

ABSTRACT
Drug resistance in fungal pathogens is of incredible importance to global health, yet the mechanisms of drug action remain only loosely defined. Antifungal compounds have been shown to trigger the intracellular accumulation of reactive oxygen species (ROS) in human-pathogenic yeasts, but the source of those ROS remained unknown. In the present study, we examined the role of endogenous ROS for the antifungal activity of the three different antifungal substances itraconazole, terbinafine, and amphotericin B, which all target the fungal cell membrane. All three antifungals had an impact on fungal redox homeostasis by causing increased intracellular ROS production. Interestingly, the elevated ROS levels induced by antifungals were abolished by inhibition of the mitochondrial respiratory complex I with rotenone. Further, evaluation of lipid peroxidation using the thiobarbituric acid assay revealed that rotenone pretreatment decreased ROS-induced lipid peroxidation during incubation of Aspergillus fumigatus with itraconazole and terbinafine. By applying the mitochondrion-specific lipid peroxidation probe MitoPerOx, we also confirmed that ROS are induced in mitochondria and subsequently cause significant oxidation of mitochondrial membrane in the presence of terbinafine and amphotericin B. To summarize, our study suggests that the induction of ROS production contributes to the ability of antifungal compounds to inhibit fungal growth. Moreover, mitochondrial complex I is the main source of deleterious ROS production in A. fumigatus challenged with antifungal compounds.
KEYWORDS: Aspergillus fumigatus, ROS, amphotericin B, itraconazole, terbinafine
INTRODUCTION
Antifungal drug resistance among strains of the human fungal pathogen A. fumigatus represents an increasing clinical problem. Heavy usage of limited antifungal drugs targeting Aspergillus fumigatus results in a high prevalence of drug-resistant isolates (1). Moreover, usage of some antifungal compounds such as azoles in European agriculture contributes to the arising number of azole-resistant environmental strains (2, 3).
Another problem is that the diverse mechanisms of drug resistance in A. fumigatus have been insufficiently investigated. The most common mechanism of resistance against azole antifungals was shown to be associated with ergosterol biosynthesis, in particular, with a mutation in the cyp51 (erg11) gene, which encodes the target protein of many azoles: the enzyme lanosterol 14-α-demethylase. However, the latest clinical surveys have revealed the involvement of other molecular pathways into azole resistance strategies of A. fumigatus. For instance, recent investigation of clinical isolates obtained from a Mycology Reference Centre in Manchester showed that more than 50% of azole-resistant strains did not possess a cyp51 mutation (4). In recent years, several mutations have been identified and further characterized (5–8).
Resistance of A. fumigatus to amphotericin B has not been detected in clinical isolates. However, intrinsic amphotericin B resistance of Aspergillus terreus was shown to be related to the increased production of antioxidant proteins such as catalase but not to the altered ergosterol content in resistant strains (9). Thus, diverse molecular strategies are important contributors to drug resistance in filamentous fungi and need to be investigated in more detail.
Recently, mitochondrial dysfunction was described to have an impact on the development of azole resistance in A. fumigatus. In particular, a mutation leading to an E180D amino acid change in a subunit of the mitochondrial complex I was found to be associated with the azole resistance of A. fumigatus isolates (10). This study also revealed that treatment with the mitochondrial complex I inhibitor rotenone led to the itraconazole resistance of A. fumigatus. The authors of that study suggested that the drastically reduced azole sensitivity due to mutation in the respiratory complex I or inhibition with rotenone could be explained by the restoration of an unbalanced hypoxic response due to the loss of complex I activity. This assumes, however, that the selective blocking of ergosterol biosynthesis by azoles activates a hypoxic response in A. fumigatus. An alternative explanation given for the azole resistance may be the reduction of cellular redox stress, which is caused by azole action on mitochondrial reactive oxygen species (ROS)-producing components.
Indeed, it has been postulated that antifungal agents like amphotericin B and azoles trigger a common oxidative-damage cellular death pathways in fungi such as Candida albicans, Saccharomyces cerevisiae, or Cryptococcus gattii (11–15). One of these studies showed that inhibition of mitochondrial activity by rotenone abolished amphotericin B-induced oxidative stress in yeast (14). In contrast to yeasts, there is little information available from human-pathogenic molds such as A. fumigatus. In order to shed light on the question to what extent ROS formation contributes to the fungicidal effect of antifungal compounds, we examined the endogenous ROS production in A. fumigatus during exposure to three different antifungal substances, namely, itraconazole, terbinafine, and amphotericin B, which all target the fungal cell membrane. Our results confirmed elevated ROS accumulation and, as a consequence, lipid peroxidation of the membrane when the fungus was treated with antifungal drugs. Inhibition of complex I greatly abolished deleterious ROS release, as well as lipid peroxidation, in A. fumigatus stressed by the tested antifungal substances. Overall, we describe here an additional mode of action of cell membrane-targeting drugs and further suggest an antifungal resistance strategy of A. fumigatus promoted by the reduced activity of the mitochondrial respiratory chain.
RESULTS
Antifungal drug susceptibility is altered by inhibition of mitochondrial complex I.
Mitochondrial respiratory complex I is one of the main sources of intracellular ROS production (16). To test changes of A. fumigatus sensitivity toward antifungal compounds in the presence or absence of the mitochondrial complex I inhibitor rotenone, a droplet growth inhibition assay on agar plates was performed (Fig. 1A). Concentrations of antifungal compounds were chosen to allow at least partial growth of the wild-type strain after several days of cultivation at 37°C. Rotenone was used in a concentration of 75 μM, which caused only partial inhibition of complex I without a detectable fungal growth defect on agar plates. Although the addition of itraconazole, terbinafine, or amphotericin B resulted in severe A. fumigatus growth inhibition, the presence of rotenone during cultivation abolished the inhibitory activity of the tested drugs (Fig. 1A). This result indicated involvement of reduced complex I activity in developing drug resistance of A. fumigatus. Interestingly, inhibition of complex III by antimycin A also had a positive impact on the growth of A. fumigatus with all tested antifungals (see Fig. S1 in the supplemental material). This observation suggested that altered activities of both complex I and complex III are related to improved drug tolerance of A. fumigatus. However, inhibition of complex I by rotenone had a more prominent effect on the antifungal drug resistance than inhibition of complex III by antimycin A. Similarly to rotenone and antimycin A, salicylhydroxamic acid (SHAM), which inhibits the non-energy-conserving alternative oxidase (AOX), improved the growth of A. fumigatus with the tested drugs as well (see Fig. S1 in the supplemental material). In contrast, inhibition of complex IV by potassium cyanide (KCN) did not change drug susceptibility of the fungus toward all antifungals (see Fig. S1 in the supplemental material).
FIG 1.

Impact of complex I inhibition and antioxidative system on growth of A. fumigatus in the presence of drugs. (A) Droplet growth inhibition assay. Aliquots (5 μl) of wild type were spotted in a serial 10-fold dilution on AMM agar plates. Mitochondrial complex I was inhibited by the addition of 75 μM rotenone. Next, 0.25 mg/liter itraconazole (ITC), 0.5 mg/liter terbinafine (TRB), and 2.5 mg/liter amphotericin B (AMB) were added to test fungal drug susceptibility. Growth differences were detected after 84 h of incubation at 37°C in the presence of itraconazole and terbinafine and after 120 h in the presence of amphotericin B. When rotenone was added, the growth was documented after 60 h of incubation with all tested drugs. (B) Northern blot analysis of cat1. A. fumigatus wild type was cultivated in AMM overnight at 37°C in the shaking flasks. Next, 4 mg/liter itraconazole, 4 mg/liter terbinafine, and 2.5 mg/liter amphotericin B were added, and mycelia were harvested after 15 min of incubation. 28S and 18S rRNA served as loading controls. (C and D) MIC determination. A total of 104 spores were diluted in AMM overlaid onto AMM agar plates. For the terbinafine MIC determination, 105 spores resuspended in AMM were incubated with different dilutions of terbinafine. N-Acetylcysteine (NAC) was added at a concentration of 3 mM. Plates were incubated at 37°C and documented after 48 h. Arrows indicate the MICs.
