

Abstract
The integrity of the distinguishing, multilaminate cell envelope surrounding mycobacteria is critical to their survival and pathogenesis. The prevalence of phosphatidylinositol mannosides in the cell envelope suggests an important role in the mycobacterial life cycle. Indeed, deletion of the pimE gene (ΔpimE) encoding the first committed step in phosphatidylinositol hexamannoside biosynthesis in Mycobacterium smegmatis results in the formation of smaller colonies than wild-type colonies on Middlebrook 7H10 agar. To further investigate potential contributors to cell-envelope mannan biosynthesis while taking advantage of this colony morphology defect, we isolated spontaneous suppressor mutants of ΔpimE that reverted to wild-type colony size. Of 22 suppressor mutants, 6 accumulated significantly shorter lipomannan or lipoarabinomannan. Genome sequencing of these mutants revealed mutations in genes involved in the lipomannan/lipoarabinomannan biosynthesis, such as those encoding the arabinosyltransferase EmbC and the mannosyltransferase MptA. Furthermore, we identified three mutants carrying a mutation in a previously uncharacterized gene, MSMEG_5785, that we designated lmeA. Complementation of these suppressor mutants with lmeA restored the original ΔpimE phenotypes and deletion of lmeA in wild-type M. smegmatis resulted in smaller lipomannan, as observed in the suppressor mutants. LmeA carries a predicted N-terminal signal peptide, and density gradient fractionation and detergent extractability experiments indicated that LmeA localizes to the cell envelope. Using a lipid ELISA, we found that LmeA binds to plasma membrane phospholipids, such as phosphatidylethanolamine and phosphatidylinositol. LmeA is widespread throughout the Corynebacteriales; therefore, we concluded that LmeA is an evolutionarily conserved cell-envelope protein critical for controlling the mannan chain length of lipomannan/lipoarabinomannan.
Keywords: bacterial genetics, carbohydrate metabolism, glycolipid, glycosyltransferase, Mycobacterium smegmatis, PimE, cell envelope, lipomannan
Introduction
The rise in multidrug-resistant Mycobacterium tuberculosis is a global concern. One avenue to search for novel drug targets is through the biosynthetic pathway of the mycobacterial cell-envelope components (1). Several enzymes in biosynthetic pathways of the key cell-envelope glycolipids, such as phosphatidylinositol mannosides (PIMs),3 lipomannan (LM), and lipoarabinomannan (LAM), are shown or predicted to be essential in M. tuberculosis (2, 3). Moreover, alterations in the LM/LAM structures make both M. tuberculosis and nonpathogenic Mycobacterium smegmatis susceptible to β-lactam antibiotics, which otherwise cannot cross the mycobacterial cell wall efficiently (4). These observations suggest that PIMs/LM/LAM play important structural roles within the cell envelope to maintain the permeability barrier. Furthermore, PIMs/LM/LAM are critical for virulence, with recognized host factor interactions (5–7).
The biosynthesis of PIMs/LM/LAM begins with phosphatidylinositol (PI) and is mediated by sequential actions of two mannosyltransferases, PimA and PimB′, followed by the acylation of the mannose residue by an acyltransferase, PatA, to produce AcPIM2 (Fig. 1A) (8–11). The sequential addition of mannoses to AcPIM2 is mediated by an undetermined mannosyltransferase(s), resulting in the production of AcPIM4 (12). The AcPIM4 intermediate serves as a branch point for PIMs/LM/LAM biosynthesis, feeding into either LM/LAM formation or AcPIM6 production (Fig. 1A). The α1–2-mannosyltransferase PimE transfers a mannose residue from polyprenol phosphate-mannose (PPM) to AcPIM4, committing the pathway to the production of AcPIM6 (13). LM/LAM biosynthesis is mediated by α1–6-mannosyltransferase MptA (MSMEG_4241) and the α1–2-mannosyltransferase MptC (MSMEG_4247) (14–18). MptA is a PPM-dependent polymerase of the α1–6-mannose backbone, and the deletion of mptA results in the accumulation of an immature LM intermediate that carries 5–20 mannose residues instead of the 21–34 mannose residues found in mature LM/LAM (14). MptC is another PPM-dependent mannosyltransferase, involved in monomannose side chain addition. Although the functional roles of monomannose side chains remain obscure, we have previously suggested that mannan chain biosynthesis requires a balance between the enzymatic activities of MptA and MptC (18).
Figure 1.
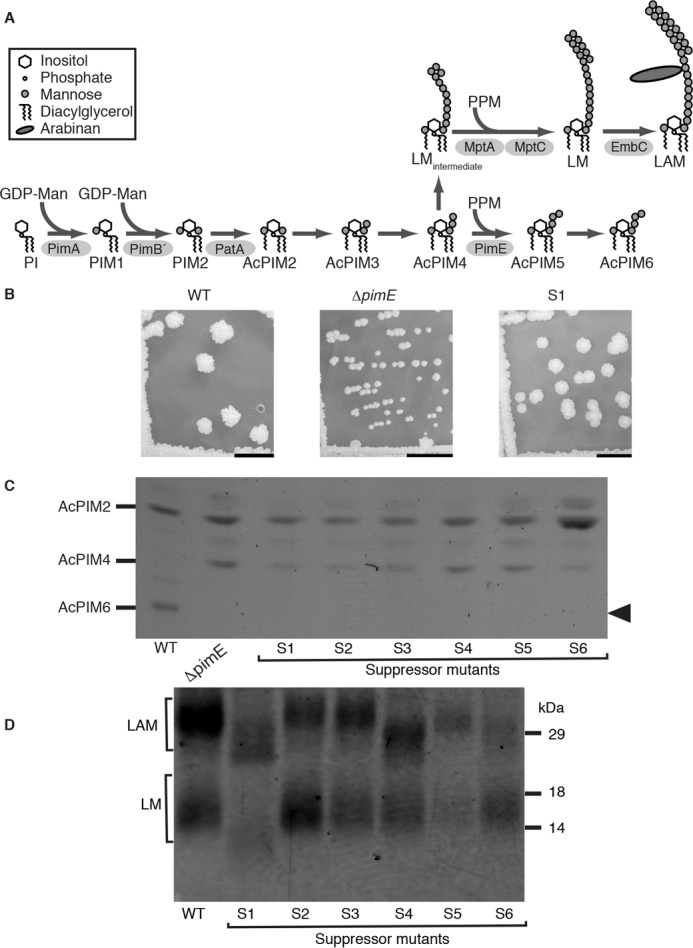
Isolation of ΔpimE suppressor mutants. A, PIMs/LM/LAM biosynthetic pathway. Mannosyltransferases involved in the biosynthesis of the LM intermediate are unknown. MptA is α1–6-mannosyltransferase involved in the mannan backbone elongation, whereas MptC is α1–2-mannosyltransferase that adds monomannose side chains. The mannose donors, GDP-mannose (GDP-Man) and PPM, are indicated for mannosyltransferase reactions. B, colony morphology of WT, ΔpimE, and the suppressor mutant S1. The suppressor mutant S1 (as well as the other 21 suppressor mutants; not shown) restored WT colony morphology. Scale bar, 1 cm. C, profile of PIMs purified from the suppressor mutants. Lipid extracts of WT, ΔpimE, and the suppressor mutants S1–S6 were separated by TLC and visualized by orcinol staining. Only a part of the TLC plate is shown. None of the suppressor mutants were able to restore the production of AcPIM6 (arrowhead). See supplemental Fig. S1 for the other suppressor mutants. D, LM/LAM profile of the suppressor mutants, showing the small LM/LAM phenotype of the mutant S1. Extracts of LM/LAM were separated on SDS-PAGE and visualized by ProQ Emerald 488 glycan staining. See supplemental Fig. S2 for the other suppressor mutants.
LpqW is a possible regulator of LM/LAM biosynthesis at the AcPIM4 branching point (19–21). Mutants lacking lpqW show defective LM/LAM biosynthesis and colony size smaller than WT. These phenotypic defects were spontaneously resolved in suppressor mutants of ΔlpqW, and mutations in pimE identified in these mutants suggest that alterations in AcPIM6 biosynthesis can phenotypically compensate for defects in LM/LAM (19). These studies highlight the complex interplay of PIMs/LM/LAM to create a functional cell envelope. In the current study, our initial observation that ΔpimE shows smaller colony morphology when grown on Middlebrook 7H10 agar led us to isolate spontaneous suppressors of ΔpimE, which restored the small colony morphology to that of WT. In some of these suppressor mutants, structural alterations in LM/LAM were caused by mutations in a previously uncharacterized protein, MSMEG_5785. Characterization of MSMEG_5785 reveals this protein to be a cell envelope–associated phospholipid-binding protein, involved in the mannan elongation of LM/LAM.
