathways inhibit the intracellular growth of both drug-susceptible and drug-resistant M. tuberculosis. In this study, we demonstrated that, unlike PA-824, 5NP is able to induce autophagy in THP-1 macrophages. Therefore, we next investigated whether 5NP is able to inhibit the intracellular growth of resistant mycobacteria (5NP-resistant strains and M. smegmatis). In support of our hypothesis, we observed that treatment with 5NP resulted in significant inhibition of the intracellular growth of both 5NP-resistant strains and M. smegmatis in comparison to that seen in untreated macrophages (P < 0.05) (Fig. 5C and D). We also demonstrated that preincubation of macrophages with 3MA reduced the killing activity of 5NP against intracellular resistant strains (P < 0.05) (Fig. 5C and D). As expected, PA-824 was unable to inhibit the growth of M. smegmatis in THP-1 macrophages (Fig. 5C). To the best of our knowledge, this is the first study to demonstrate that 5NP requires an F420-dependent nitroreductase for activation and upregulates autophagy in host macrophages (Fig. 6).
FIG 6.

Proposed model for the antimycobacterial activity of 5NP. The findings presented in this study illustrate that 5NP has a dual mechanism of action against M. tuberculosis (Mtb). In the pathogen, 5NP is activated in an unknown F420-dependent manner, resulting in the formation of two major metabolites. In addition, 5NP also activates autophagy via autophagosome formation, thereby resulting in the clearance of intracellular mycobacteria. Glucose-6-P, glucose-6-phosphate; 6-P-gluconolactone, 6-phospho-gluconolactone.
DISCUSSION
Due to the questionable long-term protective efficacy afforded by BCG vaccination, control of TB infection is primarily dependent on 6 to 24 months of chemotherapy. However, the rapid emergence of drug-resistant M. tuberculosis strains, the HIV-TB nexus, and the ability of M. tuberculosis to persist in the nonreplicating stage pose serious challenges to the fight against TB. Therefore, it is of utmost importance to identify new scaffolds with novel mechanisms of action that target both drug-resistant and metabolically less active bacteria. The combination of the results of phenotypic whole-cell screens and activity against intracellular bacteria led to the identification of NSC 125531, NSC 94945, and 5NP as lead compounds. These identified hits were active against both drug-susceptible and multidrug-resistant clinical strains, indicating that they inhibit a novel bacterial pathway. In concordance with previous reports, we observed a good correlation between the activities of these primary hits against M. bovis BCG and M. tuberculosis. The lead compounds were comparably less active against M. smegmatis, which might be due to variations in (i) the essentiality of the targets in the two strains, (ii) the affinity of these identified hits for the drug targets, or (iii) the relative intracellular concentration due to enhanced efflux or a defect in cellular uptake. 5NP has also been reported to inhibit M. tuberculosis growth in vitro and in macrophages, but its mechanism of killing is still unknown (38).
Identification of small molecules by phenotypic screening was followed by next-generation sequencing of genomic DNA isolated from revertant strains with resistance to a specific drug to identify single nucleotide polymorphisms (SNPs) associated with their mechanism of resistance. Using a combination of genetic and biochemical tools, we were able to decipher the mechanism of M. tuberculosis killing by 5NP. Using whole-genome sequencing, we identified a frameshift mutation that resulted in the inactivation of FbiB, an enzyme involved in F420 biosynthesis in resistant strains, and complementation with functional FbiB restored its susceptibility to 5NP. These findings confirm that the depletion of F420 is associated with resistance to 5NP, as has also been reported in the case of 5-nitrothiophenes and bicyclic nitroimidazoles (13, 14, 29). The mechanism was unequivocally confirmed by PA-824 cross-resistance studies using M. tuberculosis strains harboring mutations in either FGD-1 or F420 biosynthetic enzymes (13). Surprisingly, M. tuberculosis strains with a defect in Ddn enzyme activity were fully sensitive to 5NP (13). These observations suggest that 5NP is likely to be activated by an unknown nitroreductase in an F420-dependent manner. Similar to the findings for bicyclic nitroimidazoles, we observed that 1,10-phenanthroline (des-nitro) and 1,10-phenanthrolin-5-amine were the major cellular metabolites of 5NP in M. bovis BCG (15). The proposed mechanism for 5NP activation in mycobacteria would result in the generation of a reactive nitrogen intermediate which inhibits bacterial growth by affecting the electron transport chain. Similar to the findings for PA-824, exposure of M. tuberculosis to 5NP also inhibited the M. tuberculosis FAS-I/FAS-II pathways, thereby resulting in the depletion of MAMEs and FAMEs.