Oxidative stress response in drug-treated A. fumigatus.
Putative activation of the oxidative stress response system of A. fumigatus treated with antifungal compounds was evaluated by measuring the expression level of the antioxidative enzyme catalase1. Northern blot analysis showed that the transcript level of mycelial catalase1 (AFUA_3G02270) was upregulated after 15 min of incubation with itraconazole or terbinafine (Fig. 1B). Amphotericin B did not cause a high elevation of the cat1 transcript level.
We next tested the importance of intracellular ROS scavengers for A. fumigatus exposed to the antifungal drugs by measuring MICs in the presence of the antioxidant N-acetylcysteine. Before the addition of N-acetylcysteine, the MIC of itraconazole was 0.5 mg/liter, the MIC of amphotericin B was 1.5 mg/liter, and the MIC of terbinafine was 0.75 mg/liter. When the medium was supplemented with N-acetylcysteine, the MIC of itraconazole increased to 1 mg/liter, the MIC of amphotericin B increased to 12 mg/liter, and the MIC of terbinafine increased to 1.25 mg/liter. Inhibition of complex I by rotenone changed the MIC to 4 mg/liter for amphotericin B, to 1 mg/liter for itraconazole, and to 1.75 mg/liter for terbinafine. In addition, MIC determination by the EUCAST standardized broth microdilution method confirmed the results obtained by Etest strips that rotenone and N-acetylcysteine increase MICs of the antifungal drugs applied against A. fumigatus (see Table S1 in the supplemental material). Obviously, the addition of the ROS scavenger N-acetylcysteine shifted the MIC values of the tested drugs in a way similar to that for the inhibition of complex I by rotenone (Fig. 1C and D). In particular, the addition of N-acetylcysteine had a strong positive effect on the survival of A. fumigatus in the presence of amphotericin B.
Complex I inhibition reduces the accumulation of ROS in A. fumigatus exposed to drugs.
It is generally believed that complex I contributes most of the ROS generated in intact mitochondria (16). It was hypothesized that antifungal drugs influence the activity of complex I, leading to the release of an excess of ROS. Application of the oxidant-sensing fluorescent probe 2′,7′-dichlorofluorescin diacetate to the cells treated with antifungal compounds revealed an elevated production of ROS compared to untreated control cells (Fig. 2). The release of ROS was detected in a concentration-dependent manner since the addition of more itraconazole or terbinafine resulted in a higher fluorescent signal (Fig. 2). Both concentrations of amphotericin B had a strong impact on ROS induction in A. fumigatus. Inhibition of complex I significantly reduced ROS induction in cells treated with itraconazole or terbinafine to the control level (Fig. 2). The fluorescence intensity was not reduced to the control level after inhibition of complex I in cells exposed to amphotericin B but was lower than in samples treated with amphotericin B alone (Fig. 2). Possibly, the remaining activity of complex I was enough to induce high ROS induction by amphotericin B, which was the strongest oxidizing agent among all tested drugs. In addition, the cell wall-targeting compound, caspofungin, which was used at a concentration higher than the observed MIC (17), induced a strong ROS release, which could not be lowered by complex I inhibitor rotenone (Fig. 2).
FIG 2.

Amount of ROS in drug-treated A. fumigatus. ROS measurement. Different quantities of itraconazole (ITC), terbinafine (TRB), amphotericin B (AMB), or caspofungin (Cas) were added to mycelia grown in microtiter plates. Mycelia were stained with 2′,7′-dichlorofluorescin diacetate. Mitochondrial complex I was inhibited by the addition of rotenone (75 μM) to mycelia 1 h before staining. H2O2 (4 mM) was added to fungal cells and served as a positive control. Measurements were made at 37°C during 1 h. The fluorescence intensity peak was observed after 45 min of incubation with drugs. For this reason, this time point was used to illustrate the difference in ROS production between treatments. The control represents stained mycelia incubated without any stressors.
Lipid peroxidation occurs due to drug-induced ROS.
Unsaturated fatty acids are well-known targets of damage by uncontrolled oxidative stress (18). Aspergillus species possess unsaturated fatty acids, such as linoleic acid (C18:2) in their membranes (19, 20), which can be peroxidized by ROS. When lipid peroxidation occurs, secondary breakdown products of fatty acids oxidation such as malondialdehydes (MDAs) are formed. In our study, the formation of MDAs by antifungal drug-derived ROS in A. fumigatus was determined using the thiobarbituric acid (TBA) assay. The measurement revealed that itraconazole and terbinafine cause significant elevation in lipid peroxidation (Fig. 3). Membrane oxidation caused by these drugs was reduced by pretreatment of A. fumigatus with respiratory complex I inhibitor rotenone (Fig. 3).
FIG 3.

Lipid peroxidation as a consequence of the treatment with drugs as measured by a TBA (thiobarbituric acid) assay. A. fumigatus wild type was cultivated overnight at 37°C in liquid AMM. Itraconazole (ITC; 4 mg/liter), terbinafine (TRB; 4 mg/liter), and amphotericin B (AMB; 2.5 mg/liter) were added, and mycelia were harvested after 45 min of drug exposure. Mitochondrial complex I was inhibited by the addition of 75 μM rotenone. The results are expressed as TBARS normalized to the cell dry weight.
Increased protein thiol oxidation does not occur in A. fumigatus treated with drugs.
Many studies have shown that ROS may serve as an important “second messenger” to activate or inactivate protein functions by formation of reversible modifications to thiol groups of cysteines (21). Changes in thiol modifications may therefore trigger essential cellular pathways for adaptation to certain stimuli (22) or the regulation of cellular development (23). The indirect labeling of oxidized cysteines with a fluorescent dye was used to investigate whether the level of oxidized proteins was changed in A. fumigatus mycelium treated with antifungals. As a positive control, the fungus was treated with hydrogen peroxide. Accordingly, many proteins were oxidized by hydrogen peroxide and therefore showed a higher fluorescence intensity (see Fig. S2B, lane 12, in the supplemental material) than proteins in untreated cells (see Fig. S2B, lane 3). However, long-term exposure to the high dosage of hydrogen peroxide resulted in irreversible protein modifications, which could not be reversed by reduction with Tris(2-carboxyethyl)phosphine (TCEP) and thus could not be labeled and visualized. In this way, proteins, extracted from A. fumigatus cultivated in the presence of hydrogen peroxide for 1 h (see Fig. S2B, lane 11) showed a lower fluorescence signal than proteins from the control condition (see Fig. S2B, lane 3). Surprisingly, ROS derived from the treatment with antifungal drugs did not have an impact on A. fumigatus cysteine modifications, because all extracted proteins from A. fumigatus exposed to itraconazole, terbinafine, or amphotericin B in the presence or absence of complex I inhibitor showed the same labeling pattern (see Fig. S2B, lanes 5 to 10) as proteins isolated from the fungus cultivated under the unstressed conditions (see Fig. S2B, lane 3).