Results
Isolation of ΔpimE suppressor mutants
When the ΔpimE M. smegmatis mutant was grown on Middlebrook 7H10 agar plates for 4 days at 37 °C, we noticed that the colony size of the mutant was significantly smaller than WT (WT, 4.17 ± 0.91 mm; ΔpimE, 1.31 ± 0.45 mm; average ± S.D., n = 50). Interestingly, we noticed occasional appearances of spontaneous large colony mutants after subculturing (Fig. 1B). We isolated a total of 22 spontaneous large colony revertants. Because ΔpimE was generated by replacing the central 213 bp of the pimE gene with a kanamycin-resistant gene (13), it seemed unlikely that the pimE gene was restored. Indeed, all 22 isolates remained kanamycin-resistant (not shown) and failed to produce AcPIM6 (Fig. 1C and supplemental Fig. S1). These results indicated that these revertants are suppressor mutants rather than same-site revertants of the pimE gene.
Suppressor mutants show changes in LM/LAM structure
As described in the Introduction, the growth defect observed in ΔlpqW was restored by suppressor mutations in pimE (19). Inspired by these studies, we examined whether our suppressor mutants of ΔpimE show structural changes in LM/LAM. Various changes in both LM/LAM size and abundance were noted in several of the suppressor mutants (Fig. 1D and supplemental Fig. S2). For example, the suppressor mutant S4 shows smaller LAM, whereas LM appears comparable with that of the WT. Another prominent example is found in S1, S10, S20, S21, and S22, where both LM and LAM appeared smaller. We also found mutants with changes in the amount of LM/LAM (e.g. S5, S9, S12, S18, and S19). Whereas there are a number of suppressor mutants that show no apparent changes in LM/LAM, these initial observations suggested that one way for the ΔpimE mutant to revert back to the WT colony morphology is to alter the abundance or structures of LM/LAM.
Whole-genome sequencing reveals mutations in genes involved in LM/LAM biosynthesis
We used whole-genome sequencing of the parental ΔpimE mutant and the subset of suppressor mutants with apparent structural changes in LM/LAM to identify genetic alterations consistent with altered LM/LAM biosynthesis (supplemental Data Set S1). Table 1 summarizes the mutations found in the sequenced mutant strains. In the mutant S4, where smaller LAM was observed, we found a point mutation in embC, an arabinosyltransferase involved in LAM biosynthesis, which was consistent with the LAM-specific defect. For other mutants accumulating smaller LM and LAM, such as S1, S10, S20, S21, and S22, we suspected that there might be a mutation in the mptA gene, which encodes α1–6-mannosyltransferase involved in LM/LAM mannan elongation. We therefore PCR-amplified the mptA gene and determined the gene sequence by the Sanger method. As suspected, we identified a mutation in the mptA gene amplified from S20 and S21 (Table 1). These two mutants have the same 24-bp deletion, suggesting that they originated from the same parental mutant. In contrast, we found no mutation in the mptA gene from S1, S10, and S22. Therefore, we subjected S1 and S10 to whole-genome sequencing and found missense mutations (G170D and V181G, respectively) in the gene MSMEG_5785, which has no previously assigned function (Fig. 2). We asked whether the third suppressor mutant (S22) had mutations in MSMEG_5785 by PCR amplification (see supplemental Table S1 for primer sequences). Interestingly, S22 had an insertion of a 2,276-bp transposon (TnpR) after the first 14 bp of the MSMEG_5785 gene (Fig. 2). The TnpR insertion into the near 5′-end of the gene suggested that the phenotypic changes in LM/LAM are probably due to the loss of the gene function. Given these mutant characteristics and additional features described below, we termed MSMEG_5785 as LmeA (lipomannan elongation factor A). To confirm loss of LmeA function, we introduced a Phsp60-lmeA-HA expression vector into each suppressor mutant. No changes in PIMs were detected in the complemented suppressor mutants (Fig. 3A). Importantly, mature LM and LAM were restored in each mutant when complemented with the Phsp60-lmeA-HA vector (Fig. 3B). Additionally, the complemented strains restored the ΔpimE small colony morphology (Fig. 3C). Taken together, 3 of the 22 suppressor mutants of ΔpimE carried a loss-of-function mutation in the novel gene lmeA.
Table 1.
Mutations in the suppressor mutants that showed changes in LM/LAM
| Phenotype | Strain | Gene | Analysis | Mutation type |
|---|---|---|---|---|
| Small LAM | S4 | MSMEG_6385 (embC) | Whole genome | Missense (P352L) |
| Small LM/LAM | S1 | MSMEG_5785 (lmeA) | Whole genome | Missense (G170A) |
| S10 | MSMEG_5785 (lmeA) | Whole genome | Missense (V181G) | |
| S22 | MSMEG_5785 (lmeA) | PCR and Sanger | Transposon insertion | |
| S20/S21 | MSMEG_4241 (mptA) | PCR and Sanger | 24-bp deletiona |
a The deletion included 2 bp of the 3′-end of the gene, the stop codon, and 19 bp of the 3′-untranslated region.
Figure 2.

Three suppressor mutants (S1, S10, and S22) have distinct mutations in MSMEG_5785. The red lines indicate where the mutations were found. Mutated bases and amino acids are shown in red. The insertion of a 2.27-kbp transposon in S22 caused a frameshift mutation.
Figure 3.
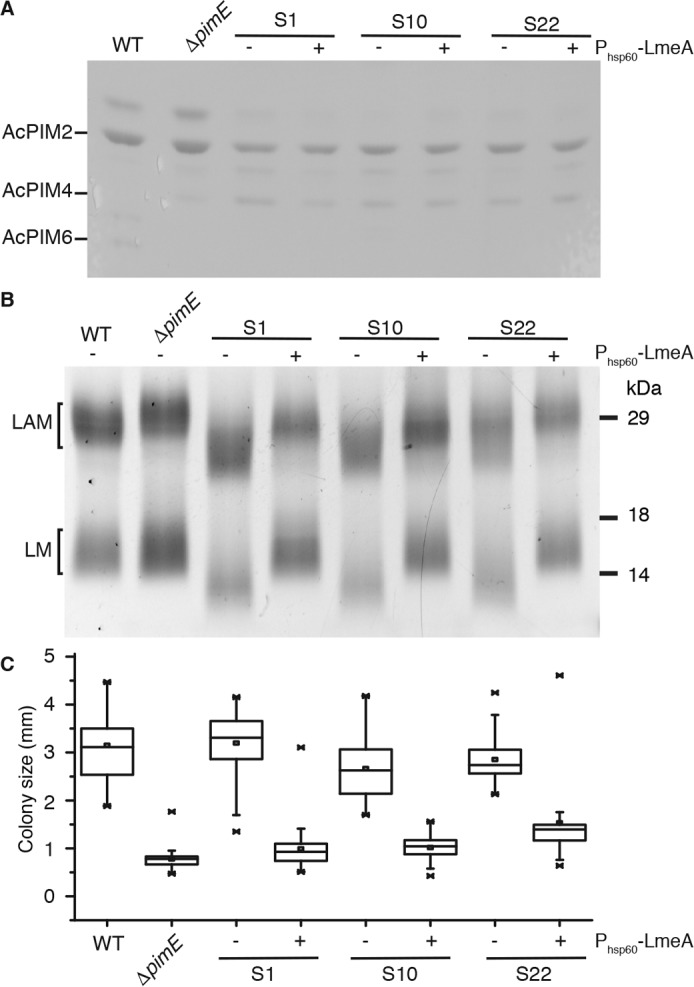
Complementation of the suppressor mutants S1, S10, and S22 by lmeA-HA restores the ΔpimE phenotype. A, AcPIM6 not restored by lmeA-HA complementation. PIMs were purified from the suppressor mutants S1, S10, and S22 with or without complementation, separated by TLC, and visualized by orcinol staining. Only a part of the TLC plate is shown. B, the normal sizes of LM/LAM restored upon complementation. LM/LAM purified from the suppressor mutants with or without complementation were separated by SDS-PAGE and visualized by ProQ Emerald 488 glycan staining. C, colony size of the suppressor mutants was restored to the smaller colony morphology of ΔpimE when complemented with LmeA-HA. Box plots of colony sizes (n = 44) are shown. Error bars, S.D.; ×, maximum and minimum data points.
