

Abstract
Affinity purification-mass spectrometry (AP-MS) has become the method of choice for discovering protein-protein interactions (PPIs) under native conditions. The success of AP-MS depends on the efficiency of trypsin digestion and the recovery of the tryptic peptides for MS analysis. Several different protocols have been used for trypsin digestion of protein complexes in AP-MS studies, but no systematic studies have been conducted on the impact of trypsin digestion conditions on the identification of PPIs. Here, we used NFκB/RelA and Bromodomain-containing protein 4 (BRD4) as baits and test five distinct trypsin digestion methods (two using “on-bead,” three using “elution-digestion” protocols). Although the performance of the trypsin digestion protocols change slightly depending on the different baits, antibodies and cell lines used, we found that elution-digestion methods consistently outperformed on-beads digestion methods. The high-abundance interactors can be identified universally by all five methods, but the identification of low-abundance RelA interactors is significantly affected by the choice of trypsin digestion method. We also found that different digestion protocols influence the selected reaction monitoring (SRM)-MS quantification of PPIs, suggesting that optimization of trypsin digestion conditions may be required for robust targeted analysis of PPIs.
Keywords: Protein-protein interaction, affinity purification, trypsin digestion, mass spectrometry, selected reaction monitoring
INTRODUCTION
Proteins rarely act alone while performing their functions in vivo.1 They are usually organized into functional modules to carry out cellular functions. These protein complexes play central roles in regulating DNA replication, transcription, translation, RNA splicing, protein secretion, cell cycle control, signal transduction, and intermediary metabolism.2,3 Protein-protein interactions (PPIs) are the key element in cellular signaling networks; for example, many cellular signals are transmitted via the interaction between the protein substrates and their posttranslational modifying enzymes such as kinases/phosphatases, ubiquitinases/deubiquitinases, and acetyltransferases/deacetylases.2,4,5 Analysis of PPIs can provide insights into the mechanism of signal transduction. Recent studies indicate that PPIs also play major roles in the invasion, replication and immune evasion of pathogens. Systems-wide analyses of PPIs between hosts and pathogens have yielded important new information on how pathogens use host systems to their own advantage.6–8
To properly understand the significance and roles of PPIs, a comprehensive method is needed to identify the PPIs. Affinity purification-mass spectrometry (AP-MS) has become the method of choice for discovering PPIs under native conditions.1,6–12 This method uses affinity purification of proteins under native conditions to preserve PPIs followed by their detection with mass spectrometry. In this method, the protein complexes were captured by antibodies specific for the bait proteins, or for tags that were introduced on the bait proteins, and pulldown onto immobilized protein A/G beads. The complexes are further digested into peptides with trypsin. The protein interactors of the bait proteins are identified by quantification of the tryptic peptides via mass spectrometry. The success of AP-MS depends on several factors: the ability of the antibody to enrich the bait protein and its associated proteins, the efficiency of trypsin digestion, and efficient recovery of the tryptic peptides for MS analysis.
A number of studies have been performed to compare the methods for trypsin digestion for global proteomic profiling in which the proteins in cell extracts or biofluids are dissolved and denatured with either chaotropic agents, surfactants or organic solvents, followed by in-solution trypsin digestion.13,14 These studies have shown that the choice of chaotropic agent, surfactant, or organic solvent has a significant impact on the efficiency, reproducibility, and completeness of trypsin digestion, and hence affects sequence coverage of protein identification by MS analysis.
The protocol for trypsin digestion of protein complexes in AP-MS studies requires additional considerations for the following reasons. First, the proteins complexes are attached to the beads, which can impose steric hindrance for trypsin to access the protein complex. Second, if the protein complexes are to be eluted from the beads before trypsin digestion, the solvents used for elution must be able to efficiently elute the protein complex from the beads and be compatible with subsequent trypsin digestion and MS analysis. Several different protocols have been used for trypsin digestion of protein complexes in AP-MS studies,9,11,12,15,16 but there have been no systematic studies on the impact of trypsin digestion conditions on the identification of PPIs via mass spectrometry.
NFκB/RelA is a pleiotropic transcription factor present in almost all cell types. It can be activated by a vast array of stimuli and initiates a series of signaling events related to many biological processes such as inflammation, immunity, differentiation, cell growth, tumorigenesis, and apoptosis.5,17 RelA is known to have interactions with many co-activators, transcription factors, co-repressors, and posttranslational modifying enzymes. Its function is controlled by various mechanisms of post-translational modification and subcellular compartmentalization, as well as by interactions with other cofactors or corepressors.5,17–19 Identification of the dynamic changes in RelA interactome has long been an interesting objective of many groups including ours.3,5,20,21 Here, we use RelA interactome as a model system and compare the coverage of RelA interactome when several different trypsin digestion protocols are used. Additional studies of BRD4 interactome were conducted to evaluate the effect of bait and cell lines on the performance to the trypsin digestion protocols. To our knowledge, our study offers the first systematic analysis on effect of trypsin digestion condition on AP-MS.
EXPERIMENTAL PROCEDURES
Regents and chemicals
Regents and chemicals - all reagents were ACS grade or higher. All solvents used, including water, were LC/MS grade. Ammonium bicarbonate (ABC), 2,2,2,-trifluoroethanol(TFE), and acetic acid were purchased from Sigma-Aldrich. Iodoacetamide (IDA), dithiothreitol (DTT), acetonitrile (ACN), formic acid, and methanol were purchased from Thermo Scientific. Urea ultra was from MP Biomedicals. Sequencing-grade modified trypsin (Promega) was used.
Cell culture
Cell culture - A549 cells (human adenocarcinomic alveolar basal epithelial cells) were grown in 10% FBS - DMEM with 1% penicillin streptomycin, 1% MEM non-essential amino acids, 1% L-glutamine and 1% sodium pyruvate.12 An immortalized primary human small airway epithelial cell line (hSAECs) was established by infecting primary hSAECs with human telomerase and cyclin dependent kinase (CDK)-4 retrovirus constructs.16 The immortalized hSAECs were grown in SAGM small airway epithelial cell growth medium (Lonza, Walkersville, MD). All cells were incubated at 37°C, 5% CO2 until confluence.
Antibodies
Antibodies - anti-RelA antibody (Rabbit polyclonal C20), IgG (Rabbit polyclonal), anti-NFKBIA antibody (mouse monoclonal H-4), anti-NFKBIE antibody (mouse monoclonal G-4), anti-NFKB1 antibody (mouse monoclonal E-10), anti-TIMM23 antibody (Mouse monoclonal H-8), and anti-Pol II antibody (Rabbit polyclonal N-20) were purchased from Santa Cruz Biotechnology. Anti-BRD4 antibody (Rabbit monoclonal E2A7X) and anti-TRIM21 antibody (Rabbit monoclonal) were purchased from Cell Signaling.
Protein extraction and RelA immunoprecipitation
Protein extraction and RelA immunoprecipitation - Before harvesting, the A549 cells were washed twice with cold phosphate-buffered saline (PBS). The cells were suspended in 1 ml of low ionic strength immunoprecipitation buffer (50 mM NaCl, 25 mM HEPES pH7.4, 1% IGEPAL CA-630, 10% glycerol, fresh 1mM DTT and protease inhibitor cocktail) and sonicated 3 times, 10 sec each time (BRANSON Sonifier 150, setting 4). The cell lysate was centrifuged for 5 min at 10K xg at 4°C, and the pellet was discarded. The supernatant was used for RelA immunoprecipitation. About 500 μg of cell lysate and 4 μg of anti-RelA antibody (Rabbit polyclonal C20, Santa Cruz) or IgG (Rabbit polyclonal, Santa Cruz) was used in each IP. The mixture was incubated overnight at 4°C, and then 30 μL of protein A magnetic beads (Dynabeads, Invitrogen) were added. After incubation at 4°C for 4h, the beads were separated from the supernatant with a magnetic stand. The beads were washed with PBS five times before trypsin digestion.
Protein extraction and BRD4 immunoprecipitation
Protein extraction and BRD4 immunoprecipitation - Before harvesting, the hSAECs were washed twice with cold phosphate-buffered saline (PBS). The cells were suspended in 2 ml of low ionic strength immunoprecipitation buffer (50 mM NaCl, 25 mM HEPES pH7.4, 1% IGEPAL CA-630, 10% glycerol, fresh 1mM DTT and protease inhibitor cocktail) and sonicated 3 times, 10 sec each time (BRANSON Sonifier 150, setting 4). The cell lysate was centrifuged for 5 min at 10K xg at 4°C, and the pellet was discarded. The supernatant was used for BRD4 immunoprecipitation. About 3 mg of cell lysate and 5 μL of anti-BRD4 antibody (Rabbit monoclonal E2A7X, Cell Signaling) or 4μg of IgG (Rabbit polyclonal, Santa Cruz) was used in each IP. The mixture was incubated overnight at 4°C, and then 40 μL of protein A magnetic beads (Dynabeads, Invitrogen) were added. After incubation at 4°C for 4h, the beads were separated from the supernatant with a magnetic stand. The beads were washed with PBS five times before trypsin digestion.
Trypsin digestion
ABC method – on-bead digestion in ammonium bicarbonate buffer
The trypsin digestion was performed as previously described.12,16 100 μL of 50 mM ammonium bicarbonate was added to each sample. The beads were suspended with gentle vortexing for 1h. The proteins on the beads were reduced with 10 mM DTT for 30 min, then alkylated with 20 mM IDA for 1h in dark. An aliquot of 4 μg of sequencing-grade trypsin was added to each sample before a 4 h incubation at 37°C with gentle shaking; the supernatant was then collected. Another 4 μg of trypsin was then added to the beads and the sample incubated at 37°C overnight with gentle shaking; the supernatant was then collected. After trypsin digestion, the beads were washed twice with 50 μL of 50% ACN and the supernatants collected. All of the supernatants were combined and dried with a SpeedVac. Three independent replicates were performed for both RelA and IgG IPs.
