

Abstract
Human DNA polymerase ν (Pol ν) is a conserved family A DNA polymerase of uncertain biological function. Physical and biochemical characterization aimed at understanding Pol ν function is hindered by the fact that, when over-expressed in E. coli, Pol ν is largely insoluble, and the small amount of soluble protein is difficult to purify. Here we describe the use of high hydrostatic pressure to refold Pol ν from inclusion bodies, in soluble and active form. The refolded Pol ν has properties comparable to those of the small amount of Pol ν that was purified from the soluble fraction. The approach described here may be applicable to other DNA polymerases that are expressed as insoluble inclusion bodies in E. coli.
Keywords: DNA polymerase, high pressure refolding, aggregation, insoluble inclusion bodies
Introduction
DNA polymerases are central players in DNA replication, recombination and the multiple DNA repair reactions that stably maintain eukaryotic nuclear genomes (1–4). Several of the many DNA polymerases encoded by the human genome have been discovered only recently, such that their biological functions are not fully understood. Included among these is DNA polymerase ν (Pol ν). Pol ν was identified (5) as a DNA polymerase that is conserved in vertebrates but not in invertebrates, yeast or fungi. Pol ν is a member of the A family of DNA polymerases, whose founding member is Escherichia coli (E. coli) DNA polymerase I, the product of the PolA gene. The polymerase domain of Pol ν shares 33% identity (48% similarity) with the equivalent domain in E. coli Pol I (5). In contrast to certain A family members that have intrinsic 3′ exonucleolytic proofreading activities and synthesize DNA accurately (e.g., E. coli Pol I, bacteriophage T7 Pol, and Pol γ), Pol ν lacks an intrinsic proofreading activity and displays considerably lower fidelity (6,7). However, Pol ν is moderately processive, suggesting that it may be kept under tight control in vivo to avoid potentially mutagenic DNA synthesis. Moreover, Pol ν has a base substitution error specificity that is somewhat different from other A family members, in that it incorporates dTMP opposite template G at a rate that is higher than for the other 11 possible base-base mismatches (7). In addition, Pol ν can perform translesion DNA synthesis in vitro (8), suggesting that it might serve this function in vivo (7,9).
The biochemical properties of Pol ν are presumably relevant to its biological function(s) in vertebrate cells. The known differences between Pol ν and other members of the A family of DNA polymerases makes Pol ν a particularly interesting candidate for structure-function studies, if sufficient protein can be isolated. To date, obtaining large quantities of Pol ν for these studies has been hindered by the fact that when recombinant Pol ν is over-expressed in E. coli, it is largely found as insoluble inclusion bodies. Moreover, protein in the soluble fraction has proven difficult to purify and is obtained in small quantities. In theory, several alternative approaches have been developed for obtaining sufficient soluble protein from E. coli for structure-function studies. These include refolding the protein from chaotrope solubilized inclusion bodies, random mutagenesis or directed evolution to improve solubility, identification and expression of soluble truncated fragments of the full length protein, or expression of chimeric tagged proteins in E. coli and other eukaryotic systems. Directed evolution and random mutagenesis result in altered proteins whose biochemical properties may differ from the native protein.
Traditional methods used to solubilize and refold recombinant proteins use high concentrations of chaotropic agents, such as 8 M urea or 6 M guanidine-HCl (GnHCl), that dissociate protein aggregates and denature proteins (10–13). Refolding is then accomplished by replacing the chaotrope with an empirically determined buffer that favors refolding into a native conformation. However, this refolding technique occurs through a series of conformational intermediates that are often susceptible to re-aggregation (14). Determination of buffer conditions that can overcome the propensity towards aggregation is time consuming and often fails to give a positive outcome. Several groups have implemented high throughput screens to identify optimal protein refolding conditions (15, 16). Recently an alternative approach to solubilizing and refolding insoluble proteins using high hydrostatic pressure was reported (17–20). Under the high pressures used in this process a protein will seek a conformation with the smallest specific volume and become a soluble monomer in the absence of chaotropic agents. The protein collapses into its most compact conformation, minimizing exposure of hydrophobic regions to the environment and maximizing intramolecular electrostatic interactions. When pressure is removed, the soluble monomer assumes its most energetically stable fold. The purpose of the present study was to determine if high pressure refolding can be used to solubilize and refold catalytically active human Pol ν from E. coli expressed inclusion bodies and to demonstrate that refolded human Pol ν exhibits biochemical properties comparable to those of purified soluble Pol ν.
Materials and Methods
Enzymes, reagents, strains
Materials for the M13mp2 fidelity assay were from sources described previously (21). [γ-32P]ATP (4500 Ci/mmol) and unlabeled deoxyribonucleoside triphosphates were from GE Healthcare Bio-sciences.
Plasmid Construction
Fragment 1 (E175-G863, 77.1 kDa, Pol ν-77) was constructed by PCR using pDEST17 vector containing human POLN (8) (kind gift from Dr. Richard D. Wood, University of Texas, MD Anderson Cancer Center) as template with primers 5′-CACCGAAGAAGATACTGATGACGCCGAAG and 5′-CTAGCCCAGGCCTCCTGCAG. The PCR product was purified and cloned into pENTR/TEV/D-TOPO (Invitrogen). The resulting plasmid, pENTR (Tev) Pol ν-77 was used as the basis for expression plasmid construction. This fragment was migrated into Gateway expression vector pDEST527 (N-terminal Hisx6) and pDEST544 (N-terminal His-NusA) (provided by Dominic Esposito, NCI-Frederick), using LR Clonase II™ (Invitrogen). These plasmid constructs were used for expression trials in Rosetta 2(DE3)pLacI (Novagen). Expression trials were run at 18°C and 30°C in LB media.
