

Abstract
Upon stimulation of toll-like receptors with various microbial ligands, induction of a variety of inflammatory genes is elicited by activation of a myeloid differentiation primary-response protein 88 (MyD88)-dependent signaling pathway. Interleukin-1 (IL-1) receptor-associated kinase 1 (IRAK1) plays an essential role in this pathway by activating nuclear factor κB (NF-κB) and mitogen-activated kinases (MAPKs). Here, we identified optineurin (OPTN) as an IRAK1-binding protein by yeast two-hybrid screening using IRAK1 as bait. A C-terminal fragment of OPTN harboring a ubiquitin-binding domain was co-immunoprecipitated with IRAK1. In reporter analyses, overexpression of OPTN inhibited IL-1β-, IRAK1-, and LPS-induced NF-κB activation. Consistently, OPTN deficiency resulted in increased NF-κB activation in response to IL-1β/LPS stimulation. To address the mechanisms underlying the inhibitory effect of OPTN on NF-κB signaling, we focused on tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), which is an adaptor protein of IRAK1 and upon polyubiquitination plays a crucial role during NF-κB activation. Overexpression of OPTN prevented TRAF6 polyubiquitination. Furthermore, OPTN H486R mutant, which is unable to recruit the deubiquitinase CYLD, failed to inhibit IRAK1-induced NF-κB activation. These results suggest that the IRAK1-binding protein OPTN negatively regulates IL-1β/LPS-induced NF-κB activation by preventing polyubiquitination of TRAF6.
Keywords: interleukin 1 (IL-1), myeloid differentiation primary response gene (88) (MYD88), NF-kappaB (NF-κB), signal transduction, yeast two-hybrid, IRAK1, TRAF6
Introduction
Innate immunity is a defense system against viral infection and bacterial invasion. Pathogen-associated molecular patterns from viruses and bacteria are recognized by toll-like receptors (TLRs), critical receptors expressed on innate immune cells (1, 2). Except for TLR3,2 all TLRs utilize a common signaling cascade dependent on the adaptor protein MyD88, which plays a crucial role in inflammatory responses (3). IL-1 receptor, which plays a crucial role in inflammatory responses after IL-1β stimulation, also recruits MyD88. When MyD88 is recruited to TLRs or IL-1 receptor via pathogen-associated molecular patterns or IL-1β stimulation, MyD88 in turn recruits IRAK1 and forms a complex with them through their respective death domains (4). After recruitment, IRAK1 is phosphorylated and dissociated from the receptor to interact with tumor necrosis factor receptor-associated factor 6 (TRAF6) (5). Activation of TRAF6 is essential for the activation of canonical and non-canonical pathways of nuclear factor (NF)-κB in the IL-1 and TLR pathways to control inflammatory gene expression (6–8). Phosphorylated IRAK1 is rapidly degraded in a proteasome-dependent manner, resulting in the down-regulation of signaling-induced inflammatory responses (9). However, the detailed mechanisms of IRAK1-dependent inflammatory responses remain unclear.
In this study we show that IRAK1 forms a complex with OPTN and that this interaction involves the C terminus (amino acids 425–577) of OPTN. We also show that OPTN negatively regulates IRAK1-dependent and IL-1β-induced NF-κB target gene expression. Further investigation reveals that OPTN prevents polyubiquitination of TRAF6, which is critical for subsequent NF-κB activation. These findings suggest that OPTN is a negative regulator of TLR/IL-1β-MyD88-IRAK1-TRAF6-NF-κB signaling cascades.
Results
Identification of OPTN as an IRAK1-binding protein by yeast two-hybrid screening
To identify proteins involved in IRAK1 signaling, we performed yeast two-hybrid screening using a human leukocyte cDNA library and obtained 410 positive clones after re-streaking from 7.85 × 106 clones. Subsequent 4 rounds of dropout selection revealed that 11 clones were able to grow on all selection plates. As each of these positive yeast clones may harbor several different plasmids, single plasmids were isolated by using Escherichia coli transformation and re-transfected into yeast cells for another round of two-hybrid screening. Thus, three positive OPTN clones were obtained (Table 1), two of which (#1 and #2) had exactly the same sequence; the third (#3) was 9 bases shorter than the others (supplemental Table 1). In addition to these OPTN clones, we also obtained eight clones containing different genes (Table 1). The OPTN clone #1 along with the eight clones was tested, and we found that only OPTN has the ability to interact with IRAK1 (Fig. 1A).
Table 1.
Identification of putative IRAK1-binding partners by yeast two-hybrid screening with four rounds of dropout selection
| Gene | Gene symbol | UniGene number | Number of clones |
|---|---|---|---|
| Optineurin | OPTN | Hs.332706 | 3 |
| Tax1 (human T-cell leukemia virus type I)-binding protein 1 | TAX1BP1 | Hs.34576 | 1 |
| FOS-like antigen 2 | FOSL2 | Hs.220971 | 1 |
| Tetratricopeptide repeat domain 14 | TTC14 | Hs.43213 | 1 |
| NF-κB-repressing factor | NKRF | Hs.437084 | 1 |
| LSM7 homolog, U6 small nuclear RNA-associated (S. cerevisiae) | LSM7 | Hs.512610 | 1 |
| Clone 48u-44 immunoglobulin heavy chain variable region | IGH | Hs.610137 | 1 |
| Eukaryotic translation initiation factor 3, subunit 7ζ, 66/67kDa | EIF3S7 | Hs.55682 | 1 |
| DnaJ (Hsp40) homolog, subfamily B, member 1 | DNAJB1 | Hs.515210 | 1 |
Figure 1.

