

ABSTRACT
The ability of an organism to replicate and segregate its genome with high fidelity is vital to its survival and for the production of future generations. Errors in either of these steps (replication or segregation) can lead to a change in ploidy or chromosome number. While these drastic genome changes can be detrimental to the organism, resulting in decreased fitness, they can also provide increased fitness during periods of stress. A change in ploidy or chromosome number can fundamentally change how a cell senses and responds to its environment. Here, we discuss current ideas in fungal biology that illuminate how eukaryotic genome size variation can impact the organism at a cellular and evolutionary level. One of the most fascinating observations from the past 2 decades of research is that some fungi have evolved the ability to tolerate large genome size changes and generate vast genomic heterogeneity without undergoing canonical meiosis.
INTRODUCTION
Cellular ploidy is the number of complete sets of chromosomes in a cell. Many eukaryotic species have two (diploid) or more than two (polyploid) sets of chromosomes (1). These diploid and polyploid states are often the result of ancient whole-genome duplication (WGD) or hybridization events that occurred throughout the evolution of plants, animals, and fungi (2–4). Ploidy changes also occur during the development of many organisms and can vary within different tissues of the same organism and between individuals of the same species. For example, ploidy changes occur during the sexual cycle of eukaryotes, from haploid gametes to diploid somatic cells. Additionally, some cells continue to increase in ploidy during development, resulting in somatic tissues that have a mixture of diploid and polyploid cells, including human hepatocytes and megakaryocytes (5–7). These ongoing, developmentally programmed changes in ploidy are important for viability and are beneficial to many organisms (8), but the mechanisms controlling ploidy and the physiological significance of each ploidy level are not well characterized.
Many clinically relevant fungi undergo ploidy changes during adaptation, especially to adverse or novel environments. Some fungi exist as stable haploid, diploid, or polyploid (e.g., triploid, tetraploid) cells, while others change ploidy under certain conditions and revert back to the original ploidy level in other conditions. Aneuploidy, an abnormal chromosome number, is observed in novel environments, during periods of cellular stress, and during ploidy level changes. An increase in ploidy can occur through mating, endoreduplication, or failure to undergo cytokinesis after replication (described in detail below). Here we refer to events that increase cellular ploidy as WGD, diploidization (e.g., haploid to diploid), or polyploidization (e.g., diploid to tetraploid). A decrease in fungal ploidy can occur through meiosis, reductional division, and random chromosome loss events (9). We will discuss only nonmeiotic ploidy decreases and refer to these events as whole-genome reduction events or haploidization events (e.g., diploid to haploid). The examples we provide here challenge the textbook definition of somatic ploidy as a consistent or defining trait of a fungal species. Instead, fungal ploidy is often context-dependent and can rapidly change from one environment to the next.
Ploidy values currently assigned to fungal species may be influenced by laboratory growth conditions and selection for traits that prove to be beneficial for conducting genetic manipulations. However, as whole-genome sequencing (WGS) and molecular assays for ploidy detection become standard in fungal research labs, the identification of ploidy variants is increasing. For example, polyploid strains of the typically haploid and/or diploid species Saccharomyces cerevisiae, Candida albicans, and Cryptococcus neoformans exist in diverse environments including desert canyons, fermentation and industrial cultures, and from human patients before and after therapeutic treatment with antifungal drugs (10–16). This suggests that many different ploidy-environment interactions may select cells with increased or decreased ploidy that provide these cells with an adaptive advantage (1, 17, 18). A mechanistic understanding of what drives environment-dependent ploidy changes remains to be discovered. Genome sequencing and phenotypic characterization of mutations in these comparatively small eukaryotic genomes (e.g., compared to human genomes) will likely place fungi at the forefront of ploidy research (19–21).
Molecular Detection of Ploidy and Aneuploidy
Ploidy is most commonly measured by flow cytometry of fluorescently labeled cells (e.g., propidium iodide), where the relative fluorescence of an unknown isolate is compared to strains of known ploidy (22). More than 30,000 single cells can be analyzed within seconds, and linear increases in ploidy are detected with great accuracy (Fig. 1A). Aneuploid isolates are detectable by flow cytometry with the caveat that isolates with a single aneuploid chromosome may not be significantly different in the fluorescent signal compared to the known ploidy control. In addition, the fluorescent signal of isolates with multiple aneuploidies (e.g., gain of one and loss of another chromosome) may not show different DNA fluorescence by flow cytometry because these specific aneuploidies cancel each other out. Instead, more quantitative methods must be used to identify the specific aneuploid chromosomes. Flow cytometry coupled with additional molecular methods such as comparative genome hybridization (aCGH), quantitative PCR, and double-digest restriction site-associated DNA sequencing (ddRADSeq) is the most comprehensive approach to detect ploidy changes and identify specific aneuploidies (23–25).
FIGURE 1.
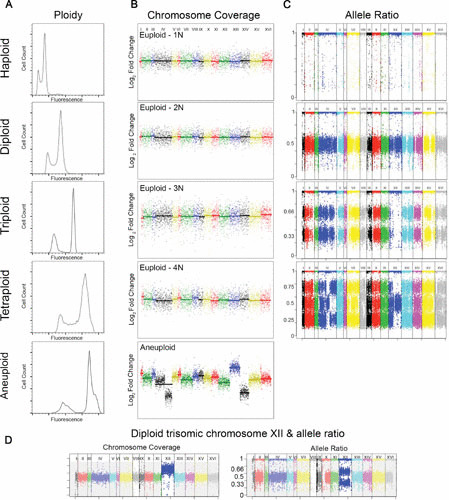
Methods for detection of ploidy and aneuploidy. (A) Ploidy is determined with flow cytometry. Total genome fluorescence, measured using a fluorescent nucleotide label (e.g., propidium iodide or Sytox Green). Cells are first fixed (in ethanol) and RNA is removed with RNase, then genomic DNA is fluorescently labeled and analyzed on a flow cytometer. Cells are passed through a laser, and the number of cells are plotted as a function of fluorescence intensity. Cells in a population typically have two fluorescent peaks, representing cells in either G1 or G2 phases of the cell cycle. Flow cytometry plots for yeast with the following ploidy levels are shown: haploid (1N), diploid (2N), triploid (3N), tetraploid (4N), and a near-tetraploid aneuploid. (B) Chromosome copy number is determined with WGS and microarray aCGH. The y axis represents a log2 fold change of sequence reads relative to the reference sequence and chromosome number increases from left to right starting with chromosome I and ending with chromosome XVI (x axis). Chromosome copy number plots for S. cerevisiae with the following ploidy levels indicate euploid genome for haploid (1N), diploid (2N), triploid (3N), and tetraploid (4N). However, the near-tetraploid isolate (bottom panel) is aneuploid for ChrXII (pentasomic) and ChrXIV (trisomic) and contains a segmental aneuploidy of ChrIV. Figures generated from data obtained in reference 20. (C) Allele frequencies obtained from WGS data also can be used to determine the ploidy of a strain. The y axis shows the heterozygous allele frequencies ranging from zero to one, plotted as a function of chromosome number starting with chromosome I and ending with chromosome XVI (x axis). Allele frequency plot of the example haploid strain with single nucleotide polymorphisms (SNPs) at allele frequencies at 1.0; a diploid strain with SNPs at allele frequencies of 0.5 and 1.0; a triploid strain with SNPs at allele frequencies of 0.33 and 0.66; and a tetraploid strain with SNPs at allele frequencies at 0.25, 0.5, 0.75, and 1.0. Images obtained from reference 16. (D) A diploid strain that is trisomic (three copies of a chromosome) for chromosome XII (left panel). Interestingly, the allele frequency plot has SNPs at allele frequencies of 0.5 and 1.0 for all chromosomes except ChrXII, which is at allele frequencies of 0.33 and 0.66, supporting that this chromosome is aneuploid (right panel).
The ploidy level and chromosome copy number of isolates can also be determined using WGS. The copy number of each chromosome is determined relative to the entire genome based on the number (depth) of aligned sequence reads. Aneuploidy is detected as an increase or decrease in read depth relative to the entire genome (Fig. 1B). Segmental chromosome aneuploidies and small gene amplifications/deletions are also detected. Additionally, WGS bioinformatics tools use allele frequencies to determine the baseline ploidy of the sequenced genome (16, 26). For example, a haploid (1N) genome has allele frequencies at 1, a diploid (2N) genome has allele frequencies at 0.5 and 1, a triploid (3N) genome has allele frequencies at 0.33, 0.67, and 1, and a tetraploid (4N) genome has allele frequencies at 0.25, 0.5, 0.75, and 1 (Fig. 1C and D). While limited to strains with significant heterozygosity, WGS simultaneously detects cellular ploidy level, chromosome copy number, and sequence polymorphisms. Further studies are needed to truly understand the extent of ploidy variation in natural populations within and between species.
Ploidy Variation in Natural Isolates
We first highlight three examples of ploidy variation found in isolates of S. cerevisiae, C. neoformans, and C. albicans. These three species have different genome sizes, haploid chromosome numbers, sexual cycles, and preferred base ploidy levels. Despite these differences, all three have been isolated from natural ecosystems and/or human and animal hosts with ploidies that range from haploid to polyploid. Furthermore, all three species can undergo somatic ploidy changes under laboratory conditions.
S. cerevisiae
The budding yeast S. cerevisiae contains 16 haploid chromosomes and reproduces by mating and meiosis or asexually via budding. Environmental isolates of S. cerevisiae include haploids, diploids, and polyploids (12, 27–30). Clinical and industrial isolates also show a wide range of ploidies and aneuploidy (16, 29). For example, WGS of 145 S. cerevisiae clinical isolates found that 34% were triploid or tetraploid and 36% were aneuploid (16). Many of these ploidy changes are thought to be adaptive, but the correlation between environmental selection and ploidy changes is not well characterized (17).
C. neoformans
Naturally found in diverse environmental niches, C. neoformans is responsible for pulmonary infections as well as cryptococcal meningoencephalitis in immunocompromised individuals (31, 32). Basidiospores are thought to be the infectious propagules, which are inhaled into the lungs followed by dissemination into other organs (33). C. neoformans is normally found in the haploid state with 14 chromosomes and reproduces both sexually and asexually, and these haploid cells can vary in chromosome copy number due to nondisjunction events (34) or due to unisexual or bisexual reproduction (35). Dramatic ploidy changes have been observed during infection. Polyploid “titan” cells that range from 4N to >64N make up ∼20% of the infectious population within the host tissue (36, 37). In conjunction with the ploidy increase, cell size, capsule structure, and cell wall structure are also modified. A haploid C. neoformans cell is generally 5 to 10 μm in diameter; titan cells can be much bigger, with some reaching upward of 50 to 100 μm in diameter (38, 39). Very little is known about the mechanism of titan cell formation; it may involve endoreplication due to alteration of cyclin proteins as is observed in Drosophila melanogaster and human hepatocytes (40, 41). Aneuploidy, in particular the amplification of chromosome 1, has also been observed in response to antifungal drug stress (42, 43).
C. albicans
The most common human fungal pathogen, C. albicans, contains eight homologous chromosome pairs. Though it was previously considered an obligate diploid organism (44, 45), alternative ploidy states have been described including haploid, triploid, and tetraploid cells (10, 15, 46–48). No meiosis has been observed in C. albicans. Rather, it undergoes a parasexual cycle in which cells of opposite mating type fuse to form tetraploids (49–53). Under nutrient starvation, tetraploid cells show an increase in genome instability that leads to the loss of individual chromosomes over time, returning progeny cells to either diploidy or (more often) near-diploidy (9, 50, 52, 54). Ploidy changes also occur in response to specific environmental conditions. For example, growth on alternative carbon sources (e.g., l-sorbose), exposure to antifungals (e.g., fluconazole), high temperature, and interactions with the host all result in ploidy changes and aneuploidy within a few cell divisions (15, 55–62). The molecular mechanisms driving these ploidy changes are currently unknown.