Terbinafine and amphotericin B cause significant oxidation of the mitochondrial membrane.
We next wanted to determine the source of ROS production. Mitochondria are the most likely source of ROS generation and, as such, we tested for mitochondrial lipid peroxidation as an early indicator of damage. The mitochondrial selective dye MitoPerOx was chosen to determine whether the mitochondrial lipid oxidation occurs when A. fumigatus hyphae are exposed to antifungals. As a mitochondrial targeting probe, MitoPerOx accumulates in the mitochondrial inner membrane and is oxidized by such reactive intermediates such as peroxy or hydroxyl radicals (24). Upon oxidation the peak of emission of the dye gets shifted from 590 to 520 nm; therefore, fluorescence detected at 520 nm reflects lipid peroxidation of mitochondria. In the presence of terbinafine and amphotericin B, the level of mitochondrial lipid oxidation appeared to be significantly higher than in unstressed mycelia (Fig. 4). Interestingly, the addition of large amounts of drugs caused immediate mitochondrial oxidation, since values obtained at the beginning of the drug treatment were higher than in the control cells (Fig. 4A). The fungal cell wall-targeting drug caspofungin did not influence lipid peroxidation of mitochondria (Fig. 4B). This suggests that only drugs that cause membrane disruption have an effect on mitochondrial lipid oxidation. Similarly, the addition of the superoxide-generation compound menadione at a concentration of 0.1 mM, which induces complete growth inhibition in A. fumigatus (25), did not result in the oxidation of MitoPerOx (Fig. 4B). On the contrary, treatment of A. fumigatus hyphae with hydrogen peroxide, a strong lipid oxidizing agent that cause hydroxyl radicals release, resulted in a strong mitochondrial lipid oxidation (Fig. 4). Inhibition of complex I could not rescue mitochondria damaged by hydrogen peroxide but lowered mitochondrial lipid oxidation in the fungus when challenged with terbinafine or amphotericin B (Fig. 4). This result additionally confirmed that rotenone protects A. fumigatus against oxidation by membrane-targeting drugs.
FIG 4.

Mitochondrial membrane peroxidation occurs in the presence of terbinafine, amphotericin B, and H2O2. Oxidation of MitoPerOx in A. fumigatus mycelia by antifungal drugs and oxidizing agents occurred after 3 min (A), 15 min (B), or 30 min (C). A. fumigatus wild type was cultivated overnight in AMM at 37°C in microtiter plates. Mitochondrial complex I was inhibited by the addition of 75 μM rotenone 1 h before treatment with stressors. Itraconazole (ITC; 4 mg/liter), terbinafine (TRB; 4 mg/liter), and amphotericin B (AMB; 2.5 mg/liter) were added after staining of mycelia with a 0.75 μM solution of MitoPerOx. Next, 10 mM H2O2, 0.1 mM menadione (Men), and 0.5 mg/liter caspofungin (Cas) were added after staining.
Ergosterol regulation is not changed by complex I inhibition.
To examine whether the ergosterol level is changed by complex I inhibition, the expression level of the erg11A (AFUA_4G06890) gene (the target of triazoles) was analyzed in A. fumigatus treated with drugs in the presence or absence of rotenone. The addition of rotenone could slightly increase the expression of the erg11 gene when rotenone was added to cultures (Fig. 5). However, the decreased complex I functionality did not affect the transcript level of this gene when both an antifungal drug and rotenone were added to A. fumigatus (Fig. 5).
FIG 5.

The expression of erg11A did not change in the presence of rotenone and antifungals, as determined by Northern blot analysis of erg11A. A. fumigatus wild type was cultivated in liquid AMM overnight at 37°C in shaking flasks. Mitochondrial complex I was inhibited by the addition of 75 μM rotenone to a fungal culture at the beginning of cultivation and 1 h before treatment with antifungal drugs. Itraconazole (ITC4; mg/liter), terbinafine (TRB; 4 mg/liter), and amphotericin B (AMB; 2.5 mg/liter) were added, and mycelia were harvested after 45 min. 28S and 18S rRNA served as a loading control.
AfYap1 accumulates in nuclei in the presence of amphotericin B.
As a mediator of the oxidative stress response in yeast and filamentous fungi, the transcription factor Yap1 is transported into the nucleus to activate antioxidant genes when cells are exposed to the oxidizing agents, such as hydrogen peroxide or menadione (26, 27). Interestingly, when the A. fumigatus yap1_eGFP strain was treated with itraconazole or terbinafine, AfYap1 mainly localized to the cytosol and did not accumulate in the nucleus. However, when the green fluorescent protein (GFP) reporter strain was exposed to amphotericin B, the AfYap1_GFP fluorescent signal showed a higher nuclear accumulation than under control conditions (Fig. 6).
FIG 6.
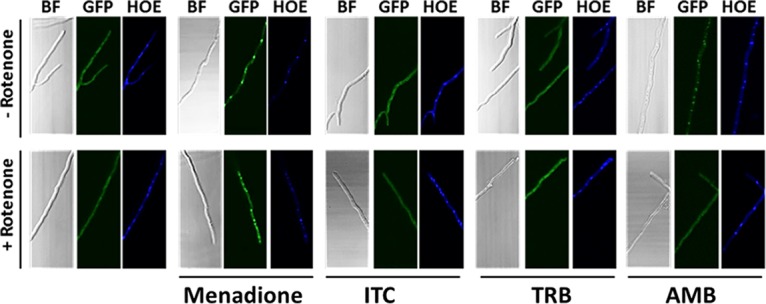
AfYap1 accumulates in the nucleus when A. fumigatus is stressed with amphotericin B but not when it is stressed with itraconazole or terbinafine. Microscopic images of the A. fumigatus yap1_eGFP strain. A total of 104 spores of the yap1_eGFP strain were cultivated in AMM at 37°C overnight. Mitochondrial complex I was inhibited by the addition of 75 μM rotenone to the fungal mycelia 1 h before the treatment with stressors. We added 5 mM menadione, 4 mg/liter itraconazole (ITC), 4 mg/liter terbinafine (TRB), and 2.5 mg/liter amphotericin B (AMB) to A. fumigatus mycelia, followed by incubation for 45 min before fixation. Exposure to menadione was used as a positive control for oxidative stress. Hoechst 34580 (HOE)-stained nuclei are shown in blue.
DISCUSSION
Recent data from human-pathogenic yeasts strongly support the idea that antifungal agents such as amphotericin B and azoles trigger intracellular ROS formation which leads to oxidative damage (11, 12, 14, 28, 29). Investigation of this concept in the most important human-pathogenic mold, A. fumigatus, has been lacking. Our research shows indeed that antifungal compounds such as itraconazole, terbinafine, or amphotericin B are also able to induce intracellular ROS production in filamentous fungi such as A. fumigatus. Moreover, we have found that the fungus can avoid this toxic effect of the drugs by altering mitochondrial activity. When different quantities of itraconazole or terbinafine were added to A. fumigatus, ROS production increased in a concentration-dependent manner. The levels of oxidative stress were similarly high in the presence of different concentrations of amphotericin B. Inhibiting the activity of mitochondrial complex I with rotenone improved the survival of A. fumigatus with all tested antifungal drugs. This is best explained by an observed decreased intracellular ROS accumulation and reduced lipid peroxidation. Furthermore, the addition of the complex I inhibitor did not change the expression level of the erg11 (cyp51) gene in the drug-treated cells, which suggests a cyp51-independent induction of drug resistance by rotenone.