TFE method – on-bead digestion in trifluoroethanol buffer
100μL of TFE buffer (10% TFE in 50 mM ABC) was added to each sample. The beads were suspended with gently shakingor 1h. The proteins on the beads were reduced with 10 mM DTT for 30min, then alkylated with 20mM IDA for 1h in the dark. An aliquot of 4 μg of sequencing-grade trypsin was added to each before a 4 h incubation at 37°C with gentle shaking; the supernatant was then collected, and another 4 μg of trypsin in TFE buffer added to the beads before an overnight incubation at 37°C with gentle shaking; the supernatant was then collected. After trypsin digestion, the beads were washed twice with 50 μL of 50% ACN and the supernatants collected. All of the supernatants were combined and dried with a SpeedVac. Three independent replicates were performed for both RelA and IgG IPs.
Urea method – two-step digestion in urea buffer
The trypsin digestion was performed as reported previously.11,22,23 Briefly, the beads were incubated in 60 μL of urea-based digestion buffer 1 (2 M urea, 50mM Tris-HCl pH7.5, 8 μg trypsin) at room temperature for 30 min with gentle vortexing. The beads were then separated from the supernatant and washed twice with 25 μL of buffer 2 (containing 2M urea, 50mM Tris-HCl pH7.5, 1mM DTT). The supernatants were combined and allowed to digest overnight at room temperature; 20ul of IDA (5mg/ml) was then added to the samples and incubated for 30 min in the dark. Three independent replicates were performed for both RelA and IgG IPs.
SDS-FASP method – SDS elution with filter-aided sample preparation (FASP)
After IP, the beads were suspended in 50 μL of Laemmli loading buffer (BIO-RAD) with 10 mM DTT and heated at 75°C for 10 min. The beads and supernatant were separated using a magnetic stand and the supernatant was collected. The beads were washed once with 50 μL of Laemmli loading buffer and the supernatants were combined and loaded onto 10 KDa filter units (Millipore) and centrifuged at 12,000 ×g for 20 min. FASP trypsin digestion was performed as described previously.24 Briefly, 100 μL of freshly made UA buffer (8M urea in 0.1M Tris-HCl pH 8.5) was added into the filter unit and centrifuged at 12,000 xg for 20 min. This step was repeated twice. About 100ul of IDA (0.05M IDA in UA buffer) was added into the filter and incubated in the dark for 20 min. After centrifugation at 12,000 xg for 20 min, 100 μL of fresh UB buffer (8M urea in 0.1M Tris-HCl pH8.0) was added into the filter unit before centrifuging at 12,000 xg or 20 min. This step was repeated twice, then 135 μL of 50 mM ABC and 8 μg of trypsin were added into the filter unit. The sample was incubated in 2 M urea overnight at room temperature. The resulting peptides were collected by centrifugation. The filter was washed with 100ul of 50mM ABC and 50 μL of 0.5M NaCl.
SDS-gel method – SDS elution with SDS-PAGE and in-gel digestion
After IP, the beads were suspended in 20 μL of Laemmli loading buffer (BIO-RAD) with 10 mM DTT and heated at 75°C for 10 min. The beads and supernatant were separated using a magnetic stand and the supernatant collected. The beads were washed once with 20 μL of Laemmli loading buffer and the supernatants combined. The proteins were separated on a 4–20% TGX gel (Bio-Rad) and then stained with a Novex colloidal blue staining kit (Invitrogen). The whole lane was excised from the gel and further cut into 1mm3 slices; in-gel digestion was performed as previously described.25,26 Briefly, the colloidal blue stain was removed with 25 mM ABC-50% MeOH and the gel further washed three times with 10% acetic acid-50% MeOH and once with water. The gels were dehydrated with ACN and dried in a SpeedVac. Then, 800 μL of 20 mM IDA in 50 mM ABC was added to cover the gel pieces and incubated in dark for 30 min. An aliquot of 8 μg of trypsin in 50 mM ABC (pH 8.0) was added to the gel pieces and incubated overnight at 37°C with gentle shaking. After trypsin digestion, the supernatant was collected and the peptides were extracted from the gels with 50% ACN/5% TFA and 75% ACN/0.1% TFA. All of the supernatants were combined and dried in a SpeedVac.
LC-MS/MS analysis
A nanoflow ultra-high performance liquid chromatography (UHPLC) instrument (Easy nLC, Thermo Fisher Scientific) was coupled on-line to a Q Exactive mass spectrometer (Thermo Fisher Scientific) with a nanoelectrospray ion source (Thermo Fisher Scientific). Peptides were loaded onto a C18-reversed phase column (25 cm long, 75 μm inner diameter) and separated with a linear gradient of 5–35% buffer B (100% acetonitrile in 0.1% formic acid) at a flow rate of 300 nL/min over 180 min. Each sample was analyzed by LC-MS/MS twice. MS data were acquired using a data-dependent Top15 method, dynamically choosing the most abundant precursor ions from the survey scan (350–1400 m/z) using HCD fragmentation. Survey scans were acquired at a resolution of 70,000 at m/z 400. Unassigned precursor ion charge states as well as singly charged species were excluded from fragmentation. The isolation window was set to 3 Da and fragmented with normalized collision energies of 27. The maximum ion injection times for the survey scan and the MS/MS scans were 20 ms and 120 ms respectively, and the ion target values were set to 3E6 and 1e5, respectively. Selected sequenced ions were dynamically excluded for 30 seconds. Data were acquired using Xcalibur software. Detail information about the parameters for LC-MS/MS analysis was tabulated in Supplemental table S1.
Data processing and Bioinformatic Analysis
Raw MS data were analyzed using MaxQuant software version 1.5.2.8 using the Andromeda search engine27,28. The initial maximum allowed mass deviation was set to 10 ppm for monoisotopic precursor ions and 20 ppm for MS/MS peaks. Enzyme specificity was set to trypsin, defined as C-terminal to arginine and lysine excluding proline, and a maximum of two missed cleavages were allowed. Carbamidomethylcysteine was set as a fixed modification, methionine oxidation as variable modifications. The spectra were searched with the Andromeda search engine against the human sequence database (containing 42,130 human protein entries, downloaded from SWISSPROT database on December 26, 2015) combined with 248 common contaminants and concatenated with the reversed versions of all sequences. Protein identification required at least one unique or razor peptide per protein group. Quantification in MaxQuant was performed using the built-in XIC-based label-free quantification (LFQ) algorithm28. The required false positive rate for identification was set to 1% at both the peptide and protein level, and the minimum required peptide length was set to 8 amino acids. Detail information about the parameters for MaxQuant analysis was tabulated in Supplemental table S1. For statistical analysis of MaxQuant output we used the Perseus platform (Version 1.5.5.3).29 Contaminants, reverse identification and proteins only identified by site were excluded from further data analysis. The LFQ values were log2-transfomed. After filtering (at least 3 valid LFQ values in at least one group), remaining missing LFQ values were imputed from a normal distribution (width, 0.3; down shift, 1.8). Student’s two-sample t test was used to assess statistical significance of protein abundances using a 5% permutation-based FDR adjustment. Venn diagram was generated by using a Web tool, InteractiVenn (http://www.interactivenn.net/).30
Scoring and Analyzing the RelA Interactome
Each MS dataset was analyzed by the mass spectrometry interaction statistics (MiST) algorithm. The possibility of each identified protein being a RelA interactor or IgG interactor was scored by the MiST algorithm.6,10,31 Two criteria were used to determine which proteins were specific RelA interactors: 1) prey proteins with a RelA-prey MiST score ≥ 0.75 and IgG-prey Mist score ≤ 0.75; 2) the enrichment of the proteins in RelA IP relative to negative control IgG was statistically significant (P value of Student’s two sample t test with permutation-based FDR adjustment <0.05).
Network analysis and visualization
The identified RelA interactors were subjected to STRING v10 (http://string-db.org) to explore the interaction between the RelA interactors.32 Only the interactions with experimentally determined interaction score ≥ 0.75 are considered to be true PPIs. The new RelA-prey interactions identified from this study and the connections between RelA interactors were combined and analyzed by Cytoscape v 3.4.0. (http://cytoscape-publications.tumblr.com/archive).33 The connectivity of RelA interactors was analyzed using the NetworkAanlyzer tool of Cytoscape. NetworkAanlyzer calculates the neighborhood connectivity of a node n as the average connectivity of all neighbors of n. The neighborhood connectivity distribution gives the average of the neighborhood connectivity of all nodes n with k neighbors for k = 0,1,….34,35
Stable isotope dilution (SID)-SRM-MS and data analysis
The SID-SRM-MS assays were developed as described previously.36,37 The three or four highest intensity y ions in MS/MS were selected to generate the SRM Q1/Q3 transition. The collision energy (CE) breakdown curve of each Q1/Q3 transition was acquired so as to select the optimal CE. The S-lens voltage breakdown curve of each precursor ion was acquired in the same fashion.The signature peptides and SRM parameters for protein RelA, nuclear factor NFκB p105 subunit (NFKB1), NFκB p100 subunit (NFKB2), NFκB inhibitor alpha (NFKBIA), NFκB inhibitor beta (NFKBIB), and NFκB inhibitor epsilon (NFKBIE) are listed in Supplemental Table S2. The peptides were chemically synthesized incorporating isotopically labeled [13C615N4] arginine or [13C615N2] lysine to a 99% isotopic enrichment (Thermo Scientific). The RelA complex was digested as described above. The tryptic digests were reconstituted in 30 μL of 5% formic acid/0.01% trifluoroacetic acid. An aliquot of 10 μL of diluted stable isotope-labeled standard (SIS) peptides was added to each tryptic digest. These samples were desalted with a ZipTip C18 cartridge. The peptides were eluted with 80% ACN and dried. The peptides were reconstituted in 30 μL of 5% formic acid/0.01% TFA and were directly analyzed by LC-SRM-MS.