1,10-Phenanthroline has been shown to mediate microbial killing by virtue of its ability to either bind DNA, chelate metal ions, or inhibit acetyl coenzyme A synthesis (30, 39–41). However, in this study, we demonstrate that the chelation of metal ions is not the mechanism by which 5NP inhibits M. tuberculosis growth in vitro. A detailed structure-activity relationship approach revealed that (i) the nitro group at the R4 position is important for antimycobacterial activity and (ii) addition of methyl and ethyl groups at the 3′ position increases the activity of the parent compound. We also showed that treatment of mice with 100 mg/kg of 3-methyl-6-nitro-1,10-phenanthroline for 28 days results in approximately 10.0- and 60.0-fold reductions in the bacterial counts in the lungs and spleens of M. tuberculosis-infected animals, respectively. Here, we also demonstrated that 5NP is able to activate autophagy in THP-1 macrophages in a dose-dependent manner and the nitro group is essential for this host modulating activity. This observed induction of autophagy could have been due to higher rates of formation of autophagosomes and was reversed in the presence of 3MA, a known inhibitor of autophagosome formation.
Several studies have shown that the treatment of mice with immunomodulatory agents in conjunction with current anti-TB drugs results in the faster clearance of MDR-TB. HDTs possess several advantages, such as the following: (i) pathogens are unable to acquire resistance to them, (ii) they have the ability to clear intracellular drug-resistant and nonreplicating bacilli, and (iii) they can lead to a shortening of the duration of chemotherapy. Autophagy is regulated by mTOR and is an essential component of the host antimicrobial machinery (35, 42). Polymorphisms in the IRGM1 (LRG47) gene have been shown to be associated with susceptibility to TB in different geographical regions (43–45). The upregulation of autophagy-related genes, like Beclin-1, P2X7, VDR, NOD2, and the Toll-like receptor 8 gene, has been reported to be a biomarker for the identification of TB-infected individuals (46–48). Several molecules, such as rapamycin, nortriptyline, prochlorperazine edisylate (PE), and tat–Beclin-1 peptide, also induce autophagy and possess antibacterial and antiviral activities (35, 49–51). In addition to these, several other immunomodulatory agents, such as metformin, imatinib, ibuprofen, zileuton, and vorinostat, are also being evaluated as treatments for individuals with MDR-TB (52, 53). Here, we show that both PA-824 and 5NP are activated in an F420-dependent manner, but unlike PA-824, 5NP is able to induce autophagy. This 5NP-dependent modulation of the host pathway also resulted in the clearance of resistant bacteria by macrophages.
In conclusion, 5NP represents a promising scaffold that possesses activity against both multidrug-resistant and susceptible M. tuberculosis strains. Mechanistic studies revealed that 5NP is a prodrug requiring F420 for activation, a process independent of Ddn. We also showed that 3-methyl-6-nitro-1,10-phenanthroline killed M. tuberculosis bacteria in mouse tissues by targeting both the host and the pathogen. To the best of our knowledge, this is the first study to report that 5NP has a dual mode of action against M. tuberculosis by inhibiting mycolic acid biosynthesis and activating host autophagy pathways. Future experiments will involve detailed mechanistic studies of the remaining identified scaffolds and the synthesis of 5NP structural analogs with better in vivo, pharmacokinetic, and pharmacodynamic properties. These findings should lead the way to the further discovery and development of scaffolds with similar antibacterial properties.
MATERIALS AND METHODS
Strains, culture conditions, and chemicals.
M. smegmatis, M. bovis BCG, and M. tuberculosis H37Rv were cultured in Middlebrook (MB) 7H9 broth supplemented with 10% albumin dextrose saline, 0.2% glycerol, and 0.05% Tween 80 or MB 7H11 agar supplemented with 10% oleic acid-albumin-dextrose-saline per standard protocols. E. coli culturing was performed in Luria-Bertani (LB) broth or agar at 37°C and 200 rpm. A chemical library consisting of approximately 2,300 compounds was obtained from National Cancer Institute Developmental Therapeutic Program (NCI-DTP; https://dtp.cancer.gov/). The compounds in this library belong to either a diversity set, a mechanistic set, or a natural product set. All other chemicals used in this study, unless mentioned otherwise, were procured from Sigma-Aldrich (St. Louis, MO, USA). For MIC determination assays, compounds were prepared as 50 mM stocks in DMSO and evaluated for antimycobacterial activity at concentrations ranging from 50 to 0.05 μM. For these assays, various strains were grown in MB 7H9 medium until the optical density at 600 nm (OD600) was 0.2, diluted 1,000 times, and added to 96-well plates containing drugs. The MIC99 value was defined as the drug concentration at which no visible bacterial growth was observed. For MIC determination, 96-well plates were incubated for either 1 day in the case of E. coli, 2 days in the case of M. smegmatis, or 14 days in the case of M. bovis BCG and M. tuberculosis per standard protocols.