Our data are in agreement with a previous report stating that amphotericin B causes oxidative damage in fungi. That study showed that the addition of exogenous catalase and/or superoxide dismutase to C. albicans cultures greatly abolished the inhibitory power of amphotericin B (29). Recently, an extensive study of different pathogenic yeast species also confirmed our findings showing mitochondrial ROS generation as an action mechanism of amphotericin B (14). Accordingly, it was revealed that fluconazole- or amphotericin B-resistant clinical isolates of C. albicans displayed high catalase activities (28). Additional evidence of the same effect of azoles and amphotericin B on fungal cells was shown in C. gattii. This fungus exhibited high ROS production and lipid peroxidation during the treatment with itraconazole or amphotericin B. Hence, the addition of a catalase inhibitor showed synergistic effects in combination with itraconazole and amphotericin B (12).
The induction of an oxidative burst as a toxic effect of antifungal drugs has not been reported for A. fumigatus yet. However, due to its intrinsic amphotericin B resistance, the impact of the oxidative stress response on the susceptibility of another filamentous fungus A. terreus to amphotericin B has been more closely studied. Amphotericin B-resistant A. terreus strains could cope better with the amphotericin B-induced oxidative stress than the susceptible ones (30). All amphotericin B-resistant A. terreus strains displayed higher basal catalase activity in comparison to amphotericin B-susceptible strains. Moreover, application of the ROS scavengers could reduce the sensitivity of the susceptible A. terreus isolates toward amphotericin B (31). Similarly, in our study we observed an increased expression of mycelial catalase cat1 in A. fumigatus upon treatment with itraconazole and terbinafine. In the same way, our research showed that N-acetylcysteine, which represents a precursor of glutathione, greatly improved the survival of A. fumigatus in the presence of all tested antifungals.
Our study showed that one of the consequences of drug-induced oxidative stress is lipid peroxidation. In particular, the TBA assay revealed that itraconazole and terbinafine increased the accumulation of lipid peroxidation by-products in A. fumigatus. Interestingly, in the presence of amphotericin B, the amount of detected lipid peroxidation by-products was not significantly changed compared to untreated cells. We cannot exclude that other lipid degradation processes took place during the treatment with amphotericin B and could not be detected by the TBA assay. For instance, 4-hydroxynonenal can also be produced by the oxidation of lipids that contain linoleic acid (32). This aldehyde can be detoxified by conjugation with glutathione. In corroboration, the addition of N-acetylcysteine, as an indirect precursor of glutathione (33), improved the growth of A. fumigatus with amphotericin B (Fig. 1C), which suggested a possible interaction of produced glutathione with amphotericin B-induced 4-hydroxynonenal (32). Furthermore, results obtained by measuring MitoPerOx oxidation in A. fumigatus treated with drugs confirmed the idea that the source of ROS production by drugs is mitochondria. Treatment with itraconazole did not cause a significant elevation of mitochondrial lipid peroxidation; however, the slight oxidation of the dye was detected in contrast to the treatment with menadione or caspofungin. Potentially, ergosterol depletion by itraconazole induced a quick disruption of the mitochondrial membrane and hence promoted diffusion of ROS throughout the cell. Therefore, in the presence of itraconazole, a portion of ROS that oxidized mitochondria was lower compared to terbinafine or amphotericin B.
In addition to causing oxidative damage, moderate levels of ROS can serve as signaling molecules to activate various adaptive cellular mechanisms (21, 22). As such, one can speculate that at the beginning of the treatment, drug-derived ROS will trigger formation of important protein oxidative modifications in A. fumigatus. In particular, we expected to observe drug-mediated oxidative modifications on cysteines. Similarly, the antibiotic nitrofurantoin caused thiol modification in Bacillus subtilis (34). As a result, proteins involved in the synthesis of amino acids and detoxification enzymes were affected (34). Surprisingly, reversible cysteine modifications were not induced in A. fumigatus exposed to the antifungal compounds. Likely, other protein modifications occurred and contributed to the stress signaling in drug-treated A. fumigatus.
Interestingly, an accumulation of the oxidative stress response regulator AfYap1 in nuclei, where it activates antioxidative genes (26), was only observed in A. fumigatus stressed with amphotericin B but not with itraconazole or terbinafine. Pretreatment of mycelia with rotenone decreased amphotericin B-induced oxidative stress and abolished AfYap1-nuclear accumulation, confirming reduction of the stress by complex I inhibition. Possibly, in contrast to amphotericin B, ROS induction by itraconazole or terbinafine did not reach the level to trigger AfYap1 activation. Nevertheless, deletion of A. fumigatus yap1 (Afyap1) did not change drug sensitivity of the knockout mutant compared to the wild type toward all tested antifungals (data not shown). An opposite phenotype was described for the pathogenic yeast Cryptococcus neoformans. Here, the deletion of the yap1 gene caused hypersensitivity of C. neoformans to the antifungal drug fluconazole (35). Similarly, the pap1 gene, a homologue of yap1, was required for the resistance of the fission yeast Schizosaccharomyces pombe against fluconazole (36). However, the importance of the yap1-mediated oxidative stress response for the drug tolerance in filamentous fungi remains unclear. Even though the transcript level of the yap1 gene was upregulated after addition of amphotericin B to the amphotericin B-susceptible A. terreus strain, the expression of this gene remained the same in the amphotericin B-resistant strain after amphotericin B exposure (31). Therefore, Yap1-dependent gene activation plays likely a minor role in the detoxification of drug-derived ROS in A. fumigatus.
In general terms, it can be noted that the induction of intracellular oxidative stress in A. fumigatus can be attributed to the cell membrane-targeting drugs. Intriguingly, caspofungin, as a cell wall-disturbing compound, was also able to cause oxidative stress, which was demonstrated by fluorescent staining with dichlorofluorescin diacetate. In agreement, a recent study of the fungal pathogen C. albicans displayed the same effect. The activities of antioxidant proteins such as catalase and superoxidase dismutase were increased in C. albicans treated with caspofungin. Also, Cap1, the homologue of the S. cerevisiae Yap1 transcription factor, displayed nuclear localization in C. albicans grown with caspofungin for 4 h (37). However, the origin of ROS in caspofungin-treated cells probably differs from the source of ROS production in cells exposed to membrane-targeting drugs. In contrast to itraconazole, terbinafine, or amphotericin B, the inhibition of respiratory complex I did not change the level of ROS production in A. fumigatus exposed to caspofungin. Also, the inhibition of complex I or complex III did not alter the susceptibility of the fungus to caspofungin (see Fig. S3 in the supplemental material). Finally, the level of mitochondria lipid oxidation in caspofungin-treated A. fumigatus remained the same as in a control condition. Taken all together, these results imply that in the presence of caspofungin there is another source of ROS production than the mitochondrial respiratory chain.
Intriguingly, our study showed that application of inhibitors of the respiratory pathways such as rotenone, antimycin A, and SHAM had a positive effect on the growth of A. fumigatus in the presence of itraconazole, terbinafine, or amphotericin B (Fig. 1; see also Fig. S1 in the supplemental material). However, potassium cyanide that inhibits complex IV of the respiratory chain did not change drug susceptibility of the fungus (see Fig. S1 in the supplemental material). This result implies that rotenone, antimycin A, and SHAM inhibit mitochondrial processes that influence the efficacy of the applied antifungals. Given that the enzymes inhibited by these chemicals use the coenzyme Q pool to transfer electrons, the metabolism of coenzyme Q could be considered a factor influencing drug sensitivity of A. fumigatus.