Expression of Hisx6 and Hisx6NusA-tagged Pol ν-77
Freshly transformed colonies of E. coli Rosetta 2 DE3 pLacI (Novagen), containing plasmid pDEST527 (N-terminal His) or pDEST544 (N-terminal His-NusA) Pol ν-77 were used in all expression experiments. For expression of soluble Hisx6-NusA Pol ν-77, soluble expression was comparable for both high density and low density expression conditions. For the high density expression a colony was grown in 5 mL LB broth supplemented with 100 μg/mL ampicillin and 25 μg/mL chloramphenicol and grown at 37°C for 6 hours. A 1 L culture of LB broth, supplemented with 100 μg/mL ampicillin and 25 μg/mL chloramphenicol, was inoculated with 100 μL of the day culture and grown overnight at 30°C. Cells harvested by centrifugation at 4700 × g for 15 min at 4°C, supernatant was decanted, and the pellet resuspended in 50 mL LB broth supplemented with 100 μg/mL ampicillin. A fresh 1 L culture was inoculated with 25 mL of the suspension and grown for 2 hours at 18°C, and induced by adding 0.5 mM IPTG (final concentration). The expression continued overnight and cells were harvested by centrifugation (4700 × g for 15 minutes at 4°C) and pellet frozen and stored at −80°C.
For expression of soluble Hisx6-Pol ν-77, a colony was grown in 5 mL of LB broth supplemented with 100 μg/mL ampicillin and 25 μg/mL chloramphenicol and grown at 37°C for approximately 6 hours. An aliquot (100 μL) was used to inoculate 50 mL LB broth supplemented with 100 μg/mL ampicillin and 25 μg/mL chloramphenicol and grown at 37°C overnight. Next morning, 5 mL of the overnight culture were used to inoculate a 1 L culture. Cultures were incubated at 37°C with shaking until cultures reached 0.4–0.6 at OD600, temperature decreased to 18°C, induced by adding IPTG to 0.2 mM, and grown overnight. Cells were harvested by centrifugation for 15 min at 5,500 × g at 4°C and cell pellets frozen at −80°C.
For expressing Hisx6-Pol ν-77 as inclusion bodies, cells from a 1 L overnight culture, grown in LB containing 100 μg/mL ampicillin and 25 μg/mL chloramphenicol, were centrifuged at 5,500 × g at 4°C and resuspended in 50 mL LB broth. Twenty five milliliters of this suspension were used to inoculate 1 L of LB containing ampicillin. Cultures were incubated at 37°C with shaking for 0.5 hour, induced by adding IPTG to 1.0 mM, and grown for an additional 3 hours. Cells were harvested by centrifugation for 15 min at 5,500 × g at 4°C.
Hisx6-Pol ν-77 inclusion body preparation
For isolation of inclusion bodies, the cells (5 grams of wet paste) were lysed using 5 mL of Bug Buster Master Mix (Novagen) containing Complete™ EDTA-free proteinase inhibitors (Roche Diagnostics) per gram of cell paste. The lysis suspension was incubated 30 min at room temperature with gentle agitation, and centrifuged for 30 min at 16,000 × g at 4°C to collect the inclusion bodies. Purification of inclusion bodies from cell debris was accomplished by washing two times with 5 mL of 0.1 X Bug Buster Master Mix and two times with 5 mL of water per gram of wet cell paste. The resulting pellets were resuspended in 50 mM Tris-HCl pH 8.0 and stored frozen at −80°C.
High Pressure Refolding
Refolding conditions were established by a two-step process using HiPER-FOLD™ Starter and Additive kits obtained from Barofold. Refolding was performed in a Barofold PreEMT-E150 pressure chamber. Conditions were set at 200 MPa, 20°C, and 20 hr with protein concentrations approximately 1 mg/mL. Samples of 0.2 mL were depressurized at a rate of 25 MPa/5 min. Upon depressurization, the samples were transferred to Eppendorf tubes and centrifuged for 15 min at 16,000 × g at 20°C. Refolding efficiency was examined by SDS-PAGE. The initial screen established the optimal pH and a positive refolding effect for 0.5 M arginine. Building upon the results of an initial screen, the effect of a range of small molecules on refolding was examined in subsequent screens. Preparative scale refolding was performed in a sealed plastic syringes containing 2 or 5 mL of refolding mixture containing inclusion body protein at 1.0 mg/mL.