Cell cytotoxicity assays.
Cell viability assays were performed using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell viability kit per the manufacturer's recommendations (Sigma-Aldrich, St. Louis, MO, USA). Briefly, 2.5 × 104 THP-1 cells were seeded in triplicate wells in 96-well plates. On the next day, the macrophages were washed and overlaid with medium containing drugs at concentrations ranging from 25 μM to 0.05 μM for 96 h. In order to determine cell viability, 10 μl of the MTT reagent was added to each well, and formazan crystals were dissolved in 100 μl of lysis solution. The absorbance at 562 nm was measured in a plate reader, and cell viability was calculated according to following formula: percent cell viability = (OD562 of the test sample/OD562 of the control sample) × 100. The 50% cytotoxic concentration (CC50) was defined as the concentration of drug at which 50% of cells were viable.
Antimycobacterial activity in vitro and in macrophages.
For in vitro experiments, M. bovis BCG cultures were grown until the OD600 was 0.2 in MB 7H9 medium and subsequently exposed to the various drugs. At 7 days postexposure, the bacterial cultures were harvested and washed once with 1× phosphate-buffered saline (PBS), and bacterial counts were enumerated by plating 100 μl of 10-fold serial dilutions on MB 7H11 plates at 37°C for 3 to 4 weeks.
For intracellular killing experiments, THP-1 macrophages were seeded at a density of 2 × 105 (for the M. bovis BCG experiment) or 5 × 105 (for the M. smegmatis experiment) in a 12-well plate and differentiated by the addition of phorbol myristate acetate (PMA). Subsequently, macrophages were infected with either M. bovis BCG or M. smegmatis at a multiplicity of infection (MOI) of 1:10 or 1:1, respectively. At 3 to 4 h postinfection, the extracellular bacteria were removed by overlaying the macrophages with RPMI medium containing 200 μg/ml amikacin. After 2 h, the infected macrophages were overlaid with RPMI medium. On the next day, the macrophages were washed twice with 1× PBS and overlaid with RPMI medium containing drugs. At designated time points (72 h and 96 h in the case of M. smegmatis and M. bovis BCG, respectively), the macrophages were lysed in 1× PBS–0.1% Triton X-100, and 100 μl of 10-fold serial dilutions was plated on MB 7H11 plates at 37°C for 3 to 4 weeks.
Generation of spontaneous resistant mutant and complemented strains.
For the generation of mutant strains spontaneously resistant to specific drugs, mid-log-phase cultures of M. bovis BCG were plated on MB 7H11 plates containing the various drugs at a concentration of 10× MIC. The colonies appearing on these drug-containing plates were grown until the OD600 was 0.2 to 0.3, and MIC values were determined as described above. For target identification, genomic DNA was isolated from both wild-type and resistant mutant strains and subjected to next-generation sequencing (Edge-Bio, Gaithersburg, MD). For complementation analysis, fbiB was PCR amplified and cloned into the mycobacterial expression vector pVV16. The complemented strain was constructed by electroporation of pVV16-FbiB into the 5NP-resistant strain, and transformants were selected on MB 7H11 plates with 25 μg/ml kanamycin.
Lipid analysis of mycolic acids from M. tuberculosis.
M. tuberculosis cultures were grown until the OD600 was 0.3 to 0.4, and 5-ml cultures were treated with the various drugs. At 1 h postincubation, M. tuberculosis was labeled using 1 μCi/ml of [14C]acetate (56 mCi/mmol; PerkinElmer, USA) for 4 h. The labeled bacilli were harvested, and the pellets were hydrolyzed by addition of 2 ml tetra-n-butyl ammonium hydroxide (TBAH) and incubated overnight at 100°C. The fatty acids were esterified by addition of 4 ml of dichloromethane, 300 μl of methyl iodide, and 2 ml of water, followed by end-to-end mixing for 1 h at room temperature. The phases were separated by centrifugation, and the lower phase was washed twice with distilled water, dried under a stream of air, resuspended in 3 ml of diethyl ether, and centrifuged. The material insoluble in the solvent was transferred to new glass tubes, dried, and resuspended in 200 μl dichloromethane. Equal volumes were loaded on silica gel 60 F254 thin-layer chromatography (TLC) plates and resolved using hexane-ethyl acetate (19:1, vol/vol; two runs). The bands corresponding to FAMEs and MAMEs were visualized by phosphorimaging and quantified using ImageQuant (version 5.2) software (GE Healthcare Life Science, UK).