LC-SRM-MS analysis was performed with a TSQ Vantage triple quadrupole mass spectrometer equipped with nanospray source (Thermo Scientific, San Jose, CA). The online chromatography were performed using an Eksigent NanoLC-2D HPLC system (AB SCIEX, Dublin, CA). Prior to LC-SRM analysis, 10 μL of stable isotope labeled peptide standards (SIS) was added to each sample. The samples were desalted with C18 ZipTip (Millipore). An aliquot of 10 μL of each of the tryptic digests was injected onto a C18 reversed-phase nano-HPLC column (PicoFrit™, 75 μm × 10 cm; tip ID 15 μm) at a flow rate of 500 nL/min with a 20-min 98% A, followed by a 15-min linear gradient from 2–30% mobile phase B (0.1 % formic acid-90% acetonitrile) in mobile phase A (0.1 % formic acid). The TSQ Vantage was operated in high-resolution SRM mode with Q1 and Q3 set to 0.2 and 0.7-Da Full Width Half Maximum. All acquisition methods used the following parameters: 1800 V ion spray voltage, a 275°C ion transferring tube temperature, a collision-activated dissociation pressure at 1.5 mTorr; the S-lens voltage used the values in the S-lens table generated during MS calibration.
All SRM data were manually inspected to ensure peak detection and accurate integration. The chromatographic retention time and the relative product ion intensities of the analyte peptides were compared to those of the stable isotope-labeled standard peptides. The analyte peptides and their stable isotopically labeled peptide standard (SIS) analogs that had same chromatographic retention times (variance below 0.05 min) and relative product ion intensities (±20% variance in the relative ratios for each fragment) were considered as true peptide identification that were free of matrix interference from co-eluting ion.36,37 The peak areas in the extract ion chromatography of the native and SIS version of each signature peptide were integrated using Xcalibur® 2.1. The default values for noise percentage and baseline subtraction window were used. The ratio between the peak areas of the native and SIS version of each peptide was calculated.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium38 via the PRIDE partner repository with the dataset identifier PXD005984 and PXD006739.
RESULTS AND DISCUSSION
Analytical workflow for the comparison of trypsin digestion methods for AP-MS
In general, AP-MS involves immunoprecipitation of a protein complex with an antibody against the bait protein, trypsin digestion of the protein complex, and mass spectrometry analysis either with shotgun proteomic approaches6,11,12, targeted selected reaction monitoring (SRM)16 or sequential window acquisition of all theoretical mass spectra (SWATH).9 Unlike global proteome profiling, in which the proteins are usually digested in solution, in AP-MS the protein complexes are either digested “on-beads” or eluted from the beads with chaotropic regents or surfactants followed by “in-solution” or “in-gel” trypsin digestion. A literature survey of AP-MS-related protein-protein interaction studies reveals that there are three widely used protocols for trypsin digestion of the protein complex isolated by affinity purification. The first (ABC method) is the most straightforward approach, where the protein complex is trypsin-digested on the beads in an ammonium bicarbonate buffer, and after the digestion, the tryptic peptides are eluted from the beads12,16. The second protocol (SDS-gel method) involves first eluting the protein complex from the beads with SDS buffer and separating the protein complex on an SDS-PAGE. The gel bands of interest or the whole lane is then subjected to in-gel digestion2,39,40. The third protocol (Urea method) is a hybrid approach11,22,23 in which the protein complex is subjected to a brief (30 min) on-bead digestion in a urea buffer and the solution is separated from the beads and the trypsin digestion is continued in solution for a longer period of time (> 16h).
In addition to these three commonly used protocols, we explored two new methods, the TFE method and the SDS-FASP method (Figure 1A). TFE can improve protein solubility and denaturation, and can be easily removed by evaporation after trypsin digestion. It has been proven that TFE can greatly improve the efficiency of in-solution trypsin digestion.14,41,42 We tested a TFE protocol in which the protein complex was digested on beads in a TFE buffer. We expected that TFE would be beneficial for on-bead digestion as it is for in-solution digestion in global proteomic or phospho-proteomic profiling. SDS has been routinely used for eluting, dissociating, and denaturing the protein complex, but must be removed before trypsin digestion to avoid inhibition of trypsin activity and interference with MS analysis. In-solution removal of SDS is preferable to in-gel removal because in-gel digestion may prevent peptide recovery and requires additional sample-handling steps. FASP has been shown to be an efficient way for in-solution depletion of SDS.24 We developed the SDS-FASP protocol by combining elution of the protein complex from beads, depleting SDS with FASP, and in-solution digestion. We divided these five protocols into two categories – on-bead digestion (ABC and TFE methods) and elution-digestion (Urea, SDS-FASP and SDS-gel methods). Our overall experimental workflow for comparing the digestion methods is shown in Figure 1A. The RelA complex was IP’ed with anti-RelA antibody from the whole lysate of A549 cells. SID-SRM-MS analysis of RelA in the starting cell lysate and flow-through after IP showed that about 93% of RelA in the whole cell lysate was pulled down by anti-RelA antibody, indicating a highly efficient affinity purification (Figure 1B). The RelA complex was subjected to trypsin digestion with one of these five methods. Pre-immune IgG was used as a negative control, and the proteins associated with IgG were processed in parallel. The tryptic peptides resulting from each digestion method were analyzed and quantified by label-free LC-MS/MS analysis. A rigorous statistical analysis was used to identify which proteins were specific RelA interactors.
Figure 1. Analytical workflow for comparison of trypsin digestion methods for AP-MS analysis of protein-protein interaction.
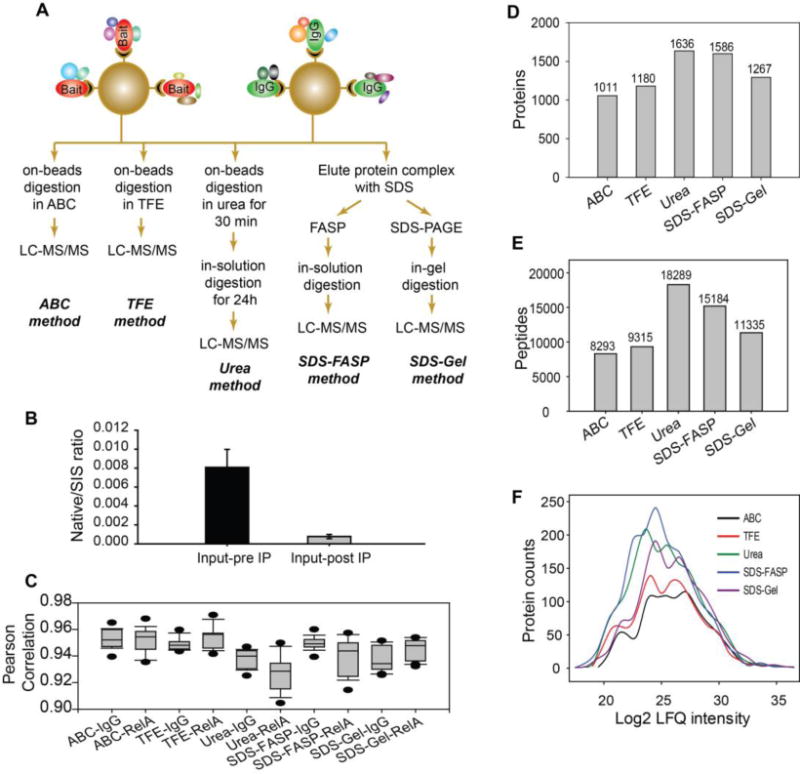
(A) Schematic workflow for five trypsin digestion methods for AP-MS analysis of RelA interactome. The protein complexes purified with RelA IP or control IgG IP were digested by five different methods, followed by label-free LC-MS/MS quantification. (B) Isolation efficiency of RelA IP. The amount of RelA in the whole cell lysate of A549 cells before and after RelA IP was measured by SID-SRM-MS. (C) Reproducibility of biological and technical replicates. The Pearson correlations (r2) of the pairwise comparison of log2-transformed protein abundance between replicates are shown for each digestion method. Note that the ranges fall between 0.9–0.97. (D) The number of proteins identified by each trypsin digestion method. (E) The number of peptides identified by each trypsin digestion method. (F) Histogram of the abundance of the proteins identified by each trypsin digestion method.
To determine the reproducibility of replicates, pairwise analysis of the log2-transformed protein abundance was analyzed. The Pearson correlations (r2) ranged from 0.90 to 0.97, indicating a high degree of concordance (Figure 1C). We noted that on-bead digestion methods have slightly better reproducibility than elution-digestion methods. The r2 was greater for the ABC (mean r2 = 0.95 ± 0.009) and TFE methods (mean r2 = 0.95 ± 0.008) vs that for the other three elution-digestion methods. The r2 was lowest for the urea methods (mean r2 = 0.93 ± 0.013). Compared to on-bead digestion, elution-digestion methods require more sample preparation steps, which may introduce some variation in quantification.
Identification of RelA-interacting proteins
The different digestion methods yielded a significantly different number of peptides and protein identifications (Figure 1D, E). A total of 1011, 1180, 1636, 1586 and 1267 proteins were quantified by the ABC, TFE, Urea, SDS-FASP, and SDS-gel methods, respectively. The proteins quantified by each method and protein-related information such as the score and q-value of identification, the fold change relative to IgG control pulldown, and statistical analysis results were tabulated in Supplemental table S3. Histograms of the number of protein identifications relative to the MS intensity of the proteins show that the five methods yielded a similar number of high-abundance proteins, but a dramatically different number of low-abundance proteins (Figure 1F). Elution-digestion methods (Urea, SDS-FASP, SDS-gel) yielded a significantly larger number of low-abundance proteins than on-bead digestion methods (ABC and TFE), suggesting that the steric interference of the beads may make the low-abundance proteins less accessible to trypsin, or, alternatively, reduces the recovery of low-abundance tryptic peptides from beads. Consequently, the SDS-FASP method is superior to the SDS-gel method both in terms of the total number of proteins and the number of low-abundance proteins identified.
