

Abstract
Obesity, primarily a consequence of poor dietary choices and an increased sedentary lifestyle, has become a global pandemic that brings with it enormous medical, social, and economic challenges. Not only does obesity increase the risk of cardiovascular disease and certain cancers, but it is also recognized as a key driver of other metabolic syndrome (MetS) components. These components include insulin resistance, hyperglycemia with prediabetes or type 2 diabetes, dyslipidemia, and hypertension, and are underlying contributors to systemic metabolic dysfunction. More recently, obesity and diet-induced metabolic dysfunction have been identified as risk factors for the development of a wide variety of neurological disorders in both the central and peripheral nervous systems. An abundance of literature has shown that obesity is associated with mild cognitive impairment and altered hippocampal structure and function, and there is a robust correlation between obesity and Alzheimer’s type dementia. Similarly, many reports show that both the autonomic and somatic components of the peripheral nervous system are impacted by obesity. The autonomic nervous system, under control of the hypothalamus, displays altered catabolic and anabolic processes in obese individuals attributed to sympathetic-parasympathetic imbalances. A close association also exists between obesity and polyneuropathy, a complication most commonly found in prediabetic and diabetic patients, and is likely secondary to a combination of obesity-induced dyslipidemia with hyperglycemia. This review will outline the pathophysiological development of obesity and dyslipidemia, discuss the adverse impact of these conditions on the nervous system, and provide evidence for lipotoxicity and metabolic inflammation as the drivers underlying the neurological consequences of obesity. In addition, this review will examine the benefits of lifestyle and surgical interventions in obesity-induced neurological disorders.
Introduction
Recent data estimate that roughly 2.1 billion individuals are overweight or obese.1 Although genetic background does predispose certain individuals/ethnicities to developing obesity,2 the current pandemic that promotes weight gain is largely driven by an environment which arose in developed Western countries in the latter part of the 20th century.3 This obesogenic environment is attributable to a number of factors, including the overconsumption of processed, affordable, heavily marketed, highly palatable, energy-dense foods combined with an increased sedentary lifestyle. Though there are indications that the exponential rise of obesity has plateaued in developed countries, numerous regions worldwide that have adopted the lifestyle of a Westernized society continue to show an increase in obesity, which is reflected by a worldwide increase.1 Currently, it is projected that over 18% of adults will be obese by 2025.4
Defined as an increase of fat mass that adversely affects health, obesity is a known risk factor for cardiovascular disease and certain cancers. Central obesity, which is accompanied by a low-grade metabolic inflammation in visceral adipose tissue, is also recognized as both a component and a driver of the metabolic syndrome (MetS).5 MetS is defined by the presence of multiple comorbidities, including dyslipidemia, decreased insulin sensitivity, hyperinsulinemia, hyperglycemia, and hypertension; thus, obesity is an underlying promoter of systemic metabolic dysfunction. Multiple tissues, including the liver, pancreas, kidney, and vasculature, exhibit dysfunction as a consequence of obesity via direct and/or indirect mechanisms. Accumulating evidence also links obesity with a wide array of neurological disorders in a manner that is complex and multifactorial (Box 1). Though the central (CNS) and peripheral (PNS) nervous systems are quite distinct in form and function, both are susceptible to obesity-driven dysfunction, suggesting that common mechanisms contributing to disease progression may be perpetuated by visceral adiposity. It is also important to recognize that obesity drives other components of the MetS which are strongly linked to neurological deficits (for example, strong associations exist between insulin resistance and cognitive decline). As a result, it is likely that multiple mechanisms may be interacting to culminate in neurological dysfunction, and ascertaining direct causality of visceral adiposity on neurological complications thus remains challenging.
Box 1. Neurological Consequences of Obesity.
Central Nervous System
Structural
Brain atrophy and reduced grey matter volume in frontal and temporal lobes and enlarged orbitofrontal white matter
Decreased hippocampus and hypothalamus integrity
Psychological and Behavioral
Altered feeding behavior and satiety control
Mild cognitive impairment (attention, learning, and memory deficits; impairments in decision making), cognitive decline and increased risk of dementia and Alzheimer’s disease
Mood disorders including anxiety and depression
Physiological
Cerebral ischemia and hypoperfusion
Decreased brain metabolism and nerve function
Peripheral Nervous System
Structural
Organ damage due to chronic activation of sympathetic nervous system
Loss of peripheral sensory neurons and small intraepidermal nerve fibers
Psychological and Behavioral
Symptoms of sensory polyneuropathy, including early onset pain, paresthesias, allodynia, and hyperalgesia, that over time converts to frank numbness and loss of perception of sensory modalities
Symptoms and signs occur in a stocking-glove distribution
Physiological
- Blunted but chronic activation of sympathetic nervous tone resulting in:
-
○Increased efferent muscle activity, cardiac output, adrenaline release from adrenal medulla, adipose tissue lipolysis, liver gluconeogenesis, and decreased release of pancreatic insulin
-
○
Gastroparesis and intestinal dysmotility
Decreased motor and sensory nerve function
Mechanistic insight provided by animal models of obesity suggests that excess dietary fat compromises hypothalamic control of energy homeostasis. This contributes to adipose tissue dysfunction, leading to elevated free fatty acids (FFA) and systemic dyslipidemia (Figure 1). Chronic caloric excess also impacts multiple organs, including the liver, and drives an increase in circulating triglycerides. This dyslipidemia can result in FFA-induced lipotoxicity – changes in lipid-induced intracellular signaling or changes in lipid utilization that result in tissue pathophysiology – that drives neurological dysfunction and neurodegeneration. While these events likely impact the CNS and PNS in a multifactorial fashion, however, recent clinical and preclinical advances have fueled improved insight into the potential neurological complications that occur with obesity. This review will outline the pathophysiological development of obesity and dyslipidemia, discuss the CNS and PNS diseases that arise as a consequence, present evidence for lipotoxicity and metabolic inflammation as the principal mechanisms underlying these neurological consequences of obesity, and address potential treatment methods.
Figure 1. Neurological consequences of obesity: an overview.
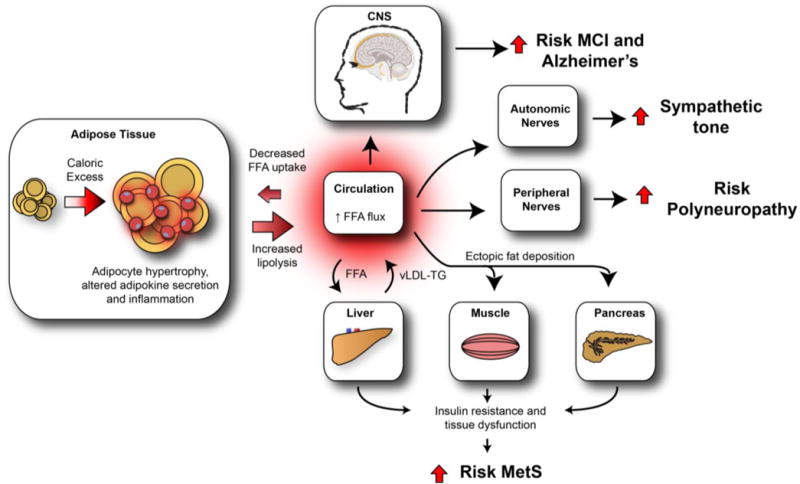
Increased caloric intake and decreased energy expenditure result in a net energy overload, leading to adipose tissue expansion (hyperplasia and hypertrophy). Sustained caloric excess in visceral adipose tissue activates resident adipose tissue macrophages, contributing to the development of adipose tissue dysfunction and metabolic inflammation. As a consequence, circulating FFAs rise, are lipotoxic to peripheral tissues, and contribute to the development of metabolic dyshomeostasis. High FFA flux into the liver increases vLDL-triglyceride production, further promoting dyslipidemia, while ectopic fat deposition in the muscle and pancreas promotes insulin resistance and pancreatic β-cell dysfunction, respectively. Collectively, these impairments lead to the MetS. Similarly, the CNS, ANS, and PNS are also detrimentally affected by obesity and obesity-induced metabolic dysfunction, which we hypothesize is driven by the lipotoxic effects of dyslipidemia and increased circulating FFA. In the CNS, dysfunction leads to MCI and Alzheimer’s disease while in the ANS and PNS, the end result is autonomic and peripheral neuropathy, respectively. FFA = free fatty acid. vLDL = very-low-density lipoprotein. TG = triglyceride. MCI = mild cognitive impairment. MetS = metabolic syndrome.
Physiological to Pathophysiological Obesity
Adipose Tissue Biology
Adipose tissue is localized into distinct anatomical regions and performs functions that include the storage and release of energy, thermal insulation, and protection of internal organs from trauma. In addition, it also possesses endocrine activity that is mediated by adipokines, a diverse set of signaling molecules which includes hormones, cytokines, acute phase reactants, and growth factors. These species are critical in regulating complex metabolic processes in metabolically active tissues involved in maintaining energy homeostasis, such as adipose tissue and the liver, pancreas, and brain. Adipose tissue is composed of adipocytes and a stromovascular compartment containing pre-adipocytes, fibroblasts, endothelial cells, resident immune cells, blood vessels, and nerve terminals. The adipocytes themselves are highly efficient fat-storing cells that stockpile energy in the form of triglyceride-rich lipid droplets. Regulation of adipocyte energy uptake and storage is primarily maintained by insulin that mediates FFA uptake and lipogenesis but inhibits lipolysis. During periods of negative energy balance, when non-adipose tissues require energy, stimulation of adipose tissue by sympathetic nerves promotes lipolysis of the stored triglycerides within adipocytes to FFA, which are released into the circulation to serve as fuel for these tissues.6 However, during prolonged periods of positive energy balance (i.e. caloric excess), adipocytes undergo hypertrophy (adipocyte enlargement), hyperplasia (adipose progenitor cells proliferate and differentiate into new adipocytes), or both, resulting in adipose tissue expansion in order to accommodate the storage of surplus calories.
