

SUMMARY
Direct interactions between pro- and anti-apoptotic BCL-2 family members form the basis of cell death decision-making at the outer mitochondrial membrane (OMM). Here we report that three antiapoptotic BCL-2 proteins (MCL-1, BCL-2, and BCL-XL) found untethered from the OMM function as transcriptional regulators of a prosurvival and growth program. Antiapoptotic BCL-2 proteins engage a BCL-2 homology (BH) domain sequence found in Suppressor of Fused (SUFU), a tumor suppressor and antagonist of the GLI DNA binding proteins. BCL-2 proteins directly promote SUFU turnover, inhibit SUFU-GLI interaction, and induce the expression of the GLI target genes BCL-2, MCL-1, and BCL-XL. Antiapoptotic BCL-2 protein/SUFU feedforward signaling promotes cancer cell survival and growth and can be disabled with BH3 mimetics – small molecules that target antiapoptotic BCL-2 proteins. Our findings delineate a chemical strategy for countering drug resistance in GLI-associated tumors and reveal unanticipated functions for BCL-2 proteins as transcriptional regulators.
INTRODUCTION
Programmed cell death or apoptosis extensively contributes to animal development and physiology by promoting the timely turnover of unwanted or compromised cells. Cell death decisions are largely governed by the localized action of B-cell lymphoma 2 (BCL-2) family of proteins at the OMM. The interaction between anti- and pro-apoptotic BCL-2 family members mediated by BH domains found in these proteins determines the activity of BAK and BAX – two proapoptotic BCL-2 proteins that permeabilize the OMM. Ensuing cytoplasmic cytochrome C leakage and the assembly of an apoptosome complex is generally recognized as the point of no return for cell death commitment1, 2. The stoichiometry of anti- and pro-apoptotic BCL-2 family members at the OMM thus dictates cellular response thresholds to intrinsic and extrinsic apoptotic signals2. Chemicals that target anti-apoptotic (henceforth prosurvival) BCL-2 proteins are in clinical development as anti-leukemic agents3.
The HH proteins play pivotal roles in body patterning and organogenesis in part by controlling expression of apoptosis regulatory proteins such as BCL-24–9. Deviant activation of HH signaling is associated with several cancers including medulloblastoma (MB) and basal cell carcinoma (BCC)10, 11. Cellular response to HH proteins is initiated upon their binding to the multi-pass protein Patched 1 (PTCH1), a suppressor of the Smoothened (SMO) seven transmembrane receptor [reviewed in12] (Fig. 1a). SMO is the target of FDA-approved anti-cancer agents used in the management of metastatic BCC13. Activated SMO promotes SUFU disassociation from GLI proteins thus allowing them to activate transcription14, 15.
Figure 1. Prosurvival BCL-2 proteins collectively promote GLI signaling by suppressing the activity of the tumor suppressor SUFU.
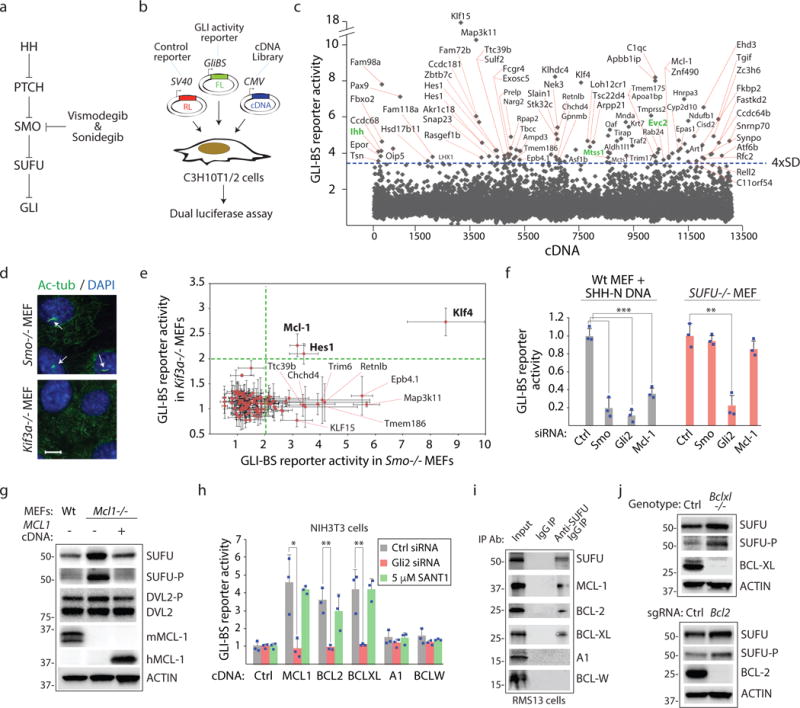
(a) Site of action for HH pathway antagonists. Mutations in PTCH1 unleash constitutive SMO activity in BCC and medulloblastoma. SMO mutations or increased GLI expression promote drug resistance. (b) A cultured cell-based screen for identifying GLI transcription regulators. FL=firefly luciferase, RL=Renilla luciferase. GLI binding enhancers in the GLI-BS reporter reports GLI activity. (c) Identification of genes that promote HH signaling using the Mammalian Gene Collection (MGC) cDNA library. C3H10T1/2 cells were transfected in duplicate experiments (biological replicates) with a single cDNA and reporters in “A”. Genes that induce FL/RL activity greater than 4xSD from the mean are labeled. Green=known HH pathway components. (d) Smo−/− or Kif3a−/− MEFs were stained with an anti-acetylated tubulin antibody to visualize primary cilia (arrows). Scale bars, 10μm. (e) MCL-1 induces HH pathway activity independently of SMO and primary cilia. Candidate HH pathway activators were evaluated for GLI inducing activity in Smo−/− or Kif3a−/− MEFs using the GLI-BS reporter (n=3 independent measurements). (f) SUFU is essential for MCL-1-dependent HH pathway response. GLI2 but not MCL-1 or SMO siRNAs reduce GLI-BS reporter activity in Sufu−/− MEFs. All three siRNAs reduced reporter activity in wt cells expressing the SHH signaling domain (SHH-N) (n=3 independent measurements). (g) MCL-1 loss increases total and phosphorylated SUFU. Mcl1−/− MEFs were transfected with control or human Mcl-1 cDNA. Lysates were subjected to Western blotting. SUFU-P antibody recognizes phospho-Ser342, a GSK3β-dependent modification (n=3 independent measurements). (h) Multiple prosurvival BCL-2 proteins induce GLI activity. DNA encoding indicated prosurvival BCL-2 proteins were evaluated for GLI activity induction using the GLI-BS reporter. SANT-1=SMO antagonist (n=3 independent measurements). (i) Prosurvival BCL-2 proteins that promote GLI activity interact with SUFU. RMS13 cell lysates were subjected to IP with control or anti-SUFU antibody. (j) Bclxl−/− and Bcl2−/− MEFs exhibit increased total and phosphorylated SUFU. All error bars represent mean ± s.d. Primary screen results are provided in Supplementary Table 1. Source data are available in Supplementary Table 4. Unprocessed blots are provided in Supplementary Fig. 7. Statistical significance was calculated using Student’s t-test, *P < 0.05, **P < 0.01 and ***P < 0.001.
Whereas much of our understanding of cell death regulation stems from studies focused on BCL-2 family proteins at the OMM1, prosurvival BCL-2 proteins have recently seen expanded functions at other organellar sites16, 17. Here, we describe a direct role of prosurvival BCL-2 proteins in transcriptional regulation. SUFU-mediated suppression of GLI activity is controlled by a BH3 sequence-dependent interaction between SUFU and three prosurvival BCL-2 gene family members (MCL-1, BCL-2, and BCL-XL). We demonstrate that these interactions occur in the cytoplasm away from the OMM and that de-repressed GLI proteins in turn induce MCL-1, BCL-2, and BCL-XL expression as well as cell growth promoting genes such as cyclin D1. Thus, the abundance of cyotaplsmic prosurvival BCL-2 proteins functions as part of a feedforward transcriptional regulatory loop that controls prosurvival BCL-2 protein expression. We demonstrate that this signaling axis can be disengaged with prosurvival BCL-2 protein inhibitors (BH3 mimetics) to achieve predictable anti-cancer effects.
RESULTS
A gain of function screen identifies the prosurvival MCL-1 protein as an instigator of GLI transcriptional activity
Murine C3H10T1/2 fibroblasts express core HH pathway components, elaborate a primary cilium which is essential for ligand-mediated signaling, and exhibit robust response to exogenously supplied HH protein18. To identify genes capable of promoting GLI activity and that may be targeted to overcome drug resistance to SMO antagonists, we seeded C3H10T1/2 cells in 96 well culture plates and transfected a single cDNA from the Mammalian Gene Collection (MGC) expression-ready cDNA library along with a HH-responsive firefly luciferase reporter (GLI-BS reporter) and a control Renilla luciferase reporter (SV40-RL)19 (Fig. 1b). The ratio of FL/RL activity was determined 36 hours later for each experiment and cDNAs inducing a signal greater than 4xSD from the mean were further considered (Fig. 1c; Supplementary Table 1). Several known HH regulators were also identified from this screen20–23. We used mouse embryonic fibroblasts (MEFs) lacking SMO or the ability to form primary cilia (Kif3a−/−) to identify direct regulators of GLI proteins (Fig. 1d,e). Three cDNAs when introduced into these cells retained their ability to induce the GLI-BS reporter: the HES Family BHLH transcription factor 1 (HES1), Kruppel-like factor 4 (KLF4), and the prosurvival protein BCL-2 family member Myeloid Cell Leukemia 1 (MCL-1). We focus here on MCL1 given that it is amplified in ~11% of all cancers24 and has no previously assigned role in GLI regulation.