To determine the statistical significance of RelA vs IgG control, we calculated fold-changes in protein abundance and the statistical significance of the differences in protein abundance using FDR adjustment of a Student’s two-sample t test. The volcano plots of proteins identified by each method (Figures 2A, C, E, G, and I) indicate very strong enrichment of proteins in the RelA IP compared to the IgG control IP.43 To access the validity of RelA•protein interactions identified by AP-MS and distinguish the true RelA-interacting proteins from false positives, we used the MiST algorithm to score the interaction between identified proteins (preys) and bait proteins (RelA or IgG). The MiST score is a composite sum of three measures: protein abundance measured by MS (Abundance), invariability of abundance over replicated experiments (Reproducibility), and uniqueness of an observed bait-prey interaction across all purifications (Specificity).6,31 For each identified protein, the MiST algorithm scored its interaction with both RelA and the negative control IgG based on MS measurement. The MiST score of the protein quantified by each method were tabulated in Supplemental table S3. As shown in Figure 2B, D, F, H, and J), the scatter plots of the RelA-prey MiST score vs the IgG-prey MiST score display a strong inverse correlation (Pearson correlation r2= −0.41 ~ −0.74), indicating that RelA and IgG IP are two distinct affinity purifications. The proteins in the bottom right quadrant have high RelA-prey MiST scores (≥ 0.75), but low IgG-prey MiST scores (≤ 0.75). We considered these proteins to be enriched in the RelA IP, and to be potential RelA interactors. To further filter the data, we adopted the second criterion that the enrichment of the proteins in RelA IP relative to the negative control IgG must be statistically significant (p value of Student’s two sample t-test with permutation-based FDR adjustment <0.05). The red dots in Figure 2 are proteins that have met these two criteria. We considered these proteins to be high-confidence RelA-interacting proteins.
Figure 2. Identification of RelA interactors.
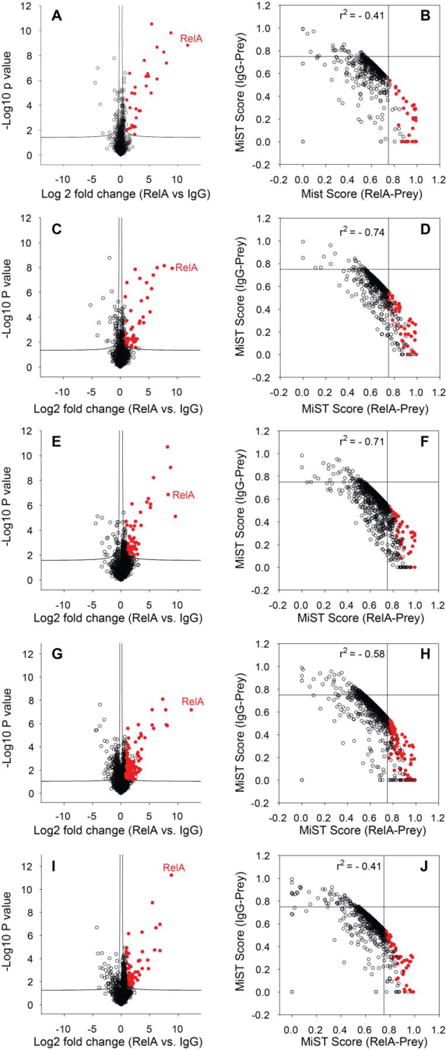
(Left panels) Volcano plots of the –log10 P-value vs. the log2-transformed intensity difference of the RelA IP vs. negative control IgG IP for proteins identified by the (A) ABC method, (C) TFE method, (E) Urea method, (G) SDS-FASP method, and (I) SDS-Gel method. The proteins in the top-right quadrant are proteins enriched with RelA IP relative to IgG IP (Student’s t-test with FDR correction P<0.05). (Right panels) Scatter plots of the MiST score of IgG-prey vs. the MiST score of RelA-prey for each protein identified by the (B) ABC method, (D) TFE method, (F) Urea method, (H) SDS-FASP method, and (J) SDS-Gel method. The red dots are the proteins that met the criteria for identification of high-confidence RelA interactors.
After applying the two-layer filtering, a total of 30, 44, 52, 114, and 58 RelA interactors were identified with the ABC, TFE, Urea, SDS-FASP, and SDS-gel methods, respectively (Figure 3A). We noted that the SDS-FASP method resulted in the largest pool of RelA interactors (114), while the ABC method produced the least (30). Histograms of the number of RelA interactors relative to the MS intensity of these proteins indicates that the ABC method identified a few more high-abundance RelA interactors, while SDS-FASP identified significantly more low-abundance RelA interactors than the other methods (Figure 3B). We identified a total of 212 RelA interactors (Supplemental Table S3). A Venn diagram analysis of the RelA interactors identified by the five different trypsin digestion methods found that 13 proteins were consistently identified by all methods (Figure 3C, Supplemental Table S3). Most of these proteins are well-characterized benchmark RelA-interacting proteins, such as NFKB1, NFKB2, Proto-oncogene c-Rel (REL), NFKBIB, and NFBKIE, indicating that our analytical workflow and rigorous statistical analysis resulted in a robust dataset of RelA interactors. We also identified some previously unknown RelA interactors, including the mitochondrial import inner membrane translocase subunit Tim23 (TIMM23) and the E3 ubiquitin-protein ligase, TRIM21. To confirm that these are true RelA interactors, we conducted reciprocal IP of TIMM23 and TRIM21, in parallel with reciprocal IP of well-known RelA interactors (NFKBIA, NFKBIE, NFKB1). IgG was used as negative control. The amount of RelA in each sample was measured by SID-SRM-MS. As shown in Figure 3D, RelA was significantly enriched with the IP of NFKBIA, NFKBIE, NFKB1, TIMM23 and TRIM21 relative to IgG, indicating that these proteins are bone fide RelA interactors.
Figure 3. RelA interactors.
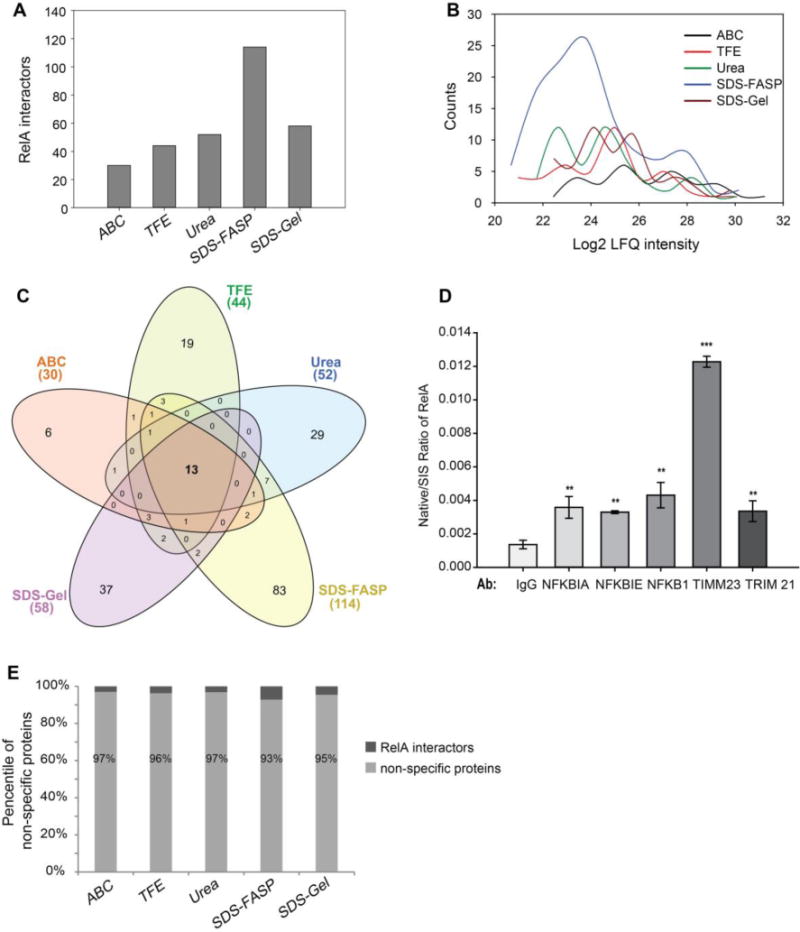
(A) The number of RelA interactors identified by each trypsin digestion method. (B) Histogram of the abundance of the RelA interactors identified by each trypsin digestion method. (C) Venn diagram of the RelA interactors identified by each trypsin digestion method. (D) SID-SRM-MS measurement of RelA in reciprocal IP of RelA interactors. (E) The percentile of non-specific binding proteins identified by each trypsin digestion method.
Comparison of the performance of different trypsin digestion methods
In elution-digestion methods, the bait protein and its interacting proteins were eluted from beads. Sometimes additional fractionation steps such as FASP or SDS-PAGE had to be done before trypsin digestion. On-bead digestion omits all these interim steps to progress straight to the digestion. The limitation of on-bead digestion is that the beads may hinder the accessibility of trypsin to the proteins. As we have demonstrated above (Figure 3A), the order of the number of RelA interactors identified by each approach was: SDS-FASP > SDS-gel > Urea > TFE > ABC. The ABC method produces the smallest pool (30) of RelA interactors, while the SDS-FASP identified the largest number of RelA interactors (114). Overall, the elution-digestion approaches gave better performance than on-bead digestion methods in terms of the depth of coverage of the RelA interactome, and SDS-FASP seems to outperform all the other methods tested.