For analysis of TDM and TMM levels, M. tuberculosis cells were treated and labeled as described above. The labeled bacilli were harvested, resuspended in chloroform-methanol (2:1, vol/vol), and incubated overnight at 55°C. The samples were centrifuged, and the supernatants were dried in new glass tubes and resuspended in 200 μl of chloroform-methanol (2:1, vol/vol). Equal volumes of the various samples were spotted on normal-phase TLC plates and resolved using chloroform-methanol-water (62:24:4, vol/vol/vol), and radioactive spots were visualized by phosphorimaging. The radioactive spots corresponding to TDM and TMM were quantified using ImageQuant (version 5.2) software (GE Healthcare Life Sciences, UK).
Intracellular metabolite analysis of M. bovis BCG treated with 5NP.
For metabolite analysis, mid-log-phase cultures of M. bovis BCG were incubated with 20 μM 5NP at 37°C with end-to-end mixing. Following 8 h of incubation, bacterial cells were harvested and lysed by addition of acetonitrile, and clarified lysates were prepared by centrifugation and filtered using 0.22-μm-pore-size filters. The filtered lysates were subjected to mass spectrometry analysis. Mass spectra were recorded with an AB Sciex triple time of flight 5600 system.
Synthesis of small molecules.
All reagents used for the chemical synthesis of 5NP structural analogs were of American Chemical Society grade and purchased from either Sigma-Aldrich, Tokyo Chemical Industry Co., or Alfa Aesar. The detailed protocol for the chemical synthesis of these structural analogs is described in the supplemental material. The chemical reactions were monitored by thin-layer chromatography using F254 glass-backed silica plates. The plates were visualized by shortwave UV light (254 nm) and stained with p-anisaldehyde and phosphomolybdic acid. The final compounds were dissolved in either CdCl3, d6-DMSO, or Cd3OD for collection of the nuclear magnetic resonance (NMR) spectra. The NMR (1H NMR, 13C NMR) spectra were recorded using a Varian Gemini 300-MHz spectrometer or a Bruker DPX 300-MHz or 500-MHz spectrometer. High-resolution mass spectrometry (time of flight mass spectrometry with electrospray or electrospray ionization) was performed using a JOEL JMS-700 mass spectrometer.
Animal ethics statement.
All animal experimental procedures were reviewed and approved by the institutional Animal Ethics Committee of the Translational Health Science and Technology Institute and the International Centre for Genetic Engineering and Biotechnology (ICGEB). Mouse efficacy experiments were performed at the Tuberculosis Aerosol Challenge Facility according to the protocol of the ICGEB Animal Care Committee.
In vivo mouse efficacy experiments.
Female BALB/c mice were procured from the National Institute of Nutrition, Hyderabad, India. In vivo mouse efficacy experiments were performed after aerosol infection of BALB/c mice with approximately 100 CFU of M. tuberculosis. At 4 weeks postinfection, the mice were treated with either 25 mg/kg or 100 mg/kg 3-methyl-6-nitro-1,10-phenanthroline or 10 mg/kg rifampin for 14 days or 28 days. These compounds were orally administered daily in 1% carboxymethyl cellulose formulations. The bacterial loads in the lungs and spleens of infected animals (5 animals per group) were determined at both 2 weeks and 4 weeks posttreatment. For bacterial enumeration, tissues were homogenized in saline, and 100 μl of 10-fold serial dilutions was plated on MB 7H11 plates at 37°C for 3 to 4 weeks.
Autophagy experiments and immunoblot analysis.