Other Fungi with Altered Ploidy Levels and Evidence of Aneuploidy
Aneuploidy has been found in many other fungi. Ashbya gossypii, a filamentous fungus, has a single syncytia with multiple nuclei, and each nucleus can have a different ploidy (63). In A. gossypii, ploidy increases with age, while stress exposure can shift a population with high ploidy variation toward a more homogenous, haploid population (63). Ploidy level variation and aneuploidy are also common in isolates of the amphibian chytrid pathogen, Batrachochytrium dendrobatidis (64), and in plant pathogens such as Fusarium oxysporum (65, 66). In fact, karyotype variability in B. dendrobatidis seems to be the norm rather than the exception; out of 22 isolates analyzed by WGS, 18 were aneuploid, with base ploidies ranging from diploid to tetraploid (64). While these are only a few examples of ploidy changes that are known to occur in the fungal kingdom, they highlight that ploidy changes play a central role in adaptive evolution and genomic diversity.
PLOIDY CHANGES IN THE CONTEXT OF LABORATORY MUTANTS: TOWARD A MECHANISM OF ASEXUAL PLOIDY CHANGE
The underlying mechanisms that drive ploidy changes are not completely understood (12). Some genes encoding ploidy regulators have been identified in yeast deletion mutant screens and gene overexpression studies (67). However, in many mutants the ploidy-altering phenotype is not 100% penetrant (see below), indicating that there are redundant mechanisms that regulate genome copy number. Alternatively, the mutant genotype may stochastically acquire fitness-associated ploidy changes, in which case the ploidy change would be secondary to the initial gene mutation.
Here we will discuss mutations that affect cell cycle, spindle pole body (SPB), kinetochore attachment, cohesion, chromatin formation, and cytokinesis, focusing on their impact on ploidy changes. Many mutations that affect chromosomal instability can also cause ploidy increases or decreases. Importantly, because chromosome aneuploidy is frequently observed in mutants that undergo ploidy change, it is difficult to determine if aneuploidy itself is driving whole-genome ploidy changes. For example, a mutation may cause aneuploidy, which then gives rise to a whole-genome ploidy change. Alternatively, this mutation may first induce a whole-genome ploidy change, and subsequent aneuploidy results from increased genome instability. The examples provided below support that the mechanisms driving changes in whole-genome ploidy and chromosome copy number are extremely complex and often involve the essential machinery of the cell.
Mutations Underlying Ploidy Amplification
Alterations in cell cycle control can cause ploidy amplification. Endoreplication, the process in which DNA replication is followed not by cytokinesis, but instead by another round of DNA replication, has been described for multiple organisms (68–70). In the fission yeast Schizosaccharomyces pombe, cyclin B (p53cdc-13) regulates the temporal order of DNA replication and mitosis. Control of cyclin B levels and the timing of this cell cycle regulator are important; formation of the p34cdc-2-p53cdc-13 complex specifies that the cell is in the G2 phase of the cell cycle. Loss of this complex redefines a cell in the G2 phase to a G1 phase cell, and the cells can then re-enter S-phase and rereplicate their genomes, causing unscheduled WGD. Re-entry into S-phase can occur multiple times and can lead to ploidy shifts from 1N to 32N (71). Interestingly, 42 uncharacterized genes were recently identified in human cells for their role in preventing endoreplication (72), supporting that DNA replication controls are still being discovered.
Under certain circumstances endoreplication is an environment-induced or programmed cell event. For example, environment-induced endoreplication likely leads to titan cell formation in C. neoformans (36, 38, 39). The exact mechanism of cell cycle alteration is not known for titan cell production, but like in S. pombe, the control of cyclin B level is a potential candidate. In the human liver, hepatocytes undergo genome replication followed by programmed cytokinesis failure to produce a binucleate daughter cell that is 4N (73). These binucleate tetraploids can then undergo another round of DNA replication followed by cytokinesis to generate mononucleate tetraploid cells. This cycle can continue to produce octaploid cells and so on. In addition, these polyploid hepatocytes can undergo mitosis with multipolar spindles, resulting in ploidy reduction and aneuploidy (8). It has been hypothesized that these aneuploid daughter cells may provide adaptive benefits during periods of cellular stress and allow for repopulation and restoration of the liver (73–75).
Accurate SPB function and attachment of the spindle microtubules to the chromosomes are required for proper chromosome segregation, and ploidy changes can occur when different components of the SPB complex are altered. During normal cell division, sister chromatids are attached to opposite SPBs and are pulled apart, thereby segregating the sister chromatids. NDC1 (nuclear division cycle 1) encodes a subunit of the nuclear pore complex in S. cerevisiae and is required for SPB duplication and insertion into the nuclear membrane (76). Mutant ndc1 cells have only a single functioning SPB to which all chromosomes attach and are then segregated into a single daughter cell, resulting in WGD.
Proper attachment of the SPB microtubules to the kinetochore is a major point of regulation during cell cycle progression. In S. cerevisiae, mutations in the essential gene IPL1 (increased ploidy level 1) can result in aneuploidy and/or elevated ploidy level. IPL1 encodes Aurora kinase, which is involved in the attachment of the spindle microtubules to the kinetochores, chromosome segregation, and checkpoints including mitotic spindle dis/assembly and DNA damage (77–79). Ipl1 is also responsible for sensing mitotic spindle attachment at the kinetochore and preventing segregation in cases where the chromosome is attached to only one SPB. Using a temperature-sensitive mutant of IPL1, Chan and Botstein (67) observed that haploid cells rapidly acquired multiple aneuploid chromosomes when grown at the restrictive temperature, and some of these mutants gained enough chromosomes to result in a ploidy increase.
In addition to SPB subunits, ploidy is affected by defects in chromosome cohesion. After replication, sister chromatids are packaged together by cohesion, a protein complex that holds the sister chromatids together until they are separated during anaphase (80). Improper loading or disassembly of the cohesion complex during the cell cycle leads to aberrant chromosome segregation that results in ploidy shifts and aneuploidy (81). Defects in the cohesion complex, as well as regulators of the complex, lead to release of sister chromatids before proper attachment to the SPB. This increases the chance that sister chromatids will be inherited together because the sensing of microtubule attachment does not occur and segregation of sister chromatids is no longer interdependent (81).
Chromatin structure and regulation are important regulators of ploidy. The dynamics of chromatin structure are determined by histone conformation and modifications. Mutations within the globular domain of histone 4 (H4) result in heterogeneous colony sizes and an increased frequency of WGD and aneuploidy in S. cerevisiae (82). Further analysis of these colonies shows that small colonies consist of a mixed population of haploid and diploid cells, while large colonies are completely diploid, suggesting extensive autodiploidization. Alterations of the amino acids in the globular domain of H4 (L97, Y98, or G99 to alanine) do not alter the ability of H4 to interact with other histones. Instead, these mutations alter the H4 interaction with the histone chaperones Rtt106 and Caf-I. Disruption of these interactions prevents the nucleosome from being loaded onto the DNA. This decrease in histone occupancy could disrupt kinetochore architecture and assembly, leading to the increase in aberrant chromosome segregation and autodiploidization (82).
Just as histone modification can affect genome stability, nucleosome stability at centromeres is necessary for kinetochore assembly and proper chromosome segregation. Nucleosome stability is achieved through proper distribution of histone variants. The histone variant H2A.Z is conserved across all fungi and higher eukaryotes and is enriched at pericentric DNA but excluded from CENP-A (Cse4) nucleosome binding sites (83). Ies6 is an essential subunit of the INO80 chromatin-remodeling complex. Loss of Ies6 causes polyploidization and increased localization of pericentric H2A.Z (84), resulting in changes to chromatin structure that inhibit centromere/kinetochore function. Furthermore, overexpressing H2A.Z in an ies6 mutant strain further increases chromosome instability, leading to more rapid polyploidization (84).
After genome replication and chromosome segregation, the cell undergoes a cleavage event controlled by the contraction of an actin ring found between the mother and daughter cell. Myo1, the sole myosin II motor in S. cerevisiae, associates with the actin ring and promotes cleavage between the mother and daughter cell (85). Haploid strains with a myo1 deletion often undergo WGD events to survive, causing ploidy level increases from 1N to 4N (86). Many of the polyploid myo1 evolved clones are mononucleate and contain multiple aneuploid chromosomes. These polyploid myo1 evolved clones are not only viable but have restored cytokinesis through a variety of mechanisms, unlike most of the myo1 cells that remain haploid. Therefore, the mutations that disrupt cell division and alter ploidy level can provide increased adaptability (86).
Mutations Underlying Ploidy Reduction
Several S. cerevisiae mutations have been identified that lead to genome reduction from diploid to haploid. For example, diploid cells with a null mutation in RAD52 (involved in strand exchange during recombination and DNA damage repair) undergo chromosome loss via sequential aneuploid transitions (87). Loss of Rad52 leads to a failure of the repair mechanism that keeps the chromosome homologs together after a double strand break (DSB). Loss of this pathway gradually leads to genome reduction toward haploidy over ∼500 generations. Interestingly, cells with higher ploidy showed increased Rad52 dependency because RAD52 is essential for growth of tetraploid cells but not isogenic diploid cells (88). This supports that increasing ploidy results in an increased frequency of DSBs that must be repaired by Rad52, and diploids, but not tetraploids, can resolve some of this damage by sequential chromosome loss and eventual ploidy reduction.
In addition to mutations in RAD52, null mutations in CTF18 (involved in sister chromatid cohesion) also lead to rapid genome reduction from diploidy to haploidy (89). This reduction in ploidy occurs over a relatively short amount of time (∼50 generations) and suggests that the haploid cells have a fitness advantage over the diploid progenitor. Surprisingly, genome duplications are also observed within the diploid ctf18 population, resulting in a heterogeneous population (haploid, diploid, aneuploid, and polyploid cells). This suggests that specific mechanisms of genome instability may simultaneously induce aneuploidy, ploidy loss, and ploidy gain.
IMPACT OF PLOIDY LEVEL ON DNA DAMAGE REPAIR
At all ploidy levels, DSBs are repaired by two main pathways: nonhomologous end-joining and homologous recombination. Nonhomologous end-joining repairs DSBs via ligation of the broken DNA ends with little or no processing of the DNA ends. Nonhomologous end-joining is considered error-prone because this process can introduce novel mutations. Alternatively, homologous recombination uses a homologous DNA sequence to serve as a donor for recombination and DSB repair (90, 91). The availability of homologous DNA sequences (sister chromatid, homologous chromosome, or an ectopic sequence) is influenced by the ploidy of the cell and phase of the cell cycle: haploid cells have homologous DNA sequences available during S/G2 only, while diploid and polyploid cells have homologous DNA sequences available throughout the cell cycle. Therefore, diploid and polyploid cells have an increased preference for homologous recombination to repair DSBs, resulting in increased frequencies of recombination, gene conversion, crossover events, and gross chromosomal rearrangements (GCR) (92–94).
Isogenic yeast strains with different ploidies exhibit different mutation rates and sensitivity to DSBs. The forward mutation rate at either the CAN1 or URA3 locus is two orders of magnitude higher in diploids than in haploids, and diploids are less sensitive to DSBs than haploids (88, 89, 95). Interestingly, the forward mutation rate at CAN1 is lower in tetraploids compared to diploids (88). Ploidy also affects sensitivity to DSBs: both haploids and tetraploids are more sensitive to DSBs than diploids. One reason for these observations may be the different requirements for DSB repair at different ploidy levels (94).