Prosurvival BCL-2 proteins disrupt SUFU repression of GLI activity
We first addressed the contribution of the GLI regulator SUFU to prosurvival BCL-2 protein-mediated GLI regulation (see Fig. 1a). In contrast to control cells, Sufu−/− MEFs exhibited little change in GLI-BS reporter activity upon introduction of Mcl-1 siRNAs suggesting that SUFU is the target of MCL-1 action (Fig. 1f). The stability and activity of SUFU is regulated by sequential phosphorylation by PKA then GSK3β25. To evaluate the effects of targeting MCL-1 on SUFU abundance and phosphorylation, we compared fibroblasts derived from wild-type (wt) and Mcl1−/− animals (Fig. 1g). Absence of Mcl1 resulted in increased total and phosphorylated SUFU but introduction of human MCL-1 into Mcl1−/− cells restored total and phosphorylated SUFU levels to those observed in wt MEFs. The change in SUFU protein abundance is a not a consequence of altered SUFU transcript levels but rather mitigation of SUFU protein turnover by a proteasome-dependent mechanism (Supplementary Fig. 1a–d).
We collected cDNAs corresponding to the remaining four prosurvival BCL-2 family members (BCL-2, BCL-XL, A1, and BCL-W) and compared their ability with that of MCL-1 to induce the GLI-BS reporter (Fig. 1h, Supplementary Fig. 1e). Notably, BCL-2 induced the GLI-BS reporter in our screen albeit not in a sufficiently robust manner to be included as a first-pass hit (see Supplementary Table 1). Overexpression of three out of five prosurvival BCL-2 proteins promoted GLI activity in a SMO-independent manner. Given the functional interaction of MCL-1 with SUFU, we surmised that the prosurvival proteins that promote GLI activity are found in a complex with SUFU. Indeed, using an immunoprecipitation (IP) approach, we observed only the three antiapoptotic proteins that promoted GLI activity to be found in a complex with SUFU (Fig. 1i; Supplementary Fig. 1f). Finally, we observed using Bcl2 or Bclxl null cells that loss of either of these proteins as in the case of MCL-1 resulted in increased abundance of total and phosphorylated SUFU thus revealing a common target for all three prosurvival BCL-2 proteins (Fig. 1j; Supplementary Fig. 1g). We also showed that the ability of an overexpressed prosurvival BCL-2 family member to interact with SUFU correlates with its ability to induce changes in SUFU abundance (Supplementary Fig. 1h).
SUFU harbors a BH3 sequence that mediates interaction with prosurvival BCL-2 proteins
Our genetically based findings suggest a direct interaction between SUFU and multiple BCL-2 proteins. To better understand the basis for the SUFU-MCL-1 interaction, we mapped the determinants in MCL-1 required for SUFU binding (Fig. 2a; Supplementary Fig. 2a,b). At the same time, we also determined the ability of the various mutant MCL-1 proteins to overcome SUFU-mediated GLI inhibition using the GLI-BS reporter (Supplementary Fig. 2c). These results taken together reveal that mutations compromising the integrity of the BCL-2 Core (BC) in MCL-1 eliminated MCL-1 ability to bind to and inactivate SUFU (see Fig. 2a). Since the BC constitutes the BH3 sequence recognition module in interactions between prosurvival and proapoptotic BCL-2 family proteins, the observation implicates a previously undefined BH3 sequence in SUFU.
Figure 2. SUFU functions as a BH3-only protein.
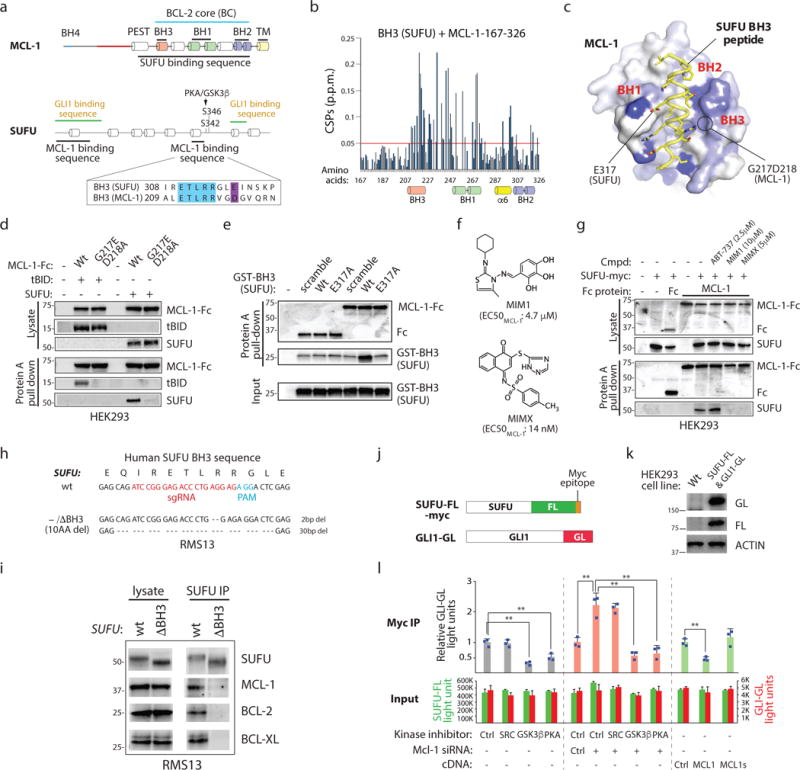
(a) The MCL-1 BC interacts with a SUFU BH3 sequence. Minimal interaction domains were identified by co-IP. Raw data is found in Supplementary Fig. 2a–e. Blue: residues identical in SUFU and MCL-1 BH3 sequences. (b) Backbone 1H–15N chemical shift perturbations (CSPs) of 15N-labeled MCL-1 induced by SUFU BH3 map within the BC groove (BH1, BH2 and BH3) and helix 6. (c) The CSPs in (a) were color coded from blue to white (large to small) onto the surface representation of a SUFU BH3–MCL-1 complex model built based on threading the SUFU BH3 onto the MCL-1 SAHB–MCL-1 complex26. SUFU E317 corresponds to an acidic residue essential to BH3/BC interactions. G217 and D218 are essential for MCL-1 interaction with BH3 sequences. (d) Residues essential for MCL-1 BC/BH3 domain interaction are required for MCL-1/SUFU binding. MCL-1-Fc = MCL-1-immunoglobulin heavy chain fusion protein. (e) The SUFU BH3 sequence is sufficient for MCL-1 interaction. Glutathione-S-transferase = GST protein. (f) Structure of chemicals targeting the MCL-1 BC groove. (g) MCL-1 inhibitors disrupt MCL-1/SUFU binding. ABT-737 = negative control. (h) SUFU genomic sequence in CRISPR-Cas9-edited RMS13 cells expressing SUFU lacking a BH3 sequence. (i) Co-IP of SUFU with prosurvival BCL-2 proteins is dependent upon the SUFU BH3 sequence. SUFU lacking an intact BH3 sequence (10AA del) fails to co-IP with prosurvival BCL-2 proteins. (j) A luciferase strategy for monitoring SUFU/GLI interactions. SUFU/GLI association is quantified by the relative FL and GL signal in antibody-isolated material. (k) Western blot analysis of a cell line expressing SUFU-FL and GLI1-GL. (l) MCL-1 regulates GLI-SUFU interaction. SUFU/GLI interaction is sensitive to PKA, GSK3β, and MCL-1 activity changes. MCL-1s = C-terminally truncated splice variant (n=3 independent measurements). All error bars represent mean ± s.d. Source data are available in Supplementary Table 4. Unprocessed blots are provided in Supplementary Fig. 7. Statistical significance was calculated using Student’s t-test, **P < 0.01.
We similarly mapped MCL-1 binding sites in SUFU (see Fig. 2a; Supplementary Fig. 2d,e). Two SUFU regions mediate MCL-1 binding: an N-terminal sequence (AAs 1-100) and a putative alpha helix adjacent to the PKA and GSK3β phosphorylation sites (AAs 302–327). The latter binding site remarkably resembled the MCL-1 BH3 sequence which constitutes a part of the MCL-1 BC and which is a high affinity ligand for MCL-1 itself26. To determine if the putative SUFU BH3 peptide indeed functions as a canonical BH3 sequence, we performed NMR titration with uniformly 15N-labeled human MCL-1 and unlabeled SUFU BH3 (Fig. 2b,c). Significant backbone 1H–15N chemical shift perturbations (CSPs) were induced by the SUFU BH3 peptide but not DMSO at the MCL-1 BC groove (Supplementary Fig. 2f,g). The chemical shifts within this minor site are likely generated as a result of the major conformational change in the BC groove as we previously observed with other BH3 complexes of BCL-2 family proteins, including the BID BH3–MCL-127 and BID BH3–BAK complexes28. Consistent with the involvement of the BH3 sequence in mediating SUFU and MCL-1 interaction, we observed stoichiometric interaction of the two proteins (Supplementary Fig. 2h). These structural studies delineate a mechanistic basis for recognition of SUFU by prosurvival BCL-2 proteins.