For autophagy experiments, THP-1 cells were differentiated by the addition of 50 ng/ml PMA and 1.5 × 106 THP-1 cells were seeded into each well of 12-well plates. On the next day, differentiated THP-1 cells were treated with the various drugs and clarified lysates were prepared by lysing macrophages in radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) containing protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). The amount of protein in the cell lysates was quantified using a bicinchoninic acid kit (Sigma-Aldrich, St. Louis, MO, USA), and 50 μg of protein was electrophoresed on 15% SDS-polyacrylamide gels and subjected to immunoblot analysis. After protein transfer, the nitrocellulose membrane was blocked in 2% skimmed milk and probed overnight with either anti-Beclin-1, anti-Atg-3, anti-Atg-7, or anti-LC3 (Cell Signaling Technology, MA, USA) antibodies at 4°C. This was followed by incubation of the nitrocellulose membrane with secondary antibody for 45 min at room temperature, and protein bands were visualized using an enhanced chemiluminescence kit (GE Healthcare Life Science, UK). In order to determine equal loading among the various lanes, the blots were stripped and probed with β-actin antibody (Cell Signaling Technology, MA, USA) following the same protocol described above.
Immunofluorescence detection of LC3.
For the confocal microscopy experiment, 5 × 105 THP-1 cells were differentiated by addition of PMA to 18-mm glass coverslips in a 12-well plate. On the next day, differentiated THP-1 cells were treated with the various drugs for 24 h, washed two times with 1× PBS, and fixed in 4% paraformaldehyde. The fixed cells were blocked in buffer containing 2% bovine serum albumin and 0.3% NP-40 for 1 h and incubated overnight with anti-human LC3 antibody (Cell Signaling Technology, MA, USA) at 4°C. The labeled cells were subsequently washed and incubated with Alexa Fluor 568-conjugated goat anti-rabbit IgG antibody (Thermo Fisher Scientific, IL, USA) for 1 h. The cells were then washed three times with 1× PBS, and the coverslips were mounted on glass slides with Fluoromount aqueous mounting medium (Sigma-Aldrich, USA) and observed under a confocal scanning laser microscope (CSLM; Leica Microsystems, Wetzlar, Germany). For quantitative analysis, images were taken at random locations and the numbers of LC3 puncta per cell in all the fields were counted by an observer who did not have any information about the experimental setup.
Statistical analysis.
Prism software (version 5.01; GraphPad Software Inc., CA, USA) was used for statistical analysis and the generation of graphs. For normally distributed data, comparisons were performed by a paired (two-tailed) t test. Differences between groups were considered significant at a P value of <0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anil K. Tyagi for access to the biosafety level 3 facility. We thank the technical staff of the Tuberculosis Aerosol Challenge Facility, ICGEB, for their help during the animal experiments. We are grateful to Clifton E. Barry and Bill Jacobs for sharing M. tuberculosis PA-824-resistant and INH-resistant M. tuberculosis strains, respectively. Sushanth Pradhan (Biotechnology and Medical Engineering Department, NIT Rourkela) is gratefully acknowledged for technical assistance with the confocal microscopy experiment. The technical help provided by R. K. Rajesh and S. K. Surender is highly acknowledged.
We declare no competing financial interests.
R.S. conceived the idea. S.K., P.T., T.P.G., P.K., and R.S. conducted the microbiology and animal experiments. C.-Y.P., M.G.J., Y.-K.H., C.S.S., A.B., and I.Y.L. performed the chemical synthesis. S.M. and R.D. conducted the macrophage experiments. R.S., I.Y.L., and R.D. supervised the study. R.S. and R.D. wrote the manuscript with inputs from P.K., I.Y.L., and A.B.
R.S. acknowledges the financial support received from THSTI and the Department of Biotechnology (BT/COE/34/15219/2015). R.S. acknowledges DBT for a National Bioscience Award (BT/HRD/NBA/37/01/2014) and a Ramalingaswami fellowship (BT/HRD/35/02/18/2009). R.D. thanks the Department of Science and Technology, Government of India, for financial support (DST/INSPIRE-Faculty award 2014/DST/INSPIRE/04/2014/01662). I.Y.L. acknowledges the funding received through the National Research Foundation of Korea from the Ministry of Science, ICT, and the Korean Research Institute of Chemical Technology. T.P.G. and S.M. are thankful to DBT and MHRD, Government of India, for their respective fellowships.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00969-17.