Ploidy-specific genome maintenance mechanisms may exist, but many questions still remain (94). For example, in S. cerevisiae there are ploidy-specific stress responses to GCR (96), and tetraploid yeasts have a greater requirement for genes involved in recombination and mitosis than haploids or diploids (88). This phenomenon, called “ploidy-specific lethality,” is used to identify the physiological alterations that accompany tetraploidy. Out of 3,740 yeast deletion mutants, only 39 genes exhibit ploidy-specific lethality. Almost all of these mutations affect genomic stability by impairing homologous recombination, sister chromatid cohesion, or mitotic spindle function (88). Why tetraploid cells have an increased requirement for these genes remains unknown (97). The rate and spectrum of mutations available to each ploidy level is remarkably different, so genome maintenance mechanisms could play an important role during adaptation.
GENOME INSTABILITY IN POLYPLOID CELLS
One of the most striking features of polyploid cells is their increased genome instability relative to diploid cells. This phenomenon is seen across many eukaryotic species, including many plant species (98–101). In fungi, polyploid cells frequently give rise to aneuploid progeny (both whole chromosome and segmental), chromosome rearrangements, and translocations. For example, tetraploid S. cerevisiae cells have a 200- to 1,000-fold increase in the rate of chromosome loss compared to isogenic diploid cells (88, 102, 103). Similarly, tetraploid C. albicans cells have a 50-fold higher rate of loss of heterozygosity (LOH) compared to isogenic diploid cells (54). Increased genome instability often results in rapid ploidy reduction during in vitro growth of most polyploid populations (9, 19, 20, 43, 54, 104), yielding progeny with high karyotypic diversity. Here we highlight examples for C. neoformans, C. albicans, and S. cerevisiae.
The dramatic ploidy reduction of C. neoformans titan cells was recently analyzed by microdissection of sequential daughter cells from a polyploid titan cell followed by colony formation assays and WGS (43). In a replete environment, polyploid titan cells produced true haploid daughter cells. In contrast, when exposed to fluconazole, titan cells frequently generated aneuploid haploid and aneuploid diploid daughter cells (43). Furthermore, daughter cells from the same titan cell parent had diverse aneuploid karyotypes, while point mutations were rarely observed. It remains to be tested whether this aneuploidy is due to an effect of fluconazole on mitotic fidelity (as was seen in C. albicans [62]) or is the result of a general stress response.
A foundational study of C. albicans tetraploid cells found that they undergo a rapid, nonmeiotic genome reduction in response to nutrient starvation, termed “concerted chromosome loss” (9). Genetic analysis of many progeny by microarray (single nucleotide polymorphism [SNP]) and aCGH arrays) found that only a very small fraction returned to a true euploid diploid state, and the majority of the progeny were aneuploid for one or more chromosomes. In addition, recombination, albeit limited to a few progeny, occurred between heterozygous loci on homologous chromosomes during tetraploid genome reduction (52). The appearance of multiple gene conversion tracts within several strains, and the general absence of gene conversion tracts in other strains, suggests that some cells become generally competent for recombination at more than one locus, while other strains do not undergo such recombination events at all. Importantly, these recombination events were dependent on Spo11, a conserved protein required for the introduction of DSBs (52). These findings suggest that at least one meiosis-specific gene has been reprogrammed to mediate genetic recombination during the alternative parasexual life cycle of C. albicans.
Recent studies have characterized the rate of concerted chromosome loss in different growth environments. For example, populations of tetraploid, triploid, and diploid C. albicans strains were grown for 28 days in yeast extract-peptone-dextrose (YPD), and chromosome loss dynamics were analyzed at different time points using flow cytometry and ddRADSeq. Initially, the tetraploid-evolved clones were highly aneuploid, and once the cells became aneuploid, further ploidy reduction accelerated, with aneuploid strains changing ploidy faster than tetraploid strains (54). Eventually, the tetraploid and highly aneuploid evolved cells converged to stable euploid levels: diploid, triploid, and (rarely) tetraploid. In addition, this study showed that chromosome loss was random; ploidy reduction in biological replicates of the same polyploid strain rarely followed the same chromosome loss trajectory.
Similar nonmeiotic ploidy reductions have been observed in tetraploid populations of S. cerevisiae during in vitro evolution (19). In this study, tetraploid S. cerevisiae strains were passaged for ∼1,800 generations in rich or high-salt medium, and genome reduction at the population level was detected with flow cytometry after ∼186 generations. Results from this study support that ploidy loss occurs as if selection is acting on multiple chromosome loss events simultaneously rather than on individual, sequential chromosome loss events (105). In addition, these in vitro evolution experiments found that ploidy loss in a tetraploid population often paused at near-euploid levels, from tetraploid to near-triploid to near-diploid (105). Furthermore, aneuploidies (detected by aCGH) present in the tetraploid progenitor were often found to be the only aneuploidy that remained in the near-diploid evolved population (19). Thus, while the mechanism of tetraploid genome reduction is unknown, the rapid ploidy loss observed in these environments supports that multiple chromosomes can be lost simultaneously.
The ploidy reduction process of S. cerevisiae tetraploid cells was further analyzed with WGS of strains from many parallel in vitro evolution experiments (20). The ploidy of tetraploid-evolved clones was determined by flow cytometry, and the copy number of every chromosome was determined by WGS. It was found that aneuploidy is more extensive during the tetraploid to diploid transition than previously appreciated. In raffinose medium, ploidy reduction did not occur by loss of a full set of chromosomes; rather, many of the tetraploid evolved cells were highly aneuploid, including individual chromosome copy numbers that ranged from 4C to 2C in the same cell (Fig. 2). This indicates that while some chromosomes are lost multiple times, others are maintained at the original tetrasomic level (e.g., 4C, 3C, and 2C). Interestingly, specific chromosome pairs were more frequently lost together and underwent multiple loss events independent of other chromosome pairs (20). These copy number differences were likely a result of the environmental selection for particular karyotypes with increased fitness; indeed, the most common aneuploid chromosome in this study, amplification of chromosome XIII, was found to provide a significant fitness benefit to tetraploid cells in raffinose medium (20).
FIGURE 2.
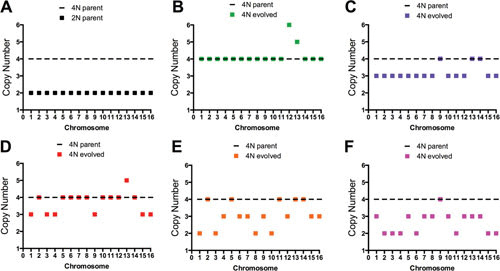
Many polyploid-evolved clones are highly aneuploid. Chromosome copy number was determined by WGS and plotted for the (A) parental diploid (2N) and tetraploid (4N) strains and different tetraploid evolved clones after 250 generations in raffinose medium. Adaptation resulted in clones with (B) increased chromosome copies, (C) approximately trisomic copies of every chromosome (∼3N), or (D-F) highly aneuploid genomes. Figures generated from data obtained from the supplementary data Table 1 in reference 20.
These studies of C. albicans and S. cerevisiae show that tetraploid-evolved clones can be highly aneuploid, with many evolved isolates reaching a near-euploid state (near-triploid or near-diploid). The rate of chromosome loss is largely dependent on the growth environment and generation time, as well as the frequency of sampling throughout the experiment. In general, the random chromosome loss that occurs during in vitro culturing of tetraploid C. albicans and S. cerevisiae populations appears mechanistically distinct from the haploid bud produced from C. neoformans titan cells. Future studies in these and other fungi may identify unique mechanisms of chromosome and genome stability and aneuploid tolerance in polyploid cells.
Given that ploidy reductions can occur over short evolutionary timescales, it is puzzling that polyploid cells are observed at all. One hypothesis is that polyploidy is simply an intermediate state to which a cell shifts for survival, for example, after cytokinesis failure due to cell wall stress (86, 106). However, another hypothesis is that the polyploid state is beneficial in specific environments and is selected upon due to increased fitness. Indeed, tetraploid S. cerevisiae strains passaged in vitro for ∼1,000 generations at 23°C stably maintain euploid tetraploid genomes (107). These evolved tetraploid cells are resistant to benomyl, a microtubule depolymerizing drug that tetraploid cells are more sensitive to than diploids (88). The stable tetraploids have increased levels of Sch9, a kinase involved in protein homeostasis, G1 cell cycle progression, nutrient signaling, and stress response, suggesting that newly formed tetraploid cells can acquire genome stability through improved cell growth processes (107).
IMPACT OF PLOIDY ON FUNGAL PHYSIOLOGY
Alterations in genome architecture have wide-reaching effects on many cellular processes. How ploidy shifts change cellular physiology is a long-standing question. A ploidy increase is associated with enlarged cell size in all eukaryotes (38, 39, 108–111). However, we lack a mechanistic understanding of how genome size controls cell size (112). Research that has compared growth rate, gene expression patterns, and cell surface area to volume ratios between haploid and diploid, or diploid and polyploid cells has provided insight into the complex, sometimes subtle, differences between isogenic fungal strains of different ploidy. Here we highlight how ploidy affects the physiological properties of cell size and gene expression.
The reduction in cell surface area to volume ratio of polyploid cells likely impacts nutrient transport, signaling pathway components, and ultimately growth rate in some environments. Upon genome duplication, polyploid cells underwent a 2-fold increase in cell volume, but only a 1.57-fold increase in cell surface area relative to their diploid progenitors (88, 113, 114). This reduced surface area in polyploid cells was expected to cause growth disadvantages in environments where nutrients are absorbed across the membrane. However, there was little evidence to support this cell surface area-to-volume ratio theory in polyploid cells (115). A robust analysis of this theory, performed with 51 haploid and diploid S. cerevisiae and S. paradoxus strains in 33 growth conditions, found that ploidy accounts for a majority (70%) of the differences in growth rate between strains (17). However, the results were inconsistent with the surface area-to-volume model. Rather, it appeared that the preference for the haploid versus diploid state was due to condition-specific effects in the strains tested, not due to differences in surface area (17). Thus, more research is needed to understand the physiological changes that occur after polyploidization and what effect the environment has on fitness and genome stability. Furthermore, identifying transcriptional changes that occur in other environments, including nutrient limitation, will provide important clues to the growth differences polyploids experience in these environments.
Gene expression studies have been used to identify molecular signatures that are unique to polyploid cells. The first gene expression microarray study on polyploid S. cerevisiae cells identified only a few genes with significantly different levels of expression in isogenic haploid and tetraploid cells when grown in rich medium (114). For example, tetraploid cells had reduced expression of genes encoding G1 cyclins (CLN1 and PCL1) and proteins involved in cytoskeletal organization (e.g., GIC2). In another study, a different S. cerevisiae strain (S288C instead of Σ1278b) was analyzed under the same growth conditions, and no genes had significantly different levels of expression in isogenic diploid and tetraploid cells (88). This suggests that either there are more transcriptional differences between haploids and tetraploids compared to diploids and tetraploids, or there are strain-specific ploidy differences (116).
Using RNA sequencing analysis, Wu et al. identified 65 transcripts with differential expression in isogenic haploid and tetraploid S. cerevisiae cells, again in rich medium (109). A majority of these genes encoded cell surface proteins, which suggests that the enlarged cell size of tetraploids caused the differential regulation of these genes, and not ploidy per se. Indeed, it was found that haploid mutants with increased cell size showed differential expression for many of the same genes as tetraploids, and the degree of changes in expression correlated with the degree of size increase (109). Importantly, gene expression of most of these cell surface proteins was repressed in tetraploid cells (and enlarged haploids), likely due to the reduced surface area relative to cell volume (88, 113, 114). These observations are consistent with proteomic studies that found reduced amounts of cell surface proteins in diploids compared to haploids (117).