We noticed that only a small fraction of protein identified (3 – 7%) were RelA interactors and the majority of proteins identified were non-specific binding proteins, which may partly be due to the high-sensitivity of MS and even a trace amount of non-specific binding proteins could be detected by MS. This highlights the advantages of quantitative proteomics analysis and rigorous statistical analysis and computational PPI scoring in isolating true interactors from a large pool of non-specific proteins. We investigated whether trypsin digestion methods would affect the identification of non-specific binding proteins. The numbers of non-specific binding proteins identified by each trypsin digestion method were different, but we found no significant difference in the percentage of non-specific proteins identified by each method (Figure 3E).
With the results of MiST analysis, we were able to compare the performance of each method in more detail. Surprisingly, the RelA interactions identified by the ABC method had the highest average MiST Score (Figure 4A); 21 out of 30 (70%) RelA interactors had a MiST score higher than 0.9. By contrast, the RelA interactors identified by SDS-FASP had the lowest average MiST Score; only 28% (32 out of 114) RelA interactors had a MiST score higher than 0.9. Similarly, RelA interactions identified by the ABC method had the highest average MiST reproducibility (Figure 4B) and MiST specificity values (Figure 4C), while the RelA interactors identified by SDS-FASP had the lowest average MiST reproducibility and specificity values among the five different methods tested. Consistent with the histogram analysis of MS intensities of RelA interactors (Figure 3B), the RelA interactors identified by the ABC method had higher average MS signals (Figure 4D) than those identified by other methods. This result indicates that the ABC method can efficiently digest moderate to high abundance proteins in the RelA complex, but the recovery of low-abundance proteins in the RelA complex was poor. The total number of tryptic peptides obtained from the ABC digestion method was much lower than with other methods (Figure 1D), a phenomenon that improved the identification of high-abundance peptides because the competition for MS/MS fragmentation during LC-MS/MS analysis was reduced. Our data indicate that the SDS-FASP method was able to recover much more low-abundance RelA interactors than other methods (Figure 3C), but these low-abundance RelA interactors often had lower MiST Score than high-abundance RelA interactors. As a consequence, the average MiST score for the RelA interactors identified by the SDS-FASP method was decreased. We plotted the MiST Score, reproducibility and specificity values against the MS intensities of the RelA interactors. We found that high-abundance proteins are more likely to have a higher MiST Score, reproducibility and specificity values than low-abundance proteins (Figures 4E–G). It appears that the lower median MiST scores of RelA interactors identified by the SDS-FASP method may be caused by a systematic bias of MiST algorithm against low-abundance interactors, not an indication that SDS-FASP method produced a lower-quality dataset.
Figure 4. Comparison of different trypsin digestion methods for AP-MS analysis of the RelA interactome.
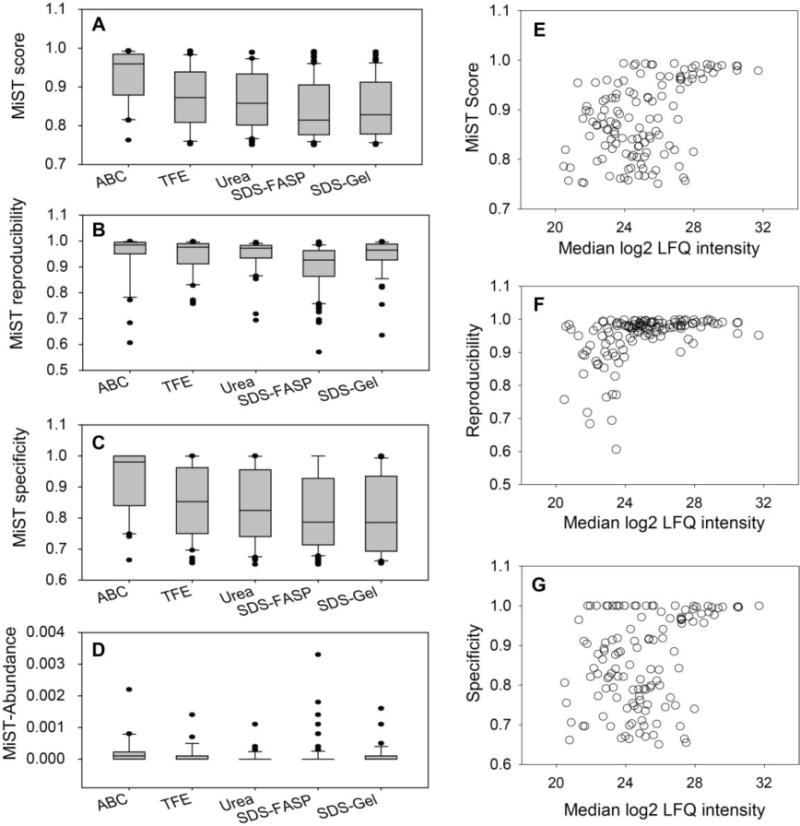
(A) Boxplot of the MiST scores of the RelA interactors identified by each trypsin digestion method. (B) Boxplot of MiST reproducibility values of the RelA interactors identified by each trypsin digestion method. (C) Boxplot of the MiST specificity values of the RelA interactors identified by each trypsin digestion method. (D) Boxplot of the MiST abundance of the RelA interactors identified by each trypsin digestion method. (E) Scatter plot of the MiST score vs. the MS intensity of proteins. (F) Scatter plot of the MiST reproducibility value vs. the MS intensity of proteins. (G) Scatter plot of the MiST specificity value vs. the MS intensity of proteins.
Several studies have indicated that TFE can greatly increase the efficiency of in-solution trypsin digestion.14,41,42 However, our data showed that the benefit of TFE for on-bead digestion is not as significant as that with in-solution digestion. The number of proteins (Fig.1C), peptides (Figure 1D), and RelA interactors (Figure 3D) identified by TFE on-bead methods is slightly higher compared to the ABC on-bead digestion method (41 to 30), suggesting that steric interference is the predominant factor for the efficiency of on-bead trypsin digestion, not the solvent used for protein denaturation.
The urea method is a hybrid method that combines a brief on-bead trypsin digestion with elution-digestion. The total number of proteins and RelA interactors identified by the urea method was higher than with on-bead digestion methods, but we found that the reproducibility of the MS intensities of the identified proteins was the lowest among the methods tested. This may be caused by incomplete and less reproducible trypsin digestion during the short on-bead digestion period.
The SDS-FASP and SDS-gel methods produced the largest number of RelA interactors, indicating that SDS is very efficient in stripping the protein complex from the beads. As expected, the data proved that FASP and in-solution digestion is superior to SDS-PAGE and in-gel digestion. The number of RelA interactors identified by the SDS-FASP method was almost double the number of RelA interactors identified by the SDS-gel method. Another advantage of the SDS-FASP method over SDS-gel is that it avoids the laborious and time-consuming steps such as SDS-PAGE, staining and destaining the gel bands, and improved peptide recovery.
We performed GO annotation enrichment analysis of the RelA interactors identified by each method (Supplemental Table S4). RelA interactors identified by the ABC, TFE and urea methods have similar GO annotation enrichment; the top biological process terms are regulation of type I interferon production; toll-like receptor 2, 5, 9, 10 signaling pathway; MyD88-dependent toll-like receptor signaling pathway; and I-kappaB kinase/NFκB signaling. These GO terms are well-known major biological functions of RelA. The RelA interactors identified by SDS-FASP had a different GO annotation enrichment; although interferon production, toll-like receptor signaling pathways, and I-kappaB kinase/NFκB signaling were still enriched, they were not the most significant terms. The top GO biological process terms for RelA interactors identified by SDS-FASP were nucleic acid metabolic process, gene expression, and chromosome organization, indicating that the ability to identify a minor component of the RelA complex was increased with the enhanced sensitivity of the SDS-FASP approach.
Comparison of the effects of different trypsin digestion methods on SID-SRM-MS analysis of PPIs
SID-SRM-MS has been used in quantifying the dynamics of PPIs.12,16 Unlike global identification of PPIs using shotgun proteomics, the SID-SRM-MS method is based on the MS analysis of pre-selected signature peptides for each targeted proteins. The quantification is usually carried out by comparing the signals of pre-selected native peptides to the signals of stable isotope labeled standard peptides.14 Therefore, when the relative quantification is performed by SRM-MS, it is the reproducible production of these pre-selected native peptides that will affect the quantification, not the completeness of digestion of the entire protein. Previous study on in-solution digestion indicated that the effect of different solvents, chaotropic agents, and surfactants on SRM quantification varied from protein to protein.14 To investigate the effects of different trypsin digestion methods on the SID-SRM-MS analysis of the protein complex, we preselected a set of RelA interactors – RelA, NFKB1, NFKB2, NFKBIA, NFKBIB, and NFKBIE from the pool of RelA interactors identified in this study, and use SID-SRM-MS to determine and compare the amounts of native peptides from these six proteins when different trypsin digestion are used. First, as can be seen in Figure 5, no matter which trypsin digestion methods were used, all six of the proteins showed significant enrichment in the RelA IP relative to the negative control IgG IP. Therefore, SRM-MS quantification provides an independent validation of the data generated through the label-free LC-MS/MS approach. Second, Figure 5 also shows that the effect of different trypsin digestion methods on the SRM-MS measurement varies from protein to protein. For RelA, NFKB2, NFKBIA, and NFBKIB, the variation of native/SIS ratio of the native peptides generated by different digestion methods is within the range from 9% to 16%. We consider that the impact of trypsin digestion protocols on SRM analysis of these proteins is minimal. However, for NFKB1 and NFKBIE the variation of native/SIS ratio of the native peptides generated by different digestion methods jumped to 46% and 64%, respectively. For NFKB1, the signals of native peptides generated by the urea method were much higher than those from other methods. For NFKBIE, the signals of native peptides generated by the urea method and SDS-FASP protocol were much higher than those from other methods; the signal from the SDS-gel method was the lowest, probably due to lower recovery the NFKBIE signature peptide after in-gel digestion. The data indicate that the effect of different trypsin digestion methods on the SRM-MS measurement varies from protein to protein. The sensitivity of SRM quantification depends on the efficiency of production of the preselected signature peptides under different trypsin digestion conditions. To maximize the sensitivity of the SRM detection of targeted protein interactors, it may be necessary to optimize trypsin digestion conditions for each targeted protein or select signature peptide to find the method that can provide the best sensitivity.