REFERENCES
- 1.Daniel TM. 2006. The history of tuberculosis. Respir Med 100:1862–1870. doi: 10.1016/j.rmed.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Barry CE III, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. 2009. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol 7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zumla A, Nahid P, Cole ST. 2013. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov 12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 4.Zumla A, Raviglione M, Hafner R, von Reyn CF. 2013. Tuberculosis. N Engl J Med 368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- 5.Udwadia ZF. 2012. MDR, XDR, TDR tuberculosis: ominous progression. Thorax 67:286–288. doi: 10.1136/thoraxjnl-2012-201663. [DOI] [PubMed] [Google Scholar]
- 6.Velayati AA, Farnia P, Masjedi MR, Ibrahim TA, Tabarsi P, Haroun RZ, Kuan HO, Ghanavi J, Farnia P, Varahram M. 2009. Totally drug-resistant tuberculosis strains: evidence of adaptation at the cellular level. Eur Respir J 34:1202–1203. doi: 10.1183/09031936.00081909. [DOI] [PubMed] [Google Scholar]
- 7.Mdluli K, Kaneko T, Upton A. 2015. The tuberculosis drug discovery and development pipeline and emerging drug targets. Cold Spring Harbor Perspect Med 5:a021154. doi: 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pym AS, Diacon AH, Tang SJ, Conradie F, Danilovits M, Chuchottaworn C, Vasilyeva I, Andries K, Bakare N, De Marez T, Haxaire-Theeuwes M, Lounis N, Meyvisch P, Van Baelen B, van Heeswijk RP, Dannemann B, TMC207-C209 Study Group. 2016. Bedaquiline in the treatment of multidrug- and extensively drug-resistant tuberculosis. Eur Respir J 47:564–574. doi: 10.1183/13993003.00724-2015. [DOI] [PubMed] [Google Scholar]
- 9.Gler MT, Skripconoka V, Sanchez-Garavito E, Xiao H, Cabrera-Rivero JL, Vargas-Vasquez DE, Gao M, Awad M, Park SK, Shim TS, Suh GY, Danilovits M, Ogata H, Kurve A, Chang J, Suzuki K, Tupasi T, Koh WJ, Seaworth B, Geiter LJ, Wells CD. 2012. Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med 366:2151–2160. doi: 10.1056/NEJMoa1112433. [DOI] [PubMed] [Google Scholar]
- 10.Diacon AH, Lounis N, Dannemann B. 2014. Multidrug-resistant tuberculosis and bedaquiline. N Engl J Med 371:2436. doi: 10.1056/NEJMc1412844. [DOI] [PubMed] [Google Scholar]
- 11.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 12.Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K. 2007. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol 3:323–324. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 13.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE III. 2006. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103:431–436. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto M, Hashizume H, Tomishige T, Kawasaki M, Tsubouchi H, Sasaki H, Shimokawa Y, Komatsu M. 2006. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med 3:e466. doi: 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE III. 2008. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiarelli LR, Mori G, Esposito M, Orena BS, Pasca MR. 2016. New and old hot drug targets in tuberculosis. Curr Med Chem 23:3813–3846. doi: 10.2174/1389557516666160831164925. [DOI] [PubMed] [Google Scholar]
- 17.Mikusova K, Ekins S. 2017. Learning from the past for TB drug discovery in the future. Drug Discov Today 22:534–545. doi: 10.1016/j.drudis.2016.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abrahams KA, Chung CW, Ghidelli-Disse S, Rullas J, Rebollo-Lopez MJ, Gurcha SS, Cox JA, Mendoza A, Jimenez-Navarro E, Martinez-Martinez MS, Neu M, Shillings A, Homes P, Argyrou A, Casanueva R, Loman NJ, Moynihan PJ, Lelievre J, Selenski C, Axtman M, Kremer L, Bantscheff M, Angulo-Barturen I, Izquierdo MC, Cammack NC, Drewes G, Ballell L, Barros D, Besra GS, Bates RH. 2016. Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat Commun 7:12581. doi: 10.1038/ncomms12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manjunatha UH, Rao SPS, Kondreddi RR, Noble CG, Camacho LR, Tan BH, Ng SH, Ng PS, Ma NL, Lakshminarayana SB, Herve M, Barnes SW, Yu W, Kuhen K, Blasco F, Beer D, Walker JR, Tonge PJ, Glynne R, Smith PW, Diagana TT. 2015. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci Transl Med 7:269ra3. doi: 10.1126/scitranslmed.3010597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee M, Cho SN, Barry CE III, Song T, Kim Y, Jeong I. 2015. Linezolid for XDR-TB—final study outcomes. N Engl J Med 373:290–291. doi: 10.1056/NEJMc1500286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gopal M, Padayatchi N, Metcalfe JZ, O'Donnell MR. 2013. Systematic review of clofazimine for the treatment of drug-resistant tuberculosis. Int J Tuberc Lung Dis 17:1001–1007. doi: 10.5588/ijtld.12.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sotgiu G, D'Ambrosio L, Centis R, Tiberi S, Esposito S, Dore S, Spanevello A, Migliori GB. 2016. Carbapenems to treat multidrug and extensively drug-resistant tuberculosis: a systematic review. Int J Mol Sci 17:373. doi: 10.3390/ijms17030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawson R, Narunsky K, Carman D, Gupte N, Whitelaw A, Efron A, Barnes GL, Hoffman J, Chaisson RE, McIlleron H, Dorman SE. 2015. Two-stage activity-safety study of daily rifapentine during intensive phase treatment of pulmonary tuberculosis. Int J Tuberc Lung Dis 19:780–786. doi: 10.5588/ijtld.14.0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sterling TR, Scott NA, Miro JM, Calvet G, La Rosa A, Infante R, Chen MP, Benator DA, Gordin F, Benson CA, Chaisson RE, Villarino ME, Tuberculosis Trials Consortium, the AIDS Clinical Trials Group for the PREVENT TB Trial (TBTC Study 26ACTG 5259). 2016. Three months of weekly rifapentine and isoniazid for treatment of Mycobacterium tuberculosis infection in HIV-coinfected persons. AIDS 30:1607–1615. doi: 10.1097/QAD.0000000000001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson R, Kumar P, Parashar V, Vilcheze C, Veyron-Churlet R, Freundlich JS, Barnes SW, Walker JR, Szymonifka MJ, Marchiano E, Shenai S, Colangeli R, Jacobs WR Jr, Neiditch MB, Kremer L, Alland D. 2013. Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat Chem Biol 9:499–506. doi: 10.1038/nchembio.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones WJ, Nagle DP Jr, Whitman WB. 1987. Methanogens and the diversity of archaebacteria. Microbiol Rev 51:135–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bleicher K, Winter J. 1991. Purification and properties of F420- and NADP(+)-dependent alcohol dehydrogenases of Methanogenium liminatans and Methanobacterium palustre, specific for secondary alcohols. Eur J Biochem 200:43–51. doi: 10.1111/j.1432-1033.1991.tb21046.x. [DOI] [PubMed] [Google Scholar]
- 28.Purwantini E, Daniels L. 1996. Purification of a novel coenzyme F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J Bacteriol 178:2861–2866. doi: 10.1128/jb.178.10.2861-2866.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartkoorn RC, Ryabova OB, Chiarelli LR, Riccardi G, Makarov V, Cole ST. 2014. Mechanism of action of 5-nitrothiophenes against Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:2944–2947. doi: 10.1128/AAC.02693-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katzianer DS, Yano T, Rubin H, Zhu J. 2014. A high-throughput small-molecule screen to identify a novel chemical inhibitor of Clostridium difficile. Int J Antimicrob Agents 44:69–73. doi: 10.1016/j.ijantimicag.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viganor L, Galdino AC, Nunes AP, Santos KR, Branquinha MH, Devereux M, Kellett A, McCann M, Santos AL. 2016. Anti-Pseudomonas aeruginosa activity of 1,10-phenanthroline-based drugs against both planktonic- and biofilm-growing cells. J Antimicrob Chemother 71:128–134. doi: 10.1093/jac/dkv292. [DOI] [PubMed] [Google Scholar]
- 32.Tay CX, Quah SY, Lui JN, Yu VS, Tan KS. 2015. Matrix metalloproteinase inhibitor as an antimicrobial agent to eradicate Enterococcus faecalis biofilm. J Endod 41:858–863. doi: 10.1016/j.joen.2015.01.032. [DOI] [PubMed] [Google Scholar]
- 33.Granato MQ, Massapust PDA, Rozental S, Alviano CS, dos Santos AL, Kneipp LF. 2015. 1,10-Phenanthroline inhibits the metallopeptidase secreted by Phialophora verrucosa and modulates its growth, morphology and differentiation. Mycopathologia 179:231–242. doi: 10.1007/s11046-014-9832-7. [DOI] [PubMed] [Google Scholar]