Identification of IRAK1-binding partners by yeast two-hybrid screening. A, AH109 cells were transfected with each of the pGADT7-based putative positive clones, which are mentioned in Table 1 and also with pGBKT7-based IRAK1 or p53. Upper and lower plates are SD/−leucine/−tryptophan and SD/−leucine/−tryptophan/−histidine/+3-AT, respectively. B, a truncated form of OPTN (OPTN425–577) was cloned and expressed in yeast cells. C, HEK293T cells were transfected with FLAG-tagged IRAK1 or IRAK1-KD along with Myc-tagged OPTN, OPTN425–577, or TRAF6. After 48 h of transfection, immunoprecipitation was performed using anti-FLAG antibody. Then Western blotting (WB) was performed using anti-Myc or anti-FLAG antibody. The data are representative of three independent experiments with similar results.
OPTN, a protein composed of 577 amino acids, is known to be a negative regulator of the TNF-α signaling pathway and has a ubiquitin-binding domain in its C-terminal region (amino acids 424–509) (10). All of the three clones of OPTN plasmids contained sequences corresponding to amino acids 425–577, thereby including most of the ubiquitin-binding domain (supplemental Table 1). Therefore, we cloned amino acids 425–577 of OPTN (OPTN425–577) and showed that IRAK1 can form a complex with OPTN through this region alone (Fig. 1B).
Next, we performed immunoprecipitation to confirm the IRAK1-OPTN interaction. Significant binding between IRAK1 and OPTN could not be detected under our experimental conditions (Fig. 1C). This is probably because IRAK1 can undergo autophosphorylation, leading to its proteasomal degradation (9, 11), Therefore, kinase-dead IRAK1 (IRAK1-KD) (12) was utilized in the following experiments to improve IRAK1 stability. IRAK1-KD was shown to bind to both full-length OPTN and OPTN425–577 (Fig. 1C). Furthermore, we confirmed by immunoprecipitation that TRAF6, an adaptor protein of IRAK1, formed a complex with IRAK1-KD.
OPTN controlled the TLR and IL-1 receptor signaling pathways
To detect the TLR/IL-1 receptor signaling, an endothelial-leukocyte adhesion molecule-1 (ELAM-1) reporter assay was utilized. The ELAM-1 reporter can be activated in HEK293 cells in an NF-κB-dependent manner in response to TNF-α or IL-1β (12, 13). Overexpression of OPTN prevented ELAM-1 reporter activation in response to TNF-α (Fig. 2A), which is consistent with a previous report that OPTN inhibited NF-κB-dependent gene transcription by TNF-α (14). Interestingly, overexpression of OPTN also prevented ELAM-1 reporter activation in response to IL-1β stimulation (Fig. 2A). To address the effect of OPTN on TLR signaling, the mouse macrophage RAW264.7 cell line was used as a responder to LPS. Indeed, overexpression of OPTN prevented LPS-induced ELAM-1 reporter activity in RAW264.7 cells (Fig. 2B). IRAK1 transfection in HEK293 cells also activated ELAM-1 reporter activity, which was effectively blocked by OPTN overexpression (Fig. 2C).
Figure 2.

Inhibition of ELAM-1 reporter activity by OPTN. A, HEK293 cells were transfected with ELAM-1 reporter vector and OPTN expression vector. After 48 h, cells were stimulated with TNF-α or IL-1β for 5 h, and the reporter activity was measured. B, RAW264.7 cells were transfected with ELAM-1 reporter vector and OPTN expression vector. After 48 h, cells were stimulated with LPS for 5 h, and the reporter activity was measured. C, HEK293 cells were transfected with ELAM-1 reporter vector and also with OPTN and IRAK1 expression vectors. After 48 h, the reporter activity was measured. The data are representative of at least three independent experiments with similar results and are shown as the mean ± SD (n = 3). **, p < 0.01.
Deficiency of OPTN led to NF-κB activation
The CRISPR/CAS9 system was used to establish an OPTN-knock-out (KO) HEK293 cell line. A four-nucleotide deletion in the OPTN-coding sequence led to a premature stop codon in both alleles (Fig. 3, A and B). As expected, OPTN protein expression was completely depleted in the KO HEK293 cells (Fig. 3C).
Figure 3.