PLOIDY CHANGES AND ANEUPLOIDY IN THE CONTEXT OF EXPERIMENTAL EVOLUTION
Ploidy Changes and Aneuploidy Occur During In Vitro Evolution and Provide a Fitness Benefit
In vitro evolution experiments provide the strongest evidence that fungi can undergo asexual ploidy changes during adaptive evolution. The environmental cues driving these genome changes are not known, and a comprehensive study of environments that induce ploidy changes (WGD and ploidy reduction) is lacking (Table 1). In this section, we will examine the different environments in which ploidy changes and acquisition of aneuploidy are documented in fungi and how these genome size changes affect organismal fitness and adaptation.
TABLE 1.
Summary of experimental evolution studies in fungi and the ploidy and aneuploidy associated with different environmental stresses. Ploidy levels of haploid (1N), diploid (2N), triploid (3N), and tetraploid (4N) are euploid states, while aneuploidy is indicated if known.
| Fungi | Stress | Timeframe | Ploidy change/aneuploidy | Reference(s) |
|---|---|---|---|---|
| S. cerevisiae | YPD, high salt | 1,800 generations | 1N → 2N and 4N → 2N | 19 |
| S. cerevisiae | YPD | 186 generations | 4N → 2N and 3N → 2N | 105 |
| S. cerevisiae | Raffinose | 250 generations | 4N → 4N–2N aneuploidies common2N → 2N and 1N → 1N (no ploidy change) | 20 |
| S. cerevisiae | Ethanol | 200 generations | 1N → 2N and 4N → 2N | 118 |
| S. cerevisiae | Low glucose (LiAc transformation) | 88–132 generations | 1N → 2N | 21, 119 |
| C. albicans | l-Sorbose, presporulation at 37°C | 5–7 days | 4N → 2N and aneuploid 2N | 9, 52 |
| C. albicans | l-Sorbose | 2 days | 2N → 2N monosomic for Chr5 | 56 |
| C. albicans | YPD | 28 days | 4N → 4N, 3N, or 2N2N → 2N | 54 |
| C. albicans | Rich media and nitrogen depletion | 140 generations | 1N → 2N and 4N → 2N | 121 |
| C. albicans | Minimal media and phosphate depletion | 140 generations | 1N → 1N and 4N → 2N | 121 |
| C. albicans | Fluconazole (2–10 μg/ml) | 8–12 h | 2N → 4N, 8N, 16N trimeras(20% of cells in the population) | 62 |
| C. albicans | Fluconazole (100 mg disk) | 12–16 h | 2N → 1N | 123 |
| C. albicans | Fluconazole (increasing MIC) | 330 generations | 2N → 2N–3N aneuploidies common: whole chromosome, isochromosomes, and telomere-telomere fusions | 46, 59, 61 |
| C. albicans | Murine host | 1–5 days | 2N → 1N and aneuploid 1N | 47 |
| C. albicans | Murine host | Single passage | 2N → 2N aneuploid | 60 |
| C. albicans | Heat shock (51°C) | 90 seconds | 4N → 2N | 55 |
| C. tropicalis | l-Sorbose | 8–10 days | 4N → 2N | 104 |
| C. tropicalis | Rich media | >120 generations | 4N → 2N | 104 |
| C. neoformans | Fluconazole (32 μg/ml) | 3–5 days | 1N → 1N aneuploidies common | 42, 124 |
| C. neoformans | Fluconazole (8 μg/ml) | 48–96 h | Polyploid titan cells → 1N and 2N aneuploidies common | 43 |
| C. neoformans | YPD, oxidative and nitrosative stress | 48 h | Polyploid titan cells → 1N | 43 |
| C. neoformans | Murine | 3 days | 1N → polyploid titan cells (20% of population) | 38 |
| C. neoformans | Unisexual reproduction | ∼2 weeks | 1N → 2N and aneuploidies common | 35 |
| A. gossypii | NaCl, ZnSO4, fluconazole (325 nM), heat (37°C) | 6 h | Polyploid and aneuploid → euploid 1N(distribution of ploidies shifted toward haploid) | 63 |
| A. gossypii | Caffeine | 6 h | Polyploid and aneuploid → polyploid and aneuploid | 63 |
We describe the in vitro studies in which whole-genome ploidy changes and chromosome copy number changes are observed. Many of these examples describe changes in ploidy that occur at the population level. If beneficial (e.g., increasing fitness), ploidy or chromosome copy number changes are selected for within the population. The exact order of events and mechanisms that drive the initial ploidy change is unknown.
Results from multiple studies showed that in S. cerevisiae, whole ploidy level changes causing diploidization are advantageous in multiple environments. For example, isogenic haploid, diploid, and tetraploid S. cerevisiae populations passaged in rich medium, medium containing high salt, or increased concentrations of ethanol all converged toward diploidy (19, 105, 118). The diploidized cells had significantly higher fitness than the isogenic progenitor strains in competition experiments (118). Furthermore, the rate of ploidy change is dependent on the specific growth conditions. For example, in high-salt medium, the rate of diploidization of haploids was faster than the rate of diploidization of isogenic tetraploids (19). Therefore, becoming diploid under these growth conditions was one of the most common and accessible routes to increased fitness.
Frequent diploidization of haploid cells also occurred during in vitro evolution experiments under glucose depletion (21, 119). In this unique study, the evolving populations consisted of ∼500,000 uniquely barcoded haploid S. cerevisiae lineages. The autodiploidized clones had a strong fitness benefit relative to their haploid progenitor, and WGS revealed that in most cases diploidy was the only adaptive mutation (21). The autodiploid lineages were traced back to their respective founding populations, suggesting that the transformation conditions (lithium acetate and heat shock) used for barcoding led to the formation of autodiploids (21). Interestingly, transformation conditions also induced ploidy loss, aneuploidy, and LOH in C. albicans (55, 120).
Convergence toward diploidy is also observed in C. albicans. Tetraploid mating products and rare haploid isolates returned to the diploid state after in vitro passage (9, 47, 54). The convergence toward diploidy occurred under different growth conditions but ultimately depended on the environment and initial genotype, not necessarily the fitness benefit of the acquired ploidy change (121). For example, during phosphorus depletion, the autodiploidization rate of haploids was reduced, while the diploidization rate of tetraploids was increased. Further studies are needed to uncover the factors that drive organisms to a “baseline” ploidy.
In contrast to the convergence toward diploidy observed in many environments, a wide array of genome changes can occur under a single growth condition. For example, growth in the fungistatic drug fluconazole often resulted in a wide variety of ploidies and aneuploid changes in C. albicans and C. neoformans (35, 43, 46, 59, 61, 62, 122–124), and just 8 to 12 hours of drug exposure resulted in both ploidy reduction and ploidy amplification in C. albicans (62, 123). Fluconazole is known to alter membrane fluidity, which causes abnormal cytokinesis and cell cycle defects leading to polyploid and aneuploid formation (62). The immediate fitness effect of these ploidy level mutations is not known, but it can be inferred by measuring how rapidly mutant cells accumulate within a population. For example, during in vitro evolution of C. albicans in the presence of fluconazole, acquisition of multiple aneuploid chromosomes was detected after just ∼3.3 generations in the majority of cells in the population (61). One of these aneuploidies, isochromosome 5L [i(5L)], conferred fluconazole resistance due to the amplification of two genes, ERG11 and TAC1 (13, 59). Due to the high fitness benefit, i(5L) accumulated rapidly in the population, and in many cases was maintained over ∼330 generations in the presence of fluconazole (61).
Many additional examples support that acquisition of aneuploid chromosomes can lead to rapid adaptation to specific environments, and like ploidy changes, these aneuploid chromosomes are reversible. For example, growth on alternative carbon sources led to loss of specific chromosomes in multiple fungal species, and the addition of glucose led to reduplication of the remaining chromosome (56, 104). Additionally, during adaptation to heat stress, S. cerevisiae rapidly duplicated chromosome III, but after continued heat stress the chromosome aneuploidy was lost while elevated expression of genes on this chromosome was maintained, suggesting that aneuploidy provided time for cells to search for optimal adaptive solutions (125).
While acquisition of aneuploidy is not always beneficial (126), aneuploidy can lead to a wide variety of fitness effects in different growth environments. Studies of the heat shock protein Hsp90 in S. cerevisiae showed that during periods of heat stress Hsp90 was titrated away from its regular clients such as kinetochore proteins (127, 128). Importantly, exposure to heat stress or inhibition of Hsp90 led to a marked increase in chromosome instability and tolerance to other stressors including hydrogen peroxide, cyclohexamide, tunicamycin, benomyl, and radicicol (128). More recently, Dunham and colleagues performed a comprehensive analysis of aneuploid genotypes in S. cerevisiae and found that each karyotype had large, but condition-dependent, changes in fitness, supporting that aneuploidy can be an important driver of adaptation (129, 130). Many more in vitro evolution experiments showed that aneuploidy and ploidy change can be beneficial during adaptation, especially during adaptation to nutrient limitation and heat stress (55, 120, 131).
While we have discussed how changes in ploidy and chromosome copy number can be adaptive in certain environments, some species of pathogenic fungi carry multiple, nonessential chromosomes called accessory, supernumerary, B, or dispensable chromosomes (132). Accessory chromosomes are characterized by highly variable regions, high mutation rates, and genes acquired via horizontal gene transfer (133–136). Many of these harbor virulence genes that allow the fungus to infect its host. The plant pathogen Nectria haematococca carries an array of genes on accessory chromosome 14 that allow the fungus to detoxify phytoalexin used by the pea plants as a defense mechanism (136, 137). In F. oxysporum, a striking 40% of the genome was found exclusively in strains that infect tomato plants. Moreover, most of the genetic material in these accessory chromosomes lacks sequence homology to any close relatives of F. oxysporum (138). Further research is needed to investigate how these accessory chromosomes evolved, are maintained, and what roles they play in virulence.
Many filamentous fungi, including the cotton pathogen A. gossypii, have evolved alternative strategies to generate genotypic diversity. This fungus has syncytia that undergo asynchronous nuclear division (139). It was originally thought to be a predominantly haploid fungus, but Gladfelter and colleagues recently demonstrated frequent ploidy variations for different nuclei within the same syncytium, including aneuploidy (63). To identify ploidy changes, they used lac operator arrays to track the copy number of individual chromosomes within the nuclei of the same syncytium and found nuclear ploidies ranging from 1N to >4N (63). Interestingly, in response to cellular stress (cell wall stress, osmotic stress, excess zinc, the antifungal fluconazole, and increased temperature), polyploid nuclei diminished and haploid nuclei predominated. These results suggest that nuclei with different ploidies are tolerated within single syncytia, and that there may be costs associated with ploidy level variation, because stress homogenizes the genome content of nuclei (63). Remarkably, ploidy reduction and homogenization of ploidy under stress is the opposite of what has been observed for the human fungal pathogens C. albicans and C. neoformans, where stress led to genomic diversification within populations and increased aneuploidy (43, 59, 61).
The above examples establish that fungal organisms can adapt rapidly to their environment by undergoing ploidy and chromosome copy number changes. It also highlights the challenge of determining the causative interactions between environment and ploidy levels (Table 1). While it is unclear what types of ploidy changes will arise in any given environment, it is a common strategy of pathogenic fungi to generate genomic variation in response to environmental perturbations.
It is important to note that in any given fungal population, one or more subpopulations may exist that have acquired viable aneuploidies or undergone ploidy shifts. This is due to background rates of chromosome missegregation and failed mitosis. It is of great interest to understand how entire populations of cells, when subjected to stress, can become polyploid or aneuploid. Are the vast majority of cells dying and selection acts upon the survivors, resulting in cells with ploidy changes beneficial for survival? Or could there be a mechanism permissive or instructive in the generation of aneuploid or polyploid organisms? Both of these questions remain open to further investigation.