Our study strongly suggests that cell membrane-targeting drugs such as itraconazole, terbinafine, and amphotericin B can cause ROS leakage in fungal mitochondria, which subsequently leads to mitochondrial lipid peroxidation. Pretreatment with mitochondrial complex I inhibitor rotenone abolished this effect on mitochondria. Therefore, the access of ROS caused by antifungal compounds could be attributed to the redox homeostasis of mitochondria and, in particular, to the electron transfer through respiratory complex I. Respectively, in our previous work we characterized a mutant of A. fumigatus with reduced complex I functionality due to depletion of coenzyme Q. Interestingly, this mutant (ΔhorA) displayed antifungal drug resistance (38). This finding supports the idea that redox cycling of coenzyme Q may be a reason for ROS release when membrane-targeting antifungal compounds are applied. As an electron carrier, coenzyme Q is involved in electron flux in the respiratory chain, which is essential for ATP production. However, one electron reduction of coenzyme Q triggers the formation of a highly reactive radical anion, referred to as the ubisemiquinone radical anion. The ubisemiquinone radical anion is involved in the reduction of molecular oxygen to a reactive superoxide anion that in turn can be dismutated into hydrogen peroxide (39). In the intact phospholipid bilayer, the rate of the superoxide anion generation due to autoxidation of the ubisemiquinone radical anion is very low (40). However, altered physical properties of the membrane can accelerate the rate of ubisemiquinone radical anion autoxidation. In particular, it was reported that altered properties of mitochondrial inner membrane, such as increased membrane fluidity, may promote autoxidation of the ubisemiquinone radical anion and subsequent superoxide production (41). Thus, we hypothesize that depletion of the ergosterol level by itraconazole or terbinafine or direct binding to ergosterol by amphotericin B can change physical characteristics of the cell, as well as the mitochondrial membrane, and which, as a consequence, leads to ROS production through coenzyme Q redox cycling (Fig. 7). When fungal complex I is inhibited, the amount of ROS released through coenzyme Q cycling becomes lower, which better supports survival of A. fumigatus when challenged with drugs (Fig. 7).
FIG 7.

Schematic model of antifungal drug-induced oxidative stress in A. fumigatus. Inhibition of ergosterol biosynthesis by itraconazole or terbinafine, as well as pore formation by amphotericin B, alters the structure and function of cellular membranes and, in particular, results in a loss of mitochondrial membrane integrity. Changes in mitochondrial membrane properties may create conditions that promote a formation of superoxide anion due to redox cycling of coenzyme Q. Superoxide anion converted into hydrogen peroxide and other oxidative species contributes to oxidative damage of cell and mitochondrial membrane. Complex I inhibitor rotenone decreases the electron transfer to coenzyme Q that presumably reduces the redox cycling of coenzyme Q and thus reduces the release of mitochondrial ROS and subsequent oxidative stress. The antioxidant N-acetylcysteine abolishes oxidative stress caused by cell membrane-targeting antifungal drugs.
In addition to the primary mode of action of itraconazole, terbinafine, and amphotericin B, which is associated with the disruption of the fungal membrane, our study implies that ROS production could be a subsequent killing mode that is connected to the described ability of antifungal drugs to damage the membrane. Overall, our research supports the hypothesis that cell membrane-targeting drugs have multiple killing strategies. Diminishing one of these cell-killing modes, for instance by the abolishment of oxidative stress due to inhibition of mitochondrial function with rotenone, reduces the efficacy of these drugs against A. fumigatus.
MATERIALS AND METHODS
Reagents.
All chemicals used in this study were purchased from Sigma-Aldrich unless otherwise noted. Rotenone (R8875) and menadione (M5, 740-5) were dissolved in acetone to concentrations of 150 or 2 mM, respectively. Rotenone was always prepared directly before usage. Itraconazole (I7000000) was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 2.5 g/liter, terbinafine (T8826) was dissolved in ethanol to a concentration of 5 g/liter, and amphotericin B was dissolved in DMSO to a concentration of 20 g/liter in. All solutions of antifungals were stored at −20°C. 2′,7′-Dichlorofluorescin diacetate (35845) and MitoPerOx (Abcam, catalog no. ab146820) solutions were prepared by dissolving them in DMSO to concentrations of 10 and 2 mM, respectively.
Growth assay and drug susceptibility testing.
Fungal growth assays were performed as previously described (38). The growth capacity of A. fumigatus wild-type ATCC 46645 in the presence of antifungal compounds was monitored for several days and was documented when at least some fungal growth was observed on the plate. To determine the MICs of itraconazole and amphotericin B, Etest strips were used according to the manufacturer's instructions (Liofilchem; bestbion, catalog nos. 01B11018 and 01B11003). Portions (5 ml) of melted Aspergillus minimal medium (AMM) (42) top agar, which each contained 104 A. fumigatus spores, were poured onto solidified AMM agar plates. Etest strips were placed on the center of the plate, and the strains were incubated at 37°C for 2 days. The MICs were determined by reading the intercept of the inhibition zone and the strip. All experiments were performed in triplicate. To assess the MIC of terbinafine, a colorimetric resazurin microtiter plate-based assay was performed (43). When the MIC of terbinafine was evaluated in the presence of N-acetylcysteine, resazurin was omitted from the medium, and inhibition was defined by the lack of visible growth. Susceptibility testing of itraconazole, terbinafine, or amphotericin B against A. fumigatus was also performed by microdilution assay according to EUCAST methodology (44). RPMI 1640 supplemented with 2% glucose was used as a medium. The final concentration of the inoculum was 2.5 × 104 spores/ml. Microdilution plates were incubated at 35°C. The MIC endpoints for all antifungal drugs were assessed visually after 2 days.
Northern blotting.
Freshly harvested A. fumigatus mycelia were frozen and disrupted in liquid nitrogen with a mortar and pestle. For Northern blot analysis, the RNA was extracted with an RNeasy minikit (Qiagen, Germany) according to the manufacturer's instructions. RNA separation, blotting, hybridization, and detection with digoxigenin-labeled DNA probes were performed as described in Kroll et al. (38). Primers used to generate the probes are shown in Table 1.
TABLE 1.
Primers used in this study for Northern blot analysis
| Primer | Sequence (5′–3′) |
|---|---|
| Cat1_F | GTTCATTCCCCTCAACCCTC |
| Cat1_R | CTAGTGATCCACGGGAAACC |
| Erg11A_F | CACACCAAGGTTGACATAAGC |
| Erg11A_R | GGACTATCTGCGCGATTCAC |
Measurement of ROS production.
Measurement of intracellular ROS production was performed by staining A. fumigatus hyphae with 2′,7′-dichlorofluorescin diacetate as previously described by Blatzer et al. (45) with slight modifications. A suspension of 104 spores/ml diluted in AMM was dispensed into a 96-well F-bottom microplate (Greiner, catalog no. 655180), followed by incubation overnight at 37°C. When necessary, a 75 μM concentration of the mitochondrial complex I inhibitor rotenone dissolved in AMM was added per well, followed by incubation for 1 h. After a washing step with AMM, the cells were stained with 3 μM 2′,7′-dichlorofluorescin diacetate at 37°C for 30 min in the dark. To remove excess unreacted fluorescent probe, cells were washed with warm phosphate-buffered saline (PBS) at least three times. Several drug concentrations that were above the MIC of the drug were prepared in AMM and added to the cells. The fluorescence intensity was measured with an excitation filter at 485 nm and an emission filter at 530 nm for 1 h in a microtiter plate reader (Infinite 200 Pro; Tecan, Switzerland) at 37°C. Unstained cells were used as a blank. The maximum fluorescence intensity values were observed after 45 min of incubation with drugs were chosen as reference to indicate relative ROS production levels in all samples.