Figure 5. SRM-MS analysis of RelA interactors with different trypsin digestion methods.
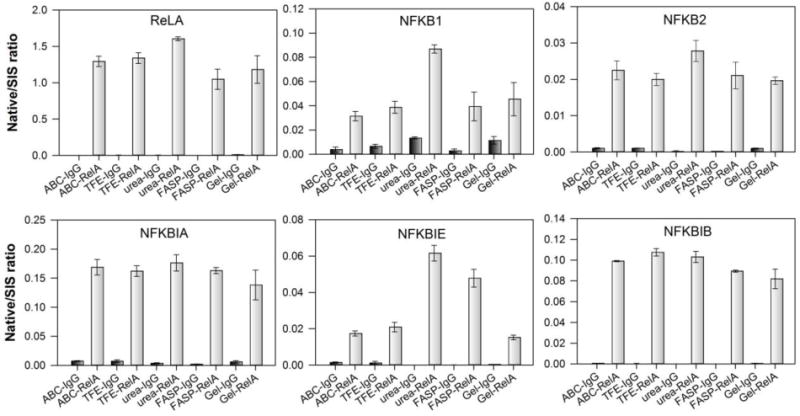
Black bars, SRM measurement of RelA interactors from negative control IgG IP; grey bars, SRM measurement of RelA interactors from RelA IP.
The RelA interactome
RelA has been shown to be predominantly cytoplasmic, but it can be translocated to subcellular organelles such as nuclear5 and mitochondria44–46 and form protein complexes with its interactors. We plotted the 212 RelA interactors identified in this study in a network representation (Figure 6A). We also introduced an additional 134 interactions between RelA interactors from the STRING protein-protein interaction database. The RelA interactors (nodes with yellow to orange color) that have known interactions with each other are clustered together to identify the function of the RelA-interactome. We found that RelA is associated with many protein complexes with important biological functions. Some examples of these complexes are the IκB/NFκB complex, RNA Pol II-mediator complex, histone methyltransferase complex, spliceosomal complex, ribonucleoprotein complex, origin recognition complex (ORC), and the calpain complex. The IκB/NFκB complex regulates the expression of genes influencing a broad range of biological processes including innate and adaptive immunity, inflammation, stress responses, and cancer.5,17 While in an inactivated state, RelA and NFKB1 form a heterodimer (NFκB) in the cytosol. Its functions are inhibited by the inhibitory protein NFKBIA/IκBα. Upon activation via a variety of extracellular signals, IKK kinases phosphorylate the NFKBIA/IκBα protein, which results in degradation and dissociation of NFKBIA/IκBα from NFκB. The activated NFκB is translocated into the nucleus, where it binds to specific DNA sequences. The DNA/NFκB complex recruits other proteins such as coactivators and RNA polymerase, which transcribes the downstream DNA into mRNA.5,17 Our finding that RelA interacts with the RNA Pol II-mediator complex has been previously reported.16 RelA interacts with P-TEFb to release paused RNA Pol II. The interaction is mediated by RelA Ser phosphorylation by a family of ribosomal S6 kinases in a stimulus-dependent manner. Specifically, the formation of phospho-Ser276 RelA is critical for activation of a subset of highly inducible “immediate-early” inflammatory genes maintained in an open chromatin conformation.19 In this pathway, phospho-Ser276 RelA binds the cyclin-dependent kinase (CDK)9 complex, responsible for phosphorylation of Ser2 in the carboxy-terminal domain (CTD) of RNA Pol II, licensing it to produce fully spliced transcripts, a process known as transcriptional elongation.18,19 Calpains are cytosolic calcium-activated neutral proteases. They mediate a parallel proteasome-independent pathway for cytokine-inducible NFKBIA/IκBα proteolysis and NFκB activation.47 The spliceosomal complex is another large group of proteins that was found to be associated with RelA. This complex includes several of the so-called “SR proteins,” ribonucleoproteins and components of the exon junction complex (EJC), such as mago nashi homolog (MAGOH), RNA-binding protein with serine-rich domain 1 (RNPS1), and pinin (PNN). The EJC complex mediates many RNA-related processing steps, such as mRNA export, transition, and decay.48,49 It also interacts with SR proteins and promotes mRNA packaging and compaction.49 Although our data indicate that RelA associates with many components of the spliceosomal complex, further studies will be required to understand how RelA regulates spliceosomal complex functions. The Origin Recognition Complex (ORC) is associated with specific loci on the chromosomal DNA where DNA replication takes place, and is the central component of cell-cycle control of the chromosomal replication pathway. During the cell cycle, CDC6/CDC18 temporally associates with ORC at the G1-S boundary and is essential for assembly of the pre-replication complex (preRC) at origins of replication before the initiation of DNA synthesis. This is followed by the loading of the minichromosome maintenance (MCM) protein complex onto the replication origins, leading to the formation of preRCs. After MCM loading, S-phase CDK phosphorylates CDC6/CDC18; this phosphorylation leads to the rapid degradation of CDC6/CDC18 to prevent re-initiation.50,51 Although it has been observed that NFκB/RelA is a regulator of the cell cycle by activating cyclin D1 expression and promoting G1-to-S progression,52 there was no report of the direct interaction between NFκB/RelA and ORC before this study.
Figure 6. RelA interactome.
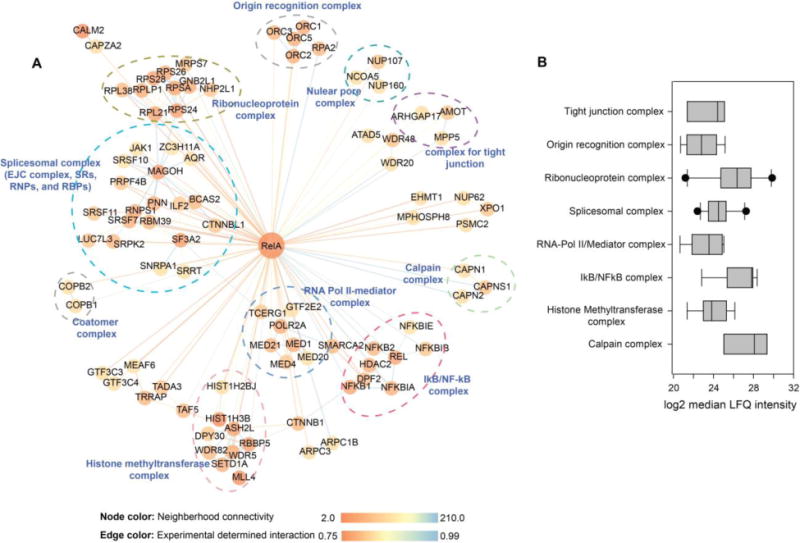
(A) Network representation of RelA interactome. In total, 212 RelA interactors (blue) are represented. The color of each node represents the neighborhood connectivity of each protein. The color of edges corresponds to the MiST score from interactions between RelA and its interactors and also to interactions between RelA interactors that were obtained from publicly available STRING database. (B) Relative abundance of protein complexes that are associated with RelA.
RelA can simultaneously interact with many individual proteins or protein complexes. We estimated the relative abundance of RelA interactors that were pulled down with RelA using the label free LFQ intensities. The boxplots of the median LFQ intensity of the components of each complexe are shown in Figure 6B. Based on this profile, we can estimate the stoichiometry of the interaction between RelA and major protein clusters. In the basal condition, the calpain complex, IκB/NFκB, and the ribonucleoprotein complex are the most abundant protein complexes that were associated with RelA. This analysis indicates that more RelA protein molecules are interacting with these three complexes than complexes such as RNA-pol II/mediator, the spliceosomal complex, or ORC.
Bromodomain-containing protein 4 (BRD4) interactome
To test whether the protocols performance might change when different baits, antibodies or cell lines are used, we conduct a BRD4 inteactome study using primary human small airway epithelial cells. BRD4 is a member of bromodomain and extra terminal domain family that functions as histone acetyltransferase, a chromatin reader protein and RNA pol II kinase. It play key role in transcription regulation of signal-inducible genes by regulating the activity of RNA polymerase II.16,53 It is also required for maintenance of higher-order chromatin structure.54 In this study, BRD4 interactors were pulled down with anti-BRD4 antibody. Different trypsin digestion methods yielded a significantly different number of protein identifications (Figure 7A). A total of 927, 864, 1008, 977 and 405 proteins were quantified by the ABC, TFE, Urea, SDS-FASP, and SDS-gel methods, respectively. The proteins quantified by each method and protein-related information such as the score and q-value of identification, the fold change relative to IgG control pulldown, and statistical analysis results were tabulated in Supplemental table S5. Similar to the results obtained from the RelA interactome, the Urea method and SDS-FASP method identified more proteins than the on-beads digestion methods (ABC and TFE). We noticed that we identified fewer proteins using SDS-Gel method, which may be caused by the low-efficiency of peptide coverage from the gel pieces. Overall, the elution of the protein complex from beads with SDS or urea before trypsin digestion increases the efficiency of trypsin digestion of protein complex, which is consistent with the conclusion that we draw from the RelA interactome study.
Figure 7. BRD4 interactome and the effects of trypsin digestion methods on the identification of BRD4 interactors.