Ploidy and Aneuploidy Changes Arise During In Vivo Evolution
Ploidy level heterogeneity and aneuploidy are frequently observed in clinical isolates of fungal species including C. albicans, C. neoformans, and Candida glabrata (10, 11, 15, 140, 141). However, very little is known about the dynamics of ploidy changes during in vivo evolution and the effects on host-fungus interactions.
To assess the types of genome alterations that C. albicans undergoes upon host encounter, single-passage in vivo experiments were conducted in a murine model of systemic infection. Ploidy changes compared to the diploid progenitor were detected in ∼3% of recovered strains and included segmental and whole chromosome aneuploidy. While this frequency is relatively low, no aneuploids were detected in strains from an in vitro control population. Results from this study suggest that conditions within the animal host affect chromosome disjunction more strongly and that this may reflect the very different population growth responses in vivo (60). More recent studies on the adaptive potential of C. albicans in response to the oral and systemic host niche show that the acquisition of aneuploidy is much more frequent than previously reported. Excitingly, the first haploids ever reported for C. albicans were found among strains recovered from an oral model of infection (47). Haploid isolates arose as early as 3 days postinfection with a frequency of ∼2 × 10−5 and not only differed in their genotypes (e.g., MTL status) but also showed phenotypic differences compared to their diploid progenitor. These phenotypes include decreased fitness in YPD at 30 and 37°C and defects in filamentation (47; A. Forche, unpublished). Importantly, haploids arose independently in six mouse hosts, and multiple haploids with different genotypes were found in the same host (A. Forche, unpublished). Overall, the frequency of phenotypic changes such as fitness at different temperatures, filamentation, or the production of hydrolytic enzymes that accompanied aneuploidy in general was much higher compared to cells that remained diploid. This suggests that some of these aneuploid states may be adaptive. Interestingly, the dramatic changes in ploidy may be specific to pathogenic host niches: passage of C. albicans using a murine commensal model found no ploidy or chromosome copy number changes by flow cytometry and WGS (142), supporting that host microenvironment impacts genome architecture.
Upon infection with C. neoformans, the host’s immune system responds by trying to combat the fungus via oxidative or nitrosative attacks by macrophages and neutrophils. The formation of polyploid titan cells inside the host (as discussed above) is accompanied by a ploidy shift from a 1N euploid state to one up to >64N (143). Current evidence points to the importance of titan cells and their ability to withstand various stresses that may be encountered within a host (38, 43). For example, among cells harvested from infected lungs, titan cells exhibited resistance to both sodium nitrate and tert-butyl hydroperoxide and survived under conditions lethal to non-titan cells (38, 43).
In summary, single passage of diploid C. albicans cells in an oropharyngeal model of infection leads to ploidy reduction to a haploid or near-haploid state (47). In addition, aneuploidy arises frequently upon encounter with the host in different models of infection (60). In contrast, haploid C. neoformans undergoes polyploidization upon host encounter (144). It is currently unknown what specific factors influence ploidy shifts in these fungi, but it is likely a combination of many variables including temperature, pH, host immune cells, and interaction/competition with other microbes (145, 146).
THE BENEFICIAL EFFECTS OF PLOIDY CHANGES
Adaptation of an organism to a novel environment is a function of the rate in which beneficial, growth-promoting mutations are acquired and spread throughout the population. The rate of adaptation is affected by multiple, interrelated properties that determine the appearance, persistence, and fixation of beneficial mutations in a population. These properties include the rate of beneficial mutations, the fitness effect of these mutations, the dominance of mutant alleles, and effective population size (147–153). While aspects of this process are still poorly understood, theoretical studies indicate that ploidy affects the rate of adaptation (2, 154, 155).
Ploidy change can impact the evolutionary trajectory of a cell by altering the rate and spectrum of beneficial mutations. For example, a ploidy increase (WGD) increases the mutational target size. A ploidy increase also provides the cell with the ability to buffer deleterious mutations due to redundant gene copy numbers (1, 156). Furthermore, some mutations and/or genome changes are only accessible to cells with specific ploidies, and this can have a profound impact on adaptation. For example, diploid and polyploid cells can undergo LOH, while haploid cells can acquire recessive mutations upon which selection can act (1, 157, 158). Similarly, polyploid cells have high rates of whole-chromosome aneuploidy, which can provide fitness benefits during adaptation (20, 86, 88), yet haploid cells rarely acquire aneuploidy during adaptation, likely due to the increased fitness cost of these mutations in most environments (126). Lastly, the fitness effect of a given mutation is assumed to be equal across all ploidy levels, but recent experimental evidence suggests that this is not the case for all mutations (20, 159–161).
IMPACT OF PLOIDY AND ANEUPLOIDY ON CANCER BIOLOGY
Ploidy changes and acquisition of aneuploid chromosome copy numbers are hallmarks of cancer cells. Tetraploid cells are frequently observed in precancerous lesions of Barrett’s esophagus (162, 163) and in preneoplastic cervical cells (164). Recently, a systematic comparison of over 5,000 human tumors found that 37% had undergone genome duplication during tumorigenesis (165). Evidence for ploidy changes in these tumors came from gene copy number and allelic ratio data obtained from SNP microarrays. Compared to diploid tumors, tumors that underwent WGD had higher rates of gene copy number alterations, including chromosome loss, resulting in ploidy levels ranging from triploid to tetraploid. WGD often preceded other gene copy number alterations in these tumors, supporting that WGD can be a driver of tumorigenesis. The idea that WGD can promote tumor development was previously shown in a mouse breast cancer model (p53–/– mammary epithelial cells [166]). Isogenic diploid and tetraploid cells were generated in vitro, mimicking a recent WGD event. When transplanted into mouse mammary epithelia, only the tetraploid cells generated tumors (166). Additional studies support that WGD resulting in tetraploidy is an intermediate step toward aneuploidy and tumorigenesis (106, 164), while other studies support that in some cancer types aneuploidy may precede the tetraploidization event (167).
Whole-genome reduction is also observed during development of rare leukemia subtypes, including acute lymphoblastic leukemia (168). These haploid leukemias are considered near-haploid because the lymphoblastic cells are often aneuploid for one or more chromosomes (1N+1), frequently involving chromosome 21 (168). Haploidization and LOH nearly across the entire genome may underlie the aggressiveness of these cancers, resulting in very poor prognosis (168). However, this haploid population appears to be unstable and undergoes WGD (autodiploidization) with some frequency. Evidence for autodiploidization was found in patients with mixed lymphoblast populations of both a near-haploid karyotype (1N+1) and a near-diploid population that was exactly twice that of the aneuploid karyotype (2N+2) (168). These data support that ploidy variation is directly linked to cancer evolution, yet the effects of ploidy change versus the effects of aneuploidy per se are difficult to distinguish.
CONCLUDING REMARKS AND OUTLOOK
There is a common theme in the examples discussed in this review: somatic ploidy changes increase the genetic heterogeneity of a population of cells (169–171). This genetic heterogeneity includes increased frequency of aneuploidy, but importantly, both ploidy and aneuploidy are reversible genomic changes. For example, a diploid cell that has undergone chromosome loss can later duplicate the remaining homologue (albeit losing heterozygous information on the lost chromosome). Similarly, WGD from a diploid to a tetraploid cell frequently results in chromosome loss and eventually ploidy is returned to the diploid state.
The initial ploidy-changing event may arise very rarely in some environments, but if there is a fitness benefit that accompanies the ploidy change, then it will spread throughout the population over time. What is surprising then is how often these ploidy-changing events occur within short-term in vitro evolution experiments and over eons of evolution. Perhaps the adaptable genome is the most successful genome, and cells with the ability to undergo rapid genome expansion and/or contraction are able to survive the most environmental insults. Additionally, it is possible that there is a subpopulation of persister cells that are primed and/or programmed to undergo ploidy changes during the response to stress. More population-level and single-cell approaches are needed to define the mechanisms and environmental conditions that cause ploidy changes in all eukaryotes (especially during fungal infections and tumorigenesis). The impressive examples of genomic heterogeneity observed in fungal pathogens support using these simple model systems to determine the mechanisms of ploidy change that occur during the somatic evolution of human cancers.
ACKNOWLEDGMENTS
We thank Hung-ji Tsai and Jun-Yi Leu for helpful comments on the manuscript, Phillip Richmond for contributing images to Fig. 1, and Kimberly Fischer for writing help and for generating Table 1.
This work is supported by Nebraska LB692 Department of Health, Nebraska LB595 Cancer and Smoking Disease Research, and Creighton University and by an NIH grant R15 AI090633 to A.F.