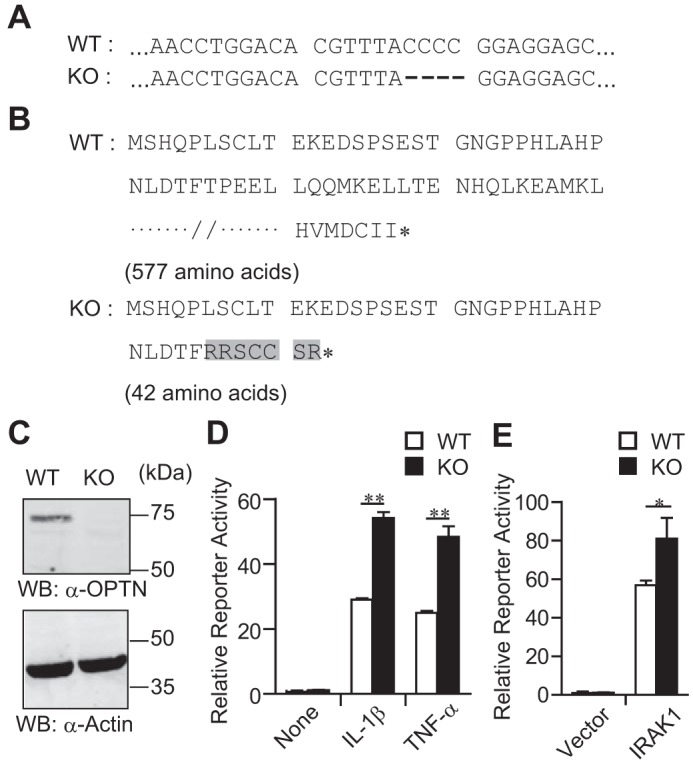
Biallelic deletion that targets the open reading frame of the OPTN gene in a HEK293 cell line. A, DNA sequences of the OPTN gene in wild-type (WT) and knock-out (KO) cells. A four-nucleotide deletion event is shown by dashes. B, amino acid sequences of OPTN in WT and KO cells. Mutated amino acids in KO cells are highlighted in gray. The asterisk (*) shows the end of an amino acid chain. C, Western blotting (WB) of OPTN and actin(internal control). Data are representative of three independent experiments with similar results. D, ELAM-1 reporter gene assay with cytokine stimulation. After 48 h of ELAM-1 reporter vector transfection, HEK293 cells were stimulated with TNF-α or IL-1β for 5 h. Then, the reporter activity was measured. Data are representative of at least three independent experiments with similar results and are shown as the mean ± SD (n = 3). E, ELAM-1 reporter gene assay with IRAK1 transfection. HEK293 cells were transfected with ELAM-1 reporter vector and IRAK1 expression vector. After 48 h, the reporter activity was measured. Data are representative of three independent experiments with similar results and are shown as the mean ± SD (n = 3). *, p < 0.05; **, p < 0.01.
To address the role of OPTN in IL-1β signaling, ELAM-1 reporter assay was performed with KO HEK293 cells. ELAM-1 reporter activity was significantly higher in KO HEK293 cells than in control (WT) HEK293 cells after either IL-1β or TNF-α stimulation (Fig. 3D). In addition, the IRAK1 transfection-induced ELAM-1 reporter activity was also significantly higher in KO HEK293 cells than in WT HEK293 cells (Fig. 3E).
As HEK293 cells do not respond to LPS stimulation in the ELAM-1 reporter assay, the role of OPTN in TLR signaling was evaluated with a newly established OPTN-knock-out (KO) RAW264.7 cell line, which has a premature stop codon due to a single nucleotide deletion in the OPTN-coding sequence in both alleles (Fig. 4, A and B). Because it was difficult to detect the OPTN protein expression by Western blotting in RAW264.7 cells under our experimental conditions (data not shown), we performed real time PCR to check the mRNA levels of OPTN. Depletion of OPTN mRNA expression was observed in the KO RAW264.7 cells (Fig. 4C), probably due to the nonsense-mediated mRNA decay (15). The ELAM-1 reporter assay demonstrated that the LPS-stimulated reporter activity of KO RAW264.7 cells was significantly up-regulated compared with that of WT RAW264.7 cells (Fig. 4D).
Figure 4.
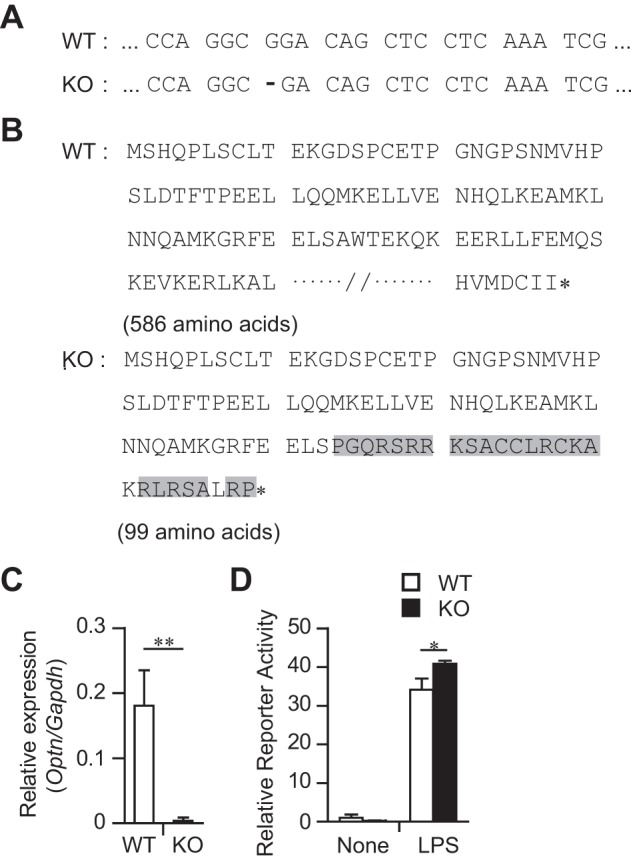
Biallelic deletion that targets the open reading frame of the Optn gene in a RAW264.7 cell line. A, DNA sequences of the Optn gene in wild-type (WT) and knock-out (KO) cells. A one-nucleotide deletion event is shown by a dash. B, amino acid sequences of OPTN in WT and KO cells. Mutated amino acids in KO cells are highlighted in gray. The asterisk (*) shows the end of an amino acid chain. C, RT-PCR for Optn mRNA expression. Data are normalized by Gapdh mRNA expression and shown as the mean ± SD of four independent experiments. D, ELAM-1 reporter gene assay with LPS. RAW264.7 cells were transfected with ELAM-1 reporter vector. After 48 h, cells were stimulated with LPS for 5 h. Then the reporter activity was measured. Data are representative of three independent experiments with similar results and are shown as the mean ± SD (n = 3). *, p < 0.05; **, p < 0.01.