Lipid peroxidation assay.
A. fumigatus strains were cultivated with shaking in liquid AMM at 37°C for 24 to 28 h. A 75 μM concentration of rotenone was added to the corresponding flasks at the beginning of cultivation and 1 h ahead of the addition of the antifungal compound. Thereafter, tested drugs were added to mycelia in concentrations above the corresponding MIC of the drug, followed by incubation for 1 h at 37°C. Subsequently, the cells were harvested through the Miracloth (Millipore), rinsed with water, and immediately frozen in liquid nitrogen. The obtained pellets were ground in liquid nitrogen with a mortar and pestle and collected. Lipid peroxidation products were determined by measuring TBARS (thiobarbituric acid reactive substances) (46). Ice-cold 1.1% (vol/vol) phosphoric acid was added to the samples, followed by incubation on ice for at least 15 min. After vigorous vortexing, 0.4-ml portions of the homogenate were transferred into 15-ml tubes containing 0.4 ml of 1% (wt/vol) thiobarbituric acid (T5500) prepared in 50 mM NaOH containing butylated hydroxytoluene. For the blank, 0.4 ml of 3 mM HCl was added to 0.4 ml of the homogenate. The remaining 0.6 ml of the homogenate was lyophilized, and the measured dry weight value was used for data normalization. Then, 0.2 ml of 7% phosphoric acid was added to the reaction, mixed properly, and incubated at 98°C for 1 h. After cooling on ice, a lipid fraction was extracted by the addition of 1.5 ml of n-butanol, followed by centrifugation at 2,000 × g for 4 min. The absorbance of the upper layer was measured at 532 nm. The nonspecific absorbance was measured at 600 nm and subtracted from the specific absorbance. The TBARS values were expressed as a net absorbance normalized to the cell dry weight.
Fluorescence thiol modification assay.
A. fumigatus mycelia were treated for 1 h with drugs and for 15 min or 1 h with hydrogen peroxide in shaking flasks. Indirect labeling of oxidized proteins extracted from A. fumigatus mycelia was performed as previously described (34). Briefly, proteins were isolated with subsequent blocking of reduced cysteines by iodoacetamide, followed by reduction with TCEP and labeling with BODIPY FL C1-IA (D6003). A flow chart of this method is depicted in Fig. S2A in the supplemental material. Effective blocking of cysteine residues was controlled by omitting the reduction step. Protein samples were separated by 4 to 12% Bis-Tris SDS-PAGE (Life Technologies, catalog no. NP0322BOX) according to the manufacturer's protocol. Staining of the gel with Coomassie brilliant blue served as a protein loading control.
Determination of mitochondrial lipid peroxidation.
Hyphae were grown in a multiwell plate as described above. Mitochondrial phospholipid peroxidation was evaluated by applying a ratiometric fluorescent probe MitoPerOx (24). Mitochondria were stained by the addition of a 0.75 μM MitoPerOx solution in AMM, followed by incubation for 30 min at 37°C in the dark. After two washing steps with PBS, drugs were added to A. fumigatus hyphae. Green fluorescence of the probe was acquired by using an excitation at 495 nm and an emission at 524 nm, red fluorescence obtained with an excitation at 495 nm and an emission was captured at 582 nm; measurements were taken every 3 min for 30 min. Subsequently, the 524-nm/582-nm ratio was calculated to assess the peroxidation-sensitive change in fluorescence at 524 nm relative to that at 582 nm.
C-terminal eGFP fusion of AfYap1.
To generate a C-terminal enhanced GFP (eGFP) fusion protein of AfYap1 under the control of the gpdA promoter, the yap1 gene was amplified from genomic DNA of A. fumigatus ATCC 46645 using the primers Yap1_gpdatail_F and Yap1_egfptail_R (Table 2). Primers were designed to amplify the Afyap1 gene, as well as contain the overlapping end sequences specific to the gfp gene and the vector. The GFP-encoding sequence was amplified from plasmid p123 (47) using the primers egfp_filler_yap1tail_F and egfp_trpCtail_R (Table 2). The PCR product contained the GFP encoding sequence and overlapping ends to the Afyap1 gene and vector. The PCR product contained the eGFP encoding sequence and overlapping ends to the Afyap1 gene and vector. Linearized vector containing gpdA promoter, trpC terminator, and pyrithiamine resistance and kanamycin resistance cassettes was amplified by using TrpC_F and Gpda_R primers (Table 2). The amplified DNA fragments and the linearized vector were produced by PCR with Phusion Flash high-fidelity polymerase (Thermo Fisher, F548S). All fragments were ligated by using a Gibson Assembly cloning kit (NEB, E5510S). Escherichia coli DH5α bacterial cells were transformed with the ligation mixture and plated on lysogeny broth (LB) agar containing 50 mg/liter kanamycin. The final plasmid was isolated from the kanamycin-resistant strain by a plasmid minikit (Omega Bio-Tek, D6942). The obtained DNA construct is depicted in Fig. S4A in the supplemental material. The plasmid was ectopically integrated into the A. fumigatus ATCC 46645 strain genome by protoplast transformation (48). The obtained transformants were validated by fluorescence microscopy. PCR analysis of the genomic DNA of the obtained strain using Yap1_F and GFP_R primers was performed to confirm the Afyap1 fusion with gfp was present in the genome (see Fig. S4B in the supplemental material).
TABLE 2.
Primers used in this study for the generation of the A. fumigatus yap1_eGFP strain
| Primer | Sequence (5′–3′) |
|---|---|
| TrpC_F | TAGTGATTTAATAGCTCCATGTC |
| Gpda_R | GTGATGTCTGCTCAAGCGGGGT |
| Yap1_gpdatail_F | TAACAGCTACCCCGCTTGAGCAGACATCACATGGCGGACTACAATACTCT |
| Yap1_egfptail_R | CTTGCTCACCATGGGATCGAATTCCTGCAGTTTGACGCGACCCATGATGT |
| egfp_filler_yap1tail_F | GGTCGCGTCAAACTGCAGGAATTCGATCCCATGGTGAGCAAGGGCGAGGA |
| egfp_trpCtail_R | TCTTGTTGACATGGAGCTATTAAATCACTATTACTTGTACAGCTCGTCCA |
| Yap1_F | CAACTCCACGTAGCGAAGC |
| GFP_R | CCATGCCGAGAGTGATCCC |
Microscopic analysis.
For fluorescence microscopy, 0.3 ml of a 104 spore suspension of A. fumigatus was inoculated into a μ-Slide 8-well coverslip (ibidi GmbH, catalog no. 80826) and cultivated overnight at 37°C. An Axio Imager M2 (Zeiss, Germany) was used for microscopic analysis. Hoechst 34580 was used to stain nuclei after fixation with formaldehyde.
Statistical analysis.
A Welch two-sample t test was used for significance testing of the two groups. Differences between the groups were considered significant if P ≤ 0.05, P ≤ 0.01, and P ≤ 0.001, and these values are respectively labeled using one, two, and three asterisks in the figures. Error bars indicate standard errors of the means. Experiments were performed on at least three independent biological replicates.
Supplementary Material
ACKNOWLEDGMENTS
We thank Silke Steinbach for excellent technical assistance. We thank Matthew Blango for critically reading the manuscript.
This research was supported by the International Leibniz Research School for Microbial and Biomolecular Interactions and the funding program Zwanzig20–Partnerschaft für Innovation, headed by the Federal Ministry of Education and Research (BMBF), Germany (FKZ 03ZZ0809A).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00978-17.