REFERENCES
- 1.Otto SP, Whitton J. 2000. Polyploid incidence and evolution. Annu Rev Genet 34:401–437 10.1146/annurev.genet.34.1.401. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Otto SP. 2007. The evolutionary consequences of polyploidy. Cell 131:452–462 10.1016/j.cell.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 3.Sémon M, Wolfe KH. 2007. Consequences of genome duplication. Curr Opin Genet Dev 17:505–512 10.1016/j.gde.2007.09.007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Albertin W, Marullo P. 2012. Polyploidy in fungi: evolution after whole-genome duplication. Proc Biol Sci 279:2497–2509 10.1098/rspb.2012.0434. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia AM. 1964. Studies on DNA in leucocytes and related cells of mammals. IV. The feulgen-DNA content of peripheral leucocytes, megakaryocytes and other bone marrow cell types of the rabbit. Acta Histochem 17:246–258. [PubMed] [PubMed] [Google Scholar]
- 6.Wheatley DN. 1972. Binucleation in mammalian liver: studies on the control of cytokinesis in vivo. Exp Cell Res 74:455–465 10.1016/0014-4827(72)90401-6. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Nadal C, Zajdela F. 1967. Hepatic polyploidy in the rat. IV. Experimental changes in the nucleolar volume of liver cells and their mechanisms of regulation. Exp Cell Res 48:518–528 10.1016/0014-4827(67)90318-7. (In Greek, Modern.) [DOI] [PubMed] [Google Scholar]
- 8.Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, Grompe M. 2010. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 467:707–710 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett RJ, Johnson AD. 2003. Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. EMBO J 22:2505–2515 10.1093/emboj/cdg235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki T, Nishibayashi S, Kuroiwa T, Kanbe T, Tanaka K. 1982. Variance of ploidy in Candida albicans. J Bacteriol 152:893–896. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki T, Hitomi A, Magee PT, Sakaguchi S. 1994. Correlation between polyploidy and auxotrophic segregation in the imperfect yeast Candida albicans. J Bacteriol 176:3345–3353 10.1128/jb.176.11.3345-3353.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ezov TK, Boger-Nadjar E, Frenkel Z, Katsperovski I, Kemeny S, Nevo E, Korol A, Kashi Y. 2006. Molecular-genetic biodiversity in a natural population of the yeast Saccharomyces cerevisiae from “Evolution Canyon”: microsatellite polymorphism, ploidy and controversial sexual status. Genetics 174:1455–1468 10.1534/genetics.106.062745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selmecki A, Gerami-Nejad M, Paulson C, Forche A, Berman J. 2008. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol 68:624–641 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- 14.Dunn B, Richter C, Kvitek DJ, Pugh T, Sherlock G. 2012. Analysis of the Saccharomyces cerevisiae pan-genome reveals a pool of copy number variants distributed in diverse yeast strains from differing industrial environments. Genome Res 22:908–924 10.1101/gr.130310.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ford CB, Funt JM, Abbey D, Issi L, Guiducci C, Martinez DA, Delorey T, Li BY, White TC, Cuomo C, Rao RP, Berman J, Thompson DA, Regev A. 2015. The evolution of drug resistance in clinical isolates of Candida albicans. eLife 4:e00662 10.7554/eLife.00662. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu YO, Sherlock G, Petrov DA. 2016. Whole genome analysis of 132 clinical Saccharomyces cerevisiae strains reveals extensive ploidy variation. G3 (Bethesda) 6:2421–2434 10.1534/g3.116.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zörgö E, Chwialkowska K, Gjuvsland AB, Garré E, Sunnerhagen P, Liti G, Blomberg A, Omholt SW, Warringer J. 2013. Ancient evolutionary trade-offs between yeast ploidy states. PLoS Genet 9:e1003388 10.1371/journal.pgen.1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dufresne F, Stift M, Vergilino R, Mable BK. 2014. Recent progress and challenges in population genetics of polyploid organisms: an overview of current state-of-the-art molecular and statistical tools. Mol Ecol 23:40–69 10.1111/mec.12581. [DOI] [PubMed] [Google Scholar]
- 19.Gerstein AC, Chun HJ, Grant A, Otto SP. 2006. Genomic convergence toward diploidy in Saccharomyces cerevisiae. PLoS Genet 2:e145 10.1371/journal.pgen.0020145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R, Pellman D. 2015. Polyploidy can drive rapid adaptation in yeast. Nature 519:349–352 10.1038/nature14187. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venkataram S, Dunn B, Li Y, Agarwala A, Chang J, Ebel ER, Geiler-Samerotte K, Herissant L, Blundell JR, Levy SF, Fisher DS, Sherlock G, Petrov DA. 2016. Development of a comprehensive genotype-to-fitness map of adaptation-driving mutations in yeast. Cell 166:1585–1596. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishan A. 1975. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol 66:188–193 10.1083/jcb.66.1.188. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. 1998. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 20:207–211 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 24.Wilhelm J, Pingoud A, Hahn M. 2003. Validation of an algorithm for automatic quantification of nucleic acid copy numbers by real-time polymerase chain reaction. Anal Biochem 317:218–225 10.1016/S0003-2697(03)00167-2. [DOI] [PubMed] [Google Scholar]
- 25.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. 2012. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7:e37135 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gompert Z, Mock KE. 2017. Detection of individual ploidy levels with genotyping-by-sequencing (GBS) analysis. Mol Ecol Resour 10.1111/1755-0998.12657. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Liti G. 2015. The fascinating and secret wild life of the budding yeast S. cerevisiae. eLife 4:4 10.7554/eLife.05835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carreto L, Eiriz MF, Gomes AC, Pereira PM, Schuller D, Santos MA. 2008. Comparative genomics of wild type yeast strains unveils important genome diversity. BMC Genomics 9:524 10.1186/1471-2164-9-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strope PK, Skelly DA, Kozmin SG, Mahadevan G, Stone EA, Magwene PM, Dietrich FS, McCusker JH. 2015. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res 25:762–774 10.1101/gr.185538.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albertin W, Marullo P, Aigle M, Bourgais A, Bely M, Dillmann C, DE Vienne D, Sicard D. 2009. Evidence for autotetraploidy associated with reproductive isolation in Saccharomyces cerevisiae: towards a new domesticated species. J Evol Biol 22:2157–2170 10.1111/j.1420-9101.2009.01828.x. [DOI] [PubMed] [Google Scholar]
- 31.Lindell RM, Hartman TE, Nadrous HF, Ryu JH. 2005. Pulmonary cryptococcosis: CT findings in immunocompetent patients. Radiology 236:326–331 10.1148/radiol.2361040460. [DOI] [PubMed] [Google Scholar]
- 32.Tien RD, Chu PK, Hesselink JR, Duberg A, Wiley C. 1991. Intracranial cryptococcosis in immunocompromised patients: CT and MR findings in 29 cases. AJNR Am J Neuroradiol 12:283–289. [PubMed] [PMC free article] [PubMed] [Google Scholar]
- 33.Velagapudi R, Hsueh YP, Geunes-Boyer S, Wright JR, Heitman J. 2009. Spores as infectious propagules of Cryptococcus neoformans. Infect Immun 77:4345–4355 10.1128/IAI.00542-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Idnurm A, Verma S, Corrochano LM. 2010. A glimpse into the basis of vision in the kingdom Mycota. Fungal Genet Biol 47:881–892 10.1016/j.fgb.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ni M, Feretzaki M, Li W, Floyd-Averette A, Mieczkowski P, Dietrich FS, Heitman J. 2013. Unisexual and heterosexual meiotic reproduction generate aneuploidy and phenotypic diversity de novo in the yeast Cryptococcus neoformans. PLoS Biol 11:e1001653 10.1371/journal.pbio.1001653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feldmesser M, Kress Y, Casadevall A. 2001. Dynamic changes in the morphology of Cryptococcus neoformans during murine pulmonary infection. Microbiology 147:2355–2365 10.1099/00221287-147-8-2355. [DOI] [PubMed] [Google Scholar]
- 37.Ene IV, Bennett RJ. 2014. The cryptic sexual strategies of human fungal pathogens. Nat Rev Microbiol 12:239–251 10.1038/nrmicro3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okagaki LH, Strain AK, Nielsen JN, Charlier C, Baltes NJ, Chrétien F, Heitman J, Dromer F, Nielsen K. 2010. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog 6:e1000953 10.1371/journal.ppat.1000953. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaragoza O, García-Rodas R, Nosanchuk JD, Cuenca-Estrella M, Rodríguez-Tudela JL, Casadevall A. 2010. Fungal cell gigantism during mammalian infection. PLoS Pathog 6:e1000945 10.1371/journal.ppat.1000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sallé J, Campbell SD, Gho M, Audibert A. 2012. CycA is involved in the control of endoreplication dynamics in the Drosophila bristle lineage. Development 139:547–557 10.1242/dev.069823. [PubMed] [DOI] [PubMed] [Google Scholar]
- 41.Dudas J, Saile B, El-Armouche H, Aprigliano I, Ramadori G. 2003. Endoreplication and polyploidy in primary culture of rat hepatic stellate cells. Cell Tissue Res 313:301–311 10.1007/s00441-003-0768-3. [DOI] [PubMed] [Google Scholar]
- 42.Sionov E, Lee H, Chang YC, Kwon-Chung KJ. 2010. Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog 6:e1000848 10.1371/journal.ppat.1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerstein AC, Fu MS, Mukaremera L, Li Z, Ormerod KL, Fraser JA, Berman J, Nielsen K. 2015. Polyploid titan cells produce haploid and aneuploid progeny to promote stress adaptation. MBio 6:e01340-15 10.1128/mBio.01340-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riggsby WS, Torres-Bauza LJ, Wills JW, Townes TM. 1982. DNA content, kinetic complexity, and the ploidy question in Candida albicans. Mol Cell Biol 2:853–862 10.1128/MCB.2.7.853. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G, Thorstenson YR, Agabian N, Magee PT, Davis RW, Scherer S. 2004. The diploid genome sequence of Candida albicans. Proc Natl Acad Sci USA 101:7329–7334 10.1073/pnas.0401648101. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selmecki A, Forche A, Berman J. 2010. Genomic plasticity of the human fungal pathogen Candida albicans. Eukaryot Cell 9:991–1008 10.1128/EC.00060-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hickman MA, Zeng G, Forche A, Hirakawa MP, Abbey D, Harrison BD, Wang YM, Su CH, Bennett RJ, Wang Y, Berman J. 2013. The ‘obligate diploid’ Candida albicans forms mating-competent haploids. Nature 494:55–59 10.1038/nature11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abbey DA, Funt J, Lurie-Weinberger MN, Thompson DA, Regev A, Myers CL, Berman J. 2014. YMAP: a pipeline for visualization of copy number variation and loss of heterozygosity in eukaryotic pathogens. Genome Med 6:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tzung KW, Williams RM, Scherer S, Federspiel N, Jones T, Hansen N, Bivolarevic V, Huizar L, Komp C, Surzycki R, Tamse R, Davis RW, Agabian N. 2001. Genomic evidence for a complete sexual cycle in Candida albicans. Proc Natl Acad Sci USA 98:3249–3253 10.1073/pnas.061628798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hull CM, Johnson AD. 1999. Identification of a mating type-like locus in the asexual pathogenic yeast Candida albicans. Science 285:1271–1275 10.1126/science.285.5431.1271. [DOI] [PubMed] [Google Scholar]
- 51.Magee BB, Magee PT. 2000. Induction of mating in Candida albicans by construction of MTLa and MTLalpha strains. Science 289:310–313 10.1126/science.289.5477.310. [PubMed] [DOI] [PubMed] [Google Scholar]
- 52.Forche A, Alby K, Schaefer D, Johnson AD, Berman J, Bennett RJ. 2008. The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS Biol 6:e110 10.1371/journal.pbio.0060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alby K, Schaefer D, Bennett RJ. 2009. Homothallic and heterothallic mating in the opportunistic pathogen Candida albicans. Nature 460:890–893 10.1038/nature08252. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hickman MA, Paulson C, Dudley A, Berman J. 2015. Parasexual ploidy reduction drives population heterogeneity through random and transient aneuploidy in Candida albicans. Genetics 200:781–794 10.1534/genetics.115.178020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hilton C, Markie D, Corner B, Rikkerink E, Poulter R. 1985. Heat shock induces chromosome loss in the yeast Candida albicans. Mol Gen Genet 200:162–168 10.1007/BF00383330. [DOI] [PubMed] [Google Scholar]