Significant increases in adipose mass, particularly in the visceral depots, contribute to adipose tissue dysfunction and promote metabolic disease via a low-grade metabolic inflammation that underlies systemic metabolic dysfunction.5 Consequences of adipose tissue inflammation include altered adipokine secretion profile, impaired insulin signaling, compromised triglyceride storage, and increased basal lipolysis. These events culminate in increased circulating adipokines and FFA and are central to obesity-induced peripheral- and nervous-tissue dysfunction (Figure 1). Metabolic inflammation occurs primarily in hypertrophied adipose tissue as a consequence of sustained caloric excess that initiates stress-signaling pathways and consists of activated resident macrophages. Local adipose tissue inflammation is characterized by increased expression of pro-inflammatory adipokines, including C-reactive protein, interleukin (IL)-1, IL-6, IL-1β, tumor necrosis factor (TNF)-α, and leptin. Furthermore, chemotactic adipokines such as monocyte chemoattractant protein (MCP)-1 can further amplify local inflammation by promoting the infiltration and accumulation of circulating leukocytes to the adipose tissue. Specifically, activation of resident macrophages can trigger the recruitment of circulating monocytes/macrophages, T cells, natural killer T cells, B lymphocytes, and mast cells.7 Moreover, chronic adipose tissue inflammation can lead to ‘spillover’ of pro-inflammatory mediators into the circulation; hence, obese patients typically display an increase in circulating pro-inflammatory adipokines that correlates with adipose mass. Metabolic inflammation also promotes adipose tissue insulin resistance, a state of decreased responsiveness to insulin that is mediated by TNF-α signaling.8 As adipose uptake and storage of circulating FFA is insulin-dependent, compromised insulin signaling within adipose tissue impairs clearance of circulating FFA,9 thus contributing to increased circulating FFA (palmitic, oleic, linoleic acid) in obesity.
Obesity and Dyslipidemia
In addition to the increase in adipose-derived FFA, both liver- and diet-derived very low density lipoprotein (vLDL)-triglycerides significantly contribute to dyslipidemia in obesity.9 FFA-induced liver dysfunction leads to an increase in production of liver-derived vLDL-triglycerides, while caloric excess increases circulating chylomicron-derived vLDL-triglycerides. These vLDL-triglycerides are hydrolyzed to their constituent long chain fatty acids (LCFA) by lipoprotein lipases present in vascular endothelium, neurons, and glia, therefore adding to the FFA load on all organ systems, including the CNS and PNS. These bioactive lipids can also become deposited along blood vessels, compromising blood flow and tissue perfusion via atherogenesis, can accumulate ectopically in tissue to compromise structure and cellular signaling, and can dysregulate local metabolism to compromise energy production. Obesity-induced dyslipidemia can therefore result in dysfunction in numerous types of tissues, including the CNS and PNS.
Effects of Obesity on the Central Nervous System
Clinical Evidence
Accumulating evidence demonstrates that the CNS and cognitive function are adversely affected by obesity. For example, a meta-analysis has shown a strong association between obesity and neurological disorders such as dementia and Alzheimer’s disease (AD).10,11 Studies indicate that obesity doubles the risk of AD when compared to individuals of normal weight10 and that a higher BMI in midlife predicts greater risk of dementia in later life.12 Furthermore, a post-mortem study showed that elderly, morbidly obese patients display higher levels of hippocampal markers associated with AD (β-amyloid, β-amyloid precursor protein, tau) than non-obese individuals.13
It is well established that mild cognitive impairment (MCI) occurs well before frank dementia, and several prospective studies (Table 1;14–18) have demonstrated that obesity confers increased risk of MCI, independent of age. Though there is some controversy (one study reported that lower weight in later life predicts MCI19 and another demonstrated no increase of MCI risk with high BMI20) the majority of studies indicate that a high BMI is associated with attention deficits, poor executive function, impaired decision-making, and decreased verbal learning and memory. Individuals with severe obesity have MCI,21 and obese children and adolescents, as well as those with MetS,22 display lower cognitive function,23 further distinguishing obesity-associated MCI from age-related dementia. Reports also suggest that obese males are more susceptible than females to MCI,14 with males also displaying greater brain atrophy,24 which may be due to estrogen-dependent protection from MetS components.25
Table 1.
Prospective clinical studies demonstrating associations between obesity and cognitive deficits, independent of dementia
| Study/Age/Sample Size | Design/duration | Cognitive tests/diagnosis | Observation | Ref. |
|---|---|---|---|---|
| Framingham Heart Study; 55–88yrs; 551 males and 872 females divided into normal, overweight or obese groups; 18 year duration; USA | Prospective 22 yr | 8 subtests: logical memory-immediate recall, visual reproductions, paired associates learning, digit span forward/backward, word fluency (Controlled Oral Word Associations), similarities, and logical memory-delayed recall | Adverse cognitive effects seen in obese men, not women | 14 |
| 32–62yr baseline; 1660 male, 1576 female; France | Prospective 5 yr | Word-list learning and recall; DSST test, attention test; delayed free recall test | Cross-section: higher BMI = lower cognitive scores; Prospective: higher BMI predicted cognitive decline | 15 |
| White men/women; Whitehall II Study; aged 35–55 at baseline; n = 5131; UK | Prospective ~36 yr | Mini-Mental State Examination and tests of memory and executive function | Cumulative effect of obesity on cognition | 16 |
| Men/women; Swedish twin registry; aged 50–60 yr at baseline;; n=417; Sweden | Prospective 30 yr | Testing of long-term memory, short-term memory, speed, verbal and spatial ability | Midlife overweight is related to lower overall cognitive function in old age | 17 |
| Men/women; Swedish Adoption/Twin Study of Aging; aged 25–50 at baseline; n=657; Sweden | Prospective ~20 yr | Verbal, spatial/fluid, memory and perceptual speed were tested | Early midlife overweight/obesity = lower cognitive function and predicts cognitive decline in late life | 18 |
DSST – Digit-Symbol Substitution Test
In both AD and MCI, impaired neurological function in the CNS may be a consequence of structural changes in the brain and decreased cerebral integrity, particularly in the hippocampus, a structure essential for learning and memory that is susceptible to aging-related atrophy.26 AD patients display brain atrophy in the medial temporal lobe, which contains the hippocampus, while patients with MCI display a similar pattern of hippocampal loss, but to a lesser degree than AD patients.27 In patients with obesity or MetS, there is also evidence for a decrease in hippocampal volume.28 Using computed tomography, a cross-sectional analysis demonstrated that women with temporal lobe atrophy had higher BMI,26 while longitudinal analysis of the same cohort revealed that BMI was a predictor for temporal lobe atrophy.26 Magnetic resonance imaging (MRI) analysis has also shown an inverse relationship between BMI and brain volume29 and neuronal viability,30 as well as gliosis in the hypothalamus that is coincident with obesity.31 Likewise, voxel-based morphometry has similarly revealed a relationship between BMI and gray matter ratio, with gray matter volume of the bilateral medial temporal lobes, anterior lobe of the cerebellum, occipital lobe, frontal lobe, precuineus, and midbrain displaying negative correlations with BMI.24,32 More recently, use of tensor-based morphometry to examine gray matter and white matter volume in a bivariate analysis of elderly patients with normal cognitive function linked increased BMI, hyperinsulinemia, and type 2 diabetes to atrophy in frontal, temporal, and subcortical brain regions.33 In the same study, an Analysis of Covariance model showed atrophy in the frontal lobes, anterior cingulate gyrus, hippocampus, and thalamus of obese patients.33 Even individuals with a high BMI and no apparent cognitive defects displayed brain atrophy in the frontal lobe, hippocampus and thalamus when compared to non-obese individuals.33 Children and adolescents with high BMI also exhibit decreased volume of frontal and limbic cerebral gray matter regions34 and decreased hippocampal volume compared to children with healthy BMI.22 The hippocampus and prefrontal cortex, which are particularly vulnerable to obesity-related changes,35 are essential for learning and memory, and changes in hippocampal volume correlate with declines in memory performance.36 In fact, increased hippocampal atrophy in MCI patients predicts the development of AD.37
Mechanisms Underlying Obesity-Mediated Cognitive Impairment: Role of the Hypothalamus
Consistent with clinical data, numerous experimental studies using animal models of high fat diet (HFD)-induced obesity reveal altered hippocampal structure and function, with animals displaying learning and memory deficits and impaired executive function.38–44 These animal models provide insight into the mechanisms underlying obesity-induced cerebral changes. For example, long term potentiation, considered to be a major mechanism that contributes to learning and memory, in the hippocampus is impaired in HFD-fed mouse models.45,46 HFD feeding also leads to a reduction in markers of neurogenesis, synaptic plasticity, and neuronal growth, including brain-derived neurotrophic factor.47,48 Interestingly, many studies that involve the use of these animal models provide strong evidence that hypothalamic inflammation is an early step in a vicious feed-forward cycle of CNS dysfunction leading ultimately to cognitive decline.31
The hypothalamus is pivotal in maintaining global energy homeostasis by regulating food intake and energy expenditure.49 To rapidly monitor and regulate metabolic homeostasis, the hypothalamus senses circulating nutrients and hormones via the median eminence, which contains a unique capillary system, and, importantly, integrates these signals of satiety and nutritional status to maintain metabolic homeostasis via neuroendocrine and autonomic signaling.50 Notably, the median eminence is not protected by the blood-brain barrier, which likely contributes to its vulnerability to circulating factors such as FFA and inflammatory adipokines.