LmeA is critical for LM/LAM mannan chain length maturation
The LM/LAM phenotype of the suppressor mutants suggested a potential role of LmeA in LM/LAM biosynthesis. However, this phenotype could be dependent on the ΔpimE background. To test a direct role of LmeA in LM/LAM biosynthesis, we generated a strain carrying a markerless deletion of lmeA (ΔlmeA) in the WT background (supplemental Fig. S3A). The deletion of lmeA was confirmed by PCR (supplemental Fig. S3B). The ΔlmeA mutant showed the production of smaller LM and more disperse LAM, which corroborates the phenotype of the suppressor mutants (Fig. 4A). Importantly, there were no apparent changes in the biosynthesis of PIMs or other phospholipids (Fig. 4B and supplemental Fig. S4A). Moreover, the ΔlmeA mutant colony morphology was similar to WT (Fig. 4C). By complementing with either Phsp60-lmeA-HA or Pnative-lmeA-HA, mature LM/LAM production was restored (Fig. 4A). We also complemented the deletion mutant with lmeA carrying the point mutations found in the suppressor mutants S1 and S10. Neither of the lmeA mutants was able to restore mature LM/LAM (supplemental Fig. S4B), further supporting our notion that the mutations are loss-of-function. Interestingly, the expression levels of the mutated LmeA proteins were lower than that of WT protein and were undetectable by Western blotting (supplemental Fig. S4C), indicating that the mutant LmeA may be unstable and susceptible to protein degradation. These results indicate that LmeA is involved in LM/LAM biosynthesis, and the lmeA mutant phenotype is independent of the pimE deletion.
Figure 4.
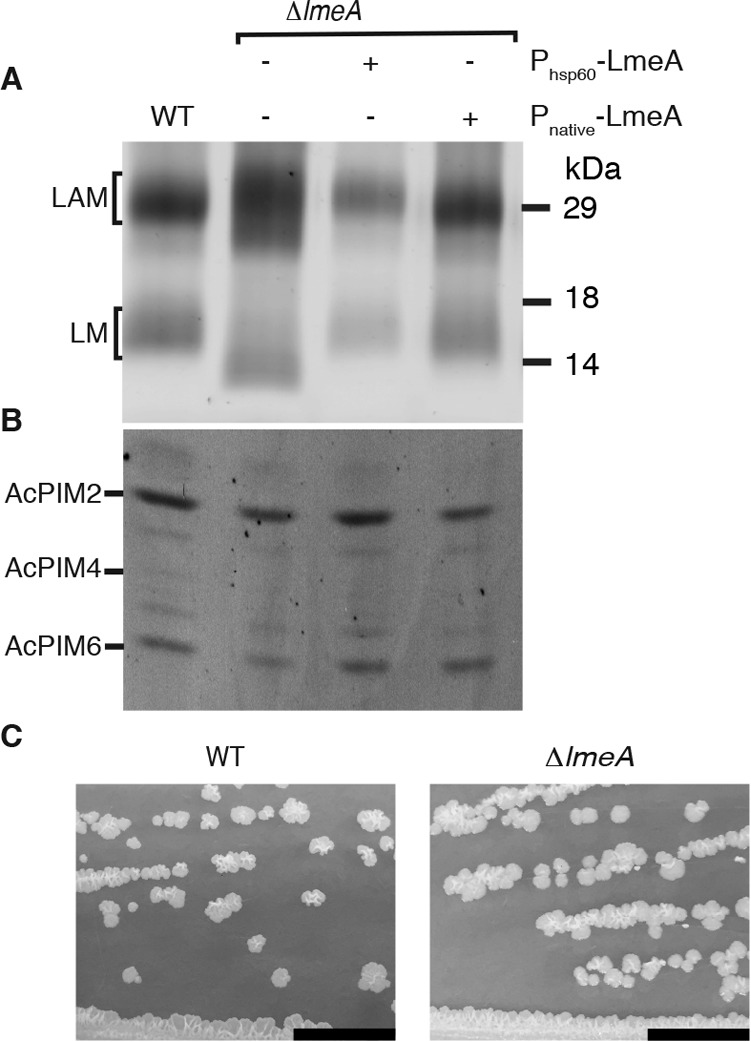
Phenotypes of lmeA deletion mutant. A, lmeA deletion in WT background, resulting in accumulation of smaller LM and diffusely distributed LAM. LM/LAM purified from ΔlmeA and its complemented strains were separated by SDS-PAGE and visualized using ProQ Emerald 488 glycan staining. Expression vectors, Pnative-lmeA-HA and Phsp60-lmeA-HA, are driven by native and strong hsp60 promoters, respectively. Pnative-lmeA-HA was more effective in restoring mature LM/LAM. B, PIMs from ΔlmeA and its complemented strains, showing that ΔMSMEG_5785 does not impact AcPIM6 biosynthesis. Only a part of the TLC plate is shown. C, colony morphology, demonstrating similar morphologies of WT and ΔlmeA colonies. Scale bar, 1 cm.
LmeA is a cell-envelope protein
LmeA is a conserved protein of unknown function that is widely present in the order Corynebacteriales (supplemental Fig. S5A) but apparently absent in other bacteria and other domains of life. Its ortholog in M. tuberculosis, Rv0817c, is 60% identical to M. smegmatis LmeA at the amino acid level, and the amino acid residues for which the missense mutations were found in the suppressor mutants S1 and S10 are both conserved in M. tuberculosis LmeA (supplemental Fig. S5B). LmeA has a conserved hydrophobic region near the amino terminus, and SignalP version 3.0, an algorithm effective for the prediction of mycobacterial signal peptides (22), predicted both Rv0817c and MSMEG_5785 to carry a signal peptide with high probabilities (0.998–1.000). Indeed, M. tuberculosis LmeA is a periplasmic protein identified by a recent secretome analysis (23). Furthermore, M. tuberculosis LmeA is found in the proteome of M. tuberculosis cell lysate but not in that of cell filtrate (24), implying cell-envelope association. In elucidating the accessibility of M. smegmatis LmeA-HA, we noticed that the protein could not be immunoprecipitated from crude cell lysate using anti-HA antibody (Fig. 5A). This is in contrast to GlnA-HA, a cytoplasmic protein readily immunoprecipitated using anti-HA antibody. When we added Triton X-100, a mild detergent, to the cell lysate, we were able to pull down the LmeA-HA protein (Fig. 5A). These data also implied that LmeA-HA is a cell-envelope protein, and the HA epitope is not exposed for the antibody recognition when detergent is not present. To confirm further, we performed sucrose density gradient sedimentation to fractionate the cell envelope, intracellular membrane domain, and cytoplasmic fractions (25, 26). Markers, MptC, PimB′, and Mpa, respectively, showed separation of these three subcellular fractions and demonstrated that LmeA-HA co-localizes to the cell-envelope fraction (Fig. 5B). These data provide further evidence that LmeA is an extracytoplasmic cell envelope-associated protein.
Figure 5.
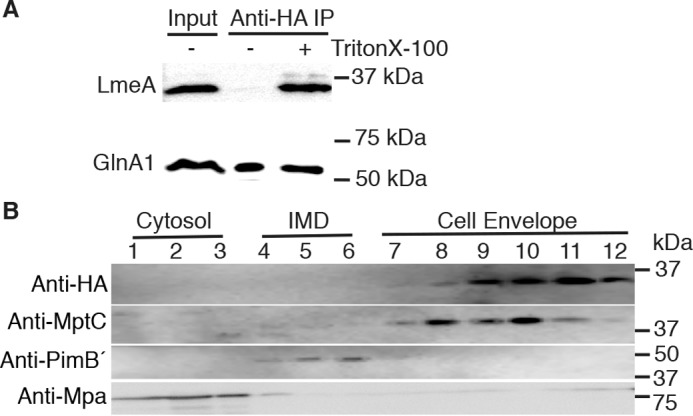
LmeA is a cell-envelope protein. A, immunoprecipitation (IP) of LmeA-HA with (+) or without (−) a mild detergent. Western blotting using anti-HA antibody showed that anti-HA immunoprecipitation of LmeA-HA was possible only when Triton X-100 was present during the incubation. Two separate strains expressing either LmeA-HA (29.5 kDa) or a cytosolic marker, GlnA1-HA (54.6 kDa), were used. B, sucrose gradient sedimentation of LmeA-HA expressed from the native promoter. Expected molecular masses were as follows: anti-HA, LmeA-HA (29.5 kDa); anti-MptC, cell-envelope marker (48.2 kDa); anti-PimB′, intracellular membrane domain (IMD) marker (41.4 kDa); and anti-Mpa, cytosolic marker (68.0 kDa).