These results suggested that OPTN is a negative regulator of IRAK1-dependent NF-κB signaling pathways.
OPTN controlled TRAF6 polyubiquitination
The IL-1β/LPS-MyD88 axis induces phosphorylation of IRAK1 (16). Then, phosphorylated IRAK1 activates TRAF6 through the K63-linked polyubiquitination, which is a prerequisite for NF-κB activation (16). We examined the effect of OPTN on TRAF6 polyubiquitination by immunoprecipitation assays. Co-transfection of HA-tagged ubiquitin and FLAG-tagged TRAF6 enabled us to detect TRAF6 polyubiquitination in HEK293T cells (Fig. 5A). Of interest, further transfection of Myc-tagged OPTN significantly decreased the level of TRAF6 polyubiquitination (Fig. 5, A and B). Thus, OPTN seems to negatively regulate NF-κB activation by preventing TRAF6 polyubiquitination.
Figure 5.
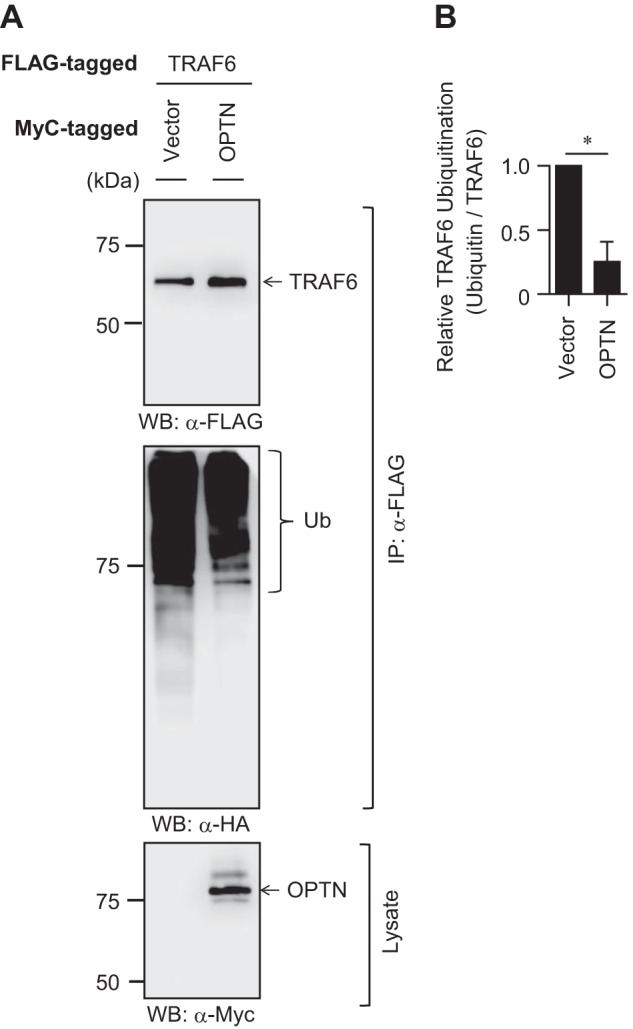
Regulation of TRAF6 polyubiquitination by OPTN. A, HEK293T cells were transfected with FLAG-tagged TRAF6 and HA-tagged ubiquitin with or without Myc-tagged OPTN. After 36 h of transfection, immunoprecipitation (IP) was performed using anti-FLAG antibody. Then Western blotting (WB) was performed using anti-FLAG, anti-HA, or anti-Myc antibody. Data are representative of three independent experiments with similar results. B, relative polyubiquitination level of TRAF6 in OPTN-transfected HEK293T cells. Data are the mean ± SD of three independent experiments and are presented relative to the vector-transfected control cells, set as 1. *, p < 0.05.
OPTN mutation H486R failed to prevent IL-1β/LPS-induced NF-κB activation
The OPTN H486R mutant loses the binding ability toward the deubiquitinase CYLD and, thus, cannot inhibit TNF-α-induced NF-κB activation (10). To confirm the involvement of CYLD recruitment in the OPTN-mediated inhibition of IL-1β/LPS-induced NF-κB activation, we performed ELAM-1 reporter assays with the OPTN H486R mutant in HEK293 cells. Consistent with the previous report (10), the H486R mutation has impaired the suppressive function of OPTN in TNF-α-induced NF-κB activation (Fig. 6A). In addition, the H486R mutant displayed a similar impairment in the prevention of IL-1β-induced NF-κB activation (Fig. 6A). In RAW264.7 cells, the H486R mutant showed a functional defect in the prevention of LPS-induced NF-κB activation (Fig. 6B). Furthermore, we found that IRAK1-induced NF-κB activation was significantly inhibited in the presence of OPTN WT but not in the presence of the OPTN H486R mutant (Fig. 6C). Of note, we found that CYLD inhibited ELAM-1 reporter activity in cooperation with OPTN WT; however, it did less well with OPTN H486R mutant (Fig. 6C). These data suggest that OPTN forms a complex with IRAK1 and inhibits the IRAK1/TRAF6-NF-κB axis by preventing TRAF6 polyubiquitination.