Early markers of inflammation, including TNF-α and microglial activation, are present in the arcuate nucleus (ARC) of the hypothalamus.51 Hypothalamic inflammation is an early acute response to elevated FFA: after one day of HFD feeding, inflammatory markers, including TNF-α, are present in the mediobasal hypothalamus,31 and HFD-induced inflammation occurs in the hypothalamus before the adipose tissue or peripheral blood.52 Glial cell activation is an early critical step in diet-induced hypothalamic inflammation, with microglial and astrocyte reactivity occurring after HFD feeding.31 In particular, microglia have been proposed as the sensors of diet-induced hypothalamic inflammation since they express toll-like receptor (TLR)-4, which recognizes LCFA.53 In animal models of obesity, activation of TLR4 signaling promotes cytokine production (TNF-α, IL-1β, and IL-6)54, activation of endoplasmic reticulum (ER) stress,55 and subsequent activation of the c-Jun N-terminal kinase (Jnk)51/inhibitor of κB kinase-β/nuclear factor-κB (IKKβ/NF-κB)53,55,56 pathways, resulting in the propagation of pro-inflammatory signaling and promotion of gliosis in the hypothalamus.31
Inflammation can drive further downstream dyslipidemia by compromising the neural circuitry governing satiety control (Figure 2a).54 In obese animals, leptin-induced satiety is blunted;57 although leptin positively correlates with adipose tissue mass, increased hypothalamic inflammatory signaling leads to leptin-resistance. This can impair appetite control mediated by hypothalamic proopiomelanocortin (POMC) neurons, which function to reduce appetite and increase energy expenditure. Moreover, HFD feeding leads to a reduction in POMC neurons, resulting in decreased inhibition of feeding.58 Dysregulation of satiety signaling promotes hyperphagia, increasing susceptibility to obesity development,57 while loss of POMC neurons contributes to weight gain.59
Figure 2. Neurological complications associated with obesity and dyslipidemia.
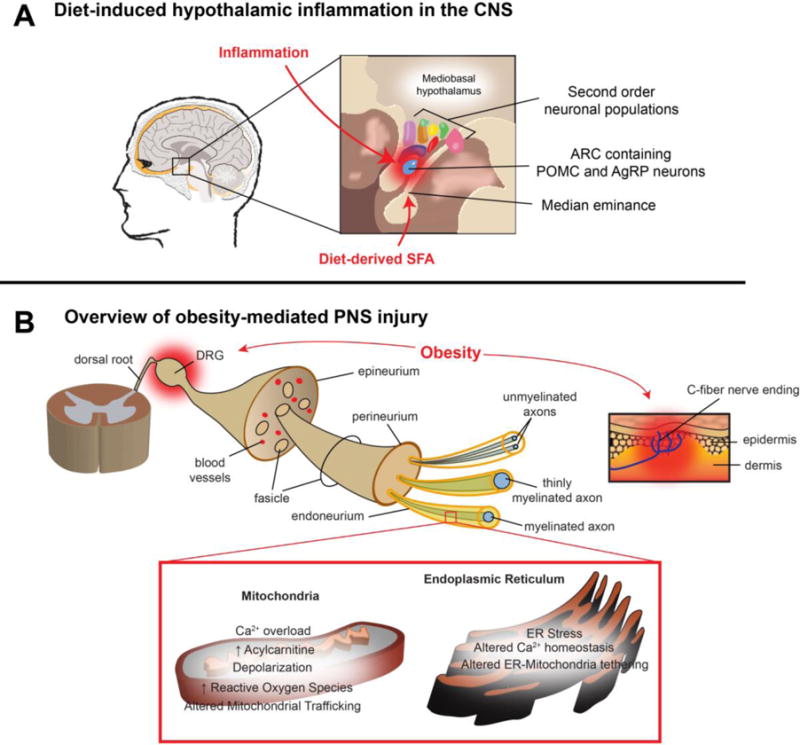
A. Diet-induced hypothalamic function in the CNS. Located in the CNS, the hypothalamus is responsible for controlling global energy balance by monitoring metabolic homeostasis. Diet-derived saturated fatty acids (SFA) can enter the CNS via the median eminence and accumulate in the mediobasal hypothalamus. In response to SFA, resident hypothalamic microglia that protect against pathogenic species become activated, resulting in inflammation, gliosis and neuronal stress. Pro-inflammatory signaling in the arcuate nucleus is associated with the development of impaired leptin signaling in pro-opiomelanocortin (POMC) and Agouti-related peptide (AgRP) neurons that in turn can affect second order neurons that govern energy balance. Hypothalamic inflammation alters satiety control, thus increasing the risk of developing obesity.
B. Overview of obesity-mediated PNS injury. As a consequence of obesity, increased levels of FFA lead to decreased neurotrophic support and increased neurodegeneration in peripheral nerves. Long-chain fatty acids and inflammatory mediators directly injure DRG neurons, C-fiber cutaneous nerve endings, and the blood-nerve-barrier. As the blood-nerve-barrier is increasingly compromised, axons and their associated Schwann cells become vulnerable to injury, leading to neurogenic inflammation, mitochondrial dysfunction and ER stress. In aggregate, these changes alter nerve function and structure, contributing to the development and progression of polyneuropathy. FFA = free fatty acids. SFA = saturated fatty acids. CNS = central nervous system. ARC = arcuate nucleus. POMC = pro-opiomelanocortin. AgRP = Agouti-related peptide. DRG = dorsal root ganglia. FFA = free fatty acid.
In addition to inflammation, altered FFA metabolism may contribute to changes in CNS function. Cerebral uptake of FFA is higher in obese subjects,60 and evidence from animal studies suggest that FFA are not completely catabolized in the hypothalamus but instead accumulate as long chain (LC)-acyl CoA esters and other bioactive lipids.56 Indeed, diacylglycerols and ceramides are present in the hypothalamus of HFD-fed mice61 and Zucker fa/fa rats,62 with direct palmitate-induced ceramide production demonstrated in hypothalamic neurons in vitro.63 These bioactive lipid species can promote ER stress and subsequent activation of IKKβ.56,61,62 Moreover, improving β-oxidation in hypothalamic neurons in vitro decreases neuronal inflammation by decreasing production of bioactive lipids63 strengthening the association between lipotoxicity and hypothalamic dysfunction.
Hippocampal Injury in Obesity
While hypothalamic injury occurs early in obesity31 as discussed above, unresolved inflammation, metabolic dysfunction, and a lack of satiety, in combination with alterations in FFAs, triglycerides, and insulin resistance, could all contribute in a multifactorial manner to establish a feed-forward cycle of injury that eventually affects other brain regions, most importantly the hippocampus. Hippocampal injury in response to increased blood-brain barrier permeability occurs in HFD-fed animals64 as a consequence of reactive glial cytokine production and circulating pro-inflammatory adipokines.65 FFA and triglycerides have also been demonstrated to directly alter hippocampal function and activity,48 and insulin resistance has been linked to neurocognitive dysfunction66 and been demonstrated to decrease hippocampal function.67 Unchecked entry of pro-inflammatory proteins into the hippocampus can initiate or amplify neuroinflammation and promote neurodegeneration.68,69 Indeed, loss of the blood-brain barrier and markers of hippocampal inflammation, including microglial activation, are present by 20 weeks of age in HFD-fed animals.70 Over time, continued increased blood-brain barrier permeability and unchecked entry of neurotoxic cytokines and chemokines, as well as immune cells,71 FFA, and triglycerides48 during obesity further potentiates HFD-induced hippocampal injury and subsequent atrophy. These findings provide a mechanistic link between the observed blood-brain barrier breakdown and impaired cognition in obesity.
Effects of Obesity on the Peripheral Nervous System
The PNS is divided into the autonomic nervous system (ANS), containing the sympathetic and parasympathetic divisions, and the somatic nervous system, composed of the sensory and motor peripheral nerves. Autonomic and sensory ganglia along with unmyelinated fibers and synaptic terminals of the PNS are not protected by the blood-brain (nerve) barrier and are therefore susceptible to the pathophysiological consequences of obesity.
The ANS plays a critical role in regulating energy balance through its control of anabolic and catabolic processes and its close relationship with the hypothalamus. The ANS is particularly sensitive to obesity-induced perturbations, and ANS dysfunction is often present in young overweight individuals.72 Obesity is associated with an imbalance between the sympathetic and parasympathetic divisions of the ANS. Increased sympathetic outflow to neuro-adipose junctions stimulates lipolysis via stimulatory G-protein coupled β-adrenoceptors. This triggers a downstream signaling pathway that ends with activation of adipose triglyceride lipase, resulting in triglyceride hydrolysis. Thus, sympathetic hyperactivation, as seen in obesity, can promote a feed-forward mechanism leading to an increased pool of circulating LCFA.73 Increased sympathetic outflow to the cardiovascular system also results in increases in heart rate, blood pressure, and microvasculature tone. It is speculated that a sustained sympathetic outflow decreases perfusion, and in turn these physiological changes also promote further neurological dysfunction in both the CNS and PNS.
Obesity and Polyneuropathy
Polyneuropathy is clinically defined as the distal-to-proximal loss of sensory perception, affecting initially the feet, but over time also the hands. Frequently referred to as a “stocking-glove” loss of sensation, polyneuropathy occurs when there is a corresponding distal-to-proximal loss of sensory axons, with motor axons only involved in very severe or end stage disease. The most common causes of polyneuropathy in Western societies are prediabetes and type 2 diabetes. However, a Cochrane systematic review of all clinical trials of patients with diabetes where polyneuropathy was measured, including several large multicenter trials, revealed that glucose lowering therapies have little effect on the prevention of polyneuropathy in patients with type 2 diabetes74. These unanticipated results suggest that other MetS components, which are present in over 75% of patients with type 2 diabetes, play a role in the pathogenesis of polyneuropathy.
In support of this idea, we recently reported that the likelihood of polyneuropathy increases as the number of MetS components increases in the 2,382 participants of the Health, Aging, and Body Composition (Health ABC) study, independent of glycemic control.75 Our study supports several reports that obesity alone is an independent risk factor for polyneuropathy,76 including multiple clinical studies linking obesity with polyneuropathy (Table 2).75–80 Indeed, lipid profiles are often abnormal early in type 2 diabetes and correlate with early polyneuropathy onset,81 and triglycerides are associated with polyneuropathy progression in diabetes.82 Moreover, elevated triglycerides are significantly higher in patients with idiopathic polyneuropathy,83 and prediabetes, which is highly prevalent in obese individuals, is also associated with polyneuropathy.77,84–86 Taken together these data suggest a role for obesity, prediabetes, and dyslipidemia in the pathophysiology of polyneuropathy.