Two residues (G217 and D218) found in the MCL-1 BC have been previously shown to be essential for recognizing BH3 ligands29 (see Fig. 2c). We compared the ability of wt and G217E/D218A MCL-1 fusion proteins to interact with a well-established BH3-only protein (tBID) as well as SUFU (Fig. 2d). Whereas wt MCL-1 protein was able to bind both tBID and SUFU, the mutant protein failed to do so. At the same time, we demonstrated that a recombinant protein expressing the SUFU BH3 sequence is able to bind to the MCL-1 fusion protein (Fig. 2e). Furthermore, this interaction was dependent upon an acidic residue in the SUFU BH3 sequence that corresponds to a residue essential to the binding capability of other BH3 sequences26. Finally, two small molecules that target the MCL-1 BC (MIM1 and MIMX)30 were able to disrupt MCL-1/SUFU interaction (Fig. 2f,g). Using CRISPR-Cas9 genome-editing, we generated from rhabdomyosarcoma-derived RMS13 cells a cell line harboring a single protein encoding SUFU allele with a 30bp deletion corresponding to the loss of 10 amino acids from the BH3 sequence (Fig. 2h). We demonstrated by using a co-IP strategy that the SUFUΔBH3 protein no longer binds to prosurvival BCL-2 proteins (Fig. 2i). Taken together, our observations provide a molecular basis for MCL-1 recognition of SUFU.
MCL-1 regulates SUFU-GLI binding in a BH3 sequence-dependent fashion
The BH3 sequence and PKA/GSK3β phosphorylation sites fall within the IDR, a region that governs overall SUFU conformation and activity but that apparently is not accessible to crystallography32, 33. Deletion of the IDR does not abolish SUFU-GLI interaction but renders SUFU unresponsive to HH signal suggesting that PKA/GSK3β phosphorylation controls SUFU activity by toggling the IDR conformation31. To test the hypothesis that MCL-1 may similarly control the IDR conformation either directly or indirectly by thwarting SUFU phosphorylation by PKA/GSK3β, we first established a cell line that provides quantitative reporting of SUFU/GLI interaction changes (Fig. 2j, k). Consistent with previous observations, we observed a decrease in SUFU/GLI association in cells treated with inhibitors of either PKA or GSK3β (Fig. 2l). On the other hand, gain or loss of MCL-1 function decreased and increased SUFU/GLI interactions, respectively. At the same time, an in vitro kinase assay using purified proteins reveals that MCL-1 prevents SUFU phosphorylation by PKA (Supplementary Fig. 2i,j). These observations suggest that prosurvival proteins like MCL-1 leverage local changes in IDR structure either directly or indirectly by blunting PKA/GSK3β phosphorylation to control SUFU responsiveness to HH.
Feedforward signaling mediated by prosurvival BCL-2 proteins is required for HH response
Our observations suggest that previously described HH-induced transcription of BCL2 may constitute an unrecognized feedforward signaling mechanism32, 33. To determine if other prosurvival BCL-2 family transcripts may be induced by GLI activation, we examined the abundance of prosurvival Bcl2 family mRNAs and protein in C3H10T1/2 cells co-transfected with SHH DNA or treated with the SMO agonist SAG (Fig. 3a,b; Supplementary Fig. 3a). With either mechanisms of inducing GLI activity we observed increases in Bcl2, Mcl1, and Bclxl mRNA and protein suggesting GLI regulates only the SUFU interacting members of the prosurvival BCL-2 family. The expression of BCL2 and MCL1 are also elevated in vivo in a comparison of medulloblastoma (MB) samples belonging to the “SHH” subgroup (predominantly driven by mutations in PTCH1) and normal cerebellar tissue (Supplementary Fig. 3b).
Figure 3. Prosurvival BCL-2 proteins promote their own expression by inducing GLI transcriptional activity.
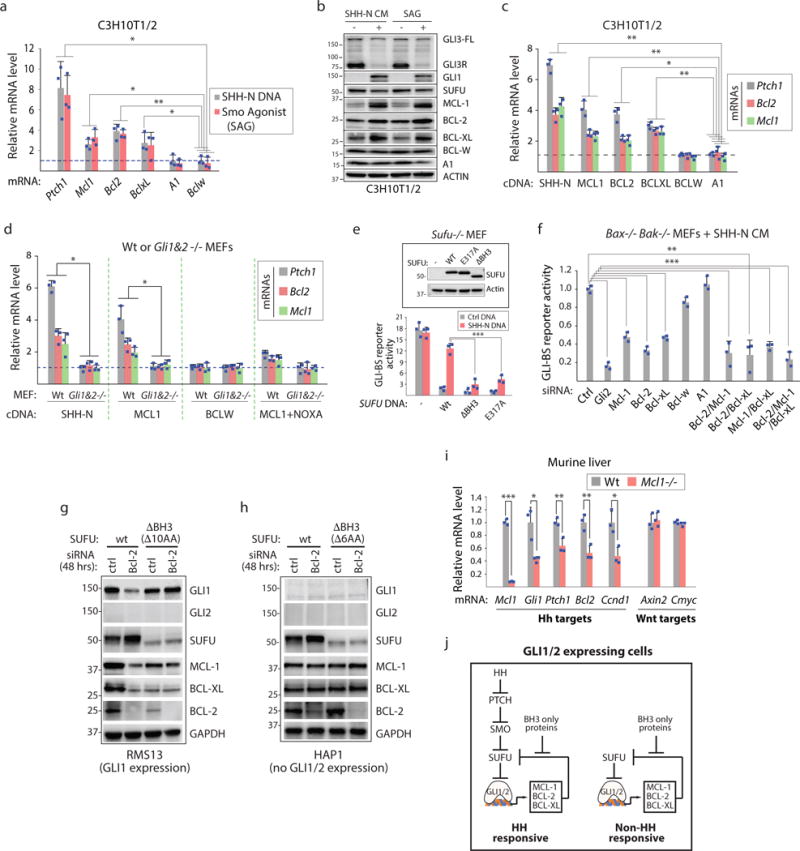
(a) MCL-1, BCL-2, and BCL-XL are GLI target genes. RT-PCR analysis of SHH or SMO agonist stimulated cells (n=3 independent measurements). (b) HH pathway activation induces expression of SUFU-interacting prosurvival BCL-2 proteins. SHH-N CM = culture cell medium containing SHH-N. SAG = SMO agonist. GLI3R= GLI3 repressor. GLI3-FL = GLI3 full length. (c) Forced expression of MCL-1, BCL-2, or BCL-XL but not BCL-W or A1 induces Ptch1, Bcl-2, and Mcl-1 mRNA (n=3 independent measurements). (d) MCL-1 overexpression-induced transcriptional changes in Ptch1, Bcl-2, and Mcl-1 are GLI1/GLI2 dependent and sensitive to NOXA co-expression. MCL-1 but not BCL-W overexpression induces gene expression changes in wt but not Gli1&2−/− MEFs. MCL-1 co-expression with the BH3 only protein NOXA (MCL-1 ligand) abrogates MCL-1 induced transcriptional changes (n=3 independent measurements). (e) Introduction of wt SUFU DNA but not DNA encoding a SUFU molecule lacking the BH3 sequence or the critical acidic residue found in BH3 sequences (E317) restores HH-mediated control of GLI activity in Sufu null cells. Sufu−/− MEFs were transfected with indicated SUFU DNAs and GLI-BS and control reporters (n=3 independent measurements; bottom). (f) Loss of any single SUFU-interacting prosurvival BCL-2 protein (MCL-1, BCL-2, BCL-XL) compromises HH response. Indicated siRNAs pools (4× different siRNAs) were evaluated in cells devoid of the pro-apoptotic effectors BAX and BAK (n=3 independent measurements). (g) Compromising the SUFU BH3 sequence abrogates SUFU stability and GLI transcriptional responses to BCL-2 expression changes. Unlike in wt cells, RMS13 cells expressing a SUFUΔBH3 protein (which harbors a 10 amino acid deletion; see Fig. 2h) exhibit little/no change in the expression of GLI target genes (Gli1, Mcl1, and Bclxl) or SUFU stability upon loss of BCL-2 induced by RNAi. (h) GLI1/GLI2 expression is required for prosurvival BCL-2-dependent changes in GLI target gene expression but not in SUFU stability. SUFU but not SUFUΔBH3 (6AA deletion; Supp. Fig. 3e) exhibit stability changes in cells transfected with BCL2 siRNAs. Both cell exhibit little change in GLI target gene expression upon loss of BCL-2. (i) MCL-1 is required for expression of GLI target genes in vivo. mRNA levels of several known HH and Wnt pathway target genes were compared from wt or MCL-1 null liver tissue (n=3 independent measurements). (j) Model of feedforward GLI signaling facilitated by MCL-1, BCL-2, and BCL-XL. All error bars represent mean ± s.d. Source data are available in Supplementary Table 4. Unprocessed blots are provided in Supplementary Fig. 7. Statistical significance was calculated using Student’s t-test, *P < 0.05, **P < 0.01 and ***P < 0.001.
The same three proteins that interact with SUFU (MCL-1, BCL-2, and BCL-XL) are able to induce expression of Ptch1, Bcl2, and Mcl1 when each prosurvival BCL-2 protein was overexpressed in C3H10T1/2 cells (Fig. 3c). We also demonstrated that this transcriptional response is GLI1/GLI2 dependent, can be suppressed by the BH3-only protein NOXA that binds to the MCL-1 BC groove2 (Fig. 3d), and requires an intact SUFU BH3 sequence (Fig. 3e). Thus, several prosurvival BCL-2 proteins can self-regulate their own abundance and their transcriptional regulation activities are required for HH responses.