Purification of Refolded Hisx6-Pol ν-77
The polymerase was purified by gel filtration chromatography on a Superdex 200 column (GE Healthcare), with a running buffer of 50 mM Tris-HCl pH 8.0, 500 mM L-arginine, and 5 mM TCEP. Fractions with Hisx6-Pol ν-77 activity were pooled and dialyzed into 1 L buffer A (20 mM Tris-HCl pH 8.0, 10% glycerol and 7 mM β-mercaptoethanol) containing 250 mM NaCl and Complete™ EDTA-free proteinase inhibitors (Roche Diagnostics). A second round of dialysis against 1 L buffer A plus 100 mM NaCl and Complete™ EDTA-free proteinase inhibitors (Roche Diagnostics) was used to prepare the sample for binding to a heparin column. The sample was then loaded onto a 1 mL HiTrap heparin column (GE Healthcare) equilibrated with 10 column volumes of dialysis buffer. Pol ν-77-containing fractions were eluted with a linear NaCl gradient from 100 mM to 1 M with peak fractions eluting at ~330 mM NaCl. Protein concentration was measured by A280. Purified, refolded Hisx6-Pol ν-77 was stored and frozen at −80°C.
Purification of soluble Hisx6-NusA Pol ν-77
Frozen cells (9 grams of wet paste) were thawed on ice and mechanically lysed (French Press 2X, ~12,000 psi) in the presence of 5 mL of buffer A plus 1 M NaCl and Complete™ EDTA-free proteinase inhibitors (Roche Diagnostics) per g of cell paste. Lysed cells were spun at 20,000 × g for 30 minutes. The viscous supernatant was collected and sonicated (2X, 20 sec at 50% duty cycle, power setting 4) to shear any remaining long strands of DNA to facilitate removal by subsequent centrifugation steps. Insoluble material was pelleted by a 30 min spin in an ultracentrifuge at 100, 000 × g. Remaining DNA in the supernatant was then precipitated with 0.3 to 0.35% polyethyleneimine (10% stock solution in water, pH 7.5) and sedimented by centrifugation for 30 min at 20,000 × g. The supernatant was supplemented with 20 mM imidazole and loaded onto a 5 mL His-trap column (GE Healthcare) equilibrated with buffer A containing 1 M NaCl and 20 mM imidazole. Pol ν-77 was eluted with a linear imidazole gradient from 20 mM to 250 mM in buffer A plus 1 M NaCl. Eluate fractions were analyzed on SDS-PAGE. Pol ν-77 containing fractions were pooled and diluted with an equal volume of buffer A (0.5 M NaCl final concentration). The diluted sample was incubated overnight at 4°C with 30 units of AcTEV (Invitrogen) to cleave the N-terminal Hisx6-NusA tag. The resulting cleaved protein was loaded onto a second 1 mL HisTrap column (GE Healthcare) equilibrated with buffer A containing 0.5 M NaCl and 25mM imidazole to remove the cleaved tag, uncut protein and the AcTEV protease. Flow-through fractions were collected and diluted in buffer A to a final concentration of 100 mM NaCl and loaded onto a 1 mL HiTrap heparin column (GE Healthcare) equilibrated with 20 column volumes buffer A plus 100 mM NaCl. Soluble Pol ν-77 was eluted with a linear NaCl gradient from 100 mM to 1M (with peak fractions eluting at ~330 mM NaCl). The fractions containing Pol ν-77 were pooled, flash-frozen and stored at −80°C.
Primer Extension
The substrate for primer extension reactions was generated by annealing a [γ-32P]ATP 5′-end labeled primer strand (5′-AATTTCTGCAGGTCGACTCCAA-3′) to a template strand (5′ CCAGCTCGGTACCGGGTTAGCCTTTGGAGTCGACCTGCAGAAATT-3′). Reaction mixtures (20 μL) contained 20 mM Tris-HCl pH 7.4, 8 mM magnesium acetate, 2 mM DTT, 4% glycerol, 0.1 mg/mL BSA, 100 μM of each dNTP and 1000 fmol of DNA. Reactions were initiated by adding 6 μL of refolded Hisx6-Pol ν-77 from the refolding screen, and were incubated at 37°C for 30 minutes. Aliquots (10 μl) were quenched by addition of an equal volume of 99% formamide, 5 mM EDTA, 0.1% xylene cyanol, 0.1% bromophenol blue, and DNA products were analyzed by electrophoresis on a 12% denaturing polyacrylamide gel.
DNA Polymerization on activated DNA
Reaction mixture (300 μl) contained 20 mM Tris-HCl pH 8.0, 8 mM magnesium acetate, 50 mM NaCl, 2 mM DTT, 4% glycerol, 80 μg/mL BSA, 200 μM of each cold dATP, dCTP, dGTP and dTTP, 82.5 nM [α-32P] dCTP, and 7.5 μg of activated calf thymus DNA (GE Healthcare). Duplicate reactions were initiated by adding Pol ν-77 and were then incubated for 30 min at 37°C. Reactions were stopped by adding EDTA to 20 mM followed by 0.5 mL of 10% trichloroacetic acid. After 15 min incubation on ice, acid-precipitable material was collected by filtration through Whatman GF/C filters. The filters were washed 3 times with 5 mL of 5% trichloroacetic acid, 1% sodium pyrophosphate and once with 5 ml of ethanol, dried, and counted in a scintillation counter.