Table 2.
Clinical studies suggesting an association between obesity and polyneuropathy
| Study/Population | Size | Aim | Method | Observations | Ref. |
|---|---|---|---|---|---|
| Utah Study; Utah, USA | 219 | Identify whether MetS features other than hyperglycemia increase polyneuropathy risk | Clinical diagnosis and nerve conduction studies/skin biopsy for measurement of epidermal nerve fiber density to confirm when necessary | Lipid abnormalities observed in patients with polyneuropathy | 80 |
| PROMISE Study; Toronto, Ontario, Canada | 467 | Determine the association of MetS components with polyneuropathy risk | Vibration perception thresholds to measure nerve dysfunction; MNSI to establish polyneuropathy | Increased waist circumference confers greater neuropathy risk | 79 |
| MONICA/KORA Study; Augsburg, Germany | 393 | Identify risk factors for polyneuropathy in patients with IGT | MNSI to establish polyneuropathy | Waist circumference associated with polyneuropathy | 77 |
| SH-DREAMS Study; Shanghai, China | 2,035 | Identify risk factors for polyneuropathy in patients with IGT | Foot examination/neuropathy symptom and neuropathy disability scores | Waist circumference associated with polyneuropathy in non-diabetic patients | 78 |
| Health ABC; Pittsburgh and Memphis, USA | 2,382 | Determine the association of MetS components with polyneuropathy | Questionnaire plus at least one of three confirmatory tests (heavy monofilament, peroneal conduction velocity and vibration threshold) | Waist circumference associated with polyneuropathy | 75 |
| EURODIAB; Europe | 1,172 | Identify risk factors for polyneuropathy in patients with type 1 diabetes | Clinical evaluation, quantitative sensory testing, and autonomic-function testing | Higher BMI, total cholesterol and vLDL cholesterol significantly associated with polyneuropathy | 76 |
PROMISE - Prospective Metabolism and Islet Cell Evaluation; MONICA/KORA - Monitoring Trends and Determinants on Cardiovascular Diseases/Cooperative Research in the Region of Augsburg; SH-DREAMS - ShangHai Diabetic neuRopathy Epidemiology and Molecular Genetics Study; EURODIAB - European Diabetes Prospective Complications Study; Health ABC - Health, Aging, and Body Composition Study; MNSI - Michigan neuropathy screening instrument; MetS – Metabolic syndrome; vLDL - very low density lipoprotein; T1D - type 1 diabetes; IGT - impaired glucose tolerance
Mechanisms Underlying Obesity-mediated Peripheral Nervous System Injury
Sensory nerves are comprised of neurons within the dorsal root ganglia (DRG), axons, and supporting Schwann cells, with each nerve covered by a connective tissue layer known as the epineurium. A second connective tissue layer known as the perineurium surrounds groups of nerves to form nerve fibers, and bundles of nerve fibers along with blood vessels are encapsulated by the final layer of connective tissue, the endoneurium, to form a pure sensory, pure motor, or mixed sensorimotor nerves. Like the blood-brain barrier, the PNS has a blood-nerve barrier consisting of the endoneurial microvessels and the perineurium. The blood-nerve barrier protects the peripheral nerve in the same fashion as the blood-brain barrier protects the CNS, except for two important exceptions: DRG and all sensory nerve endings in the skin lack the blood-nerve barrier. These two unprotected components of the PNS along with the endoneurial microvessels and the perineurium of the blood-nerve barrier are susceptible to initial obesity-mediated inflammatory injury with corresponding loss of function (Figure 2b).
To reveal the mechanisms underlying obesity-mediated PNS injury, we and others have employed HFD-fed mice that develop obesity, dyslipidemia, prediabetes, and polyneuropathy without developing frank type 2 diabetes.87 LCFA and inflammatory mediators, that are found in HFD-fed animals88, can injure and penetrate the blood-nerve barrier and activate neurogenic inflammation89. In response, DRG neurons release vasoactive and chemoattractant signals to increase vasculature permeability and attract innate and adaptive immune cells to sites of damage. While this response is initially protective, chronic dysfunction secondary to obesity-mediated inflammation can lead to changes in nerve structure and subsequent pain.89 Moreover, HFD-fed mice exhibit increased accumulation of macrophages in peripheral nerves, increased TNF-α and IL-1β mRNA, and polyneuropathy,88 all without developing type 2 diabetes.87
Studies using animal models of obesity and type 2 diabetes with microvascular complications also support a role for lipotoxicity in polyneuropathy. Gene expression analyses of peripheral nerves of ob/ob and db/db mice identified common dysregulation of inflammation and immune-related functions as early as 5 weeks.90 In db/db mice, pioglitazone treatment improved small fiber function and cutaneous innervation,91 and in Zucker fa/fa rats, acipimox treatment normalized sensory nerve conduction velocities and alleviated thermal and mechanical hypoalgesia, independently of prediabetes.92 These drugs are known to lower triglycerides and reduce FFAs, respectively; however, they also exert effects on other metabolic and endocrine measures which may contribute to their effectiveness for polyneuropathy. Together, these data suggest a role for obesity and lipid-induced inflammation in polyneuropathy.
As with the CNS, there is evidence that obesity- mediated increases in FFA and LCFA alter normal lipid metabolism in the PNS. Excess LCFA promote increased Schwann cell mitochondrial β-oxidation93,94 that is coincident with polyneuropathy.95,96 Products of LCFA metabolism, LC-acylcarnitines, accumulate in peripheral nerves of mice94 and these bioactive lipids are associated with Schwann cell dysfunction,93 axonal degeneration,94 and polyneuropathy. Accumulation of LCFA or LC-acylcarnitines in non-obesity-mediated diseases affecting mitochondrial β-oxidation also results in polyneuropathy, strengthening the association between aberrant lipid metabolism and PNS dysfunction.97
Injury to the PNS by LCFA is not limited to metabolism but also occurs when excess LCFAs in Schwann cells induce a cascade of ER dysfunction that includes a decrease in ER Ca2+ content followed by ER stress, mitochondrial depolarization, and generation of toxic reactive oxygen species.98 As anticipated, these LCFA-mediated markers of ER dysfunction are present in the PNS of fa/fa rats and HFD-fed mice with polyneuropathy, supporting their role in disease pathogenesis.99 In parallel, LCFA induced ER dysfunction also negatively impacts mitochondrial ATP production in nerves, a function required for normal nerve physiology. It is well established that tightly regulated ER Ca2+ release is critical for proper mitochondrial Ca2+ uptake and ATP production. This delicate balance is disrupted in the nerves of HFD-fed mice with polyneuropathy where disruption of ER-mitochondrial tethering results in mitochondrial Ca2+ overload and impaired ATP production100. In aggregate, these results suggest that disruption of interorganellar Ca2+ transport between the ER and mitochondria in the PNS contributes to obesity-mediated polyneuropathy and supports the idea that LCFA-induced ER stress in Schwann cells acts in a feed-forward manner to disrupt ATP mitochondrial energy production and to promote further nerve injury. Finally, mitochondrial trafficking from neuronal cell bodies along the length of the axons is required to deliver ATP for normal nerve function, and in man, this distance can be greater than one meter. Because mitochondrial trafficking is an ATP-dependent process,101 compromised ATP generation during obesity can disrupt mitochondrial trafficking in a feed-forward loop, resulting in even greater peripheral nerve damage.
Neurological Benefits of Targeting Obesity
As the neurological consequences of obesity and dyslipidemia are highly morbid, the optimal disease-modifying therapy is to target obesity itself. Lifestyle interventions, consisting of dietary modification combined with exercise, provide an accessible and inexpensive strategy that is effective in combating obesity and restoring neurological function (Table 3). The physiological benefits of lifestyle interventions, greatest when dietary and exercise regimens are used in concert, improve the systemic metabolic profile and strongly correlate with improved cognitive function and improved nerve function. Global physiological benefits include improved cellular metabolism, restoration of glucose and insulin homeostasis, decreased metabolic inflammation, and improved lipid profiles.
Table 3.