We wondered if MCL-1, BCL-2, and BCL-XL function to promote feedforward signaling in an additive or redundant fashion. We first observed that loss of a single protein was sufficient to reduce GLI activity (Fig. 3f). Loss of GLI activity here is not a collateral effect from ensuing pro-apoptotic events given these studies were performed in cells lacking the proapoptotic BAX and BAK effectors. Pairwise or combined RNAi targeting of the three BCL-2 proteins resulted in a modest enhancement of GLI reporter loss as compared to single gene focused experiments suggesting these genes mostly function in a non-redundant fashion (see Fig. 3f).
Loss of a single SUFU-interacting prosurvival BCL-2 protein results in increased SUFU stability and decreased expression of GLI target genes in RMS13 cells (Fig. 3g). However, removal of the SUFU BH3 sequence by CRISPR-Cas9 editing renders these cells non-responsive to Bcl-2 siRNAs (see Fig. 2h). On the other hand, Bcl-2 siRNA transfected HAP1 cells (which lack GLI1/GLI2 expression) nevertheless exhibit increased SUFU abundance in a SUFU BH3 sequence dependent manner but little change in the expression of GLI target genes thus affirming the requirements of pre-existing GLI1/GLI2 expression for prosurvival BCL-2-dependent transcriptional changes (Fig. 3h; Supplementary Fig. 3c–e). We also observed in C3H10T1/2 cells the contribution of the SUFU BH3 sequence to prosurvival BCL-2-dependent transcriptional regulation (Supplementary Fig. 3f–i).
Given that HH stimulation changes the abundance of MCL-1/SUFU interactions using a co-IP approach (Supplementary Fig. 3j–l), we evaluated the effects of deleting MCL-1 on HH signaling in an adult tissue (Fig. 3i). Loss of Mcl1 in the liver resulted in reduced expression of several HH target genes including Bcl2 and cyclin D1 (Ccnd1). On the other hand, expression of Wnt target genes such as Axin2 and Cmyc was untouched. Our observations taken together, reveal a feedforward GLI signaling mechanism supported by prosurvival BCL-2 protein/SUFU interactions that is sensitive to the abundance of ligands from the BCL-2 family including those that are pro-apoptotic such as NOXA (Fig. 3j).
Prosurvival BCL-2 proteins not localized to the mitochondrial membrane regulate SUFU
Prosurvival BCL-2 protein family members including MCL-1 are predominantly localized to the OMM via a hydrophobic anchoring sequence found in the C-terminus (see Fig. 2a)34, 35. Nevertheless, an MCL-1 protein lacking the anchoring sequence (MCL-1ΔTM) was able to bind to and inhibit SUFU activity suggesting that mitochondrial localization is not essential for this activity (see Supplementary Fig. 2a–c). Indeed, prosurvival BCL-2 proteins are found in other subcellular compartments away from the OMM including in the cytoplasm35, 36. We investigated the consequences of removing the TM sequence from MCL-1 or eliminating MCL-1 altogether in C3H10T1/2 cells using CRISPR-Cas9 genome editing (Fig. 4a,b; Supplementary Fig. 4a,b). Whereas the two MCL1ΔTM cell lines exhibited hyper-responsiveness to exogenously supplied SHH ligand, the Mcl1−/− cell line exhibited little response (Fig. 4c). This more robust response was also confirmed using biochemical readouts (Fig. 4d). We also evaluated HH responsiveness in a C3H10T1/2 cell line expressing BCL-2 proteins lacking an OMM tether (Fig. 4e–f; Supplementary Fig. 4c) and observed enhanced response using both the GLI-BS reporter and biochemical approaches (Fig. 4g,h). These observations taken together suggest that abundance of cytoplasmically localized BCL-2, MCL-1, and presumably BCL-XL can contribute to cell death resistance by controlling the collective transcriptional regulation of themselves at least in cells with GLI activity (Fig. 4i).
Figure 4. Extra-mitochondrial prosurvival MCL-1 protein regulates SUFU activity.
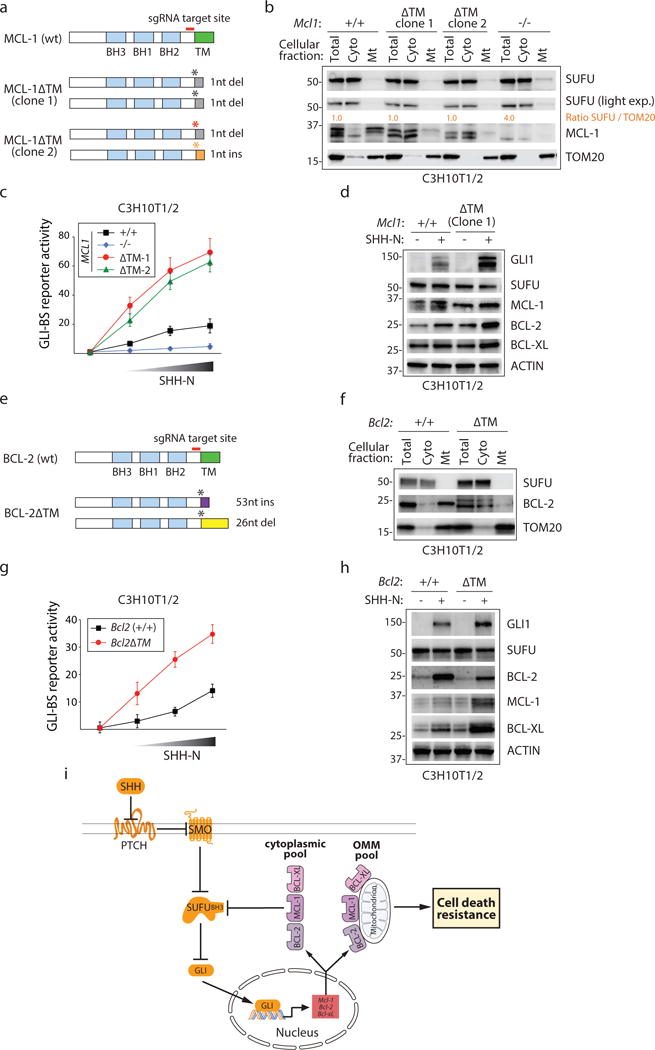
(a) CRISPR-Cas9 mediated deletion of the MCL-1 TM sequence. (b) Biochemical evaluation of MCL-1 proteins in CRISPR-Cas9 edited cells. Re-localization of MCL-1 from the OMM to the cytoplasm (MCL-1ΔTM) or MCL-1 elimination (KO) does no alter the distribution of SUFU. Quantification of relative SUFU/TOM20 levels is provided. (c) Enhanced HH response in MCL1ΔTM cells. Increasing amounts of conditioned medium (CM) containing SHH-N protein was applied to CRISPR-Cas9-edited C3H10T1/2 cells expressing wt, ΔTM or no MCL-1 (n=3 independent measurements). (d) Biochemical characterization of GLI target gene expression in CRISPR-Cas9-edited Mcl1ΔTM C3H10T1/2 cells. (e) CRISPR-Cas9 mediated deletion of the BCL-2 TM sequence. (f) Biochemical evaluation of BCL-2 proteins in CRISPR-Cas9 edited cells. Re-localization of BCL-2 to the cytoplasm (BCL-2ΔTM) does not alter the abundance of SUFU associated with the OMM. (g) Enhanced HH response in Bcl2ΔTM cells. Increasing amounts of SHH-N CM was applied to CRISPR-Cas9-edited C3H10T1/2 cells expressing wt or ΔTM BCL-2 protein (n=3 independent measurements). (h) Biochemical characterization of GLI target gene expression in Bcl2ΔTM C3H10T1/2 cells in response to SHH-N containing CM. (i) Model of extramitochondrial prosurvival BCL-2-dependent transcriptional regulation. All error bars represent mean ± s.d. Source data are available in Supplementary Table 4. Unprocessed blots are provided in Supplementary Fig. 7.
BH3 mimetics disable GLI transcriptional activity in vivo
Small molecules that target the BC groove of prosurvival BCL-2 proteins (BH3 mimetics) promote apoptosis by disrupting the interaction between prosurvival BCL-2 proteins and their BH3-only partners37. To determine if disabling GLI-mediated transcription using BH3 mimetics can impact cancerous growth, we first identified cancer-associated SUFU mutants that may be refractory to BH3 mimetic response and that could be used to distinguish cell growth effects caused by direct engagement of the pro-apoptotic machinery from effects due to GLI suppression. The overall architecture of SUFU resembles a clamp with N- and C-terminal domains that enables it to latch onto and inactivate GLI proteins31, 38 (Fig. 5a). A survey of SUFU mutants across all cancer types using cBioPortal39, 40 reveals enrichment of mutations in the N-terminal domain and in the IDR (Cluster 1 and Cluster 2 mutations, respectively; see Fig. 5a; Supplementary Fig. 5a).
Figure 5. BH3 mimetics disrupt GLI-dependent transcription in vivo.