Processivity
Reaction mixtures (30 μL) examining the processivity of Pol ν-77 was monitored using single-stranded M13mp2 DNA primed with a [γ32P]ATP 5′-end labeled DNA oligonucleotide complementary to positions +173 to +192 of the lacZα coding sequence as previously shown (7). These reactions contained 20 mM Tris-HCl pH 7.5, 8 mM magnesium acetate, 0.1 mM EDTA, 4% glycerol, 80 μg/ml bovine serum albumin and 100 μM each dATP, dCTP, dGTP and dTTP and 200 fmol DNA substrate. Reactions were initiated by adding 50 fmol of soluble and refolded Pol ν-77 and were incubated at 37°C. Ten μL aliquots were removed at 4, 8 and 12 min for both soluble and refolded Pol ν-77. Reactions were terminated and analyzed as described above for primer extension reactions. Enzyme to DNA ratios were empirically determined to assure that only a small percentage of DNA substrate was used. Under these conditions, each primer was extended only once, as determined by the observation that termination probabilities at each position remained constant over the time course of the reaction. The termination probability at each template position is defined as the ratio of products of a given length to the sum of that product plus all longer DNA products.
M13mp2 Fidelity Assay
Gap-filling reactions (25 μL) contained 20 mM Tris-HCl pH 8.8, 8 mM magnesium acetate, 0.1 mM EDTA, 4% glycerol, 80 μg/ml bovine serum albumin and 1 mM each dATP, dCTP, dGTP and dTTP and 0.2 nM M13mp2 (407-nucleotide gap) DNA substrate (from nucleotide −216 through +191 of lacZ gene). Polymerization reactions were initiated by addition of 75 nM refolded Pol ν-77, incubated at 37°C for one hour and terminated by adding 20 mM EDTA. Ten μL of the reaction mixture were mixed with 10 μl SDS buffer (20 mM Tris-HCl pH 8.0, 5 mM EDTA, 5% SDS, 0.5 % bromophenol blue and 25% glycerol) and the completeness of gap filling was monitored by agarose gel electrophoresis (21). Errors were scored as light blue or colorless mutant plaques, while correct synthesis yielded plaques that are dark blue. DNA from independent mutants was sequenced to identify the errors made during gap-filling synthesis reactions. For sequence changes that yield light blue and colorless plaques, error rates were calculated per detectable nucleotide incorporated as described previously (21).
Results and Discussion
Expression of fragments of Pol ν
The human POLN gene encodes a 100 kDa protein with template-dependent DNA polymerase activity but no 3′ exonuclease activity, as well as an N-terminal region of more than 400 amino acids and a proline-rich tail, both of unknown function (Fig. 1A). Sequence alignment with other A family members and secondary structure predictions were used to identify six fragments of Pol ν (Fig. 1A and data not shown) that might potentially enhance soluble expression in E. coli. Fragment 1 lacks amino acids 1–174 and 864–900 (the proline-rich tail), neither of which is required for polymerization in vitro (5, 8). This fragment, engineered with an N-terminal 6× histidine tag, gave the highest level of soluble protein of the fragments tested (data not shown). Following purification by affinity chromatography, the yield of purified Pol ν-77 was approximately 75 μg per liter culture (~8 μg per gram of wet cell paste). It has a predicted MW of 77.1 kDa, and was therefore designated as Pol ν-77. Pol ν-77 was expresses in E. coli at a high level (Fig. 1B, lane C), however, like full length Pol ν, it was largely insoluble (lane P). A small amount of soluble Pol ν-77 was observed (lane S), but only a fraction of that was found to bind to and elute from a Ni-NTA column (lane E), indicating that it was likely to be misfolded. Manipulating expression conditions such as temperature, media composition, concentration of induction factors and host strains failed to improve the yield of soluble Pol ν-77 (data not shown).
Figure 1. Expression and purification of Pol ν-77.


(A) Schematic representation of the H. sapiens full-length and Pol ν-77 proteins. Diagram depicts an N-terminal region (NTR), of unknown function (white) and a C-terminal portion that encodes DNA polymerase activity. Conserved motifs in the polymerase domain are numbered 1, 2a, 2b and 3–6 (white boxes). Aspartate residues located in motif 3 that are important for DNA polymerase activity are represented as black squares. The extreme C-terminal region (amino acids 864 to 900, black) contains a proline rich tail. (B) Pol ν-77 samples were prepared as described in Materials and Methods: C, crude extract; P, pellet with insoluble fraction; S, soluble fraction; E, eluate of purified soluble Pol ν-77 fraction after Ni-NTA agarose (Qiagen) chromatography; M, molecular weight markers.
Refolding Hisx6 Pol ν-77
Initial attempts to refold Pol ν-77 from chaotrope-solubilized inclusion bodies using rapid dilution plates (15) failed to yield active soluble protein. We therefore attempted to refold Pol ν-77 from inclusion bodies using high hydrostatic pressure (approximately 200 MPa). To identify starting conditions, we used the Barofold HiPer-Fold™ Starter Kit, which provides a wide range of pH conditions in the presence and absence of arginine and redox components. Figure 2A shows the quantity of protein in the soluble fraction following pressurization for 24 hr at 18°C and 200 MPa pressure. The prominent protein band observed at 15 kDa is lysozyme (derived from BugBuster Master Mix), used here to facilitate cell lysis. As shown in Figure 2A, Pol ν-77 inclusion bodies were soluble at pH 4.0 (lane 1), pH 10.0 (lane 7), and to a lesser extent at pH 9.0 (lane 6). Addition of 0.5 M arginine, which inhibits aggregation, to the pH 8.0 sample, resulted in a very low level of soluble Pol ν-77 (compare lanes 5 and 12). When the arginine concentration was increased to 1.0 M, the fraction of soluble Pol ν-77 increased significantly (lane 15). The presence of reduced and oxidized glutathione resulted in much reduced solubility (lanes 17–20).