Benefits of lifestyle intervention and surgery on neurological complications
| Method | Age (years); Sample Size | Metabolic Selection Criteria | Study Duration | Metabolic Observations | Neurological Observations | Ref. |
|---|---|---|---|---|---|---|
| Cognitive impairment, dementia risk and cerebral volume/integrity | ||||||
| Caloric restriction | 60.5±7.6; n=50 | Normal to overweight subjects (mean BMI of 28±3.7) | 3 months | Decreased BMI (0.8), CRP (0.22pg/mL) and insulin (3.5 μIU/mL) | Improved memory performance (increase in verbal memory score) | 125 |
| Caloric restriction, dietary changes and exercise | 52.3±9.6; n=124 | BMI=25–40 (mean BMI of 32.8±3.8) | 4 months | Decreased BMI (3.3), improved cardiovascular fitness and reduced blood pressure | Improved executive function-memory-learning and psychomotor speed | 126 |
| Intentional weight loss | 56.9±9.7; n=21 | BMI=30–50 (mean BMI of 36.7) | ~4 months | Decreased BMI (3.5) and fat mass (7.5kg) | Improvements in hand-grip strength and cognitive function (Mini-Mental State Examination, Short Portable Mental Status Questionnaire, and Trail-Making Test) | 127 |
| Caloric restriction and exercise | 70±4; n=28 | BMI=37.2±5.4 | 12 months | Decreased body weight (8.6±3.8kg), visceral fat (787±896cm3) and CRP (1.8±3.4mg/dL) and increased insulin sensitivity | Improvement in cognitive function (Mini-Mental State Examination, Word Fluency Test and Trail-Making Test) | 128 |
| Caloric restriction | 50±0.8; n=106 | BMI=33.7±0.4 | 12 months | Decreased body weight (13.7±1.8kg), plasma glucose (5.4mg/dL) and insulin (3.6 μIU/mL) | Improved working memory (Digit span backward test) | 129 |
| Dietary intervention | 61.1±1.6; n=20 (females only) | BMI=27–40 | 6 months | Decreased BMI (3.6), waist circumference (0.04m),plasma insulin (1.3μIU/mL) and FFA(0.13mmol/l) | Improved memory performance, increased hippocampal brain activity using MRI | 130 |
| Caloric-restriction | 60+; n=80 | BMI≥30 | 12 months | Decreased BMI (1.7 ±1.8); Improved HOMA; CRP | Improved memory, cognition and executive function | 102 |
| Diet, exercise, cognitive training | FINGER trial 69.3±4.7; n=1,260 | Baseline BMI=28.3 (no exclusions) | 24 months | Decreased BMI 0.49 (±0.05) | Improved processing speed and executive function | 103 |
| Omega-3 supplementation, exercise, cognitive stimulation | 60–80; n=22 | n/a | 6 months | No significant metabolic improvements | Preserved and increased gray matter volume in intervention group compared to controls, no improvements in cognitive function | 104 |
| Bariatric Surgery | 21–41; n=137 | Patients with morbid obesity | 12 months | Lower BMI | Improved cognitive performance | 116 |
| Polyneuropathy | ||||||
| Diet; exercise | 60±8.4; n=32 | Baseline BMI = 32.1 ±4.4 (pre-diabetics selected) | 12 months | Decreased BMI and total cholesterol | Increased intra epidermal nerve innervation; decreased neuropathic pain | 105 |
| Exercise | 30–70; n=174 | Patients with type 2 diabetes | 12 months | Increased HDL | Increased intra epidermal nerve innervation | 106 |
| Exercise | n=36 | MetS | 6 months | Increased intra epidermal nerve innervation | Increased intra epidermal nerve innervation | 107 |
| Bariatric Surgery | n=17 | Patients with morbid obesity (BMI≥40) | 12 months | Improved BMI, fasting glucose, HbA1C, C-peptide, insulin and HOMA-IR | Neuropathy incidence lower (measures not reported) | 121 |
AD - Alzheimer’s disease; BMI - body mass index; CRP - C-reactive protein; HbA1C - hemoglobin A1C; HDL - high density lipoprotein; HOMA - Homeostatic Model Assessment; FFA - free fatty acids; FINGER: Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability; IR - insulin resistance; MetS - Metabolic Syndrome; MRI – magnetic resonance imaging
For example, weight loss via a 12-month regimen of decreased caloric intake in obese elderly patients with MCI resulted in improved verbal memory, verbal fluency, executive function, and global cognition.102 Moreover, a randomized control trial consisting of dietary intervention, coupled with increased exercise and cognitive training over a two year period, prevented cognitive decline.103 Combined effects of omega-3 fatty acid supplementation, aerobic exercise, and cognitive stimulation also positively benefited brain structure and function in patients with MCI.104 In terms of polyneuropathy, obese patients placed on a diet and exercise regimen exhibited increased cutaneous nerve fibers as a marker of polyneuropathy improvement along with decreased neuropathic pain.105 Indeed, increased exercise alone can increase cutaneous nerve density in type 2 diabetic patients without neuropathy106 and in patients with the MetS.107 The sympathetic and parasympathetic nervous system imbalance that is observed in obesity is also modifiable by dietary intervention.108 Finally, animal studies support the findings in man and also demonstrate that resolving obesity improves cognitive performance.109–111 Switching mice from a HFD to a regular diet to reduce caloric intake results in improved metabolic health, with increased insulin sensitivity and improvement in cognitive function. These improvements are associated with reduced blood brain barrier-leakage, decreased glial activation, and increased vascular perfusion.109–111
While diet and exercise interventions provide the ideal therapeutic regimen, compliance is an issue supporting a role for both pharmacotherapy and bariatric surgery in the treatment of obesity-mediated neurological disorders. An observational study of 342 AD patients reported that treatment with lipid lowering agents (statins and/or fibrates) is associated with a slower cognitive decline in AD.112 In parallel, the Fremantle Diabetes Study demonstrated that statin or fibrate treatment may protect against the development of polyneuropathy,113 and the FIELD study showed that fenofibrate treatment resulted in fewer non-traumatic distal amputations, a consequence attributed to improved polyneuropathy.114 Despite these studies, more definitive clinical trials are needed, and there are no clinical guidelines on the use of statins or fibrate in the treatment of CNS or PNS disorders.
Bariatric surgical procedures, such as Roux-en-Y Gastric Bypass are a highly effective and accepted treatment for patients who are morbidly obese.115 Although many patients regain weight and medical complications are frequent, post-operative patients display profound improvements in metabolic homeostasis and exhibit improved neurocognitive function116 (Table 3) due to improvement in adipokine,117 insulin,118 and inflammation signaling.119 One study demonstrated decreased inflammation and reduced β-amyloid precursor protein expression in peripheral blood of patients following bariatric surgery, leading the authors to speculate that a similar benefit could be occurring in the hippocampus.120 Roux-en-Y Gastric Bypass surgery also improves polyneuropathy one year post-procedure.121 These surgical intervention studies support the pathogenic role of obesity in both the CNS and PNS, but are not the treatment of choice due to the high prevalence of obesity worldwide. Performing invasive surgical procedures in a large population is also not practical, so feasible alternatives focused on lifestyle intervention are urgently needed.
Clinical Implications and Future Directions
While extensive data exists to support obesity as a mediator of CNS and PNS injury, we currently do not know the ideal intervention to prevent cognitive impairment, AD, autonomic neuropathy, and polyneuropathy. Future studies are needed to determine the comparative effectiveness of pharmacologic, medical weight loss, surgical weight loss, and exercise interventions using rigorous study designs and patient oriented outcomes. These studies will also have to evaluate the cost-effectiveness of these different interventions. A combination of approaches may be ideal, but also requires multiple life-altering changes in behavior, which makes implementation more difficult. Given the available data to date, clinicians should advise obese individuals, especially those that already have early evidence of CNS and PNS injury, to pursue at least one of the obesity interventions listed above.
Conclusion
In conclusion, increased visceral adiposity is a risk factor for the development of a wide range of neurological conditions, and obesity and the resulting metabolic disorders will be one of the greatest medical challenges of the 21st century. Though the precise mechanisms of obesity-induced nerve and cognitive dysfunction need to be fully elucidated, it is clear that obesity, insulin resistance, and dyslipidemia converge at the CNS and PNS as FFA-induced lipotoxicity, resulting in inflammation, neurological dysfunction, and neurodegeneration. And while inflammation may play a critical role in the development of neurological dysfunction, it is also likely that it impacts and can be influenced by the other proposed mechanistic pathways linked to obesity, such as oxidative stress, ER stress, sympathetic alterations, and metabolic dysfunction. Additional research is needed to identify the best treatment targets and most efficient and practical combination of approaches to ensure sustained positive health outcomes. While pharmacotherapy and bariatric surgery have shown promising results, more research is necessary, and in the case of bariatric surgery large-scale application is not feasible. Based on published reports, we maintain that lifestyle intervention consisting of dietary changes and increased activity should be adopted, as it is both inexpensive and highly accessible.
Panel 1: Obesity Screening
In clinical research, accurate and absolute methods for obesity quantification include dual energy x-ray absorptiometry (DEXA), magnetic resonance imaging (MRI), and computed tomography (CT). However, as it is not feasible to perform these routinely in the clinic and for large population studies, body mass index (BMI) and, to a lesser degree, waist-hip ratio (WHR) are used to assess the degree of obesity as they are inexpensive, easy to perform, and act as adequate surrogate markers. For Caucasian males, a BMI of 25–29 kg/m2 is defined as overweight, a BMI > 30kg/m2 is defined as obese, and a BMI > 40 kg/m2 is defined as morbidly obese (as adipose distribution varies depending on age, sex, and ethnicity, different criteria are used for children, women, and ethnic groups). Though routinely used to screen for obesity, BMI does not reveal differences in fat distribution, and therefore BMI can give an inaccurate assessment of disease risk. For example, a subgroup of individuals who are obese according to BMI criteria (BMI >30) still remain relatively metabolically healthy with low fasting insulin, glucose, and triglycerides and exhibit protection from diseases related to metabolic dysfunction.122 This protection can likely be attributed to increased adiposity in lower fat depots than in abdominal regions; these depots (e.g. gluteal, femoral, subcutaneous, and intramuscular) exhibit distinct physiological features from abdominal depots and are comprised of adipocytes which have greater capacity to deal with an increased metabolic load.123 Moreover, both obese and non-obese individuals124 with excess fat in the intra-abdominal region (visceral fat comprised of omental and mesenteric depots) are more susceptible to obesity-related diseases. For these reasons, clinical assessment of abdominal waist circumference, a surrogate marker of abdominal adipose fat mass, is a better marker for disease risk than BMI.
Acknowledgments
Funding support
Funding support is provided by the National Institutes of Health (R24 DK082841 and R01 DK107956 to ELF; K23 NS079417 to BCC), the Novo Nordisk Foundation (NNF14SA0006 to ELF and BCC), the Program for Neurology Research & Discovery, and the A. Alfred Taubman Medical Research Institute.
Dr. Callaghan reports grants from National Institutes of Health during the conduct of the study; other from Imepto Medical Inc, personal fees from Advance Medical, personal fees from Medical Legal consulting, and grants and personal fees from PCORI outside the submitted work; Dr. Feldman reports grants from National Institutes of Health during the conduct of the study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Review criteria
We searched Pubmed and Google Scholar for articles published in English up to July 2016 and examined original articles and review articles. The following terms were used in various combinations: “obesity”, “dyslipidemia”, “adipose”, “adipocyte”, “inflammation”, “brain”, “Alzheimer’s disease”, “cognitive impairment”, “hypothalamus”, “hippocampus”, “high fat diet”, “high fat diet”, “autonomic nervous system”, “peripheral nervous system”, “peripheral neuropathy”, “lifestyle intervention”, “exercise” and “bariatric surgery”. We used our judgment to select articles to provide a summary of the causes of neurological complications of obesity rather than aiming to provide an exhaustive list of all research.