LmeA acts on a biosynthetic step genetically hypostatic to MptA-mediated LM mannan elongation
Small intermediates of LM have been previously identified in the knockdown or knockout of MptA, the mannosyltransferase that elongates the mannan chain during LM/LAM biosynthesis (14, 27). This immature LM intermediate is known to carry 5–20 mannoses. Whereas LmeA has no homology to known mannosyltransferases, the small LM produced in the ΔlmeA mutant suggested that LmeA plays a role in the elongation of LM. At least two mannosyltransferases are thought to be involved in the elongation step: an unknown α1–6-mannosyltransferase(s) that extends the α1–6-mannose chain on AcPIM4 to produce the LM intermediates seen in mptA deletion or knockdown strains, and MptA, the α1–6-mannosyltransferase that extends the chain generated by the unknown polymerase(s) (14, 18). We hypothesize that LmeA may facilitate either the unknown polymerase or MptA. To determine the genetic interaction of LmeA with either polymerase, we introduced a tet-off mptA knockdown system (27) into the ΔlmeA mutant and suppressed the expression of mptA by the addition of anhydrotetracycline (atc). As shown in Fig. 6A, the small LM accumulating in ΔlmeA (−atc, lane 3) became even smaller on SDS-PAGE upon suppression of mptA expression (+atc, lane 4). The small LM accumulating in the ΔlmeA strain under mptA knockdown conditions was apparently no different from the small LM accumulating in the mptA knockdown under the WT background (compare lanes 2 and 4). To confirm further, we analyzed the size distribution of LM by MALDI-TOF using a previously established method (27). We observed that the ΔlmeA LM carried 10–22 mannose residues, which was larger than the LM carrying 11–17 mannoses produced under mptA knockdown (Fig. 6B). Nevertheless, ΔlmeA LM was still significantly smaller than WT LM (20–31 mannose residues). Therefore, these analyses by MALDI-TOF were consistent with the SDS-PAGE migration patterns. Importantly, the LM intermediates accumulating in the mptA knockdown strains were identical regardless of lmeA deletion. Taken together, these data suggest that MptA is epistatic to LmeA.
Figure 6.

Effect of MptA depletion on ΔlmeA. A, LM/LAM profile. LM/LAM were extracted after either 48-h incubation with (+) or without (−) atc to induce mptA knockdown in ΔlmeA mutant and WT control. B, MALDI-TOF mass spectra of LM for WT, ΔlmeA, mptA knockdown, and mptA knockdown in ΔlmeA. Mannose residues are indicated for their corresponding peaks. LM peaks are detected as sinapinic acid adducts. KD, knockdown. a.u., arbitrary unit.
The hypostatic nature of LmeA suggested that it might facilitate the function of MptA or other downstream enzymes. We therefore wondered whether the overexpression of LmeA-HA could restore the small colony morphology in other suppressor mutants, such as S4 and S21, which have mutations in embC (S4) and mptA (S21), respectively. However, the additional expression of LmeA had no obvious impact on the colony size (supplemental Table S2), indicating that LmeA does not have dominant effects on these other mutants.
LmeA binds to phospholipids
Although LmeA is widely conserved in Corynebacteriales, standard BLAST analyses did not reveal any homologs with a protein of known function. Using the RaptorX structure prediction algorithm (28), we were able to predict the β-rich structure of LmeA with a p value of 4.90 × 10−4 using several templates including the bactericidal/permeability-increasing protein (BPI). BPI is a lipid-binding protein produced by lymphocytes and specifically binds to the lipid portion of bacterial lipopolysaccharides (29). Given this homology, we were prompted to test the possibility that LmeA binds the plasma membrane phospholipids or LM intermediates directly to exert its function in the LM biosynthesis. We created an Escherichia coli strain transformed with an IPTG-inducible His-LmeA expression vector and prepared a cell lysate of E. coli heterologously expressing His-LmeA. We tested the E. coli lysate by anti-His Western blotting and found a single band with the expected molecular mass of 29 kDa (Fig. 7A). We modified an established LAM-binding assay (30) and designed a lipid ELISA assay to test whether His-LmeA binds to any lipids. Commercially available lipids were first separated by thin-layer chromatography (TLC) and stained by cupric acetate to evaluate their purities (supplemental Fig. S6, A–C). We also purified the small LM intermediates from the mptA-knockdown strain. His-LmeA binding to the LM intermediates was minimal (Fig. 7B and supplemental Fig. S6D). In contrast, we observed more robust binding of His-LmeA to phospholipid species, such as PI and phosphatidylethanolamine (PE) (Fig. 7B), which are major structural components of M. smegmatis plasma membrane (31, 32). The binding of His-LmeA to PI and PE was dose-dependent and saturated at ∼2 μm (supplemental Fig. S6, E and F). Although our current assay system has limitations that make the accurate determination of Kd values difficult, we determined the apparent Kd values from the available data shown in supplemental Fig. S6 to compare the relative binding affinities of LmeA with different lipid species. The Kd values for PI and PE were 0.242 and 0.487 μm, respectively, showing that the binding affinity of LmeA to PI was comparable with, but slightly higher than, that of PE. Because LmeA bound both PI and PE effectively, we used phosphatidic acid (PA), a “headless” phospholipid, to examine whether the phospholipid headgroup is relevant to the binding (Fig. 7B and supplemental Fig. S6G). The binding of LmeA to PA was comparable with PI with a Kd value of 0.297 μm, indicating that the phospholipid headgroups do not affect the binding affinity. We also tested triacylglycerol (TAG), but LmeA did not bind to this glycerolipid (Fig. 7B and supplemental Fig. S6H), indicating that the phosphate residue is critical for the binding. Additionally, because mannan polymerization during LM biosynthesis utilizes PPM as the mannose donor, we tested whether LmeA binds to polyprenol phosphate with an affinity comparable with PA. However, binding of LmeA to geranylgeranyl monophosphate (GGP) was minimal, with an apparent Kd value of 8.65 μm (supplemental Fig. S6I). Finally, we tested whether LmeA binding to PE could be inhibited by the addition of soluble mannose 1-phosphate or GDP-mannose. However, up to a 10 mm concentration of these compounds had no effect on the binding of LmeA to PE, implying that mannose-containing molecules are not involved in the substrate recognition of LmeA (supplemental Fig. S7). Taken together, we conclude that the binding of LmeA is specific to glycerophospholipids.
Figure 7.
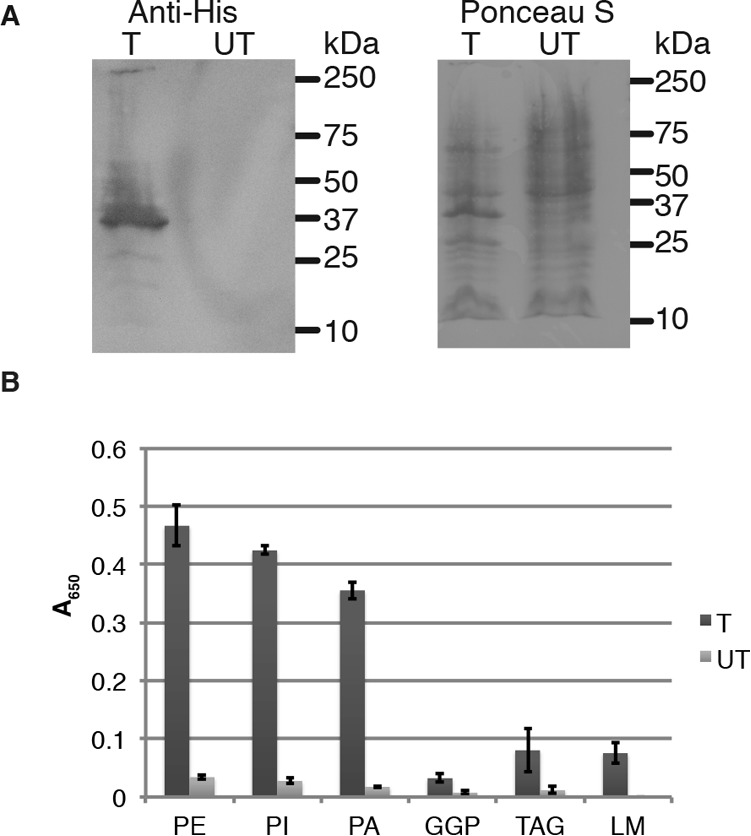
LmeA binds to phospholipids. A, anti-His Western blotting of E. coli lysate upon 3-h IPTG induction. T, transformed with the His-LmeA expression vector; UT, untransformed. Ponceau S staining shows protein loading. B, binding assay using E. coli lysate expressing His-LmeA. Microtiter plates were coated with 2.5 μm TAG, PI, PE, PA, GGP, and small LM intermediates, and a fixed amount (8 μl) of E. coli lysate, transformed (T) with the His-LmeA expression vector or untransformed (UT), was added. Error bars, S.D.