REFERENCES
- 1.Bueid A, Howard SJ, Moore CB, Richardson MD, Harrison E, Bowyer P, Denning DW. 2010. Azole antifungal resistance in Aspergillus fumigatus: 2008 and 2009. J Antimicrob Chemother 65:2116–2118. doi: 10.1093/jac/dkq279. [DOI] [PubMed] [Google Scholar]
- 2.Verweij PE, Snelders E, Kema GH, Mellado E, Melchers WJ. 2009. Azole resistance in Aspergillus fumigatus: a side-effect of environmental fungicide use? Lancet Infect Dis 9:789–795. doi: 10.1016/S1473-3099(09)70265-8. [DOI] [PubMed] [Google Scholar]
- 3.Bowyer P, Denning DW. 2014. Environmental fungicides and triazole resistance in Aspergillus. Pest Manag Sci 70:173–178. doi: 10.1002/ps.3567. [DOI] [PubMed] [Google Scholar]
- 4.Fraczek MG, Bromley M, Buied A, Moore CB, Rajendran R, Rautemaa R, Ramage G, Denning DW, Bowyer P. 2013. The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus. J Antimicrob Chemother 68:1486–1496. doi: 10.1093/jac/dkt075. [DOI] [PubMed] [Google Scholar]
- 5.Gsaller F, Eisendle M, Lechner BE, Schrettl M, Lindner H, Muller D, Geley S, Haas H. 2012. The interplay between vacuolar and siderophore-mediated iron storage in Aspergillus fumigatus. Metallomics 4:1262–1270. doi: 10.1039/c2mt20179h. [DOI] [PubMed] [Google Scholar]
- 6.Bowyer P, Mosquera J, Anderson M, Birch M, Bromley M, Denning DW. 2012. Identification of novel genes conferring altered azole susceptibility in Aspergillus fumigatus. FEMS Microbiol Lett 332:10–19. doi: 10.1111/j.1574-6968.2012.02575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camps SM, Dutilh BE, Arendrup MC, Rijs AJ, Snelders E, Huynen MA, Verweij PE, Melchers WJ. 2012. Discovery of a HapE mutation that causes azole resistance in Aspergillus fumigatus through whole-genome sequencing and sexual crossing. PLoS One 7:e50034. doi: 10.1371/journal.pone.0050034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gsaller F, Hortschansky P, Furukawa T, Carr PD, Rash B, Capilla J, Muller C, Bracher F, Bowyer P, Haas H, Brakhage AA, Bromley MJ. 2016. Sterol biosynthesis and azole tolerance is governed by the opposing actions of SrbA and the CCAAT binding complex. PLoS Pathog 12:e1005775. doi: 10.1371/journal.ppat.1005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blum G, Perkhofer S, Haas H, Schrettl M, Wurzner R, Dierich MP, Lass-Florl C. 2008. Potential basis for amphotericin B resistance in Aspergillus terreus. Antimicrob Agents Chemother 52:1553–1555. doi: 10.1128/AAC.01280-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bromley M, Johns A, Davies E, Fraczek M, Mabey Gilsenan J, Kurbatova N, Keays M, Kapushesky M, Gut M, Gut I, Denning DW, Bowyer P. 2016. Mitochondrial complex I is a global regulator of secondary metabolism, virulence and azole sensitivity in fungi. PLoS One 11:e0158724. doi: 10.1371/journal.pone.0158724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belenky P, Camacho D, Collins JJ. 2013. Fungicidal drugs induce a common oxidative-damage cellular death pathway. Cell Rep 3:350–358. doi: 10.1016/j.celrep.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferreira GF, Baltazar Lde M, Santos JR, Monteiro AS, Fraga LA, Resende-Stoianoff MA, Santos DA. 2013. The role of oxidative and nitrosative bursts caused by azoles and amphotericin B against the fungal pathogen Cryptococcus gattii. J Antimicrob Chemother 68:1801–1811. doi: 10.1093/jac/dkt114. [DOI] [PubMed] [Google Scholar]
- 13.Guirao-Abad JP, Sanchez-Fresneda R, Alburquerque B, Hernandez JA, Arguelles JC. 2017. ROS formation is a differential contributory factor to the fungicidal action of amphotericin B and micafungin in Candida albicans. Int J Med Microbiol 307:241–248. doi: 10.1016/j.ijmm.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Mesa-Arango AC, Trevijano-Contador N, Roman E, Sanchez-Fresneda R, Casas C, Herrero E, Arguelles JC, Pla J, Cuenca-Estrella M, Zaragoza O. 2014. The production of reactive oxygen species is a universal action mechanism of amphotericin B against pathogenic yeasts and contributes to the fungicidal effect of this drug. Antimicrob Agents Chemother 58:6627–6638. doi: 10.1128/AAC.03570-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sangalli-Leite F, Scorzoni L, Mesa-Arango AC, Casas C, Herrero E, Gianinni MJ, Rodriguez-Tudela JL, Cuenca-Estrella M, Zaragoza O. 2011. Amphotericin B mediates killing in Cryptococcus neoformans through the induction of a strong oxidative burst. Microbes Infect 13:457–467. doi: 10.1016/j.micinf.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Hirst J, King MS, Pryde KR. 2008. The production of reactive oxygen species by complex I. Biochem Soc Trans 36:976–980. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 17.Arikan S, Lozano-Chiu M, Paetznick V, Rex JH. 2001. In vitro susceptibility testing methods for caspofungin against Aspergillus and Fusarium isolates. Antimicrob Agents Chemother 45:327–330. doi: 10.1128/AAC.45.1.327-330.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Girotti AW. 1985. Mechanisms of lipid peroxidation. J Free Radic Biol Med 1:87–95. doi: 10.1016/0748-5514(85)90011-X. [DOI] [PubMed] [Google Scholar]
- 19.Nemec T, Jernejc K, Cimerman A. 1997. Sterols and fatty acids of different Aspergillus species. FEMS Microbiol Lett 149:201–205. doi: 10.1111/j.1574-6968.1997.tb10329.x. [DOI] [Google Scholar]
- 20.Fraga ME, Santana DM, Gatti MJ, Direito GM, Cavaglieri LR, Rosa CA. 2008. Characterization of Aspergillus species based on fatty acid profiles. Mem Inst Oswaldo Cruz 103:540–544. doi: 10.1590/S0074-02762008000600005. [DOI] [PubMed] [Google Scholar]
- 21.Miki H, Funato Y. 2012. Regulation of intracellular signaling through cysteine oxidation by reactive oxygen species. J Biochem 151:255–261. doi: 10.1093/jb/mvs006. [DOI] [PubMed] [Google Scholar]
- 22.Ray PD, Huang BW, Tsuji Y. 2012. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguirre J, Rios-Momberg M, Hewitt D, Hansberg W. 2005. Reactive oxygen species and development in microbial eukaryotes. Trends Microbiol 13:111–118. doi: 10.1016/j.tim.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Prime TA, Forkink M, Logan A, Finichiu PG, McLachlan J, Li Pun PB, Koopman WJ, Larsen L, Latter MJ, Smith RA, Murphy MP. 2012. A ratiometric fluorescent probe for assessing mitochondrial phospholipid peroxidation within living cells. Free Radic Biol Med 53:544–553. doi: 10.1016/j.freeradbiomed.2012.05.033. [DOI] [PubMed] [Google Scholar]