Contributors
All authors contributed equally to the concept and design of the Review. ELF had full access to all the data and had final responsibility for the decision to submit for publication. POB and LMH completed the literature search. POB and LMH drafted and, with ELF, finalized the Review. BBC and ELF provided critical review of the manuscript. POB created the figures. All authors have approved the final version.
Conflicts of Interest
Dr. O’Brien and Dr. Hinder have nothing to disclose.
References
- 1.Ng M, Fleming T, Robinson M, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384(9945):766–81. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grarup N, Sandholt CH, Hansen T, Pedersen O. Genetic susceptibility to type 2 diabetes and obesity: from genome-wide association studies to rare variants and beyond. Diabetologia. 2014;57(8):1528–41. doi: 10.1007/s00125-014-3270-4. [DOI] [PubMed] [Google Scholar]
- 3.Swinburn BA, Sacks G, Hall KD, et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet. 2011;378(9793):804–14. doi: 10.1016/S0140-6736(11)60813-1. [DOI] [PubMed] [Google Scholar]
- 4.Collaboration NCDRF. Di Cesare M, Bentham J, et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet. 2016;387(10026):1377–96. doi: 10.1016/S0140-6736(16)30054-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kloting N, Bluher M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord. 2014;15(4):277–87. doi: 10.1007/s11154-014-9301-0. [DOI] [PubMed] [Google Scholar]
- 6.Bartness TJ, Shrestha YB, Vaughan CH, Schwartz GJ, Song CK. Sensory and sympathetic nervous system control of white adipose tissue lipolysis. Mol Cell Endocrinol. 2010;318(1–2):34–43. doi: 10.1016/j.mce.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson EK, Gutierrez DA, Hasty AH. Adipose tissue recruitment of leukocytes. Curr Opin Lipidol. 2010;21(3):172–7. doi: 10.1097/MOL.0b013e3283393867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 9.Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60(10):2441–9. doi: 10.2337/db11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anstey KJ, Cherbuin N, Budge M, Young J. Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes Rev. 2011;12(5):e426–37. doi: 10.1111/j.1467-789X.2010.00825.x. [DOI] [PubMed] [Google Scholar]
- 11.Pedditizi E, Peters R, Beckett N. The risk of overweight/obesity in mid-life and late life for the development of dementia: a systematic review and meta-analysis of longitudinal studies. Age Ageing. 2016;45(1):14–21. doi: 10.1093/ageing/afv151. [DOI] [PubMed] [Google Scholar]
- 12.Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71(14):1057–64. doi: 10.1212/01.wnl.0000306313.89165.ef. [DOI] [PubMed] [Google Scholar]
- 13.Mrak RE. Alzheimer-type neuropathological changes in morbidly obese elderly individuals. Clin Neuropathol. 2009;28(1):40–5. doi: 10.5414/npp28040. [DOI] [PubMed] [Google Scholar]
- 14.Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Obesity, diabetes and cognitive deficit: The Framingham Heart Study. Neurobiol Aging. 2005;26(Suppl 1):11–6. doi: 10.1016/j.neurobiolaging.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 15.Cournot M, Marquie JC, Ansiau D, et al. Relation between body mass index and cognitive function in healthy middle-aged men and women. Neurology. 2006;67(7):1208–14. doi: 10.1212/01.wnl.0000238082.13860.50. [DOI] [PubMed] [Google Scholar]
- 16.Sabia S, Kivimaki M, Shipley MJ, Marmot MG, Singh-Manoux A. Body mass index over the adult life course and cognition in late midlife: the Whitehall II Cohort Study. Am J Clin Nutr. 2009;89(2):601–7. doi: 10.3945/ajcn.2008.26482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hassing LB, Dahl AK, Pedersen NL, Johansson B. Overweight in midlife is related to lower cognitive function 30 years later: a prospective study with longitudinal assessments. Dement Geriatr Cogn Disord. 2010;29(6):543–52. doi: 10.1159/000314874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahl AK, Hassing LB, Fransson EI, Gatz M, Reynolds CA, Pedersen NL. Body mass index across midlife and cognitive change in late life. Int J Obes (Lond) 2013;37(2):296–302. doi: 10.1038/ijo.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alhurani RE, Vassilaki M, Aakre JA, et al. Decline in Weight and Incident Mild Cognitive Impairment: Mayo Clinic Study of Aging. JAMA Neurol. 2016;73(4):439–46. doi: 10.1001/jamaneurol.2015.4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qizilbash N, Gregson J, Johnson ME, et al. BMI and risk of dementia in two million people over two decades: a retrospective cohort study. Lancet Diabetes Endocrinol. 2015;3(6):431–6. doi: 10.1016/S2213-8587(15)00033-9. [DOI] [PubMed] [Google Scholar]
- 21.Rochette AD, Spitznagel MB, Sweet LH, et al. Gender Differences in Cognitive Test Performance in Adults With Heart Failure. J Cardiovasc Nurs. 2016 doi: 10.1097/JCN.0000000000000330. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yau PL, Castro MG, Tagani A, Tsui WH, Convit A. Obesity and metabolic syndrome and functional and structural brain impairments in adolescence. Pediatrics. 2012;130(4):e856–64. doi: 10.1542/peds.2012-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang J, Matheson BE, Kaye WH, Boutelle KN. Neurocognitive correlates of obesity and obesity-related behaviors in children and adolescents. Int J Obes (Lond) 2014;38(4):494–506. doi: 10.1038/ijo.2013.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taki Y, Kinomura S, Sato K, et al. Relationship between body mass index and gray matter volume in 1,428 healthy individuals. Obesity (Silver Spring) 2008;16(1):119–24. doi: 10.1038/oby.2007.4. [DOI] [PubMed] [Google Scholar]
- 25.Mauvais-Jarvis F. Sex differences in metabolic homeostasis, diabetes, and obesity. Biol Sex Differ. 2015;6:14. doi: 10.1186/s13293-015-0033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gustafson D, Lissner L, Bengtsson C, Bjorkelund C, Skoog I. A 24-year follow-up of body mass index and cerebral atrophy. Neurology. 2004;63(10):1876–81. doi: 10.1212/01.wnl.0000141850.47773.5f. [DOI] [PubMed] [Google Scholar]
- 27.De Santi S, de Leon MJ, Rusinek H, et al. Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiol Aging. 2001;22(4):529–39. doi: 10.1016/s0197-4580(01)00230-5. [DOI] [PubMed] [Google Scholar]
- 28.Jagust W, Harvey D, Mungas D, Haan M. Central obesity and the aging brain. Arch Neurol. 2005;62(10):1545–8. doi: 10.1001/archneur.62.10.1545. [DOI] [PubMed] [Google Scholar]
- 29.Ward MA, Carlsson CM, Trivedi MA, Sager MA, Johnson SC. The effect of body mass index on global brain volume in middle-aged adults: a cross sectional study. BMC Neurol. 2005;5:23. doi: 10.1186/1471-2377-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gazdzinski S, Kornak J, Weiner MW, Meyerhoff DJ. Body mass index and magnetic resonance markers of brain integrity in adults. Ann Neurol. 2008;63(5):652–7. doi: 10.1002/ana.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thaler JP, Yi CX, Schur EA, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122(1):153–62. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pannacciulli N, Del Parigi A, Chen K, Le DS, Reiman EM, Tataranni PA. Brain abnormalities in human obesity: a voxel-based morphometric study. Neuroimage. 2006;31(4):1419–25. doi: 10.1016/j.neuroimage.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 33.Raji CA, Ho AJ, Parikshak NN, et al. Brain structure and obesity. Hum Brain Mapp. 2010;31(3):353–64. doi: 10.1002/hbm.20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alosco ML, Stanek KM, Galioto R, et al. Body mass index and brain structure in healthy children and adolescents. Int J Neurosci. 2014;124(1):49–55. doi: 10.3109/00207454.2013.817408. [DOI] [PubMed] [Google Scholar]
- 35.Gomez-Pinilla F. Brain foods: the effects of nutrients on brain function. Nature reviews Neuroscience. 2008;9(7):568–78. doi: 10.1038/nrn2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kramer JH, Mungas D, Reed BR, et al. Longitudinal MRI and cognitive change in healthy elderly. Neuropsychology. 2007;21(4):412–8. doi: 10.1037/0894-4105.21.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henneman WJ, Sluimer JD, Barnes J, et al. Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology. 2009;72(11):999–1007. doi: 10.1212/01.wnl.0000344568.09360.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molteni R, Barnard RJ, Ying Z, Roberts CK, Gomez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112(4):803–14. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- 39.Jurdak N, Lichtenstein AH, Kanarek RB. Diet-induced obesity and spatial cognition in young male rats. Nutr Neurosci. 2008;11(2):48–54. doi: 10.1179/147683008X301333. [DOI] [PubMed] [Google Scholar]
- 40.Ledreux A, Wang X, Schultzberg M, Granholm AC, Freeman LR. Detrimental effects of a high fat/high cholesterol diet on memory and hippocampal markers in aged rats. Behav Brain Res. 2016;312:294–304. doi: 10.1016/j.bbr.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winocur G, Greenwood CE. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol Aging. 2005;26(Suppl 1):46–9. doi: 10.1016/j.neurobiolaging.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Kanoski SE, Davidson TL. Different patterns of memory impairments accompany short- and longer-term maintenance on a high-energy diet. J Exp Psychol Anim Behav Process. 2010;36(2):313–9. doi: 10.1037/a0017228. [DOI] [PubMed] [Google Scholar]
- 43.McNeilly AD, Williamson R, Sutherland C, Balfour DJ, Stewart CA. High fat feeding promotes simultaneous decline in insulin sensitivity and cognitive performance in a delayed matching and non-matching to position task. Behav Brain Res. 2011;217(1):134–41. doi: 10.1016/j.bbr.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 44.Kosari S, Badoer E, Nguyen JC, Killcross AS, Jenkins TA. Effect of western and high fat diets on memory and cholinergic measures in the rat. Behav Brain Res. 2012;235(1):98–103. doi: 10.1016/j.bbr.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 45.Stranahan AM, Norman ED, Lee K, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18(11):1085–8. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karimi SA, Salehi I, Komaki A, Sarihi A, Zarei M, Shahidi S. Effect of high-fat diet and antioxidants on hippocampal long-term potentiation in rats: an in vivo study. Brain Res. 2013;1539:1–6. doi: 10.1016/j.brainres.2013.09.029. [DOI] [PubMed] [Google Scholar]