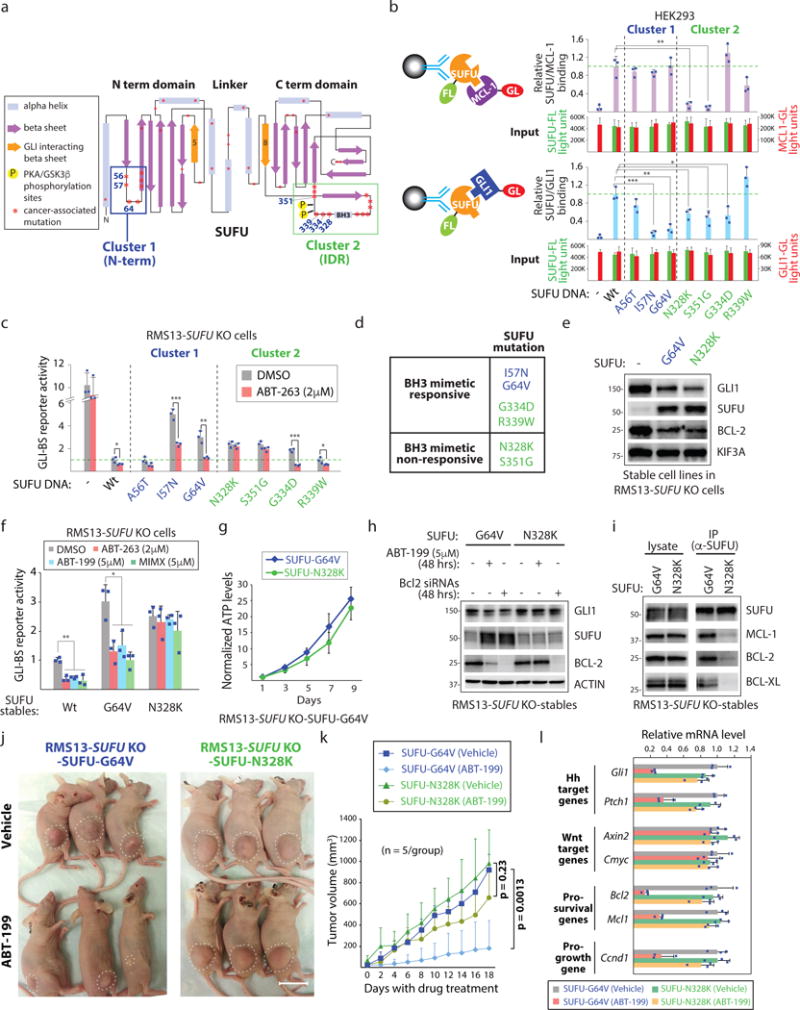
(a) SUFU mutants in cancer with potential differential sensitivities to BH3 mimetics. Cancer-associated missense SUFU mutations (cBioPortal) reveal two mutation clusters centered on the N-terminus (Cluster 1) and the intrinsically disorganized region (IDR; Cluster 2). Both mutation sets are distal to β5 and β8 which harbor the GLI1 interaction residues31. (b) The majority of SUFU Cluster 2 mutations affect SUFU binding to MCL-1. Relative MCL-1/SUFU and GLI1/SUFU interactions were determined using Gaussia luciferase (GL)/Firefly (FL) activity ratios in the IPed material (n=3 independent measurements). (c) SUFU mutants that retain responsiveness to a BH3 mimetic. RMS13 SUFU−/− cells transfected with indicated SUFU DNAs and the GLI-BS and control reporters were cultured with/without ABT-263 (n=3 independent measurements). (d) SUFU Class I and II mutants and their responsiveness to a BH3 mimetic. (e) RMS13 SUFU−/− cells stably expressing SUFU-G64V or SUFU-N328K exhibit similar ability to suppress GLI expression and activity. (f) BH3 mimetics with distinct targeting profiles for MCL-1, BCL-2, and BCL-XL exhibit similar activity profiles in cells expressing either SUFU-G64V or -N328K (n=3 independent measurements). (g) SUFU-G64V and -N328K cells exhibit similar growth rates in vitro (n=3 independent measurements). (h) Reducing BCL-2 expression using RNAi mimics the effects of ABT-199 in SUFU-G64V and -N328K expressing cell lines. (i) SUFU-N328K exhibits poor ability to bind to prosurvival BCL-2 proteins. (j) ABT-199 prevents growth of SUFU-G64V but not SUFU-N328K cells when subcutaneously transplanted in nude mice (n=5 mice/group). Scale bar: 20mm. (k) Quantification of in vivo tumor growth (n=5 mice/group). (l) ABT-199 suppresses GLI activity in vivo. SUFU-G64V and SUFU-N328K expressing tumors from control or ABT-199 treated animals were subjected to qPCR analysis (n=3 mice/group). All error bars represent mean ± s.d. Source data are available in Supplementary Table 4. Unprocessed blots are provided in Supplementary Fig. 7. Statistical significance was calculated using Student’s t-test, *P < 0.05, **P < 0.01 and ***P < 0.001.
We directly evaluated the ability of several SUFU Cluster 1 and 2 mutants to bind MCL-1 using a quantitative co-IP strategy that allows us to normalize interactions with the abundance of associated bait protein (Fig. 5b; top). Three out of four IDR mutations compromised MCL-1 binding to SUFU thus confirming the importance of this region for SUFU interaction with prosurvival BCL-2 proteins. However, the majority of SUFU proteins with mutations in either cluster retained to different degrees binding to GLI1 (see Fig. 5b; bottom). These biochemical results were largely matched by functional tests of mutant SUFU proteins in RMS13 SUFU−/− cells for their ability to suppress GLI activity and to respond to a BH3 mimetic (Fig. 5c; Supplementary Figure 5b).
We next generated cell lines expressing either SUFU-G64V (BH3 mimetic responsive) or -N328K (BH3 mimetic non-responsive) in RMS13 SUFU−/− cells so that we can evaluate the efficacy of BH3 mimetics in blunting cell growth in vivo (Fig. 5d, e). RMS13 cells exhibit GLI1 overexpression as a consequence of GLI1 gene amplification thus affording a robust platform for interrogating the mechanistic basis of BH3 mimetic activity against cancer-associated forms of HH pathway induction41 (Supplementary Figure 5c). Both cell lines exhibited similar reduction in GLI1 and BCL-2 proteins thereby demonstrating their shared capacity to repress GLI (see Fig. 5e). We also confirmed that other BH3 mimetics such as those targeting BCL-2 (ABT-199) or MCL-1 (MIMX) exhibit a similar GLI response profile in these cell lines and that the cell lines exhibit similar in vitro growth rates (Fig. 5f,g). We biochemically confirmed that a BH3 mimetic (ABT-199, which targets BCL-2) or BCL2 siRNAs are able to increase the abundance of the G64V but not the N328K protein and also each protein’s ability to interact with prosurvival BCL-2 proteins (Fig. 5h,i).
We implanted the SUFU-G64V and -N328K cell lines in nude mice and allowed tumors to develop to 30mm2 before dosing with the FDA-approved ABT-199/Venetoclax37. Whereas we observed greatly diminished growth of SUFU-G64V cells, the SUFU-N328K cells failed to respond to the drug (Fig. 5j,k; Supplementary Fig. 5d,e). These observations reveal a previously unappreciated mechanism associated with the anti-cancer activity of BH3 mimetics At the same time, they reveal that a BH3 mimetic targeting a single prosurvival BCL-2 protein can have an effect on the collective expression of the gene class. Indeed, qPCR analysis of RMS13 SUFU-G64V and -N328K tumors in control and ABT-199 treated animals reveals differential responses to ABT-199 exposure with respect to HH pathway target, prosurvival, and pro-growth genes but not Wnt pathway responsive genes (Fig. 5l). These in vivo observations taken together reveal an ability of BH3 mimetics to inhibit cancerous growth by disrupting GLI transcription.
BH3 mimetics disrupt GLI activity driven by genetic mutations in HH pathway regulators
Despite the recent clinical approval of SMO antagonists for metastatic BCC, anti-cancer strategies premised upon targeting SMO suffer from rapidly acquired drug resistance42. Many of these mechanisms converge upon the emergence of SMO mutations that abrogate drug interaction43, 44, or cellular mechanisms that buttress GLI activity even in the presence of SMO antagonists45–47. We evaluated the ability of various BH3 mimetics to disrupt GLI activity induced by cancer-associated alterations of HH pathway components48 as well as drug-resistance associated mechanisms49 (Fig. 6a,b; Supplementary Fig. 6a–h). Although SUFU mutations are infrequent in adolescent/adult MBs, their prevalence in infant MBs is common (~40%)11 (see Supplementary Fig. 6a). Whereas Vismodegib exhibited an activity profile consistent with SMO targeting, the BH3 mimetics retained similar activity against all mechanisms of GLI activation evaluated (see Fig. 6a). We demonstrated that the BH3 mimetics selectivity profile could be used to predictably disable prosurvival BCL-2 protein/SUFU interactions (Fig. 6c). Given their attack within the HH pathway at the level of transcriptional regulation, BH3 mimetics may be useful in countering HH pathway-associated oncogenic changes including those induced by long-term exposure to SMO antagonists.
Figure 6. BH3 mimetics disable deviant signaling induced by cancer-associated mutations in core HH pathway components.