Discussion
In this study, we revealed that the previously uncharacterized protein LmeA encoded by MSMEG_5785 is involved in LM/LAM biosynthesis through a forward genetic screen of the ΔpimE mutant. We propose that LmeA is a cell envelope–associated phospholipid-binding protein that facilitates the maturation of mannan chain length.
Several pieces of evidence suggest that LmeA is directly involved in mannan elongation of LM/LAM biosynthesis. First, the deletion of the lmeA gene resulted in the production of LM/LAM with a shorter chain length. Second, LmeA is an extracytoplasmic cell envelope–associated protein. Third, LmeA is genetically hypostatic to MptA. Together, these observations are consistent with the idea that LmeA regulates the mannan chain polymerization by controlling the catalytic activity or processivity of MptA (Fig. 8). However, the precise function of LmeA remains unknown. One possibility is that LmeA controls the availability of the mannose donor substrate PPM or polyprenol phosphate–based carbohydrate donors in general. Such a function of LmeA could rescue other suppressor mutants, such as S4 and S21, by increasing the general availability of polyprenol phosphate–based carbohydrate donors. However, we did not observe such effects of LmeA on these other suppressor mutants. Furthermore, lipid-binding assays showed that LmeA preferentially binds glycerophospholipids over geranylgeranyl phosphate, and that this binding is not competitively inhibited by GDP-mannose or mannose 1-phosphate. Therefore, we suggest that LmeA is not involved in the metabolism and trafficking of polyprenol phosphate–based substrates.
Figure 8.
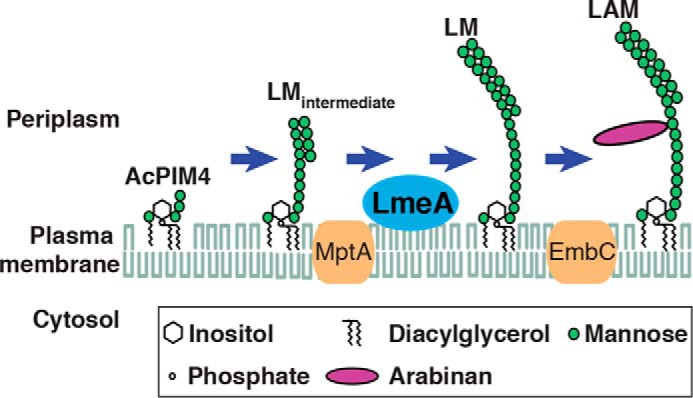
A model of LmeA function and localization in LM/LAM biosynthesis. LmeA potentially binds to plasma membrane phospholipids on the periplasmic side and functions downstream of MptA. MptC is not shown.
Based on the fact that LmeA is extracytoplasmic and binds to phospholipids such as PI and PE, we propose that LmeA is a protein peripherally bound to the plasma membrane. Nikaido and colleagues (33, 34) have taken the approach of reverse micelle extraction to show that major phospholipid species, such as PI and PE, are found predominantly in the plasma membrane and relatively depleted in the mycobacterial or corynebacterial outer membrane. Therefore, the ability of LmeA to bind to phospholipid species suggests that LmeA is a periplasmic plasma membrane-associated protein (Fig. 8). In Mycobacterium marinum, the LmeA ortholog (MMAR_4866) was relatively resistant to a differential detergent extraction, which was effective in selectively extracting known outer membrane proteins (35), further supporting the periplasmic location of LmeA. Nevertheless, the M. tuberculosis homolog of LmeA (Rv0817c) is predicted to be an outer membrane β-barrel (36). These contradictory observations highlight that further studies are needed to determine functional and structural features of LmeA. Although our attempts have so far been unsuccessful, an important next step is the purification of LmeA in its active form.
Complex interplay of PIMs/LM/LAM in the integrity of the mycobacterial cell envelope is an emerging theme. Previous studies showed that mutations in pimE can compensate for the growth defect of the lpqW mutant that produces reduced levels of LM/LAM (19, 21). Our current study showed an opposite biological response, where the growth defect of ΔpimE was compensated by additional structural changes in LM/LAM. It appears that the balance between the levels of AcPIM6 and LM/LAM is critical for fitness, and mycobacteria regain optimal growth by genetic changes that warrant the homeostasis of the glycocalyx. A conceptually similar glycan compensation is well known as chitin emergency response in Saccharomyces cerevisiae (37, 38). In this stress response, compromised cell wall integrity due to defects in surface glycans, such as β-glucan, mannan, O-linked glycans, or glycosylphosphatidylinositol anchors, is compensated by the up-regulation of chitin synthesis. More recently, it was reported that the lack of one particular glycan structure in mammalian cells is compensated by the production of bioequivalent glycans in the Golgi apparatus (39). Such principles of glycan homeostasis may apply to the maintenance of cell surface glycocalyx in mycobacteria as well. However, completely different scenarios are also possible. For example, it has not been explored whether PIMs play a role as signaling molecules to facilitate colony growth. We have previously reported that M. smegmatis, as well as Corynebacterium glutamicum, produce PI 3-phosphate (40), but nothing is known about whether inositol polyphosphates are released by a phospholipase. Similarly, it is completely unknown whether there are phospholipases that can act on PIMs to release the glycan headgroups and whether such released molecules can function as signaling molecules. Interestingly, a recent report suggested that synthetic lipid–linked arabinomannan heptasaccharide can effectively inhibit biofilm formation in M. smegmatis (41), implying that comparable molecules like phospholipase-digested AcPIM6 may have similar biological activities. Finally, lipid-free d-arabino-d-mannan, d-mannan, and their phosphorylated counterparts are found in the extracellular capsules of M. tuberculosis and other mycobacteria, implying the presence of phospholipases that can act on larger PI-anchored glycolipids, such as LM and LAM (42–46). We speculate that such phospholipases may promiscuously act on PIMs as well. The ΔpimE suppressor mutants that we identified include many mutants that do not show changes in LM/LAM structures, suggesting that there are multiple pathways to rescue the small colony morphology of ΔpimE. Analysis of other suppressor mutants may further reveal the potential molecular mechanisms behind the biological responses of PIMs/LM/LAM mutants.
The enzymes for the synthesis of AcPIM2, such as PimA and PimB′, are essential for the viability of M. smegmatis (8, 9). AcPIM2 is a mature product but also serves as a precursor for the synthesis of AcPIM6, LM, and LAM. Therefore, it remains unknown whether AcPIM2 or any of the downstream products are essential. Interestingly, a recent study revealed that a corynebacterial membrane protein, NCgl2760, is involved in LM biosynthesis, probably at a step before the MptA-mediated mannan elongation (47). The orthologous gene is predicted to be essential in M. tuberculosis and cannot be deleted from the endogenous locus in M. smegmatis (MSMEG_0317) unless an extra copy of the gene is present (47). These data imply that defects in the early stage of LM/LAM biosynthesis might be lethal to M. smegmatis. In contrast, mild structural defects in LM/LAM, such as the one caused by the mptA depletion, can be tolerated in M. smegmatis. Therefore, the non-essential nature of LmeA further supports our hypothesis that this protein acts on the downstream of MptA-mediated mannan elongation.
In contrast to M. smegmatis, the requirement of PIMs/LM/LAM appears to be more stringent in M. tuberculosis. EmbC, an arabinosyltransferase involved in LAM biosynthesis, has been experimentally shown to be essential for viability in M. tuberculosis (3). Genome-wide transposon mutagenesis studies further predict that many genes involved in PIMs/LM/LAM biosynthesis, such as pimA, pimB′, pimE, and mptA, are essential in M. tuberculosis (2). We have previously created a mutant strain of M. tuberculosis overexpressing MptC (Rv2181) (27). This mutant produces aberrant LM/LAM with a truncated mannan chain, and such mild modifications were sufficient to make M. tuberculosis defective in establishing infection in mice. Indeed, the lmeA ortholog (Rv0817c) is predicted to be essential in M. tuberculosis (2). The cell-envelope localization of LmeA, its predicted essentiality in M. tuberculosis, and the absence of its homologs in humans make this protein a potentially attractive drug target. To this end, we are currently generating a conditional lmeA mutant in M. tuberculosis to demonstrate that lmeA is an essential gene.