- 47.Lindqvist A, Mohapel P, Bouter B, et al. High-fat diet impairs hippocampal neurogenesis in male rats. Eur J Neurol. 2006;13(12):1385–8. doi: 10.1111/j.1468-1331.2006.01500.x. [DOI] [PubMed] [Google Scholar]
- 48.Farr SA, Yamada KA, Butterfield DA, et al. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149(5):2628–36. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richard D. Cognitive and autonomic determinants of energy homeostasis in obesity. Nat Rev Endocrinol. 2015;11(8):489–501. doi: 10.1038/nrendo.2015.103. [DOI] [PubMed] [Google Scholar]
- 50.Fekete C, Lechan RM. Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocr Rev. 2014;35(2):159–94. doi: 10.1210/er.2013-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Souza CT, Araujo EP, Bordin S, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146(10):4192–9. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- 52.Maric T, Woodside B, Luheshi GN. The effects of dietary saturated fat on basal hypothalamic neuroinflammation in rats. Brain Behav Immun. 2014;36:35–45. doi: 10.1016/j.bbi.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 53.Milanski M, Degasperi G, Coope A, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci. 2009;29(2):359–70. doi: 10.1523/JNEUROSCI.2760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thaler JP, Schwartz MW. Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010;151(9):4109–15. doi: 10.1210/en.2010-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Posey KA, Clegg DJ, Printz RL, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296(5):E1003–12. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21(11):643–51. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Tang Y, Cai D. IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol. 2012;14(10):999–1012. doi: 10.1038/ncb2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gropp E, Shanabrough M, Borok E, et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8(10):1289–91. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 60.Karmi A, Iozzo P, Viljanen A, et al. Increased brain fatty acid uptake in metabolic syndrome. Diabetes. 2010;59(9):2171–7. doi: 10.2337/db09-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Borg ML, Omran SF, Weir J, Meikle PJ, Watt MJ. Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J Physiol. 2012;590(17):4377–89. doi: 10.1113/jphysiol.2012.233288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Contreras C, Gonzalez-Garcia I, Martinez-Sanchez N, et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 2014;9(1):366–77. doi: 10.1016/j.celrep.2014.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McFadden JW, Aja S, Li Q, et al. Increasing fatty acid oxidation remodels the hypothalamic neurometabolome to mitigate stress and inflammation. PLoS One. 2014;9(12):e115642. doi: 10.1371/journal.pone.0115642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hargrave SL, Davidson TL, Zheng W, Kinzig KP. Western diets induce blood-brain barrier leakage and alter spatial strategies in rats. Behav Neurosci. 2016;130(1):123–35. doi: 10.1037/bne0000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cellular and molecular neurobiology. 2000;20(2):131–47. doi: 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stoeckel LE, Arvanitakis Z, Gandy S, et al. Complex mechanisms linking neurocognitive dysfunction to insulin resistance and other metabolic dysfunction [version 2] F1000Res. 2016;5:353. doi: 10.12688/f1000research.8300.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grillo CA, Piroli GG, Lawrence RC, et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes. 2015;64(11):3927–36. doi: 10.2337/db15-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sims-Robinson C, Bakeman A, Glasser R, Boggs J, Pacut C, Feldman EL. The role of endoplasmic reticulum stress in hippocampal insulin resistance. Exp Neurol. 2016;277:261–7. doi: 10.1016/j.expneurol.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cai M, Wang H, Li JJ, et al. The signaling mechanisms of hippocampal endoplasmic reticulum stress affecting neuronal plasticity-related protein levels in high fat diet-induced obese rats and the regulation of aerobic exercise. Brain, behavior, and immunity. 2016;57:347–59. doi: 10.1016/j.bbi.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 70.Jeon BT, Jeong EA, Shin HJ, et al. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes. 2012;61(6):1444–54. doi: 10.2337/db11-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buckman LB, Hasty AH, Flaherty DK, et al. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain, behavior, and immunity. 2014;35:33–42. doi: 10.1016/j.bbi.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lambert E, Sari CI, Dawood T, et al. Sympathetic nervous system activity is associated with obesity-induced subclinical organ damage in young adults. Hypertension. 2010;56(3):351–8. doi: 10.1161/HYPERTENSIONAHA.110.155663. [DOI] [PubMed] [Google Scholar]
- 73.Zeng W, Pirzgalska RM, Pereira MM, et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell. 2015;163(1):84–94. doi: 10.1016/j.cell.2015.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Callaghan BC, Little AA, Feldman EL, Hughes RA. Enhanced glucose control for preventing and treating diabetic neuropathy. The Cochrane database of systematic reviews. 2012;6:CD007543. doi: 10.1002/14651858.CD007543.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Callaghan BC, Xia R, Banerjee M, et al. Metabolic Syndrome Components Are Associated With Symptomatic Polyneuropathy Independent of Glycemic Status. Diabetes Care. 2016;73(12):1468–76. doi: 10.2337/dc16-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tesfaye S, Chaturvedi N, Eaton SE, et al. Vascular risk factors and diabetic neuropathy. The New England journal of medicine. 2005;352(4):341–50. doi: 10.1056/NEJMoa032782. [DOI] [PubMed] [Google Scholar]
- 77.Ziegler D, Rathmann W, Dickhaus T, Meisinger C, Mielck A, Group KS. Prevalence of polyneuropathy in pre-diabetes and diabetes is associated with abdominal obesity and macroangiopathy: the MONICA/KORA Augsburg Surveys S2 and S3. Diabetes Care. 2008;31(3):464–9. doi: 10.2337/dc07-1796. [DOI] [PubMed] [Google Scholar]
- 78.Lu B, Hu J, Wen J, et al. Determination of peripheral neuropathy prevalence and associated factors in Chinese subjects with diabetes and pre-diabetes - ShangHai Diabetic neuRopathy Epidemiology and Molecular Genetics Study (SH-DREAMS) PloS one. 2013;8(4):e61053. doi: 10.1371/journal.pone.0061053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee CC, Perkins BA, Kayaniyil S, et al. Peripheral Neuropathy and Nerve Dysfunction in Individuals at High Risk for Type 2 Diabetes: The PROMISE Cohort. Diabetes Care. 2015;38(5):793–800. doi: 10.2337/dc14-2585. [DOI] [PubMed] [Google Scholar]
- 80.Smith AG, Rose K, Singleton JR. Idiopathic neuropathy patients are at high risk for metabolic syndrome. Journal of the neurological sciences. 2008;273(1–2):25–8. doi: 10.1016/j.jns.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clemens A, Siegel E, Gallwitz B. Global risk management in type 2 diabetes: blood glucose, blood pressure, and lipids–update on the background of the current guidelines. ExpClinEndocrinolDiabetes. 2004;112:493–503. doi: 10.1055/s-2004-821306. [DOI] [PubMed] [Google Scholar]
- 82.Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes. 2009;58(7):1634–40. doi: 10.2337/db08-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hughes RA, Umapathi T, Gray IA, et al. A controlled investigation of the cause of chronic idiopathic axonal polyneuropathy. Brain: a journal of neurology. 2004;127(Pt 8):1723–30. doi: 10.1093/brain/awh192. [DOI] [PubMed] [Google Scholar]
- 84.Callaghan BC, Xia R, Reynolds E, et al. Association Between Metabolic Syndrome Components and Polyneuropathy in an Obese Population. JAMA Neurol. 2016;73(12):1468–76. doi: 10.1001/jamaneurol.2016.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Papanas N, Vinik AI, Ziegler D. Neuropathy in prediabetes: does the clock start ticking early? Nat Rev Endocrinol. 2011;7(11):682–90. doi: 10.1038/nrendo.2011.113. [DOI] [PubMed] [Google Scholar]
- 86.Papanas N, Ziegler D. Polyneuropathy in impaired glucose tolerance: is postprandial hyperglycemia the main culprit? A mini-review. Gerontology. 2013;59(3):193–8. doi: 10.1159/000343988. [DOI] [PubMed] [Google Scholar]