Figure 6.
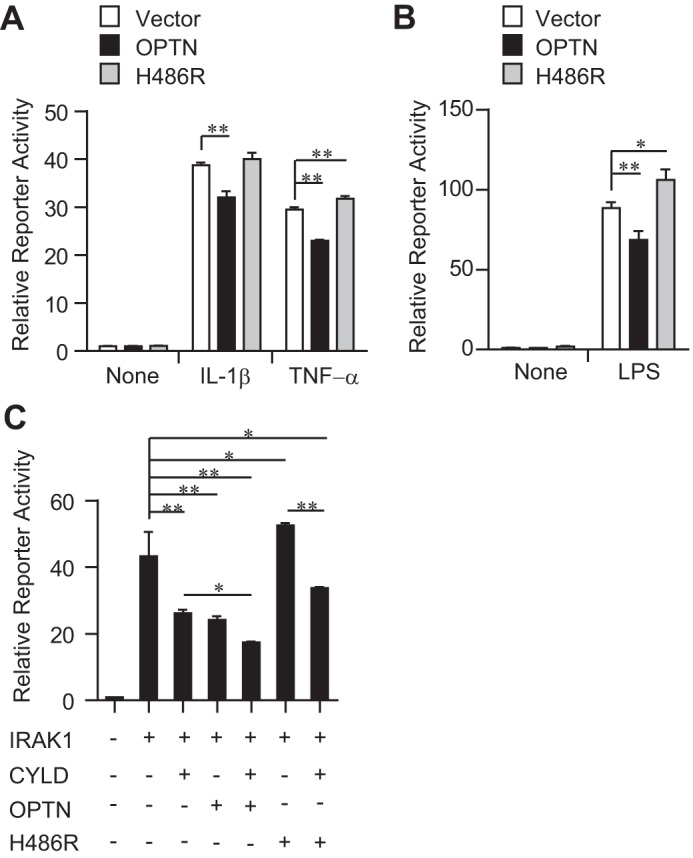
Effects of the OPTN H486R mutant on ELAM-1 reporter activity. A, HEK293 cells were transfected with ELAM-1 reporter vector and with either OPTN WT or H486R mutant expression vector. After 48 h, cells were stimulated with TNF-α or IL-1β for 5 h. Then, the reporter activity was measured. B, RAW264.7 cells were transfected with ELAM-1 reporter vector and with either OPTN WT or H486R mutant expression vector. After 48 h, cells were stimulated with LPS for 5 h. Then, the reporter activity was measured. C, HEK293 cells were transfected with ELAM-1 reporter vector along with other expression vectors as indicated. After 24 h, the reporter activity was measured. All data are representative of three independent experiments with similar results and are shown as the mean ± SD (n = 3). *, p < 0.05; **, p < 0.01.
Discussion
OPTN negatively regulates TNF-α receptor signaling by localizing in the cytoplasm (17). Mechanistically, OPTN forms a complex with CYLD and polyubiquitinated receptor-interacting protein (RIP). As a member of the deubiquitinating enzyme family, CYLD deubiquitinates RIP and thereby prevents binding of nuclear factor κB essential modulator (NEMO) to RIP (10), resulting in the inhibition of TNF-α-induced NF-κB activation (17, 18). In this study, we reveal that OPTN is a novel binding partner of IRAK1 and prevents the IRAK1-TRAF6-NF-κB signaling axis. It is worth noting that the IRAK1 is involved in IL-1 receptor and TLR signaling, but not in TNF-α receptor signaling (1, 19).
Considering the data of the OPTN H486R mutant that is deficient in the binding to CYLD (10), OPTN is likely to exert an inhibitory effect on IL-1 receptor and TLR signaling by recruiting CYLD to polyubiquitinated molecules. Thus, the potential target molecule(s) of CYLD is likely to be not only associated with IRAK1 but also essential for IL-1 receptor and TLR signaling, similar to RIP in TNF-α receptor signaling. We assumed that the polyubiquitinated TRAF6 exhibits the requirements to be considered as a CYLD target (supplemental Fig. S1). Indeed, our results consistently demonstrate that OPTN reduces the polyubiquitination level of TRAF6, although other molecular mechanisms may also be involved in the OPTN inhibition of the IRAK1-TRAF6-NF-κB signaling axis.
OPTN knockdown in a mouse neuroblastoma cell line promoted cell death in response to TNF-α, which was associated with an anomalous increase in NF-κB activity (20). In humans, OPTN mutations that lack the ability to inhibit NF-κB activity were reported to be involved in the pathogenesis of amyotrophic lateral sclerosis (ALS) (18), a fatal neurodegenerative disorder affecting large motor neurons of the brain and spinal cord. These reports indicate that aberrant activation of NF-κB can underlie ALS. Here, we propose a novel mechanism for the ALS pathogenesis in which the aberrant NF-κB activation by ALS-related OPTN mutations is partly due to enhanced signaling from IL-1 receptor and TLR (21, 22). Indeed, the spinal cord of ALS patients contained higher levels of IL-1β (23), which induced neural inflammation and accelerated ALS pathogenesis (24). In the case of TLR, high expression of neuronal TLR4 was reported in the spinal cords of ALS patients (25). TLR4 was shown to be stimulated by high mobility group box1, which is also extensively present in the spinal cord of ALS patients and regulates neurotoxicity (26). Therefore, inhibition of NF-κB activation by targeting IL-1 receptor and TLR4 signaling via OPTN may represent a potential therapeutic strategy for treatment of ALS patients.