Experimental procedures
Mycobacterial growth conditions
M. smegmatis mc2155 and derived mutants were grown at 37 °C on Middlebrook 7H10 agar (BD Biosciences) supplemented with 0.2% glucose (w/v) and 15 mm NaCl as described (13). Liquid cultures were at 30 °C in Middlebrook 7H9 broth (BD Biosciences) supplemented with 0.2% glycerol (v/v), 0.05% Tween 80 (v/v), 0.2% glucose (w/v), and 15 mm NaCl (13). Knockdown of mptA was induced with 40 ng/ml atc (Acros) for 48 h. Antibiotic concentrations used were 20 μg/ml kanamycin (MP Biomedicals), 50 μg/ml streptomycin (Fisher), and 100 μg/ml hygromycin (Wako).
DNA purification, whole-genome sequencing, and mutation analysis
Genomic DNA from select suppressor mutants was purified as described previously (49). Whole-genome sequencing was performed on the Illumina MiSeq platform with 251-bp paired-end sequencing. Each genomic DNA (300 ng) was sheared to an average size of 600 bp with the Covaris S220 (Covaris). The DNA library was prepared using the KAPA Library Preparation Kit (Kapa Biosystems) and TruSeq adapters (Illumina) according to the manufacturer's instructions. Sequences were filtered and trimmed based on quality score using Quick Read Quality Control (Bioconductor, Seattle, WA; available at www.bioconductor.org/packages/release/bioc/html/qrqc.html)4 (54, 55), Sickle (GitHub, San Francisco; available at https://github.com/najoshi/sickle)4 (56), and Scythe (GitHub, San Francisco; available at https://github.com/ucdavis-bioinformatics/scythe)4 and were aligned to the reference M. smegmatis NC_008596.1 genome using Bowtie2 (50), and variant calls were made using Samtools (51, 52). The sequence data were analyzed by the Integrative Genomics Viewer (53). Additional suppressor mutants were analyzed specifically for pimE (A115 and A118), mptA (A193 and A194), and MSMEG_5785 (A197 and A198) by amplifying the genes by PCR (see supplemental Table S1) and sequencing the amplified products by standard Sanger sequencing.
Lipid extraction and analysis
Crude lipids were extracted as described, and the delipidated pellet was incubated with phenol/water (1:1) for 2 h at 55 °C to extract LM/LAM (18). PIMs were separated by high-performance TLC silica gel 60 (EMD Merck) using chloroform, methanol, 13 m ammonia, 1 m ammonium acetate, water (180:140:9:9:23) as a mobile phase and visualized by orcinol staining as described (18). LM/LAM samples were separated by SDS-PAGE (15% gel) and visualized using the ProQ Emerald 488 glycan staining kit (Life Technologies). For the mass spectrometric analysis and lipid-binding assay (see below), LM/LAM were purified using an octyl-Sepharose column (GE Healthcare) as before (18).
Construction of plasmids
Knockout and expression vectors were constructed as detailed below.
pMUM57
The upstream and downstream region of lmeA were amplified using A217/A218 and A219/A220 primer sets (supplemental Table S1), respectively. These two fragments were then digested with Van91I and ligated into Van91I-digested pCOM1 (26), resulting in the MSMEG_5785 knockout construct.
pMUM54
lmeA was amplified using primers A215 and A216. The fragment was then digested by NdeI and ScaI and ligated into pMUM12 (26), which was digested with NdeI and ScaI, to generate pMUM54, an expression vector for Phsp60-lmeA-HA.
pMUM107, pMUM125, and pMUM126
Primers A470 and A472 were designed to amplify lmeA, including 165 bp of upstream native promoter region from either WT (pMUM107), S1 (pMUM125), or S10 (pMUM126). The PCR fragment was then digested by KpnI and XbaI and ligated into pMV306 that was digested by KpnI and XbaI, resulting in pMUM107, pMUM125, and pMUM126, expression vectors for Pnative-lmeA-HA, Pnative-lmeA(G170D)-HA, and Pnative-lmeA(V181G)-HA, respectively.
pMUM121
Primers A552 and A553 were designed to amplify lmeA, excluding the sequence coding for the predicted N-terminal signal sequence. A TOPO cloning kit (Invitrogen) was used to insert the fragment into pET100, creating an IPTG-inducible His-LmeA expression construct.
MALDI-TOF MS analysis
Octyl-Sepharose–purified LM/LAM preparation (0.5 μl) was mixed with an equal volume of matrix solution (20 mg/ml sinapinic acid (Millipore-Sigma), 30% acetonitrile (Fisher), 1% trifluoroacetic acid (Fisher) in water). Samples were analyzed on a Bruker Microflex MALDI-TOF instrument (Bruker Daltonics) using linear mode and positive ion detection. The data were analyzed using the Microflex software.
Bead-beating cell lysis
Cell pellets were washed in 50 mm HEPES/NaOH (pH 7.4) twice and resuspended in a lysis buffer containing 25 mm HEPES/NaOH (pH 7.4), 15% glycerol, 2 mm EGTA and a protease inhibitor mix. Four times the pellet weight of acid-washed glass beads (Millipore-Sigma) was added, and cells were lysed by a BeadBug microtube homogenizer (Benchmark Scientific) at 4 °C with beating at 4,000 rpm for 30 s. Bead beating was repeated 5 times with 1-min intervals on ice. Beads and cell debris were removed by centrifugation.
SDS-PAGE and Western blotting
Protein samples (12 μl) were mixed with reducing sample loading buffer, denatured on ice for 30 min or boiled for 5 min, and separated on SDS-PAGE (12% gel). After Western blot transfer, the PVDF membrane was incubated with a primary antibody at a 1:2,000 dilution (mouse anti-HA (Millipore-Sigma), rabbit anti-MptC (18), mouse anti-penta-His (Qiagen), rabbit anti-PimB′ (18), or rabbit anti-Mpa (48)), followed by incubation with a horseradish peroxidase-conjugated secondary antibody, either anti-rabbit or anti-mouse IgG (GE Healthcare), at a 1:2,000 dilution. Bands were visualized by chemiluminescence and recorded using ImageQuant LAS 4000mini (GE Healthcare).
Sucrose density gradient sedimentation and fractionation
Sucrose density gradient fractionation was performed as before (25, 26).
Immunoprecipitation
Anti-HA-agarose beads (Millipore-Sigma) were washed with a buffer containing 25 mm HEPES-NaOH (pH 7.4), 2 mm EGTA, and 150 mm NaCl (HES), or another buffer containing 25 mm HEPES-NaOH (pH 7.4), 2 mm EGTA, 150 mm NaCl, and 1% Triton X-100 (HEST). Bead-beating cell lysate (36 μl) was added to the prewashed beads (10-μl bed volume) and incubated with HES or HEST buffer at 4 °C overnight under gentle rotation at 5 rpm. Beads were washed with 1 ml of HES or HEST buffer before elution with 10 μl of 1 mg/ml HA peptide (AnaSpec) twice at 30 °C. Eluates were separated on SDS-PAGE and visualized via Western blotting as described above.
E. coli cell transformation and lysis
E. coli BL21 strain was transformed with pMUM121 by heat shock. The transformed strain was grown in terrific broth to log phase and induced with 1 mm IPTG (Fisher) for 3 h at 37 °C. Untransformed E. coli was grown under the same conditions. Cells were incubated with 1 mg/ml lysozyme (Fisher) for 20 min at room temperature in a buffer containing 50 mm HEPES-NaOH (pH 7.4), 200 mm NaCl, and 1× FastBreak Cell Lysis Reagent (Promega) and were lysed by sonication. The resulting lysate was centrifuged, and the supernatant was used for all binding assays or Western blot analysis.