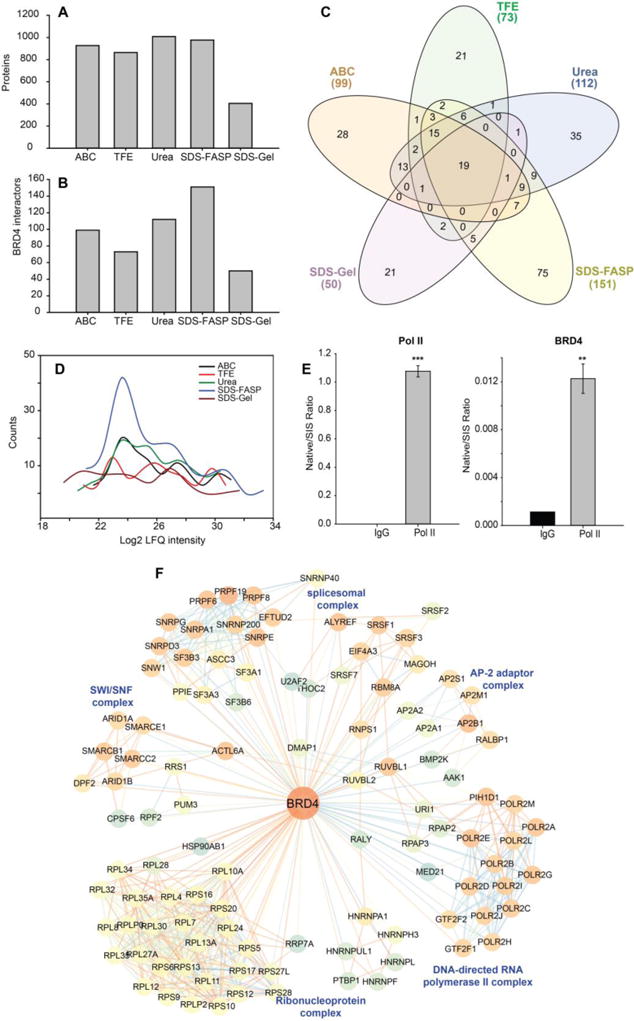
BRD4 complex was IPed with anti-BRD4 antibody and digested with trypsin with five different protocols. (A) The number of proteins identified by each trypsin digestion method. (B) the number of BRD4 interactors identified by each trypsin digestion method. (C) Venn diagram of BRD4 interactors identified by each trypsin digestion method. (D) Histogram of the abundance of the BRD4 interactors identified by each trypsin digestion method. (E) SID-SRM-MS measurement of Pol II and BRD4 in reciprocal IP of Pol II. (F) Network representation of BRD4 interactome. The color of each node represents the neighborhood connectivity of each protein. The color of edges corresponds to the MiST score from interactions between RelA and its interactors and also to interactions between RelA interactors that were obtained from publicly available STRING database.
The high-confidence BRD4 interactors were determined using the same two-layer filter that was used in the RelA interactome study. The MiST score of the protein quantified by each method were tabulated in Supplemental table S5, After applying the two-layer filtering, a total of 99, 73, 112, 151, and 50 BRD4 interactors were identified with the ABC, TFE, Urea, SDS-FASP, and SDS-gel methods, respectively (Figure 7B). SDS-FASP method and Urea method resulted in the larger pool of BRD4 interactors than ABC method and TFE method, a finding that is consistent with RelA interactome study. We noticed that the protocols perform slightly differently in BRD4 interactome study, for example, TFE method identified less number of BRD4 interactors than ABC method and SDS-gel method yield the smallest number of BRD4 interactors. A Venn diagram analysis of the BRD4 interactors identified by the five different trypsin digestion methods found that 19 proteins were consistently identified by all methods (Figure 7C, Supplemental table S5). Consistent with the conclusion obtained from RelA interactome study, elution-digestion methods identified more low-abundance BRD4 interactors (Figure 7D).
In this BRD4 interactome study, we identified 16 core members of RNA pol II complex including DNA-directed RNA polymerase II subunit RPB1 (POLR2A). We conducted a reciprocal IP of pol II. The amount of POLR2A and BRD4 were measured by SID-SRM-MS. As shown in Figure 7E, BRD4 was significantly enriched with the IP of POLRA2 relative to IgG, confirming the interaction between BRD4 and POLR2A.
We plotted the 277 BRD4 interactors identified in this study in a network representation (Figure 7F). We also introduced an additional 596 interactions between BRD4 interactors from the STRING protein-protein interaction database. The BRD4 interactors (nodes with blue to orange color) that have known interactions with each other are clustered together to identify the function of the BRD4-interactome. We found that BRD4 is associated with many protein complexes with important biological functions. Some examples of these complexes are DNA-directed RNA polymerase II complex, spliceosomal complex, ribonucleoprotein complex, SWI/SNF complex, and AP-2 adaptor complex.
CONCLUSIONS
In this study, we used RelA as a model bait protein to determine the trypsin digestion conditions under which the highest possible PPI coverage can be achieved. We found that the identification of the high-abundance RelA interactors is not affected by the choice of trypsin digestion protocols, but trypsin digestion methods have a significant impact on the identification of low-abundance RelA interactors. Overall, eluting the RelA complex from beads before trypsin digestion produces higher coverage of the RelA interactome compared to direct on-bead digestion, and SDS is preferable as the elution buffer vs. urea. To remove SDS before trypsin digestion, two approaches were used, FASP/in-solution digestion and SDS-PAGE/in-gel digestion. Our data suggested that FASP/in-solution digestion is superior to SDS-PAGE/in-gel digestion in terms of identification of low-abundance of RelA interactors. With rigorous statistical and PPI scoring analysis, we identified 212 RelA interactors; many of the PPIs are known and have been studied extensively, while some of the RelA interactors have not been reported before. The functions of these PPI require further study.
From the RelA interactome and BRD4 interactome studies, we can conclude that elution-digestion methods (SDS-FASP and Urea methods) yield higher interactome coverage than on-beads digestions methods; and SDS-FASP method is slightly better than Urea method. We also notice that the performance of the trypsin digestion protocols may change depending on the different baits, antibodies or cell lines used. The binding affinity between bait and interactors, the orientation of the interactors within the complex, and the recovery of tryptic peptides may also affect the performance of the trypsin digestion protocols.
It is also worth noting that different digestion protocols can affect the SRM-MS quantification of PPIs. Even in the relatively small set of proteins that we quantified by SRM-MS, wide variations caused by different digestion protocols were observed, and there is no common procedure that is best for all of the proteins, so optimization of trypsin digestion conditions for each targeted protein may be required.
Supplementary Material
Supplemental table S1: The parameters for LC-MS/MS and Maxquant data analysis. The table contains three spreadsheets.
Supplemental table S2: The parameters for LC-SRM analysis of six RelA interactors.
Supplemental table S3: Proteins that were quantified in the RelA interactome study and the MiST scores of RelA-Prey and IgG-Prey. This table contains six speadsheets as listed below.
Spreadsheet 1. Proteins that were quantified by ABC method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 2. Proteins that were quantified by TFE method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 3. Proteins that were quantified by urea method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 4. Proteins that were quantified by SDS-FASP method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 5. Proteins that were quantified by SDS-gel method and the MiST scores of RelA-Prey and IgG-Prey.
Speadsheet 6. RelA interactors.
Supplemental Table S4. GO annotation enrichment of the RelA interactors identified by each method.
Supplemental table S5: Proteins quantified in the BRD4 interactome study and the MiST scores of RelA-Prey and IgG-Prey. This table contains six speadsheets as listed below.
Spreadsheet 1. Proteins that were quantified by ABC method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 2. Proteins that were quantified by TFE method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 3. Proteins that were quantified by urea method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 4. Proteins that were quantified by SDS-FASP method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 5. Proteins that were quantified by SDS-gel method and the MiST scores of RelA-Prey and IgG-Prey.
Speadsheet 6. BRD4 interactors.
Acknowledgments
We thank Dr. David Konkel for critically editing the manuscript.
Funding
This work was supported by National Institutes of Health Grants NCATS UL1TR001439 (to ARB), DMS-1361411/DMS-1361318 (ARB, YXZ), NIAID AI062885 (ARB), NIEHS P30 ES006676 (ARB) and pilot funding from the Sealy Center for Molecular Sciences (SCMM-SysBio-2016).
Abbreviations
- AP
Affinity purification
- BRD4
Bromodomain-containing protein 4
- CDK9
Cyclin dependent kinase 9
- DTT
Dithiothreitol
- FASP
filter-aided sample preparation
- FDR
False discovery rate
- IDA
Iodoacetamide
- IgG
Immunoglobin G
- IP
Immunoprecipitation
- LC
Liquid chromatography
- LFQ
Label free quantification
- MS
Mass spectrometry
- ORC
Origin recognition complex
- PBS
Phosphate-buffered saline
- PPI
Protein-protein interaction
- RelA
transcription factor p65
- SDS
sodium dodecyl sulfate
- SID
Stable isotope dilution
- SIS
Stable isotope labeled internal standard
- SRM
Selected reaction monitoring
- SWATH
Sequential window acquisition of all theoretical mass spectra
- TFA
Trifluoroacetic acid
- TFE
2,2,2,-trifluoroethanol
- UHPLC
Ultra high performance liquid chromatography
Footnotes
Conflict of interest disclosure
The authors declare no competing financial interests.