Figure 2. Solubility and activity assays on Pol ν-77 refolding screens.



(A) Primary refolding screen. Samples (200 μL) at designated pH values were sealed in 1.0 mL syringes and placed in the Barofold EMT-150 pressure chamber. The chamber was sealed and pressurized to 200 MPa at 18°C. Samples were incubated under pressure for 24 hr. The pressure was reduced in 25 MPa increments, pausing 5 min between each reduction. When the pressure reached 0 MPa the samples were transferred into microfuge tubes and centrifuged at 15 k × g for 5 min at 4°C to remove any remaining insoluble and/or re-precipitated protein. Sample aliquots were analyzed by SDS-PAGE. Lanes 1–16 contain 5 mM TCEP, lanes 17–20 contain 5 mM reduced and oxidized glutathione and lane 16 contains 500 mM NaCl, “+” = 500 mM arginine, “++” equals 1 M arginine. (B) Additive refolding screen. Samples (200 μL) at pH 8.0 with L-arginine (left gel) or at pH 9.0 without L-arginine (right gel) included the following additives: (1) 10 mM β-octyl glucoside; (2) 0.05 mM Brij-35; (3) 0.03 mM Tween-20; (4) 0.02 mM Pluronic F-68; (5) 5 mM Na-cholate; (6) 3 mM CHAPS; (7). 0.1 mM Triton X-100; (8) 500 mM NDSB201; (9) 0.1 mM PEG 3350; (10) 5% hexylene glycol; (11) 5% sucrose; (12) 5% glycerol; (13) 500 mM ammonium sulfate; (14) 500 mM sarcosine; (15) 375 mM NDSB256. Refolding was performed as described above. Sample aliquots were analyzed by SDS-PAGE. (C) Primer extension reactions with refolded Pol ν-77. Reactions were performed as described in Materials and Methods. Representative phosphor image of the primer extension reaction products resolved on a 12% denaturing polyacrylamide gel. Lanes 1–15 from the gel to the left represent additive screen reactions performed at pH 8.0 in the presence of arginine. Lanes 1–15 from the gel on the right are refolding reactions performed at pH 9.0. Reaction details are described in Materials and Methods. Full-length (FL), primer and degradation products are designated on the gel.
Based upon the results of this primary refolding screen, refolding conditions were further examined using a wide range of small molecule additives present in the Barofold HiPer-Fold™ Additive Screening Kit. Two basic sets of conditions were chosen for the additive screen: pH 8.0, 0.5 M arginine plus additive (Fig. 2B, left lanes 1–15), and pH 9.0 plus additive (Fig. 2B, right lanes 16–30). Slight improvements in solubility were observed with several additives, and complete solubilization of inclusion bodies was observed under both sets of conditions when the non-detergent sulfobetaine (NDSB256) was included (Fig. 2B, lane 15).
Since solubility does not necessarily equate to proper folding and therefore enzyme activity, refolded Pol ν-77 solubilized in each condition described above in the additive screen was further tested for polymerase activity, as measured by the ability to extend an oligonucleotide primer-template (Fig. 2C). The protein preparations that were refolded at pH 9.0 exhibited little polymerization activity, and instead primarily exhibited 3′ exonuclease activity (presumably an E. coli exonuclease) that degraded the 5′-end labeled primer strand. Several of the samples that refolded at pH 8.0 in the presence of arginine did extend the primer. The samples supplemented with NDSB201 (lane 8), with hexylene glycol (lane 10), with glycerol (lane 12) and particularly with NDSB256 (lane 15) exhibited more polymerase activity.
Purification of refolded Pol ν-77
Based on the results from the high pressure refolding screens, we attempted to purify active Pol ν-77 from inclusion bodies that were refolded in the presence of 50 mM Tris-HCl, pH 8.0, 5 mM TCEP, 500 mM arginine, 375 mM NDSB256 and Complete™ protease inhibitors. When refolded Pol ν-77 was applied to a Superdex 200 column, it eluted as a broad peak across 15 fractions (Fig. 3A). Using activated DNA as substrate to monitor incorporation of a radiolabeled dNTP into DNA, polymerase activity was primarily detected in the trailing half of the peak (Fig. 3B). The observation that earlier fractions were soluble but much less active suggested that they contained soluble aggregates of Pol ν-77 that may not be folded properly. When the fractions that exhibited polymerase activity were pooled and loaded onto a heparin column, a portion of the 77 kDa protein and most of the lower molecular weight material flowed through the column without binding (Fig. 3C, lane 3, designated FT). When the bound protein was eluted with a linear NaCl gradient, fractions 7 through 13 contained Pol ν-77 (Fig. 3C, lanes 5–10). Although the earliest eluting of these fractions contained minor amounts of higher mobility proteins, fractions 11 and 12 were highly purified. When analyzed by SDS-PAGE, the purity of refolded Pol ν-77 was comparable to that of Pol ν-77 purified from the soluble extract (Fig. 4A). Starting with approximately 8 mg of inclusion body protein, the yield of highly purified Pol ν-77 eluted from the heparin column was approximately 270 μg (see Table 1). The yield of highly purified protein from refolded inclusion bodies was approximately 1 mg per g of wet cell paste, or about 125-fold greater than the yield obtained when starting with protein derived from the soluble extract. In the future, it may be possible to improve yield by investigating additional refolding conditions and/or changing subsequent manipulations of the solubilized protein. It should also be possible to scale up the procedure in order to obtain greater quantities of Pol ν-77.