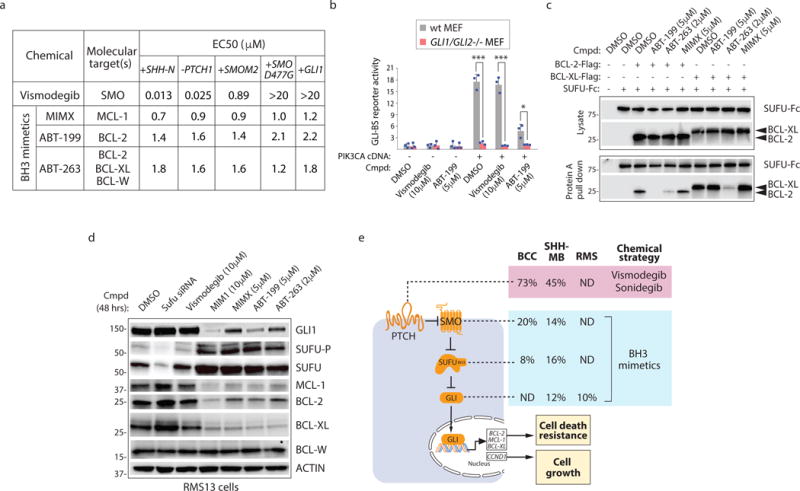
(a) Activity profile of SMO and BH3 mimetics in cancer-associated HH signaling contexts. The effects of chemicals on the GLI-BS reporter in C3H10T1/2 cells with forced expression of SHH-N, GLI1, the constitutively active SMO-M2, and the drug resistant SMO-D477G proteins, or in Ptch1−/− MEFs. (b) ABT-199 inhibits PIK3CA-induced GLI activity. C3H10T1/2 cells transfected with the GLI-BS reporter were treated with ABT-199 for 48 hrs before luciferase activity was measured from cell lysate (n=3 independent measurements). (c) BH3 mimetic selectivity correlates with its ability to disrupt SUFU interaction with BCL-2 and BCL-XL (n=3 independent measurements). (d) BH3 mimetics decrease GLI1, MCL-1, and BCL-2 expression regardless of their prosurvival BCL-2 protein target(s). SUFU abundance increases in the presence of BH3 mimetics but not Vismodegib. (e) Break down of mutations found in HH pathway components in basal cell carcinoma (BCC), SHH subgroup of medulloblastoma (MB), and rhabdomyosarcoma (RMS) and their known sensitivity to SMO antagonists Vismodegib and Sonidegib, and potential sensitivity to BH3 mimetics. Data source: BCC10, MB11, and RMS60. Source data are available in Supplementary Fig. 6 and 7.
Consistent with our previous observations, GLI1, BCL-2, MCL-1, and BCL-XL protein abundance was diminished upon drug exposure whereas levels of total and phosphorylated SUFU increased (Fig. 6d). Notably, chemically induced reduction of BCL-2, MCL-1, and BCL-XL proteins was independent of which prosurvival BCL-2 protein(s) were targeted by the BH3 mimetic applied (see Fig. 6d) thus confirming the importance of feedforward signaling by interaction between a single prosurvival BCL-2 protein and SUFU to the collective regulation of prosurvival BCL-2 proteins. This feedforward signaling mechanism thus reveals opportunities for disabling cancerous GLI activity using BH3 mimetics that target either a single or multiple prosurvival BCL-2 proteins that interact with SUFU (Fig. 6e).
DISCUSSION
Evasion of programmed cell death by heightened expression of prosurvival BCL-2 protein families is a hallmark of cancer50. The inhibition of OMM permeabilization by prosurvival BCL-2 proteins has provided the mechanistic rationale for the development of BH3 mimetics as anti-cancer agents, and has served as the major cellular end point assay for monitoring the activity of BCL-2 family proteins. Our identification of a prosurvival BCL-2 recognition sequence in the transcriptional regulator SUFU not only extends the potential utility of BH3 mimetics to include cancers typically associated with mutations in HH pathway components, but also enables the use of GLI targets genes as biomarkers of BCL-2 family protein activity in homeostatic and cancerous contexts (Supplementary Fig. 6i). Our observations that prosurvival BCL-2 protein/SUFU interactions occur away from the OMM also expands the subcellular reach of BCL-2 proteins and may provide mechanistic insight into the anti-apoptotic activity of prosurvival BCL-2 proteins not found on the OMM51–53. Given the GLI proteins also control the expression of cell proliferation genes such as cyclin D154, our findings also reveal unappreciated pro-growth activities of BCL-2 family proteins.
Our findings also reveal that a subset of the prosurvival BCL-2 proteins (MCL-1, BCL-2, and BCL-XL) collectively self-regulate their own expression levels using the abundance of competing BH3 sequences from BCL-2 family ligands and SUFU as a rheostat in cell death decision-making. It is important to note that this feedforward system is likely to be relevant only in GLI1/2 expressing cells. Indeed, other compensatory transcriptional regulatory loops may control the expression of prosurvival BCL-2 proteins such as in the case of drug resistance to BH3 mimetics55.
We have demonstrated that loss of MCL-1, BCL-2, or BCL-XL results in SUFU accumulation and an increase in SUFU ability to bind to GLI proteins. The region that encompasses the PKA and GSK3β phosphorylation sites and the BH3 sequence is intrinsically disorganized but somehow functions as a switch for toggling SUFU conformation31, 38. The inability to resolve the structure of the BH3 encoding region in SUFU is reminiscent of canonical BH3 sequences which are similarly unstructured in solution but organize upon binding to BCs56, 57.
Although ABT-199/Venetoclax has recently been approved for the treatment of chronic lymphocytic leukemia58 its utility in solid tumors is currently limited59. Our study delineates an unconventional path for patient selection in clinical testing of BH3 mimetics predicated upon understanding the mutational status of HH pathway components including those associated with resistance to SMO antagonists, and those found in SUFU where these chemicals may function as indirect pharmacoperones. Further studies should reveal the extent to which deviant GLI target gene expression can serve as a predictive biomarker of BH3 mimetic sensitivity in cancers not typically associated with HH signaling.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of this paper.
Methods
Cell culture and chemical reagents
NIH3T3, C3H10T1/2, HEK293, Bax−/− Bak−/−, and RMS13 cell lines were purchased from the American Type Culture Collection (ATCC). HAP1 cell was purchased from Horizon Discovery. The following cell lines were provided: Kif3a−/− MEFs (P.T. Chuang, UCSF), Smo−/− MEFs (J.K. Chen, Stanford University), Sufu−/− MEFs (R. Toftgård, Karolinska Institutet), Mcl1−/− MEFs (J.T. Opferman, St. Jude’s Children’s Research Hospital), Bclxl−/− MEFs (C. Li, University of Louisville), Gli1/2−/− MEFs (J. Gipp, University of Wisconsin-Madison). All cell lines were authenticated by short tandem repeat profiling and further tested to show no mycoplasma contamination.
Sources of chemical reagents are: SAG (Cat. No. ALX-270-426-M001, Alexis Biochemicals); SANT1 and GSK3β inhibitor SB216763 (Cat. No. S4572 and S3442, Sigma); PKA inhibitor H89 (Cat. No. 2910, Tocris); MG132, ABT-263, ABT-737, and Src inhibitor SU6656 (Cat. No. S8410, S1001, S1002, and S7774, Selleckchem), MIMX (ChemBridge), ABT-199 (Cat. No. HY-15531, Medchemexpress), MIM1 was previously described30. Vismodegib was synthesized by Chuo Chen (UT Southwestern Medical Center).
Plasmids, expression constructs, and generation of mutants
Human MCL-1-Flag DNA was a gift from X. Wang (National Institute of Biological Sciences, Beijing). Human SUFU, BCL2, BCLXL, BCLW DNA was purchased from Origene. All wild type DNA fusion constructs were generated using PCR-based cloning. Mutants were generated using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). Construct sequences were confirmed by DNA sequencing.
cDNA library screen
The Mammalian Gene Collection (MGC) library (ThermoFisher) were screened by co-transfecting individual cDNAs into C3H10T1/2 cells seeded in 96-well white opaque plates (Greiner) with the HH-responsive firefly luciferase reporter (GLI-BS) and a control Renilla luciferase reporter (SV40-RL) using Fugene 6 (Roche) and a Biomek FX Liquid Handler (Beckman Coulter). Transfected DNA ratios: 2/4/1 (cDNA/Gli-BS/SV40-RL). 24 hrs after transfection, cells were switched to low serum media (0.5% FBS), and grown for another 48 hrs. in 5% CO2. FL and RL activities in lysate generated using Passive Lysis Buffer (Promega) were then assessed using the Dual-Luciferase kit (Promega) and a 96-well plate reading luminometer (BMG). The ratio of FL/RL was calculated and averaged ratios from the duplicate experiments were used to rank order cDNAs with respect to magnitude of GLI activation. cDNAs selected for subsequent testing were sequence verified. Primary screen results are provided in Supplementary Table 1.
RNAi and overexpression studies
siRNAs used in the study were purchased from GE Dharmacon. For RNAi experiments, cells were transfected with DharmaFECT 3 (GE Dharmacon), Lipofectamine RNAiMAX (Thermo Fisher), or Effectene (when cDNA and siRNA were co-delivered)(Qiagen). For overexpression studies, cells were transfected with Lipofectamine 2000 (Invitrogen), FuGENE 6 (Roche), or Effectene (Qiagen). Detailed information of siRNAs is provided in Supplemental Table 3.
qPCR
RNA was isolated using the RNeasy Plus Mini kit from Qiagen (Cat. No. 74134). cDNA was prepared using ProtoScript® First Strand cDNA Synthesis Kit (NEB). qPCR analysis was performed with a Lightcycler 480 machine (Roche). mRNAs levels was normalized to that of GAPDH control. Primer sequences can be found in Supplementary Table 3.
Immunofluorescence
Primary cilia were visualized in cells grown to high density, serum starved overnight (0.5% serum), and stained with anti-acetylated tubulin antibody (T6793, 1:1000, Sigma) diluted in PBS/0.2% Triton X-100/5% goat serum after fixation in 4% paraformaldehyde (Sigma). Images were collected using a Nikon microscope with a 63× objective.