References
- 1.Berggard T, Linse S, James P. Methods for the detection and analysis of protein-protein interactions. Proteomics. 2007;7:2833–42. doi: 10.1002/pmic.200700131. [DOI] [PubMed] [Google Scholar]
- 2.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O’Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–43. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 3.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksoz E, Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–68. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 4.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–8. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 5.Brasier AR. The NF-kappaB regulatory network. Cardiovasc Toxicol. 2006;6:111–30. doi: 10.1385/ct:6:2:111. [DOI] [PubMed] [Google Scholar]
- 6.Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D’Orso I, Fernandes J, Fahey M, Mahon C, O’Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ. Global landscape of HIV-human protein complexes. Nature. 2011;481:365–70. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah PS, Wojcechowskyj JA, Eckhardt M, Krogan NJ. Comparative mapping of host-pathogen protein-protein interactions. Curr Opin Microbiol. 2015;27:62–8. doi: 10.1016/j.mib.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins BC, Gillet LC, Rosenberger G, Rost HL, Vichalkovski A, Gstaiger M, Aebersold R. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14–3–3 system. Nat Methods. 2013;10:1246–53. doi: 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- 10.Morris JH, Knudsen GM, Verschueren E, Johnson JR, Cimermancic P, Greninger AL, Pico AR. Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat Protoc. 2014;9:2539–54. doi: 10.1038/nprot.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J Cell Biol. 2010;189:739–54. doi: 10.1083/jcb.200911091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Zhao Y, Kalita M, Li X, Jamaluddin M, Tian B, Edeh CB, Wiktorowicz JE, Kudlicki A, Brasier AR. Systematic Determination of Human Cyclin Dependent Kinase (CDK)-9 Interactome Identifies Novel Functions in RNA Splicing Mediated by the DEAD Box (DDX)-5/17 RNA Helicases. Mol Cell Proteomics. 2015;14:2701–21. doi: 10.1074/mcp.M115.049221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leon IR, Schwammle V, Jensen ON, Sprenger RR. Quantitative assessment of in-solution digestion efficiency identifies optimal protocols for unbiased protein analysis. Mol Cell Proteomics. 2013;12:2992–3005. doi: 10.1074/mcp.M112.025585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Proc JL, Kuzyk MA, Hardie DB, Yang J, Smith DS, Jackson AM, Parker CE, Borchers CH. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J Proteome Res. 2010;9:5422–37. doi: 10.1021/pr100656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Zhao Y, Tian B, Jamaluddin M, Mitra A, Yang J, Rowicka M, Brasier AR, Kudlicki A. Modulation of gene expression regulated by the transcription factor NF-kappaB/RelA. J Biol Chem. 2014;289:11927–44. doi: 10.1074/jbc.M113.539965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian B, Zhao Y, Sun H, Zhang Y, Yang J, Brasier AR. BRD4 mediates NF-kappaB-dependent epithelial-mesenchymal transition and pulmonary fibrosis via transcriptional elongation. Am J Physiol Lung Cell Mol Physiol. 2016;311:L1183–L1201. doi: 10.1152/ajplung.00224.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 18.Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, Brasier AR. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol Cell Biol. 2008;28:3623–38. doi: 10.1128/MCB.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brasier AR, Tian B, Jamaluddin M, Kalita MK, Garofalo RP, Lu M. RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J Virol. 2011;85:11752–69. doi: 10.1128/JVI.05360-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tieri P, Termanini A, Bellavista E, Salvioli S, Capri M, Franceschi C. Charting the NF-kappaB pathway interactome map. PLoS One. 2012;7:e32678. doi: 10.1371/journal.pone.0032678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feldman I, Feldman GM, Mobarak C, Dunkelberg JC, Leslie KK. Identification of proteins within the nuclear factor-kappa B transcriptional complex including estrogen receptor-alpha. Am J Obstet Gynecol. 2007;196:394 e1–11. doi: 10.1016/j.ajog.2006.12.033. discussion 394 e11–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Thun A, Preisinger C, Rath O, Schwarz JP, Ward C, Monsefi N, Rodriguez J, Garcia-Munoz A, Birtwistle M, Bienvenut W, Anderson KI, Kolch W, von Kriegsheim A. Extracellular signal-regulated kinase regulates RhoA activation and tumor cell plasticity by inhibiting guanine exchange factor H1 activity. Mol Cell Biol. 2013;33:4526–37. doi: 10.1128/MCB.00585-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turriziani B, Garcia-Munoz A, Pilkington R, Raso C, Kolch W, von Kriegsheim A. On-beads digestion in conjunction with data-dependent mass spectrometry: a shortcut to quantitative and dynamic interaction proteomics. Biology (Basel) 2014;3:320–32. doi: 10.3390/biology3020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–62. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y, Zhang W, White MA, Zhao Y. Capillary high-performance liquid chromatography/mass spectrometric analysis of proteins from affinity-purified plasma membrane. Anal Chem. 2003;75:3751–7. doi: 10.1021/ac034184m. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y, Zhang W, Kho Y, Zhao Y. Proteomic analysis of integral plasma membrane proteins. Anal Chem. 2004;76:1817–23. doi: 10.1021/ac0354037. [DOI] [PubMed] [Google Scholar]
- 27.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 28.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–26. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–40. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 30.Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015;16:169. doi: 10.1186/s12859-015-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verschueren E, Von Dollen J, Cimermancic P, Gulbahce N, Sali A, Krogan NJ. Scoring Large-Scale Affinity Purification Mass Spectrometry Datasets with MiST. Curr Protoc Bioinformatics. 2015;49:8 19 1–16. doi: 10.1002/0471250953.bi0819s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–2. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doncheva NT, Assenov Y, Domingues FS, Albrecht M. Topological analysis and interactive visualization of biological networks and protein structures. Nat Protoc. 2012;7:670–85. doi: 10.1038/nprot.2012.004. [DOI] [PubMed] [Google Scholar]
- 35.Maslov S, Sneppen K. Specificity and stability in topology of protein networks. Science. 2002;296:910–3. doi: 10.1126/science.1065103. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y, Widen SG, Jamaluddin M, Tian B, Wood TG, Edeh CB, Brasier AR. Quantification of activated NF-kappaB/RelA complexes using ssDNA aptamer affinity-stable isotope dilution-selected reaction monitoring-mass spectrometry. Mol Cell Proteomics. 2011;10:M111 008771. doi: 10.1074/mcp.M111.008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y, Tian B, Edeh CB, Brasier AR. Quantitation of the dynamic profiles of the innate immune response using multiplex selected reaction monitoring-mass spectrometry. Mol Cell Proteomics. 2013;12:1513–29. doi: 10.1074/mcp.M112.023465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–6. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A, Lawo S, Pelletier L, Stark H, Nasmyth K, Ellenberg J, Durbin R, Buchholz F, Mechtler K, Hyman AA, Peters JM. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science. 2010;328:593–9. doi: 10.1126/science.1181348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 41.Dickhut C, Feldmann I, Lambert J, Zahedi RP. Impact of digestion conditions on phosphoproteomics. J Proteome Res. 2014;13:2761–70. doi: 10.1021/pr401181y. [DOI] [PubMed] [Google Scholar]
- 42.Russell WK, Park ZY, Russell DH. Proteolysis in mixed organic-aqueous solvent systems: applications for peptide mass mapping using mass spectrometry. Anal Chem. 2001;73:2682–5. doi: 10.1021/ac001332p. [DOI] [PubMed] [Google Scholar]
- 43.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bottero V, Rossi F, Samson M, Mari M, Hofman P, Peyron JF. Ikappa b-alpha, the NF-kappa B inhibitory subunit, interacts with ANT, the mitochondrial ATP/ADP translocator. J Biol Chem. 2001;276:21317–24. doi: 10.1074/jbc.M005850200. [DOI] [PubMed] [Google Scholar]
- 45.Cogswell PC, Kashatus DF, Keifer JA, Guttridge DC, Reuther JY, Bristow C, Roy S, Nicholson DW, Baldwin AS., Jr NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J Biol Chem. 2003;278:2963–8. doi: 10.1074/jbc.M209995200. [DOI] [PubMed] [Google Scholar]
- 46.Johnson RF, Perkins ND. Nuclear factor-kappaB, p53, and mitochondria: regulation of cellular metabolism and the Warburg effect. Trends Biochem Sci. 2012;37:317–24. doi: 10.1016/j.tibs.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 47.Han Y, Weinman S, Boldogh I, Walker RK, Brasier AR. Tumor necrosis factor-alpha-inducible IkappaBalpha proteolysis mediated by cytosolic m-calpain. A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-kappab activation. J Biol Chem. 1999;274:787–94. doi: 10.1074/jbc.274.2.787. [DOI] [PubMed] [Google Scholar]
- 48.Giorgi C, Moore MJ. The nuclear nurture and cytoplasmic nature of localized mRNPs. Semin Cell Dev Biol. 2007;18:186–93. doi: 10.1016/j.semcdb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 49.Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151:750–64. doi: 10.1016/j.cell.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- 51.Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–12. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–99. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–34. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 54.Wang R, Li Q, Helfer CM, Jiao J, You J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J Biol Chem. 2012;287:10738–52. doi: 10.1074/jbc.M111.323493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental table S1: The parameters for LC-MS/MS and Maxquant data analysis. The table contains three spreadsheets.
Supplemental table S2: The parameters for LC-SRM analysis of six RelA interactors.
Supplemental table S3: Proteins that were quantified in the RelA interactome study and the MiST scores of RelA-Prey and IgG-Prey. This table contains six speadsheets as listed below.
Spreadsheet 1. Proteins that were quantified by ABC method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 2. Proteins that were quantified by TFE method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 3. Proteins that were quantified by urea method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 4. Proteins that were quantified by SDS-FASP method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 5. Proteins that were quantified by SDS-gel method and the MiST scores of RelA-Prey and IgG-Prey.
Speadsheet 6. RelA interactors.
Supplemental Table S4. GO annotation enrichment of the RelA interactors identified by each method.
Supplemental table S5: Proteins quantified in the BRD4 interactome study and the MiST scores of RelA-Prey and IgG-Prey. This table contains six speadsheets as listed below.
Spreadsheet 1. Proteins that were quantified by ABC method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 2. Proteins that were quantified by TFE method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 3. Proteins that were quantified by urea method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 4. Proteins that were quantified by SDS-FASP method and the MiST scores of RelA-Prey and IgG-Prey.
Spreadsheet 5. Proteins that were quantified by SDS-gel method and the MiST scores of RelA-Prey and IgG-Prey.
Speadsheet 6. BRD4 interactors.