Experimental procedures
Cells and plasmids
HEK293T, HEK293, and RAW264.7 cells were cultured in DMEM (Wako Pure Chemical Industries, Osaka, Japan) supplemented with 10% heat-inactivated FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. Yeast strain AH109 was obtained from Clontech Laboratories (Mountain View, CA). To delete the Optn gene, RAW264.7 cells were transfected with CRISPR/CAS9 KO plasmid (sc-427990: Santa Cruz Biotechnology) using Neon transfection system (Thermo Fisher Scientific, Waltham, MA). After a couple of days, GFP-positive RAW264.7 cells were sorted by using a cell sorter SH800 (SONY, Tokyo, Japan). Sorted cells were plated on 10-cm dishes at 100–400 cells/dish. Isolated single colonies grown on the plates were picked up manually. OPTN-KO RAW264.7 cell lines were identified by DNA sequencing for the target sites. HEK293 cells were transfected with CRISPR/Cas9 KO plasmid, (PX458: Addgene), which was designed to target the human OPTN gene, using PEI-MAX reagent (Polysciences, Warrington, PA). Single clones were isolated, and KO lines were identified as described above for RAW264.7 cells.
We used the plasmids pEF6-FLAG, pEF6-Myc, pEF6-IRAK1-FLAG, pEF6-IRAK1-KD-FLAG, pEF6-TRAF6-FLAG, and pELAM1-Luc, which were described previously (12, 27). pcDNA3-HA-ubiquitin was obtained from Addgene (Cambridge, MA) (28). pcDNA3-FLAG-CYLD (Isoform 2) was a kind gift from Dr. Fuminori Tokunaga (Osaka City University) (29). Human IRAK1 was cloned into the BamHI/EcoRI site in pGBKT7 (Clontech Laboratories). phRLTK-Luc was obtained from Promega Corp. (Madison, WI). The expression plasmids for full-length OPTN and truncated (amino acids 425–577) OPTN were created by using human cDNA fragments that were amplified by PCR and subcloned into the BamHI/EcoRI site in the pEF6-Myc vector. The H486R mutation was introduced into pEF6-OPTN-Myc by changing the codon CAT into CGT.
Yeast two-hybrid screening
Yeast AH109 clones were cultured on YPDA-agarose plates (20 g/liter Bacto Peptone, 10 g/liter yeast extract, 20 g/liter agar, 0.003% adenine hemisulfate) at 30 °C for 3 days. Then, yeast AH109 colonies were picked and cultured in YPDA broth at 30 °C for 16–20 h. Cultured broth was centrifuged for 5 min at 850 × g at room temperature, and the supernatant was discarded. The pellet was washed using TE (10 mm Tris-HCl (pH 7.5) and 1 mm EDTA) solution and then resuspended in TE containing 0.1 m LiAc (pH 7.5) (competent yeast AH109 cell solution).
Two-hybrid screening was performed using a human leukocyte cDNA library (Clontech Laboratories) according to the manufacturer's instructions. In brief, pGBKT7-IRAK1, the human leukocyte cDNA library (Clontech Laboratories) preincubated (100 °C for 20 min) salmon testis carrier DNA (Sigma) were mixed at 1:0.5:20 ratio and added to the competent yeast AH109 cell solution and PEG/LiAc solution. After vortexing for 1 min, the mixture was incubated in a shaking water bath (200 rpm, 30 °C) for 30 min, and DMSO was added up to 10% of the solution. The solution was further incubated at 42 °C for 15 min and then on ice for 1 min. The solution of transformed yeast AH109 cells was centrifuged at 10,000 × g for 10 s. The supernatant was removed, and the pellet was resuspended in YPDA broth and spread on synthetically defined (SD) media lacking leucine/tryptophan/histidine + 25 mm 3-amino-1,2,4-triazole (3-AT) plates (Sigma). After 6–8 days of culture at 30 °C, positive blue colonies were picked and spread on SD/−leucine/−tryptophan/−histidine/−adenine + 25 mm 3-AT plates. The picking and spreading of colonies was then repeated using SD/−leucine/−tryptophan/−histidine + 25 mm 3-AT plates and SD/−leucine/−tryptophan + 25 mm 3-AT plates.
Plasmid isolation from yeast cells
Yeast AH109 cells, cultured on SD/−leucine/−tryptophan/−histidine + 25 mm 3-AT broth at 30 °C, were centrifuged. The pellet was then resuspended in TE solution containing 5 units/μl lyticase (Sigma) and incubated in a shaking water bath (230 rpm, 37 °C) for 60 min. After SDS up to 14.3% was added, the solution was subjected to 1 freeze-thaw cycle (from −20 °C to room temperature). Freeze-thaw reagents and other unwanted substances were removed from the solution using phenol/chloroform/isoamyl alcohol (25:24:1) solution, and plasmids were purified according to the standard ethanol precipitation method (30).
Immunoprecipitation
Immunoprecipitation was carried out as described previously (31). Briefly, HEK293T cells were transfected with the indicated expression vectors by the calcium phosphate method (32). Two days after transfection, cells were washed with PBS, lysed in radioimmune precipitation assay buffer (10 mm sodium phosphate (pH 7.2), 150 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 50 mm NaF, and 0.2 mm Na3VO4), and incubated on ice for 15 min. The cell lysate was clarified by centrifugation and then subjected to immunoprecipitation with anti-FLAG (M2) monoclonal antibody and M2-agarose beads (Sigma).