- 34.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE III, Boshoff HI. 2012. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother 56:1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 36.Levine B, Deretic V. 2007. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khare G, Kumar P, Tyagi AK. 2013. Whole-cell screening-based identification of inhibitors against the intraphagosomal survival of Mycobacterium tuberculosis. Antimicrob Agents Chemother 57:6372–6377. doi: 10.1128/AAC.01444-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khorasani-Motlagh M, Noroozifar M, Moodi A, Niroomand S. 2013. Biochemical investigation of yttrium(III) complex containing 1,10-phenanthroline: DNA binding and antibacterial activity. J Photochem Photobiol B 120:148–155. doi: 10.1016/j.jphotobiol.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Li X, Zhang Z, Dong Y, Liu P, Zhang C. 2013. Studies on antibacterial mechanisms of copper complexes with 1,10-phenanthroline and amino acid on Escherichia coli. Biol Trace Elem Res 154:150–155. doi: 10.1007/s12011-013-9707-7. [DOI] [PubMed] [Google Scholar]
- 41.Zhu X, Li T, Gu X, Zhang S, Liu Y, Wang Y, Tan X. 2013. Structural and functional investigation into acetyl-coenzyme A synthase and methyltransferase from human pathogen Clostridium difficile. Metallomics 5:551–558. doi: 10.1039/c3mt20257g. [DOI] [PubMed] [Google Scholar]
- 42.Kumar D, Nath L, Kamal MA, Varshney A, Jain A, Singh S, Rao KV. 2010. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 140:731–743. doi: 10.1016/j.cell.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 43.Intemann CD, Thye T, Niemann S, Browne EN, Amanua Chinbuah M, Enimil A, Gyapong J, Osei I, Owusu-Dabo E, Helm S, Rusch-Gerdes S, Horstmann RD, Meyer CG. 2009. Autophagy gene variant IRGM −261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5:e1000577. doi: 10.1371/journal.ppat.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Che N, Li S, Gao T, Zhang Z, Han Y, Zhang X, Sun Y, Liu Y, Sun Z, Zhang J, Ren W, Tian M, Li Y, Li W, Cheng J, Li C. 2010. Identification of a novel IRGM promoter single nucleotide polymorphism associated with tuberculosis. Clin Chim Acta 411:1645–1649. doi: 10.1016/j.cca.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 45.King KY, Lew JD, Ha NP, Lin JS, Ma X, Graviss EA, Goodell MA. 2011. Polymorphic allele of human IRGM1 is associated with susceptibility to tuberculosis in African Americans. PLoS One 6:e16317. doi: 10.1371/journal.pone.0016317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Songane M, Kleinnijenhuis J, Alisjahbana B, Sahiratmadja E, Parwati I, Oosting M, Plantinga TS, Joosten LA, Netea MG, Ottenhoff TH, van de Vosse E, van Crevel R. 2012. Polymorphisms in autophagy genes and susceptibility to tuberculosis. PLoS One 7:e41618. doi: 10.1371/journal.pone.0041618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Songane M, Kleinnijenhuis J, Netea MG, van Crevel R. 2012. The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis 92:388–396. doi: 10.1016/j.tube.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 48.Yu Q, Zhu X, Di W, Jiang Y. 2016. High Beclin-1 expression in human alveolar macrophage significantly correlate with the bacteriologic sterilization in pulmonary tuberculosis patients. Clin Lab 62:1003–1007. [DOI] [PubMed] [Google Scholar]
- 49.Floto RA, Sarkar S, Perlstein EO, Kampmann B, Schreiber SL, Rubinsztein DC. 2007. Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington's disease models and enhance killing of mycobacteria by macrophages. Autophagy 3:620–622. doi: 10.4161/auto.4898. [DOI] [PubMed] [Google Scholar]
- 50.Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV, Levine B. 2013. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494:201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sundaramurthy V, Barsacchi R, Samusik N, Marsico G, Gilleron J, Kalaidzidis I, Meyenhofer F, Bickle M, Kalaidzidis Y, Zerial M. 2013. Integration of chemical and RNAi multiparametric profiles identifies triggers of intracellular mycobacterial killing. Cell Host Microbe 13:129–142. doi: 10.1016/j.chom.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 52.Zumla A, Maeurer M. 2016. Host-directed therapies for multidrug resistant tuberculosis. Int J Mycobacteriol 5(Suppl 1):S21–S22. doi: 10.1016/j.ijmyco.2016.09.044. [DOI] [PubMed] [Google Scholar]
- 53.Hawn TR, Shah JA, Kalman D. 2015. New tricks for old dogs: countering antibiotic resistance in tuberculosis with host-directed therapeutics. Immunol Rev 264:344–362. doi: 10.1111/imr.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. 2000. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.