- 25.Qiao J, Liu W, Li R. 2010. Truncated Afyap1 attenuates antifungal susceptibility of Aspergillus fumigatus to voriconazole and confers adaptation of the fungus to oxidative stress. Mycopathologia 170:155–160. doi: 10.1007/s11046-010-9309-2. [DOI] [PubMed] [Google Scholar]
- 26.Lessing F, Kniemeyer O, Wozniok I, Loeffler J, Kurzai O, Haertl A, Brakhage AA. 2007. The Aspergillus fumigatus transcriptional regulator AfYap1 represents the major regulator for defense against reactive oxygen intermediates but is dispensable for pathogenicity in an intranasal mouse infection model. Eukaryot Cell 6:2290–2302. doi: 10.1128/EC.00267-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuge S, Jones N, Nomoto A. 1997. Regulation of yAP-1 nuclear localization in response to oxidative stress. EMBO J 16:1710–1720. doi: 10.1093/emboj/16.7.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linares CEB, Giacomelli SR, Altenhofen D, Alves SH, Morsch VM, Schetinger MRC. 2013. Fluconazole and amphotericin B resistance are associated with increased catalase and superoxide dismutase activity in Candida albicans and Candida dubliniensis. Rev Soc Bras Med Trop 46:752–758. doi: 10.1590/0037-8682-0190-2013. [DOI] [PubMed] [Google Scholar]
- 29.Sokol-Anderson ML, Brajtburg J, Medoff G. 1986. Amphotericin B-induced oxidative damage and killing of Candida albicans. J Infect Dis 154:76–83. doi: 10.1093/infdis/154.1.76. [DOI] [PubMed] [Google Scholar]
- 30.Blum G, Hortnagl C, Jukic E, Erbeznik T, Pumpel T, Dietrich H, Nagl M, Speth C, Rambach G, Lass-Florl C. 2013. New insight into amphotericin B resistance in Aspergillus terreus. Antimicrob Agents Chemother 57:1583–1588. doi: 10.1128/AAC.01283-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blatzer M, Jukic E, Posch W, Schopf B, Binder U, Steger M, Blum G, Hackl H, Gnaiger E, Lass-Florl C, Wilflingseder D. 2015. Amphotericin B resistance in Aspergillus terreus is overpowered by coapplication of pro-oxidants. Antioxid Redox Signal 23:1424–1438. doi: 10.1089/ars.2014.6220. [DOI] [PubMed] [Google Scholar]
- 32.Ayala A, Munoz MF, Arguelles S. 2014. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dringen R, Hamprecht B. 1999. N-Acetylcysteine, but not methionine or 2-oxothiazolidine-4-carboxylate, serves as cysteine donor for the synthesis of glutathione in cultured neurons derived from embryonal rat brain. Neurosci Lett 259:79–82. doi: 10.1016/S0304-3940(98)00894-5. [DOI] [PubMed] [Google Scholar]
- 34.Hochgrafe F, Mostertz J, Albrecht D, Hecker M. 2005. Fluorescence thiol modification assay: oxidatively modified proteins in Bacillus subtilis. Mol Microbiol 58:409–425. doi: 10.1111/j.1365-2958.2005.04845.x. [DOI] [PubMed] [Google Scholar]
- 35.Paul S, Doering TL, Moye-Rowley WS. 2015. Cryptococcus neoformans Yap1 is required for normal fluconazole and oxidative stress resistance. Fungal Genet Biol 74:1–9. doi: 10.1016/j.fgb.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veal EA, Toone WM, Jones N, Morgan BA. 2002. Distinct roles for glutathione S-transferases in the oxidative stress response in Schizosaccharomyces pombe. J Biol Chem 277:35523–35531. doi: 10.1074/jbc.M111548200. [DOI] [PubMed] [Google Scholar]
- 37.Kelly J, Rowan R, McCann M, Kavanagh K. 2009. Exposure to caspofungin activates Cap and Hog pathways in Candida albicans. Med Mycol 47:697–706. doi: 10.3109/13693780802552606. [DOI] [PubMed] [Google Scholar]
- 38.Kroll K, Shekhova E, Mattern DJ, Thywissen A, Jacobsen ID, Strassburger M, Heinekamp T, Shelest E, Brakhage AA, Kniemeyer O. 2016. The hypoxia-induced dehydrogenase HorA is required for coenzyme Q10 biosynthesis, azole sensitivity and virulence of Aspergillus fumigatus. Mol Microbiol 101:92–108. doi: 10.1111/mmi.13377. [DOI] [PubMed] [Google Scholar]
- 39.Turrens JF, Alexandre A, Lehninger AL. 1985. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 40.James AM, Smith RA, Murphy MP. 2004. Antioxidant and prooxidant properties of mitochondrial coenzyme. Q Arch Biochem Biophys 423:47–56. doi: 10.1016/j.abb.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 41.Nohl H, Gille L, Schonheit K, Liu Y. 1996. Conditions allowing redox-cycling ubisemiquinone in mitochondria to establish a direct redox couple with molecular oxygen. Free Radic Biol Med 20:207–213. doi: 10.1016/0891-5849(95)02038-1. [DOI] [PubMed] [Google Scholar]
- 42.Bergh KT, Litzka O, Brakhage AA. 1996. Identification of a major cis-acting DNA element controlling the bidirectionally transcribed penicillin biosynthesis genes acvA (pcbAB) and ipnA (pcbC) of Aspergillus nidulans. J Bacteriol 178:3908–3916. doi: 10.1128/jb.178.13.3908-3916.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monteiro MC, de la Cruz M, Cantizani J, Moreno C, Tormo JR, Mellado E, De Lucas JR, Asensio F, Valiante V, Brakhage AA, Latge JP, Genilloud O, Vicente F. 2012. A new approach to drug discovery: high-throughput screening of microbial natural extracts against Aspergillus fumigatus using resazurin. J Biomol Screen 17:542–549. doi: 10.1177/1087057111433459. [DOI] [PubMed] [Google Scholar]
- 44.Arendrup M, Guinea J, Cuenca-Estrella M, Meletiadis J, Mouton J, Lagrou K, Howard S. 2015. Method for the determination of broth dilution minimum inhibitory concentrations of antifungal agents for conidia forming moulds. EUCAST definitive document E. Def 9.3. http://www.aspergillus.org.uk/sites/default/files/pictures/Lab_protocols/EUCAST_E_Def_9_3_Mould_testing_definitive_0.pdf. [Google Scholar]
- 45.Blatzer M, Binder U, Haas H. 2011. The metalloreductase FreB is involved in adaptation of Aspergillus fumigatus to iron starvation. Fungal Genet Biol 48:1027–1033. doi: 10.1016/j.fgb.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peever TL, Higgins VJ. 1989. Electrolyte leakage, lipoxygenase, and lipid peroxidation induced in tomato leaf tissue by specific and nonspecific elicitors from Cladosporium fulvum. Plant Physiol 90:867–875. doi: 10.1104/pp.90.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aichinger C, Hansson K, Eichhorn H, Lessing F, Mannhaupt G, Mewes W, Kahmann R. 2003. Identification of plant-regulated genes in Ustilago maydis by enhancer-trapping mutagenesis. Mol Genet Genomics 270:303–314. doi: 10.1007/s00438-003-0926-z. [DOI] [PubMed] [Google Scholar]
- 48.Szewczyk E, Nayak T, Oakley CE, Edgerton H, Xiong Y, Taheri-Talesh N, Osmani SA, Oakley BR. 2006. Fusion PCR and gene targeting in Aspergillus nidulans. Nat Protoc 1:3111–3120. doi: 10.1038/nprot.2006.405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.