Lipid binding assay
PI from soybeans, PE from egg yolk, and TAG (glycerol trioleate) were purchased from Millipore-Sigma. PA from chicken eggs was purchased from Avanti. GGM was purchased from Larodan. PI was a crude preparation with many contaminants and was further purified by preparative TLC. The purity of each lipid was examined by TLC using two solvent systems: hexane/diethyl ether/formic acid (40:10:1) for TAG or chloroform, methanol, 13 m ammonia, 1 m ammonium acetate, water (180:140:9:9:23) for PI, PE, PA, and GGM. For lipid ELISA, lipids were serially diluted in isopropyl alcohol 2-fold from an initial amount of 0.4 nmol/well, and the solvent was evaporated in 96-well immunoplates (Brand GmbH). Once the wells were completely dried, they were blocked at 4 °C overnight with 200 μl of 5% milk in PBS with 0.05% Tween 20 (PBST20). A mixture of 8 μl of cell lysate and 32 μl of PBST20 was added to each well and incubated at 37 °C for 2 h. After washing with PBST20, 40 μl of mouse anti-penta-His IgG (1:2,000 dilution; Qiagen) was added and incubated at 4 °C overnight. After washing with PBST20, 40 μl of horseradish peroxidase-conjugated anti-mouse IgG (1:4,000 dilution; GE Healthcare) was added and incubated at room temperature for 1 h. After washing with PBST20, 100 μl of 3,3′,5,5′-tetramethylbenzidine (BD Biosciences) was added, and the colorimetric changes were read at 650 nm after a 1-h incubation at room temperature. Kd values were calculated using a nonlinear regression function of Prism version 7 (GraphPad Software), assuming that LmeA has one lipid binding site. For competition assays, microtiter plates were coated with 0.05 nmol of PE (corresponding to 1.25 μm). E. coli lysate was preincubated with 10 mm mannose 1-phosphate (Millipore-Sigma) or GDP-mannose (Millipore-Sigma) at room temperature for 30 min before being added to the PE-coated well.
Author contributions
K. C. R. conducted most of the experiments, analyzed the results, and wrote the paper. S. A. H. conducted a part of the experiments shown in Figs. 5 and 6. D. M., J. A. M., and S. N. sequenced and analyzed the whole genome of the suppressor mutants (Fig. 2 and Table 1). L. R. B. and A. P. T.-O. conducted some initial experiments shown in Fig. 1. J. N. S. contributed to the experiments shown in Fig. 7. Y. S. M. conceived the idea, designed the study, and wrote the paper.
Supplementary Material
Acknowledgments
We thank Dr. Stephen Eyles (Institute for Applied Life Sciences, University of Massachusetts Amherst) for help with mass spectrometry and Dr. Heran Darwin (New York University) for the gift of anti-Mpa antibody. We also thank Julia Puffal, William Eagen, and Sarah Osman for discussion and critical reading of the manuscript.
This work was supported by Biomedical Research Grant RG-414805 from the American Lung Association and a Research Grant from the Pittsfield Anti-Tuberculosis Association (to Y. S. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1 and S2, Figs. S1–S7, and Data Set S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PIM
- phosphatidylinositol mannoside
- LM
- lipomannan
- LAM
- lipoarabinomannan
- PI
- phosphatidylinositol
- PPM
- polyprenol phosphate-mannose
- atc
- anhydrotetracycline
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- PE
- phosphatidylethanolamine
- PA
- phosphatidic acid
- TAG
- triacylglycerol
- GGP
- geranylgeranyl monophosphate
- TLC
- thin-layer chromatography.
References
- 1. Kaur D., Guerin M. E., Skovierová H., Brennan P. J., and Jackson M. (2009) Chapter 2: Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69, 23–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griffin J. E., Gawronski J. D., Dejesus M. A., Ioerger T. R., Akerley B. J., and Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goude R., Amin A. G., Chatterjee D., and Parish T. (2008) The critical role of embC in Mycobacterium tuberculosis. J. Bacteriol. 190, 4335–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jarlier V., and Nikaido H. (1990) Permeability barrier to hydrophilic solutes in Mycobacterium chelonei. J. Bacteriol. 172, 1418–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hmama Z., Peña-Díaz S., Joseph S., and Av-Gay Y. (2015) Immunoevasion and immunosuppression of the macrophage by Mycobacterium tuberculosis. Immunol. Rev. 264, 220–232 [DOI] [PubMed] [Google Scholar]
- 6. Vergne I., Gilleron M., and Nigou J. (2014) Manipulation of the endocytic pathway and phagocyte functions by Mycobacterium tuberculosis lipoarabinomannan. Front. Cell Infect. Microbiol. 4, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ishikawa E., Mori D., and Yamasaki S. (2017) Recognition of mycobacterial lipids by immune receptors. Trends Immunol. 38, 66–76 [DOI] [PubMed] [Google Scholar]
- 8. Korduláková J., Gilleron M., Mikusova K., Puzo G., Brennan P. J., Gicquel B., and Jackson M. (2002) Definition of the first mannosylation step in phosphatidylinositol mannoside synthesis: PimA is essential for growth of mycobacteria. J. Biol. Chem. 277, 31335–31344 [DOI] [PubMed] [Google Scholar]
- 9. Guerin M. E., Kaur D., Somashekar B. S., Gibbs S., Gest P., Chatterjee D., Brennan P. J., and Jackson M. (2009) New insights into the early steps of phosphatidylinositol mannoside biosynthesis in mycobacteria: PimB′ is an essential enzyme of Mycobacterium smegmatis. J. Biol. Chem. 284, 25687–25696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lea-Smith D. J., Martin K. L., Pyke J. S., Tull D., McConville M. J., Coppel R. L., and Crellin P. K. (2008) Analysis of a new mannosyltransferase required for the synthesis of phosphatidylinositol mannosides and lipoarbinomannan reveals two lipomannan pools in Corynebacterineae. J. Biol. Chem. 283, 6773–6782 [DOI] [PubMed] [Google Scholar]
- 11. Korduláková J., Gilleron M., Puzo G., Brennan P. J., Gicquel B., Mikusová K., and Jackson M. (2003) Identification of the required acyltransferase step in the biosynthesis of the phosphatidylinositol mannosides of Mycobacterium species. J. Biol. Chem. 278, 36285–36295 [DOI] [PubMed] [Google Scholar]
- 12. Mishra A. K., Alderwick L. J., Rittmann D., Wang C., Bhatt A., Jacobs W. R. Jr., Takayama K., Eggeling L., and Besra G. S. (2008) Identification of a novel α(1→6) mannopyranosyltransferase MptB from Corynebacterium glutamicum by deletion of a conserved gene, NCgl1505, affords a lipomannan- and lipoarabinomannan-deficient mutant. Mol. Microbiol. 68, 1595–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morita Y. S., Sena C. B. C., Waller R. F., Kurokawa K., Sernee M. F., Nakatani F., Haites R. E., Billman-Jacobe H., McConville M. J., Maeda Y., and Kinoshita T. (2006) PimE is a polyprenol-phosphate-mannose-dependent mannosyltransferase that transfers the fifth mannose of phosphatidylinositol mannoside in mycobacteria. J. Biol. Chem. 281, 25143–25155 [DOI] [PubMed] [Google Scholar]
- 14. Kaur D., McNeil M. R., Khoo K.-H., Chatterjee D., Crick D. C., Jackson M., and Brennan P. J. (2007) New insights into the biosynthesis of mycobacterial lipomannan arising from deletion of a conserved gene. J. Biol. Chem. 282, 27133–27140 [DOI] [PubMed] [Google Scholar]
- 15. Mishra A. K., Alderwick L. J., Rittmann D., Tatituri R. V. V., Nigou J., Gilleron M., Eggeling L., and Besra G. S. (2007) Identification of an α(1→6) mannopyranosyltransferase (MptA), involved in Corynebacterium glutamicum lipomanann biosynthesis, and identification of its orthologue in Mycobacterium tuberculosis. Mol. Microbiol. 65, 1503–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaur D., Berg S., Dinadayala P., Gicquel B., Chatterjee D., McNeil M. R., Vissa V. D., Crick D. C., Jackson M., and Brennan P. J. (2006) Biosynthesis of mycobacterial lipoarabinomannan: role of a branching mannosyltransferase. Proc. Natl. Acad. Sci. U.S.A. 103, 13664–13669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaur D., Obregón-Henao A., Pham H., Chatterjee D., Brennan P. J., and Jackson M. (2008) Lipoarabinomannan of Mycobacterium: mannose capping by a multifunctional terminal mannosyltransferase. Proc. Natl. Acad. Sci. U.S.A. 105, 17973–17977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sena C. B. C., Fukuda T., Miyanagi K., Matsumoto S., Kobayashi K., Murakami Y., Maeda Y., Kinoshita T., and Morita Y. S. (2010) Controlled expression of branch-forming mannosyltransferase is critical for mycobacterial lipoarabinomannan biosynthesis. J. Biol. Chem. 285, 13326–13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crellin P. K., Kovacevic S., Martin K. L., Brammananth R., Morita Y. S., Billman-Jacobe H., McConville M. J., and Coppel R. L. (2008) Mutations in pimE restore lipoarabinomannan synthesis and growth in a Mycobacterium smegmatis lpqW mutant. J. Bacteriol. 190, 3690–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rainczuk A. K., Yamaryo-Botte Y., Brammananth R., Stinear T. P., Seemann T., Coppel R. L., McConville M. J., and Crellin P. K. (2012) The lipoprotein LpqW is essential for the mannosylation of periplasmic glycolipids in corynebacteria. J. Biol. Chem. 287, 42726–42738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kovacevic S., Anderson D., Morita Y. S., Patterson J., Haites R., McMillan B. N. I., Coppel R., McConville M. J., and Billman-Jacobe H. (2006) Identification of a novel protein with a role in lipoarabinomannan biosynthesis in mycobacteria. J. Biol. Chem. 281, 9011–9017 [DOI] [PubMed] [Google Scholar]