Covalently coupling antibody to Protein A agarose beads
100 ul of anti-SUFU antibody from Cell Signaling Technologies (Cat. No. 2522) was diluted with 10 ml PBS, and mixed with 500 ul Protein A agarose beads by gentle agitation for 60 min at room temperature. Beads were washed twice with 0.2 M sodium borate (pH 9.0) pre-heated to 50°C, and suspended into 10 ml 0.2 M sodium borate (pH 9.0). Crosslinking was initiated by the addition of DMP (dimethyl pimelimidate, D8388, Sigma) at a final concentration of 20 mM, and the mixture was incubated 30 min at RT with gentle agitation. The reaction was terminated with wash using 0.2 M ethanolamine (pH 8.0)(E9508, Sigma), and followed by 2 hrs incubation at RT. Antibody crosslinked beads was then washed 3 times with PBS and stored in PBS with 0.01% sodium azide (S2002, Sigma).
Immunoblotting and immunoprecipitation
For immunoblotting, cells were lysed in 1× protein sample loading buffer, and proteins were separated on SDS-PAGE (BioRad Criterion TGX Precast Gels). For immunoprecipitation or IgG pull-down studies, cells were lysed in lysis buffer (PBS with 1% NP-40). Cleared lysates were mixed with Protein A agarose beads in the presence or absence of 2 μg of desired antibody and rotated for 4 hrs at 4°C. For immunoprecipitation of endogenous SUFU proteins, cleared lysate from 10 × 15 cm2 dish cells were mixed with 100 ul Protein A agarose beads covalently coupled with anti-SUFU antibody, and rotated overnight at 4°C. Beads were then washed three times with lysis buffer. Bound proteins were eluted using 2× protein sample loading buffer and separated on SDS-PAGE. Detailed information for all antibodies used in immunoblotting and immunoprecipitation is provided in Supplementary Table 2. SUFU-P antibody was provided by Steven Cheng (Nanjing Medical University).
In vitro kinase assay
The coding region of human SUFU (residues 27–476) was cloned into a modified pET-28(a) vector (Novagen) that encodes an N-terminal His6-tag followed by a recognition site for human rhinovirus 3C protease. The plasmid was transformed into the E. coli strain BL21(DE3), and protein expression was induced by 0.2 mM IPTG at 16°C overnight. The protein was purified by using a 1 mL HisTrap column (GE Healthcare) and treated with recombinant human rhinovirus 3C protease at 4°C overnight to remove the N-terminal tag. The protein was further purified by using Resource Q (GE Healthcare) anion-exchange chromatography with a 10 mM to 1000 mM NaCl gradient elution, followed by gel filtration chromatography using Superdex 200 HR 10/30 (GE Healthcare) equilibrated with the buffer containing 20 mM Tris (pH 8.0), 150 mM NaCl, 10% glycerol, and 2 mM DTT. The purified protein was concentrated and stored at −80°C. 100 ng of purified SUFU protein was mixed with Protein A agarose beads bound with control and MCL-1 IgG Fc fusion proteins produced in a 10cm2 dish of HEK293 cells. The mixtures were incubated for 4 hrs at room temperature with gentle agitation. Mixtures were then brought up to 1× kinase reaction buffer (50mM Tris-HCl, 10mM MgCl2, pH 7.5) and 5 μCi of [γ-32P]ATP (3000 Ci/mmol) and 5000 units of PKAc (NEB) or BSA (Sigma). Mixtures were incubated at 30°C for 30 min. Samples were then resolved on SDS-PAGE and radiolabeled protein visualized by autoradiography.
MCL-1 protein expression, SUFU BH3 peptide synthesis, and NMR analysis
Homogeneously 15N labeled human MCL-1 protein was produced in E. coli based on the standard minimal M9 medium protocol using 15N ammonium chloride61. MCL-1 purification and processing with calpain were performed as previously reported to generate cMCL-1 (herein referred to as MCL-1), the stable BC of MCL-1 produced by calpain proteolysis27. We previously assigned the backbone 1H and 15N resonances of MCL-14. The human SUFU BH3 peptide, RRLSGKDTEQIRETLRRGLEINSKPVLPPINPQ, was synthesized, HPLC purified, and its molecular weight was confirmed by mass spectrometry (Supplementary Fig. 2f) at the peptide synthesis facility of the Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital. 15N/1H TROSY NMR titrations of 15N-labeled cMCL-1 with unlabeled SUFU BH3 peptide were performed at 25°C in a buffer containing 20 mM phosphate, pH 6.8, and 10% D20 (Cambridge Isotope). The SUFU BH3 peptide stock was prepared at 25 mM in deuterated dimethyl sulfoxide (Chembridge Isotope). The DMSO control titration was performed under the same conditions excluding the peptide. 15N–1H chemical-shift perturbations were calculated as CSPs = [(Δ1H p.p.m.)2 + (Δ15N p.p.m./5)2)]1/2.
Mitochondrial fractionation
Cells from a 10cm2 culture dish were scraped into microcentrifuge tubes, washed with cold PBS and collected by low speed centrifugation. Cells were incubated for 10 min in 500 μl of cold hypotonic buffer (10 mM HEPES [pH 7.5], 1 mM EDTA, 70 mM sucrose, 200 mM mannitol) containing protease inhibitors (Roche). Cells were then passed through a 25 gauge needle twenty four times on ice and resulting lysate was cleared by centrifugation (4°C, 10 min, 700 g). The supernatant was separated by a second centrifugation step (4°C, 10 min, 9,000 g). The supernatant contains the cytosolic material while the pellet contains mitochondrial membranes and their associated proteins.
CRISPR Cas9 genome editing
Genome editing was achieved using CRISPR Cas9 technique in the following cell lines: MEF (Bcl2 KO), C3H10T1/2 (Bcl2-∆TM, Mcl1-∆TM, Mcl1 KO, SufuΔBH3, Sufu KO), HAP1 (SUFUΔBH3 and SUFU KO), and RMS13 (SUFUΔBH3 and SUFU KO). In brief, cells were transfected with Cas9 DNA and a sgRNA expressing plasmid. Cells were clonally selected using puromycin (0.5ug/ml) for 4 days followed by another 4 days without selection for expansion. Isolated and further expanded clones were used to generate lysates for Western blot analysis. Candidate clones were subjected to genomic sequencing using amplicons flanking sgRNA-targeting site. sgRNA sequences used in the study are available in Supplementary Table 3.
Mcl1 deletion in liver tissue
Liver tissue was isolated from Mx1-Cre1; Mcl1 fl/fl mice 11 days after three doses of pIpC as previously described5.
Xenograft studies
Female J:NU nude mice (5 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and housed in accordance with the Institutional Animal Care guidelines. All animal studies were performed in accordance to approved protocols by UT Southwestern Medical Center Institutional Animal Care and Use Committee (IACUC). Mice were divided into four groups of five mice each using power calculation derived from the outcomes of Eichenmuller et al63. 1×107 exponentially growing RMS13 cells were injected subcutaneously into the right flank above the hindlimb of each mouse. Two groups of mice were injected with RMS13-SUFU-G64V cells, and the other 2 groups with RMS13-SUFU-N328K cells. 10 days post RMS13 cell injection animals with tumors averaging 4×4 mm2 were randomized into two groups with each group subsequently receiving on a daily basis either 100mg/kg of ABT-199 or vehicle for 18 consecutive days. ABT-199 was formulated for oral dosing in 60% phosal 50 propylene glycol (PG), 30% polyethylene glycol (PEG) 400 and 10% ethanol as described in64. Tumor size was measured every other day using a caliper, and tumor volume was calculated using the formula X × Y × Y × 3.1415/6. Body weight of control and ABT-199 treated animals were recorded every two days for the duration of the study. p values were calculated using two-sided Student’s t-test analysis. All data are shown as mean + standard deviation (s.d.). Study participants were aware of control and ABT-199 treated animal group allocation during outcome assessment.
SHH subgroup medulloblastoma gene expression profiling
The gene expression analysis shown for candidate genes in medulloblastoma and normal cerebellum tissues was compiled from multiple gene expression profiling studies11, 61–66.
Code availability
The MAS5.0 algorithm of the GCOS program (Affymetrix Inc.) was used to normalize the medulloblstoma gene expression data. Data were analyzed using the R2 software for analysis and visualization of microarray data (see http://r2.amc.nl).
Statistics and reproducibility
All experiments were repeated at least twice unless otherwise indicated. N numbers are indicated in the figure legends. Sample sizes were not pre-determined on the basis of statistical power calculation. No formal randomization technique was used. Investigators were not blinded to the group allocation during the experiments. Quantitative data are presented as mean ± standard deviation (s.d.) from at least three independent measurements. Statistical testing was performed using the two-sided Student’s t-test. A P value of 0.05 was considered as a borderline for statistical significance.
Data availability
Published microarray data that was re-analyzed in this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE4924311, GSE1299261, GSE1032766, GSE3741864, GSE352665, and EGAS0000100195363. The source data for statistical analysis for Figures 1e–f, 1h, 2l, 3a, 3c–f, 3i, 4c, 4g, 5b–c, 5f–g, 5k–l, 6b, and Supplementary Figures 1a, 2c, 3a, 3h, 3k–l, and 5d–e is provided as Supplementary Table 4. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
We thank C. Chen, R. Toftgård, X. Wang, S. Skypek, M.P. Scott, and J.K. Chen for reagents. This work was supported in part by the Welch Foundation (I-1665, L.L.), CPRIT (RP130212, L.L). Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA168761 (LL), R01CA196851 (LL), and P50-CA70907 (Minna). Q. Barrett was supported by a T32 training grant (5T32CA124334). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. SYC was supported by the National Basic Research Program of China (2012CB945003).