Western blotting
Cells were lysed in radioimmune precipitation assay buffer as described above. Equal amounts of proteins from the cell lysate samples or the immunoprecipitated samples were subjected to 10% SDS-PAGE and analyzed by immunoblotting with peroxidase-conjugated anti-FLAG (M2, Sigma), anti-c-Myc (9E10, Roche Diagnostics), or anti-HA (3F10, Roche Diagnostics). Anti-OPTN (sc-271549) was obtained from Santa Cruz Biotechnology. Bound antibodies were visualized by chemiluminescence after incubation with Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA).
Evaluation of TRAF6 polyubiquitination level
After the Western blot analysis of immunoprecipitated samples was performed as described above, the band intensities of HA-tagged ubiquitin and FLAG-tagged TRAF6 were quantitated by densitometry using a C-DiGit blot scanner (LI-COR Biotechnology, Lincoln, NE). To evaluate the levels of TRAF6 polyubiquitination, the intensity ratio of immunoprecipitated ubiquitin to immunoprecipitated TRAF6 was calculated. For normalization, the ratio of pEF6-Myc (vector control) transfection sample was set as 1 in each experiment.
Luciferase assay
To perform transfection with the indicated expression vectors along with the pELAM1-Luc reporter and phRLTK-Luc vectors, HEK293 cells and RAW264.7 cells were transfected with calcium phosphate and Hillymax (Dojindo, Kumamoto, Japan), respectively. After 48 h of transfection, luciferase activity was measured by the Dual-Luciferase Reporter Assay System according to the manufacturer's instructions (Promega). For cell stimulation, IL-1β, TNF-α, or LPS was used at a concentration of 10 ng/ml, after which cells were incubated for another 5 h.
Real-time RT-PCR
Total RNA was prepared using RNAiso plus (Takara Bio Inc., Otsu, Japan). mRNA levels were quantified by real time RT-PCR with High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) and SYBR Premix EX TaqII (Takara Bio Inc.) using a LightCycler 3302 (Roche Diagnostics). The primer sequences used were as follows: murine OPTN FW, (5′-ATGTCCCATCAACCTCTGAGC-3′) and REV (5′-TCAAATCGCCCTTTCATAGCTTG-3′); murine GAPDH FW (5′-AGAGCTGAACGGGAAG-3′) and REV, (5′-GAAGTCGCAGGAGACA-3′).
Statistical analysis
Student's t test (two-tailed) was used to compare between 2 groups. For more than three groups, one-way analysis of variance with Tukey's test was used to compare among all groups. One-way analysis of variance with Dunnett's test was performed to compare with the control group. *, p < 0.05; **, p < 0.01.
Author contributions
M. T. designed the research project and performed the experiments. S. T. and A. H. generated the KO cell line using CRISPR/CAS9 systems and performed the experiments. D. Y. and T. T. performed the experiments. S. I. designed the research project and wrote the manuscript. T. M. directed the research, performed experiments, and wrote the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Dr. Muta Tatsushi (Tohoku University) for providing the expression vectors and suggestions. We are grateful to Daiki Kobayashi (Akita University) for technical assistance.
This work was supported by JSPS KAKENHI Grant JP 16H05136 (to S. I.). The authors declare that they have no conflicts of interest regarding the contents of this article.
This article contains supplemental Table 1 and Fig. S1.
- TLR
- toll-like receptor
- ALS
- amyotrophic lateral sclerosis
- 3-AT
- 3-amino-1,2,4-triazole
- ELAM-1
- endothelial-leukocyte adhesion molecule-1
- IRAK1
- interleukin-1 receptor-associated kinase 1
- KD
- kinase dead
- NF-κB
- nuclear factor κB
- OPTN
- optineurin
- TRAF
- tumor necrosis factor receptor associated factor
- MyD88
- myeloid differentiation primary-response protein 88
- RIP
- receptor-interacting protein
- SD
- synthetically defined.