- 22. Leversen N. A., de Souza G. A., Målen H., Prasad S., Jonassen I., and Wiker H. G. (2009) Evaluation of signal peptide prediction algorithms for identification of mycobacterial signal peptides using sequence data from proteomic methods. Microbiology 155, 2375–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perkowski E. F., Zulauf K. E., Weerakoon D., Hayden J. D., Ioerger T. R., Oreper D., Gomez S. M., Sacchettini J. C., and Braunstein M. (2017) The EXIT strategy: an approach for identifying bacterial proteins exported during host infection. mBio 8, e00333–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kelkar D. S., Kumar D., Kumar P., Balakrishnan L., Muthusamy B., Yadav A. K., Shrivastava P., Marimuthu A., Anand S., Sundaram H., Kingsbury R., Harsha H. C., Nair B., Prasad T. S. K., Chauhan D. S., et al. (2011) Proteogenomic analysis of Mycobacterium tuberculosis by high resolution mass spectrometry. Mol. Cell. Proteomics 10, M111.011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morita Y. S., Velasquez R., Taig E., Waller R. F., Patterson J. H., Tull D., Williams S. J., Billman-Jacobe H., and McConville M. J. (2005) Compartmentalization of lipid biosynthesis in mycobacteria. J. Biol. Chem. 280, 21645–21652 [DOI] [PubMed] [Google Scholar]
- 26. Hayashi J. M., Luo C.-Y., Mayfield J. A., Hsu T., Fukuda T., Walfield A. L., Giffen S. R., Leszyk J. D., Baer C. E., Bennion O. T., Madduri A., Shaffer S. A., Aldridge B. B., Sassetti C. M., et al. (2016) Spatially distinct and metabolically active membrane domain in mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 113, 5400–5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fukuda T., Matsumura T., Ato M., Hamasaki M., Nishiuchi Y., Murakami Y., Maeda Y., Yoshimori T., Matsumoto S., Kobayashi K., Kinoshita T., and Morita Y. S. (2013) Critical roles for lipomannan and lipoarabinomannan in cell wall integrity of mycobacteria and pathogenesis of tuberculosis. mBio 4, e00472–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Källberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., and Xu J. (2012) Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alva V., and Lupas A. N. (2016) The TULIP superfamily of eukaryotic lipid-binding proteins as a mediator of lipid sensing and transport. Biochim. Biophys. Acta 1861, 913–923 [DOI] [PubMed] [Google Scholar]
- 30. Yonekawa A., Saijo S., Hoshino Y., Miyake Y., Ishikawa E., Suzukawa M., Inoue H., Tanaka M., Yoneyama M., Oh-Hora M., Akashi K., and Yamasaki S. (2014) Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity 41, 402–413 [DOI] [PubMed] [Google Scholar]
- 31. Subramoniam A., and Subrahmanyam D. (1982) Light-induced changes in the phospholipid composition of Mycobacterium smegmatis ATCC 607. J. Gen. Microbiol. 128, 419–421 [DOI] [PubMed] [Google Scholar]
- 32. Khuller G. K., Taneja R., and Nath N. (1983) Effect of fatty acid supplementation on the lipid composition of Mycobacterium smegmatis ATCC 607, grown at 27 degrees and 37 degrees C. J. Appl. Bacteriol. 54, 63–68 [DOI] [PubMed] [Google Scholar]
- 33. Bansal-Mutalik R., and Nikaido H. (2011) Quantitative lipid composition of cell envelopes of Corynebacterium glutamicum elucidated through reverse micelle extraction. Proc. Natl. Acad. Sci. U.S.A. 108, 15360–15365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bansal-Mutalik R., and Nikaido H. (2014) Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc. Natl. Acad. Sci. U.S.A. 111, 4958–4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van der Woude A. D., Mahendran K. R., Ummels R., Piersma S. R., Pham T. V., Jiménez C. R., de Punder K., van der Wel N. N., Winterhalter M., Luirink J., Bitter W., and Houben E. N. G. (2013) Differential detergent extraction of Mycobacterium marinum cell envelope proteins identifies an extensively modified threonine-rich outer membrane protein with channel activity. J. Bacteriol. 195, 2050–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Song H., Sandie R., Wang Y., Andrade-Navarro M. A., and Niederweis M. (2008) Identification of outer membrane proteins of Mycobacterium tuberculosis. Tuberculosis 88, 526–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orlean P. (2012) Architecture and biosynthesis of the Saccharomyces cerevisiae cell wall. Genetics 192, 775–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levin D. E. (2011) Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics 189, 1145–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mkhikian H., Mortales C.-L., Zhou R. W., Khachikyan K., Wu G., Haslam S. M., Kavarian P., Dell A., and Demetriou M. (2016) Golgi self-correction generates bioequivalent glycans to preserve cellular homeostasis. eLife 5, e14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morita Y. S., Yamaryo-Botte Y., Miyanagi K., Callaghan J. M., Patterson J. H., Crellin P. K., Coppel R. L., Billman-Jacobe H., Kinoshita T., and McConville M. J. (2010) Stress-induced synthesis of phosphatidylinositol 3-phosphate in mycobacteria. J. Biol. Chem. 285, 16643–16650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jayaraman N., Maiti K., Syal K., and Chatterji D. (2017) Synthetic arabinomannan heptasaccharide glycolipids inhibit biofilm growth and supplements isoniazid effects in Mycobacterium smegmatis. Chembiochem 10.1002/cbic.201700247 [DOI] [PubMed] [Google Scholar]
- 42. Lemassu A., and Daffé M. (1994) Structural features of the exocellular polysaccharides of Mycobacterium tuberculosis. Biochem. J. 297, 351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ortalo-Magné A., Dupont M. A., Lemassu A., Andersen A. B., Gounon P., and Daffé M. (1995) Molecular composition of the outermost capsular material of the tubercle bacillus. Microbiology 141, 1609–1620 [DOI] [PubMed] [Google Scholar]
- 44. Ortalo-Magné A., Andersen A. B., and Daffé M. (1996) The outermost capsular arabinomannans and other mannoconjugates of virulent and avirulent tubercle bacilli. Microbiology 142, 927–935 [DOI] [PubMed] [Google Scholar]
- 45. Maes E., Coddeville B., Kremer L., and Guérardel Y. (2007) Polysaccharide structural variability in mycobacteria: identification and characterization of phosphorylated mannan and arabinomannan. Glycoconj. J. 24, 439–448 [DOI] [PubMed] [Google Scholar]
- 46. Lemassu A., Ortalo-Magné A., Bardou F., Silve G., Laneélle M. A., and Daffé M. (1996) Extracellular and surface-exposed polysaccharides of non-tuberculous mycobacteria. Microbiology 142, 1513–1520 [DOI] [PubMed] [Google Scholar]
- 47. Cashmore T. J., Klatt S., Yamaryo-Botte Y., Brammananth R., Rainczuk A. K., McConville M. J., Crellin P. K., and Coppel R. L. (2017) Identification of a membrane protein required for lipomannan maturation and lipoarabinomannan synthesis in Corynebacterineae. J. Biol. Chem. 292, 4976–4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Darwin K. H., Lin G., Chen Z., Li H., and Nathan C. F. (2005) Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol. Microbiol. 55, 561–571 [DOI] [PubMed] [Google Scholar]
- 49. Larsen M. H., Biermann K., and Tandberg S. (2007) Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. 10.1002/9780471729259.mc10a02s6 [DOI] [PubMed] [Google Scholar]
- 50. Langmead B., and Salzberg S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li H. (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thorvaldsdóttir H., Robinson J. T., and Mesirov J. P. (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinformatics 14, 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huber W., Carey V. J., Gentleman R., Anders S., Carlson M., Carvalho B. S., Bravo H. C., Davis S., Gatto L., Girke T., Gottardo R., Hahne F., Hansen K. D., Irizarry R. A., Lawrence M., et al. (2015) Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Joshi N. A., and Fass J. N. (2011) Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (version 1.33) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.