- 56.Janbon G, Sherman F, Rustchenko E. 1998. Monosomy of a specific chromosome determines L-sorbose utilization: a novel regulatory mechanism in Candida albicans. Proc Natl Acad Sci USA 95:5150–5155 10.1073/pnas.95.9.5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kabir MA, Ahmad A, Greenberg JR, Wang YK, Rustchenko E. 2005. Loss and gain of chromosome 5 controls growth of Candida albicans on sorbose due to dispersed redundant negative regulators. Proc Natl Acad Sci USA 102:12147–12152 10.1073/pnas.0505625102. (Erratum, doi:10.1073/pnas.0507247102.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forche A, May G, Magee PT. 2005. Demonstration of loss of heterozygosity by single-nucleotide polymorphism microarray analysis and alterations in strain morphology in Candida albicans strains during infection. Eukaryot Cell 4:156–165 10.1128/EC.4.1.156-165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Selmecki A, Forche A, Berman J. 2006. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313:367–370 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Forche A, Magee PT, Selmecki A, Berman J, May G. 2009. Evolution in Candida albicans populations during a single passage through a mouse host. Genetics 182:799–811 10.1534/genetics.109.103325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J. 2009. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 5:e1000705 10.1371/journal.pgen.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harrison BD, Hashemi J, Bibi M, Pulver R, Bavli D, Nahmias Y, Wellington M, Sapiro G, Berman J. 2014. A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol 12:e1001815 10.1371/journal.pbio.1001815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson CA, Roberts S, Zhang H, Kelly CM, Kendall A, Lee C, Gerstenberger J, Koenig AB, Kabeche R, Gladfelter AS. 2015. Ploidy variation in multinucleate cells changes under stress. Mol Biol Cell 26:1129–1140 10.1091/mbc.E14-09-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farrer RA, Henk DA, Garner TW, Balloux F, Woodhams DC, Fisher MC. 2013. Chromosomal copy number variation, selection and uneven rates of recombination reveal cryptic genome diversity linked to pathogenicity. PLoS Genet 9:e1003703 10.1371/journal.pgen.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vlaardingerbroek I, Beerens B, Schmidt SM, Cornelissen BJ, Rep M. 2016. Dispensable chromosomes in Fusarium oxysporum f. sp. lycopersici. Mol Plant Pathol 17:1455–1466 10.1111/mpp.12440. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kasuga T, Bui M, Bernhardt E, Swiecki T, Aram K, Cano LM, Webber J, Brasier C, Press C, Grünwald NJ, Rizzo DM, Garbelotto M. 2016. Host-induced aneuploidy and phenotypic diversification in the sudden oak death pathogen Phytophthora ramorum. BMC Genomics 17:385 10.1186/s12864-016-2717-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan CS, Botstein D. 1993. Isolation and characterization of chromosome-gain and increase-in-ploidy mutants in yeast. Genetics 135:677–691. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Porter AC. 2008. Preventing DNA over-replication: a Cdk perspective. Cell Div 3:3 10.1186/1747-1028-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edgar BA, Orr-Weaver TL. 2001. Endoreplication cell cycles: more for less. Cell 105:297–306 10.1016/S0092-8674(01)00334-8. [DOI] [PubMed] [Google Scholar]
- 70.Neiman M, Beaton MJ, Hessen DO, Jeyasingh PD, Weider LJ. 2017. Endopolyploidy as a potential driver of animal ecology and evolution. Biol Rev Camb Philos Soc 92:234–247 10.1111/brv.12226. [DOI] [PubMed] [Google Scholar]
- 71.Hayles J, Fisher D, Woollard A, Nurse P. 1994. Temporal order of S phase and mitosis in fission yeast is determined by the state of the p34cdc2-mitotic B cyclin complex. Cell 78:813–822 10.1016/S0092-8674(94)90542-8. [PubMed] [DOI] [PubMed] [Google Scholar]
- 72.Vassilev A, Lee CY, Vassilev B, Zhu W, Ormanoglu P, Martin SE, DePamphilis ML. 2016. Identification of genes that are essential to restrict genome duplication to once per cell division. Oncotarget 7:34956–34976. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duncan AW. 2013. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin Cell Dev Biol 24:347–356 10.1016/j.semcdb.2013.01.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 74.Guidotti JE, Brégerie O, Robert A, Debey P, Brechot C, Desdouets C. 2003. Liver cell polyploidization: a pivotal role for binuclear hepatocytes. J Biol Chem 278:19095–19101 10.1074/jbc.M300982200. [DOI] [PubMed] [Google Scholar]
- 75.Margall-Ducos G, Celton-Morizur S, Couton D, Brégerie O, Desdouets C. 2007. Liver tetraploidization is controlled by a new process of incomplete cytokinesis. J Cell Sci 120:3633–3639 10.1242/jcs.016907. [DOI] [PubMed] [Google Scholar]
- 76.Winey M, Hoyt MA, Chan C, Goetsch L, Botstein D, Byers B. 1993. NDC1: a nuclear periphery component required for yeast spindle pole body duplication. J Cell Biol 122:743–751 10.1083/jcb.122.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Biggins S, Severin FF, Bhalla N, Sassoon I, Hyman AA, Murray AW. 1999. The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev 13:532–544 10.1101/gad.13.5.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Francisco L, Chan CS. 1994. Regulation of yeast chromosome segregation by Ipl1 protein kinase and type 1 protein phosphatase. Cell Mol Biol Res 40:207–213. [PubMed] [PubMed] [Google Scholar]
- 79.He X, Rines DR, Espelin CW, Sorger PK. 2001. Molecular analysis of kinetochore-microtubule attachment in budding yeast. Cell 106:195–206 10.1016/S0092-8674(01)00438-X. [DOI] [PubMed] [Google Scholar]
- 80.Guacci V, Koshland D, Strunnikov A. 1997. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 91:47–57 10.1016/S0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Covo S, Puccia CM, Argueso JL, Gordenin DA, Resnick MA. 2014. The sister chromatid cohesion pathway suppresses multiple chromosome gain and chromosome amplification. Genetics 196:373–384 10.1534/genetics.113.159202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu Y, Srinivasan M, Nakanishi S, Leatherwood J, Shilatifard A, Sternglanz R. 2011. A conserved patch near the C terminus of histone H4 is required for genome stability in budding yeast. Mol Cell Biol 31:2311–2325 10.1128/MCB.01432-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. 2007. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446:572–576 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 84.Chambers AL, Ormerod G, Durley SC, Sing TL, Brown GW, Kent NA, Downs JA. 2012. The INO80 chromatin remodeling complex prevents polyploidy and maintains normal chromatin structure at centromeres. Genes Dev 26:2590–2603 10.1101/gad.199976.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Watts FZ, Shiels G, Orr E. 1987. The yeast MYO1 gene encoding a myosin-like protein required for cell division. EMBO J 6:3499–3505. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling-Hampton K, Seidel CW, Li R. 2008. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135:879–893 10.1016/j.cell.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Song W, Petes TD. 2012. Haploidization in Saccharomyces cerevisiae induced by a deficiency in homologous recombination. Genetics 191:279–284 10.1534/genetics.111.138180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Storchová Z, Breneman A, Cande J, Dunn J, Burbank K, O’Toole E, Pellman D. 2006. Genome-wide genetic analysis of polyploidy in yeast. Nature 443:541–547 10.1038/nature05178. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.Alabrudzinska M, Skoneczny M, Skoneczna A. 2011. Diploid-specific [corrected] genome stability genes of S. cerevisiae: genomic screen reveals haploidization as an escape from persisting DNA rearrangement stress. PLoS One 6:e21124 10.1371/journal.pone.0021124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haber JE. 1992. Exploring the pathways of homologous recombination. Curr Opin Cell Biol 4:401–412 10.1016/0955-0674(92)90005-W. [DOI] [PubMed] [Google Scholar]
- 91.Mehta A, Beach A, Haber JE. 2017. Homology requirements and competition between gene conversion and break-induced replication during double-strand break repair. Mol Cell 65:515–526. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen C, Kolodner RD. 1999. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet 23:81–85 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 93.Hiraoka M, Watanabe K, Umezu K, Maki H. 2000. Spontaneous loss of heterozygosity in diploid Saccharomyces cerevisiae cells. Genetics 156:1531–1548. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Skoneczna A, Kaniak A, Skoneczny M. 2015. Genetic instability in budding and fission yeast-sources and mechanisms. FEMS Microbiol Rev 39:917–967 10.1093/femsre/fuv028. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ohnishi G, Endo K, Doi A, Fujita A, Daigaku Y, Nunoshiba T, Yamamoto K. 2004. Spontaneous mutagenesis in haploid and diploid Saccharomyces cerevisiae. Biochem Biophys Res Commun 325:928–933 10.1016/j.bbrc.2004.10.120. [DOI] [PubMed] [Google Scholar]
- 96.Jung PP, Fritsch ES, Blugeon C, Souciet JL, Potier S, Lemoine S, Schacherer J, de Montigny J. 2011. Ploidy influences cellular responses to gross chromosomal rearrangements in Saccharomyces cerevisiae. BMC Genomics 12:331 10.1186/1471-2164-12-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Storchova Z, Kuffer C. 2008. The consequences of tetraploidy and aneuploidy. J Cell Sci 121:3859–3866 10.1242/jcs.039537. [DOI] [PubMed] [Google Scholar]
- 98.Matzke MA, Mittelsten Scheid O, Matzke AJ. 1999. Rapid structural and epigenetic changes in polyploid and aneuploid genomes. BioEssays 21:761–767 10.1002/(SICI)1521-1878(199909)21:9<761::AID-BIES7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 99.Comai L. 2005. The advantages and disadvantages of being polyploid. Nat Rev Genet 6:836–846 10.1038/nrg1711. [DOI] [PubMed] [Google Scholar]
- 100.Comai L, Tyagi AP, Winter K, Holmes-Davis R, Reynolds SH, Stevens Y, Byers B. 2000. Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 12:1551–1568 10.1105/tpc.12.9.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hufton AL, Panopoulou G. 2009. Polyploidy and genome restructuring: a variety of outcomes. Curr Opin Genet Dev 19:600–606 10.1016/j.gde.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 102.Mayer VW, Aguilera A. 1990. High levels of chromosome instability in polyploids of Saccharomyces cerevisiae. Mutat Res 231:177–186 10.1016/0027-5107(90)90024-X. [PubMed] [DOI] [PubMed] [Google Scholar]
- 103.Andalis AA, Storchova Z, Styles C, Galitski T, Pellman D, Fink GR. 2004. Defects arising from whole-genome duplications in Saccharomyces cerevisiae. Genetics 167:1109–1121 10.1534/genetics.104.029256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seervai RN, Jones SK Jr, Hirakawa MP, Porman AM, Bennett RJ. 2013. Parasexuality and ploidy change in Candida tropicalis. Eukaryot Cell 12:1629–1640 10.1128/EC.00128-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gerstein AC, McBride RM, Otto SP. 2008. Ploidy reduction in Saccharomyces cerevisiae. Biol Lett 4:91–94 10.1098/rsbl.2007.0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Levine DS, Sanchez CA, Rabinovitch PS, Reid BJ. 1991. Formation of the tetraploid intermediate is associated with the development of cells with more than four centrioles in the elastase-simian virus 40 tumor antigen transgenic mouse model of pancreatic cancer. Proc Natl Acad Sci USA 88:6427–6431 10.1073/pnas.88.15.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu YJ, Swamy KB, Leu JY. 2016. Experimental evolution reveals interplay between Sch9 and polyploid stability in yeast. PLoS Genet 12:e1006409 10.1371/journal.pgen.1006409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miettinen TP, Pessa HK, Caldez MJ, Fuhrer T, Diril MK, Sauer U, Kaldis P, Björklund M. 2014. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr Biol 24:598–608 10.1016/j.cub.2014.01.071. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wu CY, Rolfe PA, Gifford DK, Fink GR. 2010. Control of transcription by cell size. PLoS Biol 8:e1000523 10.1371/journal.pbio.1000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Watanabe T, Tanaka Y. 1982. Age-related alterations in the size of human hepatocytes. A study of mononuclear and binucleate cells. Virchows Arch B Cell Pathol Incl Mol Pathol 39:9–20 10.1007/BF02892832. [DOI] [PubMed] [Google Scholar]
- 111.Conlon I, Raff M. 1999. Size control in animal development. Cell 96:235–244 10.1016/S0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 112.Tsukaya H. 2013. Does ploidy level directly control cell size? Counterevidence from Arabidopsis genetics. PLoS One 8:e83729 10.1371/journal.pone.0083729. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weiss RL, Kukora JR, Adams J. 1975. The relationship between enzyme activity, cell geometry, and fitness in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 72:794–798 10.1073/pnas.72.3.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Galitski T, Saldanha AJ, Styles CA, Lander ES, Fink GR. 1999. Ploidy regulation of gene expression. Science 285:251–254 10.1126/science.285.5425.251. [DOI] [PubMed] [Google Scholar]
- 115.Mable BK, Otto SP. 2001. Masking and purging mutations following EMS treatment in haploid, diploid and tetraploid yeast (Saccharomyces cerevisiae). Genet Res 77:9–26 10.1017/S0016672300004821. [DOI] [PubMed] [Google Scholar]