References
- 1. Akira S., and Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 [DOI] [PubMed] [Google Scholar]
- 2. Akira S., Uematsu S., and Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 3. Deguine J., and Barton G. M. (2014) MyD88: a central player in innate immune signaling. F1000Prime Rep. 6, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Loiarro M., Gallo G., Fantò N., De Santis R., Carminati P., Ruggiero V., and Sette C. (2009) Identification of critical residues of the MyD88 death domain involved in the recruitment of downstream kinases. J. Biol. Chem. 284, 28093–28103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao Z., Xiong J., Takeuchi M., Kurama T., and Goeddel D. V. (1996) TRAF6 is a signal transducer for interleukin-1. Nature 383, 443–446 [DOI] [PubMed] [Google Scholar]
- 6. Lomaga M. A., Yeh W. C., Sarosi I., Duncan G. S., Furlonger C., Ho A., Morony S., Capparelli C., Van G., Kaufman S., van der Heiden A., Itie A., Wakeham A., Khoo W., Sasaki T., Cao Z., Penninger J. M., et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13, 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Naito A., Azuma S., Tanaka S., Miyazaki T., Takaki S., Takatsu K., Nakao K., Nakamura K., Katsuki M., Yamamoto T., and Inoue J. (1999) Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 4, 353–362 [DOI] [PubMed] [Google Scholar]
- 8. Thomas R. (2005) The TRAF6-NFκB signaling pathway in autoimmunity: not just inflammation. Arthritis Res. Ther. 7, 170–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamin T. T., and Miller D. K. (1997) The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J. Biol. Chem. 272, 21540–21547 [DOI] [PubMed] [Google Scholar]
- 10. Nagabhushana A., Bansal M., and Swarup G. (2011) Optineurin is required for CYLD-dependent inhibition of TNFα-induced NF-κB activation. PLoS ONE 6, e17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cao Z., Henzel W. J., and Gao X. (1996) IRAK: a kinase associated with the interleukin-1 receptor. Science 271, 1128–1131 [DOI] [PubMed] [Google Scholar]
- 12. Ohba T., Ariga Y., Maruyama T., Truong N. K., Inoue J., and Muta T. (2012) Identification of interleukin-1 receptor-associated kinase 1 as a critical component that induces post-transcriptional activation of IκBζ. FEBS J 279, 211–222 [DOI] [PubMed] [Google Scholar]
- 13. Matsuo S., Yamazaki S., Takeshige K., and Muta T. (2007) Crucial roles of binding sites for NF-κB and C/EBPs in IκBζ-mediated transcriptional activation. Biochem. J. 405, 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sudhakar C., Nagabhushana A., Jain N., and Swarup G. (2009) NF-κB mediates tumor necrosis factor α-induced expression of optineurin, a negative regulator of NF-κB. PLoS ONE 4, e5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang Y. F., Imam J. S., and Wilkinson M. F. (2007) The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 76, 51–74 [DOI] [PubMed] [Google Scholar]
- 16. Hu H., and Sun S. C. (2016) Ubiquitin signaling in immune responses. Cell Res. 26, 457–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu G., Wu C. J., Zhao Y., and Ashwell J. D. (2007) Optineurin negatively regulates TNFα-induced NF-κB activation by competing with NEMO for ubiquitinated RIP. Curr. Biol. 17, 1438–1443 [DOI] [PubMed] [Google Scholar]
- 18. Maruyama H., Morino H., Ito H., Izumi Y., Kato H., Watanabe Y., Kinoshita Y., Kamada M., Nodera H., Suzuki H., Komure O., Matsuura S., Kobatake K., Morimoto N., Abe K., et al. (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226 [DOI] [PubMed] [Google Scholar]
- 19. Brenner D., Blaser H., and Mak T. W. (2015) Regulation of tumour necrosis factor signalling: live or let die. Nat. Rev. Immunol. 15, 362–374 [DOI] [PubMed] [Google Scholar]
- 20. Akizuki M., Yamashita H., Uemura K., Maruyama H., Kawakami H., Ito H., and Takahashi R. (2013) Optineurin suppression causes neuronal cell death via NF-κB pathway. J. Neurochem. 126, 699–704 [DOI] [PubMed] [Google Scholar]
- 21. O'Neill L. A., and Kaltschmidt C. (1997) NF-κ B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 20, 252–258 [DOI] [PubMed] [Google Scholar]
- 22. Grilli M., and Memo M. (1999) Nuclear factor-κB/Rel proteins: a point of convergence of signalling pathways relevant in neuronal function and dysfunction. Biochem. Pharmacol. 57, 1–7 [DOI] [PubMed] [Google Scholar]
- 23. Li M., Ona V. O., Guégan C., Chen M., Jackson-Lewis V., Andrews L. J., Olszewski A. J., Stieg P. E., Lee J. P., Przedborski S., and Friedlander R. M. (2000) Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 288, 335–339 [DOI] [PubMed] [Google Scholar]
- 24. Meissner F., Molawi K., and Zychlinsky A. (2010) Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 13046–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Casula M., Iyer A. M., Spliet W. G., Anink J. J., Steentjes K., Sta M., Troost D., and Aronica E. (2011) Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience 179, 233–243 [DOI] [PubMed] [Google Scholar]
- 26. Maroso M., Balosso S., Ravizza T., Liu J., Aronica E., Iyer A. M., Rossetti C., Molteni M., Casalgrandi M., Manfredi A. A., Bianchi M. E., and Vezzani A. (2010) Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16, 413–419 [DOI] [PubMed] [Google Scholar]
- 27. Motoyama M., Yamazaki S., Eto-Kimura A., Takeshige K., and Muta T. (2005) Positive and negative regulation of nuclear factor-κB-mediated transcription by IκBζ, an inducible nuclear protein. J. Biol. Chem. 280, 7444–7451 [DOI] [PubMed] [Google Scholar]
- 28. Kamitani T., Kito K., Nguyen H. P., and Yeh E. T. (1997) Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 272, 28557–28562 [DOI] [PubMed] [Google Scholar]
- 29. Tokunaga F., Nishimasu H., Ishitani R., Goto E., Noguchi T., Mio K., Kamei K., Ma A., Iwai K., and Nureki O. (2012) Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-κB regulation. EMBO J. 31, 3856–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sambrook J., and Russell D. W. (2006) Standard ethanol precipitation of DNA in microcentrifuge tubes. CSH Protoc. 10.1101/pdb.prot4456 [DOI] [PubMed] [Google Scholar]
- 31. Fujimoto T., Yamazaki S., Eto-Kimura A., Takeshige K., and Muta T. (2004) The amino-terminal region of toll-like receptor 4 is essential for binding to MD-2 and receptor translocation to the cell surface. J. Biol. Chem. 279, 47431–47437 [DOI] [PubMed] [Google Scholar]
- 32. Chen C., and Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7, 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.