Figure 3. Refolded Pol ν-77 purification using size exclusion and heparin affinity chromatography.


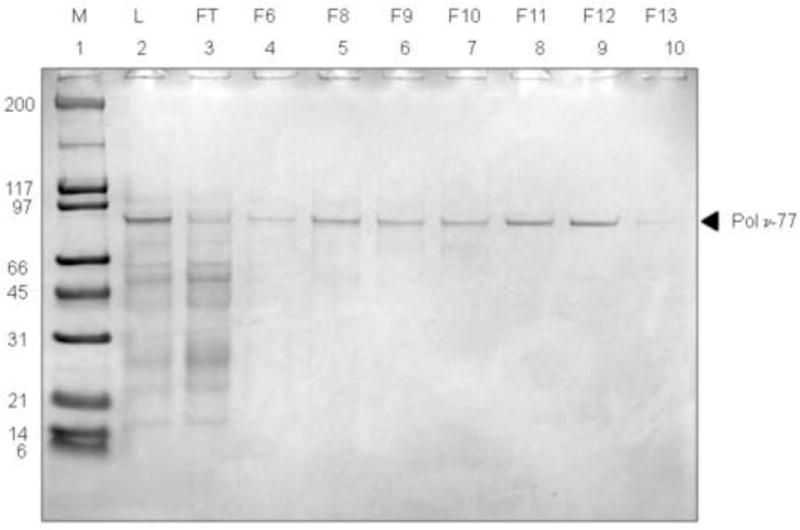
(A) Size exclusion chromatography was performed using a Superdex 200 (GE Healthsciences) 10/30 HR column at 4°C. Pressure refolded inclusion body protein (IB) was eluted in 50 mM Tris-HCl, pH 8.0, 5 mM TCEP, 500 mM L-arginine. Lanes 1–15 represent elution fractions. Pol ν-77 fractions (1.0 mL) were collected and 20 μL of each fraction was resolved on a 4–12% Bis-Tris SDS gel. Lanes 1–15 represent eluted fractions. Fractions 7–13 were pooled for affinity chromatography see section C, below. (B) DNA polymerization on activated DNA by fractions eluted from Superdex 200 10/30 HR column. (C) Affinity chromatography was performed on a 1 mL heparin column (GE Healthsciences) at 4°C. Refolded Pol ν-77 was eluted with a linear NaCl gradient (100 mM to 1.0 M) in 20 mM Tris-HCl, pH 8.0, 10% glycerol, 7 mM β-mercaptoethanol. Fractions were collected in 1 mL increments during the elution and 15 μL aliquots were resolved on a 4–12% Bis-Tris SDS gel. Lanes 1–10 are as designated on the figure. L, load and FT, flow-through.
Figure 4. Comparison of soluble and refolded Pol ν-77.

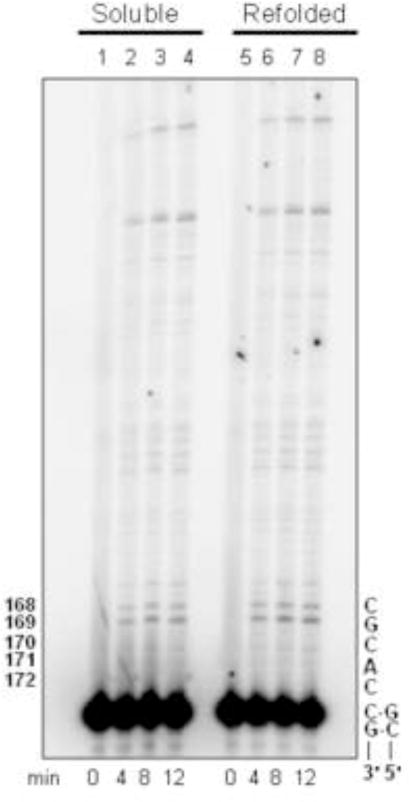
(A) Equal amounts of refolded and soluble Pol ν-77 proteins were resolved by SDS-PAGE. Lane 1, soluble Pol ν-77 and lane 2, refolded Hisx6-Pol ν-77. Note that the Hisx6/NusA tag was cleaved from the soluble Pol ν-77, while the Hisx6 remains on the refolded protein. (B) Processive synthesis by soluble and refolded Pol ν-77. Reactions were performed as described in Materials and Methods. Representative phosphor image of the reaction products of processive DNA synthesis were resolved on a 12% denaturing polyacrylamide gel. Lanes 2–4 represent primer extension products by soluble Pol ν-77 at 4, 8, and 12 min and lanes 6–8 represent primer extension products by refolded Pol ν-77, also at 4, 8, and 12 min. Lanes 1 and 5 represent negative control reactions, no enzyme. Numbers to the left indicate the nucleotide position along the lacZ template. Nomenclature on the right represents template primer sequence as described in Materials and Methods.