- 87.O’Brien PD, Sakowski SA, Feldman EL. Mouse models of diabetic neuropathy. ILAR journal/National Research Council, Institute of Laboratory Animal Resources. 2014;54(3):259–72. doi: 10.1093/ilar/ilt052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jayaraman A, Lent-Schochet D, Pike CJ. Diet-induced obesity and low testosterone increase neuroinflammation and impair neural function. Journal of neuroinflammation. 2014;11:162. doi: 10.1186/s12974-014-0162-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chiu IM, von Hehn CA, Woolf CJ. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci. 2012;15(8):1063–7. doi: 10.1038/nn.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.O’Brien PD, Hur J, Hayes JM, Backus C, Sakowski SA, Feldman EL. BTBR ob/ob mice as a novel diabetic neuropathy model: Neurological characterization and gene expression analyses. Neurobiology of disease. 2014;73C:348–55. doi: 10.1016/j.nbd.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hur J, Dauch JR, Hinder LM, et al. The Metabolic Syndrome and Microvascular Complications in a Murine Model of Type 2 Diabetes. Diabetes. 2015;64(9):3294–304. doi: 10.2337/db15-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lupachyk S, Watcho P, Hasanova N, Julius U, Obrosova IG. Triglyceride, nonesterified fatty acids, and prediabetic neuropathy: role for oxidative-nitrosative stress. Free Radic Biol Med. 2012;52(8):1255–63. doi: 10.1016/j.freeradbiomed.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hinder LM, Figueroa-Romero C, Pacut C, et al. Long-chain acyl coenzyme A synthetase 1 overexpression in primary cultured Schwann cells prevents long chain fatty acid-induced oxidative stress and mitochondrial dysfunction. Antioxidants & redox signaling. 2014;21(4):588–600. doi: 10.1089/ars.2013.5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Viader A, Sasaki Y, Kim S, et al. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron. 2013;77(5):886–98. doi: 10.1016/j.neuron.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pande M, Hur J, Hong Y, et al. Transcriptional profiling of diabetic neuropathy in the BKS db/db mouse: a model of type 2 diabetes. Diabetes. 2011;60(7):1981–9. doi: 10.2337/db10-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Viader A, Golden JP, Baloh RH, Schmidt RE, Hunter DA, Milbrandt J. Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. J Neurosci. 2011;31(28):10128–40. doi: 10.1523/JNEUROSCI.0884-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rector RS, Payne RM, Ibdah JA. Mitochondrial trifunctional protein defects: clinical implications and therapeutic approaches. Adv Drug Deliv Rev. 2008;60(13–14):1488–96. doi: 10.1016/j.addr.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Padilla A, Descorbeth M, Almeyda AL, Payne K, De Leon M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Res. 2011;1370:64–79. doi: 10.1016/j.brainres.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lupachyk S, Watcho P, Obrosov AA, Stavniichuk R, Obrosova IG. Endoplasmic reticulum stress contributes to prediabetic peripheral neuropathy. Exp Neurol. 2012;247:342–8. doi: 10.1016/j.expneurol.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 100.Arruda AP, Pers BM, Parlakgul G, Guney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20(12):1427–35. doi: 10.1038/nm.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sajic M. Mitochondrial dynamics in peripheral neuropathies. Antioxidants & redox signaling. 2014;21(4):601–20. doi: 10.1089/ars.2013.5822. [DOI] [PubMed] [Google Scholar]
- 102.Horie NC, Serrao VT, Simon SS, et al. Cognitive Effects of Intentional Weight Loss in Elderly Obese Individuals With Mild Cognitive Impairment. The Journal of clinical endocrinology and metabolism. 2016;101(3):1104–12. doi: 10.1210/jc.2015-2315. [DOI] [PubMed] [Google Scholar]
- 103.Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255–63. doi: 10.1016/S0140-6736(15)60461-5. [DOI] [PubMed] [Google Scholar]
- 104.Kobe T, Witte AV, Schnelle A, et al. Combined omega-3 fatty acids, aerobic exercise and cognitive stimulation prevents decline in gray matter volume of the frontal, parietal and cingulate cortex in patients with mild cognitive impairment. NeuroImage. 2015;131:226–38. doi: 10.1016/j.neuroimage.2015.09.050. [DOI] [PubMed] [Google Scholar]
- 105.Smith AG, Russell J, Feldman EL, et al. Lifestyle intervention for pre-diabetic neuropathy. Diabetes Care. 2006;29(6):1294–9. doi: 10.2337/dc06-0224. [DOI] [PubMed] [Google Scholar]
- 106.Singleton JR, Marcus RL, Jackson JE, M KL, Graham TE, Smith AG. Exercise increases cutaneous nerve density in diabetic patients without neuropathy. Annals of clinical and translational neurology. 2014;1(10):844–9. doi: 10.1002/acn3.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Singleton JR, Marcus RL, Lessard MK, Jackson JE, Smith AG. Supervised exercise improves cutaneous reinnervation capacity in metabolic syndrome patients. Annals of neurology. 2015;77(1):146–53. doi: 10.1002/ana.24310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.de Jonge L, Moreira EA, Martin CK, Ravussin E, Pennington CT. Impact of 6-month caloric restriction on autonomic nervous system activity in healthy, overweight, individuals. Obesity. 2010;18(2):414–6. doi: 10.1038/oby.2009.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Johnson LA, Zuloaga KL, Kugelman TL, et al. Amelioration of Metabolic Syndrome-Associated Cognitive Impairments in Mice via a Reduction in Dietary Fat Content or Infusion of Non-Diabetic Plasma. EBioMedicine. 2016;3:26–42. doi: 10.1016/j.ebiom.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim H, Kang H, Heo RW, et al. Caloric restriction improves diabetes-induced cognitive deficits by attenuating neurogranin-associated calcium signaling in high-fat diet-fed mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2016;36(6):1098–110. doi: 10.1177/0271678X15606724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sims-Robinson C, Bakeman A, Bruno E, et al. Dietary Reversal Ameliorates Short- and Long-Term Memory Deficits Induced by High-fat Diet Early in Life. PloS one. 2016;11(9):e0163883. doi: 10.1371/journal.pone.0163883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Masse I, Bordet R, Deplanque D, et al. Lipid lowering agents are associated with a slower cognitive decline in Alzheimer’s disease. Journal of neurology, neurosurgery, and psychiatry. 2005;76(12):1624–9. doi: 10.1136/jnnp.2005.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Davis TM, Yeap BB, Davis WA, Bruce DG. Lipid-lowering therapy and peripheral sensory neuropathy in type 2 diabetes: the Fremantle Diabetes Study. Diabetologia. 2008;51(4):562–6. doi: 10.1007/s00125-007-0919-2. [DOI] [PubMed] [Google Scholar]
- 114.Ansquer JC, Foucher C, Aubonnet P, Le Malicot K. Fibrates and microvascular complications in diabetes–insight from the FIELD study. Current pharmaceutical design. 2009;15(5):537–52. doi: 10.2174/138161209787315701. [DOI] [PubMed] [Google Scholar]
- 115.Sjostrom L, Peltonen M, Jacobson P, et al. Bariatric surgery and long-term cardiovascular events. Jama. 2012;307(1):56–65. doi: 10.1001/jama.2011.1914. [DOI] [PubMed] [Google Scholar]
- 116.Miller LA, Crosby RD, Galioto R, et al. Bariatric surgery patients exhibit improved memory function 12 months postoperatively. Obesity surgery. 2013;23(10):1527–35. doi: 10.1007/s11695-013-0970-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alosco ML, Spitznagel MB, Strain G, et al. Improved serum leptin and ghrelin following bariatric surgery predict better postoperative cognitive function. Journal of clinical neurology. 2015;11(1):48–56. doi: 10.3988/jcn.2015.11.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Plum L, Ahmed L, Febres G, et al. Comparison of glucostatic parameters after hypocaloric diet or bariatric surgery and equivalent weight loss. Obesity. 2011;19(11):2149–57. doi: 10.1038/oby.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vazquez LA, Pazos F, Berrazueta JR, et al. Effects of changes in body weight and insulin resistance on inflammation and endothelial function in morbid obesity after bariatric surgery. The Journal of clinical endocrinology and metabolism. 2005;90(1):316–22. doi: 10.1210/jc.2003-032059. [DOI] [PubMed] [Google Scholar]
- 120.Ghanim H, Monte SV, Sia CL, et al. Reduction in inflammation and the expression of amyloid precursor protein and other proteins related to Alzheimer’s disease following gastric bypass surgery. The Journal of clinical endocrinology and metabolism. 2012;97(7):E1197–201. doi: 10.1210/jc.2011-3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zeve JL, Tomaz CA, Nassif PA, Lima JH, Sansana LR, Zeve CH. Obese patients with diabetes mellitus type 2 undergoing gastric bypass in Roux-en-Y: analysis of results and its influence in complications. Arquivos brasileiros de cirurgia digestiva: ABCD = Brazilian archives of digestive surgery. 2013;26(Suppl 1):47–52. doi: 10.1590/s0102-67202013000600011. [DOI] [PubMed] [Google Scholar]
- 122.van Vliet-Ostaptchouk JV, Nuotio ML, Slagter SN, et al. The prevalence of metabolic syndrome and metabolically healthy obesity in Europe: a collaborative analysis of ten large cohort studies. BMC Endocr Disord. 2014;14:9. doi: 10.1186/1472-6823-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tchoukalova YD, Votruba SB, Tchkonia T, Giorgadze N, Kirkland JL, Jensen MD. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(42):18226–31. doi: 10.1073/pnas.1005259107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Acosta JR, Douagi I, Andersson DP, et al. Increased fat cell size: a major phenotype of subcutaneous white adipose tissue in non-obese individuals with type 2 diabetes. Diabetologia. 2016;59(3):560–70. doi: 10.1007/s00125-015-3810-6. [DOI] [PubMed] [Google Scholar]
- 125.Witte AV, Fobker M, Gellner R, Knecht S, Floel A. Caloric restriction improves memory in elderly humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(4):1255–60. doi: 10.1073/pnas.0808587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331–8. doi: 10.1161/HYPERTENSIONAHA.109.146795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Siervo M, Nasti G, Stephan BC, et al. Effects of intentional weight loss on physical and cognitive function in middle-aged and older obese participants: a pilot study. J Am Coll Nutr. 2012;31(2):79–86. doi: 10.1080/07315724.2012.10720012. [DOI] [PubMed] [Google Scholar]
- 128.Napoli N, Shah K, Waters DL, Sinacore DR, Qualls C, Villareal DT. Effect of weight loss, exercise, or both on cognition and quality of life in obese older adults. The American journal of clinical nutrition. 2014;100(1):189–98. doi: 10.3945/ajcn.113.082883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brinkworth GD, Buckley JD, Noakes M, Clifton PM, Wilson CJ. Long-term effects of a very low-carbohydrate diet and a low-fat diet on mood and cognitive function. Arch Intern Med. 2009;169(20):1873–80. doi: 10.1001/archinternmed.2009.329. [DOI] [PubMed] [Google Scholar]
- 130.Boraxbekk CJ, Stomby A, Ryberg M, et al. Diet-Induced Weight Loss Alters Functional Brain Responses during an Episodic Memory Task. Obes Facts. 2015;8(4):261–72. doi: 10.1159/000437157. [DOI] [PMC free article] [PubMed] [Google Scholar]