- 116.Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B, Nislow C, Giaever G, Phillips PC, Fink GR, Gifford DK, Boone C. 2010. Genotype to phenotype: a complex problem. Science 328:469 10.1126/science.1189015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.de Godoy LM, Olsen JV, Cox J, Nielsen ML, Hubner NC, Fröhlich F, Walther TC, Mann M. 2008. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455:1251–1254 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- 118.Voordeckers K, Kominek J, Das A, Espinosa-Cantú A, De Maeyer D, Arslan A, Van Pee M, van der Zande E, Meert W, Yang Y, Zhu B, Marchal K, DeLuna A, Van Noort V, Jelier R, Verstrepen KJ. 2015. Adaptation to high ethanol reveals complex evolutionary pathways. PLoS Genet 11:e1005635 10.1371/journal.pgen.1005635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Levy SF, Blundell JR, Venkataram S, Petrov DA, Fisher DS, Sherlock G. 2015. Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature 519:181–186 10.1038/nature14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bouchonville K, Forche A, Tang KE, Selmecki A, Berman J. 2009. Aneuploid chromosomes are highly unstable during DNA transformation of Candida albicans. Eukaryot Cell 8:1554–1566. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gerstein AC, Lim H, Berman J, Hickman MA. 2017. Ploidy tug-of-war: evolutionary and genetic environments influence the rate of ploidy drive in a human fungal pathogen. Evolution 71:1025–1038 10.1111/evo.13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Coste A, Selmecki A, Forche A, Diogo D, Bougnoux ME, d’Enfert C, Berman J, Sanglard D. 2007. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell 6:1889–1904 10.1128/EC.00151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chang FM, Ou TY, Cheng WN, Chou ML, Lee KC, Chin YP, Lin CP, Chang KD, Lin CT, Su CH. 2014. Short-term exposure to fluconazole induces chromosome loss in Candida albicans: an approach to produce haploid cells. Fungal Genet Biol 70:68–76 10.1016/j.fgb.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 124.Ngamskulrungroj P, Chang Y, Hansen B, Bugge C, Fischer E, Kwon-Chung KJ. 2012. Characterization of the chromosome 4 genes that affect fluconazole-induced disomy formation in Cryptococcus neoformans. PLoS One 7:e33022 10.1371/journal.pone.0033022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, Kupiec M, Pilpel Y, Dahan O. 2012. Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci USA 109:21010–21015 10.1073/pnas.1211150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317:916–924 10.1126/science.1142210. [PubMed] [DOI] [PubMed] [Google Scholar]
- 127.Lingelbach LB, Kaplan KB. 2004. The interaction between Sgt1p and Skp1p is regulated by HSP90 chaperones and is required for proper CBF3 assembly. Mol Cell Biol 24:8938–8950 10.1128/MCB.24.20.8938-8950.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen G, Bradford WD, Seidel CW, Li R. 2012. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482:246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sunshine AB, Payen C, Ong GT, Liachko I, Tan KM, Dunham MJ. 2015. The fitness consequences of aneuploidy are driven by condition-dependent gene effects. PLoS Biol 13:e1002155 10.1371/journal.pbio.1002155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Payen C, Sunshine AB, Ong GT, Pogachar JL, Zhao W, Dunham MJ. 2016. High-throughput identification of adaptive mutations in experimentally evolved yeast populations. PLoS Genet 12:e1006339 10.1371/journal.pgen.1006339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gresham D, Desai MM, Tucker CM, Jenq HT, Pai DA, Ward A, DeSevo CG, Botstein D, Dunham MJ. 2008. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet 4:e1000303 10.1371/journal.pgen.1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Croll D, McDonald BA. 2012. The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog 8:e1002608 10.1371/journal.ppat.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wittenberg AH, van der Lee TA, Ben M’barek S, Ware SB, Goodwin SB, Kilian A, Visser RG, Kema GH, Schouten HJ. 2009. Meiosis drives extraordinary genome plasticity in the haploid fungal plant pathogen Mycosphaerella graminicola. PLoS One 4:e5863 10.1371/journal.pone.0005863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Van de Wouw AP, Cozijnsen AJ, Hane JK, Brunner PC, McDonald BA, Oliver RP, Howlett BJ. 2010. Evolution of linked avirulence effectors in Leptosphaeria maculans is affected by genomic environment and exposure to resistance genes in host plants. PLoS Pathog 6:e1001180 10.1371/journal.ppat.1001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Raffaele S, Farrer RA, Cano LM, Studholme DJ, MacLean D, Thines M, Jiang RH, Zody MC, Kunjeti SG, Donofrio NM, Meyers BC, Nusbaum C, Kamoun S. 2010. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science 330:1540–1543 10.1126/science.1193070. [DOI] [PubMed] [Google Scholar]
- 136.Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, Schmutz J, Taga M, White GJ, Zhou S, Schwartz DC, Freitag M, Ma LJ, Danchin EG, Henrissat B, Coutinho PM, Nelson DR, Straney D, Napoli CA, Barker BM, Gribskov M, Rep M, Kroken S, Molnár I, Rensing C, Kennell JC, Zamora J, Farman ML, Selker EU, Salamov A, Shapiro H, Pangilinan J, Lindquist E, Lamers C, Grigoriev IV, Geiser DM, Covert SF, Temporini E, Vanetten HD. 2009. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet 5:e1000618 10.1371/journal.pgen.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Miao VP, Covert SF, VanEtten HD. 1991. A fungal gene for antibiotic resistance on a dispensable (“B”) chromosome. Science 254:1773–1776 10.1126/science.1763326. [DOI] [PubMed] [Google Scholar]
- 138.Ma LJ, et al. 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464:367–373 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Roper M, Ellison C, Taylor JW, Glass NL. 2011. Nuclear and genome dynamics in multinucleate ascomycete fungi. Curr Biol 21:R786–R793 10.1016/j.cub.2011.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rustchenko-Bulgac EP. 1991. Variations of Candida albicans electrophoretic karyotypes. J Bacteriol 173:6586–6596 10.1128/jb.173.20.6586-6596.1991. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ahmad KM, Kokošar J, Guo X, Gu Z, Ishchuk OP, Piškur J. 2014. Genome structure and dynamics of the yeast pathogen Candida glabrata. FEMS Yeast Res 14:529–535 10.1111/1567-1364.12145. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sem X, Le GT, Tan AS, Tso G, Yurieva M, Liao WW, Lum J, Srinivasan KG, Poidinger M, Zolezzi F, Pavelka N. 2016. Beta-glucan exposure on the fungal cell wall tightly correlates with competitive fitness of Candida species in the mouse gastrointestinal tract. Front Cell Infect Microbiol 6:186 10.3389/fcimb.2016.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zaragoza O, Nielsen K. 2013. Titan cells in Cryptococcus neoformans: cells with a giant impact. Curr Opin Microbiol 16:409–413 10.1016/j.mib.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lin X, Patel S, Litvintseva AP, Floyd A, Mitchell TG, Heitman J. 2009. Diploids in the Cryptococcus neoformans serotype A population homozygous for the alpha mating type originate via unisexual mating. PLoS Pathog 5:e1000283 10.1371/journal.ppat.1000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zaragoza O, Fries BC, Casadevall A. 2003. Induction of capsule growth in Cryptococcus neoformans by mammalian serum and CO(2). Infect Immun 71:6155–6164 10.1128/IAI.71.11.6155-6164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kronstad JW, Attarian R, Cadieux B, Choi J, D’Souza CA, Griffiths EJ, Geddes JM, Hu G, Jung WH, Kretschmer M, Saikia S, Wang J. 2011. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat Rev Microbiol 9:193–203 10.1038/nrmicro2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Paquin C, Adams J. 1983. Frequency of fixation of adaptive mutations is higher in evolving diploid than haploid yeast populations. Nature 302:495–500 10.1038/302495a0. [DOI] [PubMed] [Google Scholar]
- 148.DeLuna A, Vetsigian K, Shoresh N, Hegreness M, Colón-González M, Chao S, Kishony R. 2008. Exposing the fitness contribution of duplicated genes. Nat Genet 40:676–681 10.1038/ng.123. [PubMed] [DOI] [PubMed] [Google Scholar]
- 149.Desai MM, Fisher DS, Murray AW. 2007. The speed of evolution and maintenance of variation in asexual populations. Curr Biol 17:385–394 10.1016/j.cub.2007.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gerrish PJ, Lenski RE. 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102-103:127–144 10.1023/A:1017067816551. [DOI] [PubMed] [Google Scholar]
- 151.Zeyl C, DeVisser JA. 2001. Estimates of the rate and distribution of fitness effects of spontaneous mutation in Saccharomyces cerevisiae. Genetics 157:53–61. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Otto SP, Yong P. 2002. The evolution of gene duplicates. Adv Genet 46:451–483 10.1016/S0065-2660(02)46017-8. [DOI] [PubMed] [Google Scholar]
- 153.Zeyl C. 2005. The number of mutations selected during adaptation in a laboratory population of Saccharomyces cerevisiae. Genetics 169:1825–1831 10.1534/genetics.104.027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Adams J, Hansche PE. 1974. Population studies in microorganisms. I. Evolution of diploidy in Saccharomyces cerevisiae. Genetics 76:327–338. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gerstein AC, Otto SP. 2009. Ploidy and the causes of genomic evolution. J Hered 100:571–581 10.1093/jhered/esp057. [PubMed] [DOI] [PubMed] [Google Scholar]
- 156.Korona R. 1999. Unpredictable fitness transitions between haploid and diploid strains of the genetically loaded yeast Saccharomyces cerevisiae. Genetics 151:77–85. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Orr HA, Otto SP. 1994. Does diploidy increase the rate of adaptation? Genetics 136:1475–1480. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Anderson JB, Sirjusingh C, Ricker N. 2004. Haploidy, diploidy and evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics 168:1915–1923 10.1534/genetics.104.033266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gerstein AC. 2013. Mutational effects depend on ploidy level: all else is not equal. Biol Lett 9:20120614 10.1098/rsbl.2012.0614. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sellis D, Callahan BJ, Petrov DA, Messer PW. 2011. Heterozygote advantage as a natural consequence of adaptation in diploids. Proc Natl Acad Sci USA 108:20666–20671 10.1073/pnas.1114573108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sellis D, Kvitek DJ, Dunn B, Sherlock G, Petrov DA. 2016. Heterozygote advantage is a common outcome of adaptation in Saccharomyces cerevisiae. Genetics 203:1401–1413 10.1534/genetics.115.185165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barrett TB, Sampson P, Owens GK, Schwartz SM, Benditt EP. 1983. Polyploid nuclei in human artery wall smooth muscle cells. Proc Natl Acad Sci USA 80:882–885 10.1073/pnas.80.3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Reid BJ, Blount PL, Rubin CE, Levine DS, Haggitt RC, Rabinovitch PS. 1992. Flow-cytometric and histological progression to malignancy in Barrett’s esophagus: prospective endoscopic surveillance of a cohort. Gastroenterology 102:1212–1219 10.1016/0016-5085(92)90758-Q. [DOI] [PubMed] [Google Scholar]
- 164.Olaharski AJ, Sotelo R, Solorza-Luna G, Gonsebatt ME, Guzman P, Mohar A, Eastmond DA. 2006. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 27:337–343 10.1093/carcin/bgi218. [DOI] [PubMed] [Google Scholar]
- 165.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, Meyerson M, Beroukhim R. 2013. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45:1134–1140 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. 2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437:1043–1047 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 167.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, Lander ES, Meyerson M, Getz G. 2012. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30:413–421 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brodeur GM, Williams DL, Look AT, Bowman WP, Kalwinsky DK. 1981. Near-haploid acute lymphoblastic leukemia: a unique subgroup with a poor prognosis? Blood 58:14–19. [PubMed] [PubMed] [Google Scholar]
- 169.Farabegoli F, Santini D, Ceccarelli C, Taffurelli M, Marrano D, Baldini N. 2001. Clone heterogeneity in diploid and aneuploid breast carcinomas as detected by FISH. Cytometry 46:50–56 10.1002/1097-0320(20010215)46:1<50::AID-CYTO1037>3.0.CO;2-T. [PubMed] [DOI] [PubMed] [Google Scholar]
- 170.Torres L, Ribeiro FR, Pandis N, Andersen JA, Heim S, Teixeira MR. 2007. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res Treat 102:143–155 10.1007/s10549-006-9317-6. [DOI] [PubMed] [Google Scholar]
- 171.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472:90–94 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]