Table 1.
Purification Summary Table for Pol ν-77
| Fraction | Volume (ml) | Total Protein (mg) | Activity (units/ml)b | Total Activity (units)b | Specific Activity (units/mg)b | Fold Purification | % Yield |
|---|---|---|---|---|---|---|---|
| Inclusion bodies | 2.0 | 8.46 | N/Aa | N/A | N/A | N/A | N/A |
| Refolded soluble | 2.0 | 5.12 | 30.4 | 60.8 | 11.9 | 1.0 | 100 |
| Superdex 200 eluate | 3.5 | 1.49 | 7.2 | 25.1 | 16.8 | 1.4 | 41 |
| Heparin eluate | 2.0 | 0.27 | 7.7 | 15.3 | 56.7 | 4.8 | 25 |
N/A – This fraction was not assayed due to the insolubility of Pol ν-77 at this stage in the purification.
Unit = 1 pmol dCTP incorporated/min/mg protein in the DNA polymerization assay described in Materials and Methods.
Comparison of refolded Hisx6 Pol ν-77 and purified soluble Pol ν-77
When assayed using activated DNA as a substrate, fraction 11 of refolded Pol ν-77 and purified soluble Pol ν-77 exhibited a specific activity of 0.395 vs. 0.330 pmol of dNTP incorporated/pmol of enzyme/min, respectively. Under conditions allowing a single cycle of processive DNA synthesis, the processivity of refolded Pol ν-77 was similar to that of Pol ν-77 purified from the soluble fraction (Fig. 4B) or from that of Pol ν (amino acids 1–863) purified earlier from the soluble protein fraction by a different procedure (7). Neither refolded nor soluble Pol ν-77 exhibited exonuclease activity (data not shown).
As an additional measure of comparability, we determined the fidelity and the error specificity of refolded Pol ν-77 during synthesis to copy a 407-nucleotide single-stranded template encoding the lacZ α-complementation gene within gapped M13mp2 DNA (7). In this assay, polymerization errors are detected as light blue and colorless lacZ mutant plaques among dark blue plaques resulting from correct synthesis. Since the assay monitors loss of a gene function that is not essential for M13 plaque formation, it scores a broad range of errors, including all 12 possible single-base substitutions in a variety of sequence contexts, as well as additions and deletions of many different template nucleotides (14). Refolded Pol ν-77 filled the gapped substrate to completion, and while doing so generated a lacZ mutant frequency that was similar to that reported earlier for Pol ν-863 (Table 2 and see ref. 7). LacZ mutants were sequenced and the results were used to calculate specific error rates. The type of errors observed and calculated error rates were comparable to a previous study (Table 2, see ref. 7). Of the total errors generated by refolded Pol ν-77, 160 were substitutions known to individually result in reduced plaque color (those depicted by the gray letters in Fig. 5). Refolded His Pol ν-77 also generated a high rate of errors reflecting template G-dTMP mismatches (Table 2). These results are similar to those obtained in an earlier study (7) of Pol ν (amino acids 1–863) purified from the soluble protein fraction by a different procedure (8).
Table 2.
Error Rates for Refolded Pol ν-77
| Pol ν-77 | Pol ν 1–863 | ||
|---|---|---|---|
|
| |||
| Refoldeda | Solubleb | ||
| LacZ Mutant Frequency | 0.18 | 0.18 | |
| Total mutants sequenced | 93 | 162 | |
| Total bases sequenced | 37, 851 | 65, 934 | |
|
| |||
| Detectable changesc | # | Error Rate (× 10−4)c | Error Rate (× 10−4)b |
|
| |||
| Base substitutions | 160 | 40 | 35 |
| Frameshifts (−1) | 10 | 1.6 | 1.6 |
| Frameshifts (+1) | 8 | 1.3 | 0.7 |
| G · dTMP | 130 | 190 | 170 |
| T · dGMP | 24 | 28 | 13 |
Error rates and mutant frequencies were calculated using data from two independent experiments. Experiment 1: Mutant frequency was 16.3% (452 mutant plaques from a total of 2779). Experiment 2: Mutant frequency was 20.0% (896 mutant plaques from a total of 4386).
Previously published (7).
Error rates calculated from detectable changes (see Materials and Methods).
Figure 5. Spectrum of errors generated by refolded Pol ν-77.

The 407 template nucleotides within the single-strand gap of the M13mp2 substrate are shown in black. Letters above the target sequence indicate base substitutions. Deletion of a base is depicted by an open triangle whereas addition of a base is represented by a closed inverted triangle above the target sequence. Gray characters represent phenotypically detectable changes in the gap region while black characters represent phenotypically undetectable changes found in association with detectable changes. Nucleotide +1 represents the first transcribed nucleotide of the lacZα-complementation region.
Overall, these results clearly demonstrate that human recombinant DNA polymerase ν can be expressed as insoluble protein in E. coli, and then refolded into active protein by exposure to high hydrostatic pressure. The refolded protein is an active polymerase with properties indistinguishable from the small amount of soluble protein purified from the soluble fraction. This approach may be applicable to other DNA polymerases, many of which have been found to be highly insoluble when expressed in E. coli.