Footnotes
Note: Supplementary Information is available in the online version of the paper.
AUTHOR CONTRIBUTIONS
X.W., L-s.Z., J.T. Y-c.K., J.T.P., R.T., Q.B., C-w.F., T.M., and L.L. performed the experiments. X.W., L.D.W., M.K., S.Y.C., R.B., J.T.O, D.R.G, and L.L. helped with data analysis and discussions. X.W., L-s.Z., R.B., T.M., and L.L. conceived the study and experimental design. X.W. and L.L. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Bhola PD, Letai A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol Cell. 2016;61:695–704. doi: 10.1016/j.molcel.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarosiek KA, Letai A. Directly targeting the mitochondrial pathway of apoptosis for cancer therapy with BH3 mimetics: recent successes, current challenges and future promise. FEBS J. 2016 doi: 10.1111/febs.13714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charrier JB, Lapointe F, Le Douarin NM, Teillet MA. Anti-apoptotic role of Sonic hedgehog protein at the early stages of nervous system organogenesis. Development. 2001;128:4011–4020. doi: 10.1242/dev.128.20.4011. [DOI] [PubMed] [Google Scholar]
- 5.Chiang C, et al. Manifestation of the limb prepattern: limb development in the absence of sonic hedgehog function. Dev Biol. 2001;236:421–435. doi: 10.1006/dbio.2001.0346. [DOI] [PubMed] [Google Scholar]
- 6.Dierks C, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–249. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Hutchin ME, et al. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005;19:214–223. doi: 10.1101/gad.1258705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Machold R, et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- 9.te Welscher P, Fernandez-Teran M, Ros MA, Zeller R. Mutual genetic antagonism involving GLI3 and dHAND prepatterns the vertebrate limb bud mesenchyme prior to SHH signaling. Genes Dev. 2002;16:421–426. doi: 10.1101/gad.219202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonilla X, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. 2016 doi: 10.1038/ng.3525. [DOI] [PubMed] [Google Scholar]
- 11.Kool M, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–812. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sekulic A, Von Hoff D. Hedgehog Pathway Inhibition. Cell. 2016;164:831. doi: 10.1016/j.cell.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 14.Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010;24:670–682. doi: 10.1101/gad.1902910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191:415–428. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Llambi F, et al. BOK Is a Non-canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell. 2016 doi: 10.1016/j.cell.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perciavalle RM, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai CK, et al. Functional characterization of putative cilia genes by high-content analysis. Molecular biology of the cell. 2011;22:1104–1119. doi: 10.1091/mbc.E10-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacob LS, et al. Genome-wide RNAi screen reveals disease-associated genes that are common to Hedgehog and Wnt signaling. Science signaling. 2011;4:ra4. doi: 10.1126/scisignal.2001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bershteyn M, Atwood SX, Woo WM, Li M, Oro AE. MIM and cortactin antagonism regulates ciliogenesis and hedgehog signaling. Dev Cell. 2010;19:270–283. doi: 10.1016/j.devcel.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blair HJ, et al. Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC biology. 2011;9:14. doi: 10.1186/1741-7007-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorn KV, Hughes CE, Rohatgi R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev Cell. 2012;23:823–835. doi: 10.1016/j.devcel.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang C, Chen W, Chen Y, Jiang J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012;22:1593–1604. doi: 10.1038/cr.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, et al. Dual Phosphorylation of suppressor of fused (Sufu) by PKA and GSK3beta regulates its stability and localization in the primary cilium. The Journal of biological chemistry. 2011;286:13502–13511. doi: 10.1074/jbc.M110.217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Q, et al. Apoptotic regulation by MCL-1 through heterodimerization. The Journal of biological chemistry. 2010;285:19615–19624. doi: 10.1074/jbc.M110.105452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moldoveanu T, et al. BID-induced structural changes in BAK promote apoptosis. Nature structural & molecular biology. 2013;20:589–597. doi: 10.1038/nsmb.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clohessy JG, Zhuang J, de Boer J, Gil-Gomez G, Brady HJ. Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. The Journal of biological chemistry. 2006;281:5750–5759. doi: 10.1074/jbc.M505688200. [DOI] [PubMed] [Google Scholar]
- 30.Cohen NA, et al. A Competitive Stapled Peptide Screen Identifies a Selective Small Molecule that Overcomes MCL-1-Dependent Leukemia Cell Survival. Chem Biol. 2012;19:1175–1186. doi: 10.1016/j.chembiol.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cherry AL, et al. Structural basis of SUFU-GLI interaction in human Hedgehog signalling regulation. Acta crystallographica. Section D, Biological crystallography. 2013;69:2563–2579. doi: 10.1107/S0907444913028473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bigelow RL, et al. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. The Journal of biological chemistry. 2004;279:1197–1205. doi: 10.1074/jbc.M310589200. [DOI] [PubMed] [Google Scholar]
- 33.Regl G, et al. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004;64:7724–7731. doi: 10.1158/0008-5472.CAN-04-1085. [DOI] [PubMed] [Google Scholar]
- 34.Krajewski S, et al. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- 35.Yang T, Kozopas KM, Craig RW. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J Cell Biol. 1995;128:1173–1184. doi: 10.1083/jcb.128.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez D, Rojas-Rivera D, Hetz C. Integrating stress signals at the endoplasmic reticulum: The BCL-2 protein family rheostat. Biochim Biophys Acta. 2011;1813:564–574. doi: 10.1016/j.bbamcr.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 37.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16:273–284. doi: 10.1038/nrd.2016.253. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, et al. Structural insight into the mutual recognition and regulation between Suppressor of Fused and Gli/Ci. Nature communications. 2013;4:2608. doi: 10.1038/ncomms3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts WM, Douglass EC, Peiper SC, Houghton PJ, Look AT. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989;49:5407–5413. [PubMed] [Google Scholar]
- 42.Metcalfe C, de Sauvage FJ. Hedgehog fights back: mechanisms of acquired resistance against Smoothened antagonists. Cancer Res. 2011;71:5057–5061. doi: 10.1158/0008-5472.CAN-11-0923. [DOI] [PubMed] [Google Scholar]
- 43.Atwood SX, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342–353. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharpe HJ, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015;27:327–341. doi: 10.1016/j.ccell.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013;494:484–488. doi: 10.1038/nature11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metcalfe C, et al. PTEN loss mitigates the response of medulloblastoma to Hedgehog pathway inhibition. Cancer Res. 2013;73:7034–7042. doi: 10.1158/0008-5472.CAN-13-1222. [DOI] [PubMed] [Google Scholar]
- 47.Zhao X, et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway-Dependent Tumors. Cancer Res. 2015;75:3623–3635. doi: 10.1158/0008-5472.CAN-14-2999-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teglund S, Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805:181–208. doi: 10.1016/j.bbcan.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 49.Yauch RL, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–574. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 51.Borner C, et al. The protein bcl-2 alpha does not require membrane attachment, but two conserved domains to suppress apoptosis. J Cell Biol. 1994;126:1059–1068. doi: 10.1083/jcb.126.4.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Germain M, Duronio V. The N terminus of the anti-apoptotic BCL-2 homologue MCL-1 regulates its localization and function. The Journal of biological chemistry. 2007;282:32233–32242. doi: 10.1074/jbc.M706408200. [DOI] [PubMed] [Google Scholar]
- 53.Zhu W, et al. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- 54.Oliver TG, et al. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci U S A. 2003;100:7331–7336. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin KH, et al. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci Rep. 2016;6:27696. doi: 10.1038/srep27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hinds MG, et al. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell death and differentiation. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- 57.Zheng JH, Viacava Follis A, Kriwacki RW, Moldoveanu T. Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS J. 2015 doi: 10.1111/febs.13527. [DOI] [PubMed] [Google Scholar]
- 58.Green DR. A BH3 Mimetic for Killing Cancer Cells. Cell. 2016;165:1560. doi: 10.1016/j.cell.2016.05.080. [DOI] [PubMed] [Google Scholar]
- 59.Bai L, Wang S. Targeting apoptosis pathways for new cancer therapeutics. Annu Rev Med. 2014;65:139–155. doi: 10.1146/annurev-med-010713-141310. [DOI] [PubMed] [Google Scholar]
- 60.Shern JF, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer discovery. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fattet S, Haberler C, Legoix P, et al. Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. The Journal of pathology. 2009;218(1):86–94. doi: 10.1002/path.2514. [DOI] [PubMed] [Google Scholar]
- 62.Kool M, Jones DT, Jager N, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25(3):393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Northcott PA, Buchhalter I, Morrissy AS, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488(7409):43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roth RB, Hevezi P, Lee J, et al. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics. 2006;7(2):67–80. doi: 10.1007/s10048-006-0032-6. [DOI] [PubMed] [Google Scholar]
- 66.Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE. 2008;3(8):e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Published microarray data that was re-analyzed in this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE4924311, GSE1299261, GSE1032766, GSE3741864, GSE352665, and EGAS0000100195363. The source data for statistical analysis for Figures 1e–f, 1h, 2l, 3a, 3c–f, 3i, 4c, 4g, 5b–c, 5f–g, 5k–l, 6b, and Supplementary Figures 1a, 2c, 3a, 3h, 3k–l, and 5d–e is provided as Supplementary Table 4. All other data supporting the findings of this study are available from the corresponding author on reasonable request.