Acknowledgments
The authors thank Dr. Lars C. Pedersen and Andrea F. Moon for critical review of this manuscript. We thank the NIEHS DNA sequencing and Protein Microcharacterization core facilities for expert technical assistance. This work was supported in part by Project Z01 ES065070 to TAK from the Division of Intramural Research of the National Institutes of Health, National Institute of Environmental Health Sciences.
References
- 1.Hanawalt P. Paradigms for the three rs: DNA replication, recombination, and repair. Mol Cell. 2007;28:702–707. doi: 10.1016/j.molcel.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2nd. ASM Press; 2006. [Google Scholar]
- 3.Shcherbakova PV, Fijalkowska IJ. Translesion synthesis DNA polymerases and control of genome stability. Front Biosci. 2006;11:2496–2517. doi: 10.2741/1985. [DOI] [PubMed] [Google Scholar]
- 4.Bebenek K, Kunkel TA. Functions of DNA polymerases. Adv Protein Chem. 2004;69:137–165. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 5.Marini F, Kim N, Schuffert A, Wood RD. POLN, a nuclear PolA family DNA polymerase homologous to the DNA cross-link sensitivity protein Mus308. J Biol Chem. 2003;278:32014–32019. doi: 10.1074/jbc.M305646200. [DOI] [PubMed] [Google Scholar]
- 6.Longley MJ, Nguyen D, Kunkel TA, Copeland WC. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J Biol Chem. 2001;276:38555–38562. doi: 10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- 7.Arana ME, Takata K, Garcia-Diaz M, Wood RD, Kunkel TA. A unique error signature for human DNA polymerase nu. DNA Repair (Amst) 2007;6:213–223. doi: 10.1016/j.dnarep.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takata KI, Shimizu T, Iwai S, Wood RD. Human DNA polymerase N (POLN) is a low-fidelity enzyme capable of error-free bypass of 5S-thymine glycol. J Biol Chem. 2006;281:23445–23455. doi: 10.1074/jbc.M604317200. [DOI] [PubMed] [Google Scholar]
- 9.Fischhaber PL, Gerlach VL, Feaver WJ, Hatahet Z, Wallace SS, Friedberg EC. Human DNA polymerase kappa bypasses and extends beyond thymine glycols during translesion synthesis in vitro, preferentially incorporating correct nucleotides. J Biol Chem. 2002;277:37604–37611. doi: 10.1074/jbc.M206027200. [DOI] [PubMed] [Google Scholar]
- 10.Middelberg APJ. Preparative protein refolding. Trends Biotechnol. 2002;20:437–443. doi: 10.1016/s0167-7799(02)02047-4. [DOI] [PubMed] [Google Scholar]
- 11.Rudolh R, Lilie H. In vitro folding of inclusion body proteins. FASEB J. 1996;10:49–56. [PubMed] [Google Scholar]
- 12.De Bernardez-Clark E. Refolding of recombinant proteins. Curr Opin Biotechnol. 1998;9:157–163. doi: 10.1016/s0958-1669(98)80109-2. [DOI] [PubMed] [Google Scholar]
- 13.Tsumoto K, Ejima D, Kumagai L, Arakawa T. Practical considerations in refolding proteins from inclusion bodies. Protein Expr Purif. 2003;28:1–8. doi: 10.1016/s1046-5928(02)00641-1. [DOI] [PubMed] [Google Scholar]
- 14.Qoronfleh MW, Hesterberg LK, Seefeldt MB. Confronting high-throughput protein refolding using high pressure and solution screens. Protein Expr Purif. 2007;55:209–224. doi: 10.1016/j.pep.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 15.Vincentelli R, Canaan S, Campanacci V, Valencia C, Maurin D, Frassinetti F, Scappucini-Calvo L, Bourne Y, Cambillau C, Bignon C. High-throughput automated refolding screening of inclusion bodies. Protein Sci. 2004;13:2782–2792. doi: 10.1110/ps.04806004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willis MS, Hogan MJK, Prabhakar P, Liu X, Tsai K, Wei Y, Fox T. Investigation of protein refolding using a fractional factorial screen: a study of reagent effects and interactions. Protein Sci. 2005;14:1818–1826. doi: 10.1110/ps.051433205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.St John RJ, Carpenter JF, Randolph TW. High pressure fosters protein refolding from aggregates at high concentrations. Proc Natl Acad Sci U S A. 1999;96:13029–13033. doi: 10.1073/pnas.96.23.13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Randolph TW, Carpenter JF, St John RJ. 7,064,192 and 6,489,450. USA patent no. 2002
- 19.Lee SH, Carpenter JF, Chang BS, Randolph TW, Kim YS. Effects of solutes on solubilization and refolding of proteins from inclusion bodies with high hydrostatic pressure. Protein Sci. 2006;15:304–313. doi: 10.1110/ps.051813506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crisman RL, Randolph TW. Refolding of proteins from inclusion bodies is favored by a diminished hydrophobic effect at elevated pressures. Biotechnol Bioeng. 2008;102:483–492. doi: 10.1002/bit.22082. [DOI] [PubMed] [Google Scholar]
- 21.Bebenek K, Kunkel TA. Analyzing fidelity of DNA polymerases. Methods Enzymol. 1995;262:217–232. doi: 10.1016/0076-6879(95)62020-6. [DOI] [PubMed] [Google Scholar]