

SUMMARY
Mammals have evolved neurophysiologic reflexes such as coughing and scratching to expel invading pathogens and noxious environmental stimuli. It is well established that these responses are also associated with chronic inflammatory diseases such as asthma and atopic dermatitis. However, the mechanisms by which inflammatory pathways promote sensations such as itch remain poorly understood. Here, we show that type 2 cytokines directly activate sensory neurons in both mice and humans. Further, we demonstrate that chronic itch is dependent on neuronal IL-4Rα and JAK1 signaling. We also observe that patients with recalcitrant chronic itch that failed other immunosuppressive therapies markedly improve when treated with JAK inhibitors. Thus, signaling mechanisms previously ascribed to the immune system may represent novel therapeutic targets within the nervous system. Collectively, this study reveals an evolutionarily conserved paradigm in which the sensory nervous system employs classical immune signaling pathways to influence mammalian behavior.
Keywords: Atopic dermatitis, itch, pruritus, pruriceptor, type 2 cytokines, IL-4, IL-13, IL-4Rα, JAK1
eTOC
Type 2 cytokines directly stimulate itch sensory neurons and blocking this pathway is effective in a proof-of-concept study in patients with recalcitrant chronic itch
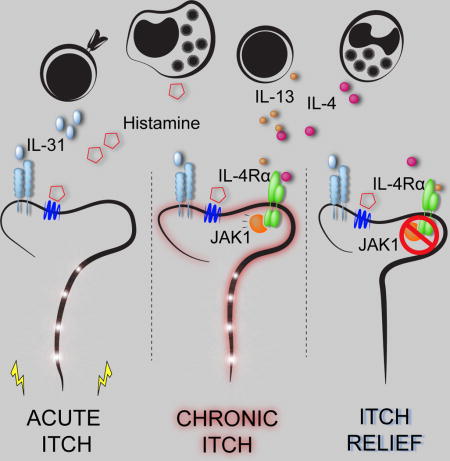
INTRODUCTION
The nervous and immune systems work in concert to sense potential pathogens and provide host-protective responses (Ordovas-Montanes et al., 2015; Veiga-Fernandes and Mucida, 2016). Emerging evidence has demonstrated extensive crosstalk between these systems at multiple barrier surfaces including the gut (Gabanyi et al., 2016), lung (Talbot et al., 2015), and skin (Kashem et al., 2015; Riol-Blanco et al., 2014). These interactions orchestrate multifaceted responses including neurophysiologic reflexes such as coughing and scratching to expel invading pathogens and noxious environmental stimuli (Bautista et al., 2014; Veiga-Fernandes and Mucida, 2016). Although evidence suggests that neuroimmunologic processes have coevolved to promote host survival, whether dysregulation of these responses can drive sensory dysfunction such as chronic itch remains poorly understood.
Chronic itch, defined as symptoms of itch lasting longer than six weeks, affects up to 15% of the population and has a profoundly negative impact on quality of life (Kini et al., 2011; Matterne et al., 2011; Yosipovitch and Bernhard, 2013). While chronic itch is a central feature of many primary inflammatory skin disorders such as atopic dermatitis (AD), it also presents secondarily to a variety of medical conditions and numerous neuropathies (Mollanazar et al., 2016; Yosipovitch and Bernhard, 2013). Further, chronic itch can present in the absence of any known disease process, and, in this setting, has been described as chronic idiopathic pruritus (CIP) (Berger and Steinhoff, 2011; Xu et al., 2016). Alarmingly, despite its high incidence and enormous burden on quality of life, there are currently no medications specifically indicated for the treatment of chronic itch.
It is widely appreciated that skin inflammation can lead to itch (Bautista et al., 2014). For example, the pruritogen histamine is associated with acute itch in the setting of urticaria (Kaplan, 2004), and interleukin (IL)-31, a previously identified cytokine pruritogen, has been linked to AD-associated itch (Cevikbas et al., 2014; Dillon et al., 2004; Ruzicka et al., 2017; Sonkoly et al., 2006). However, the precise mechanisms by which proinflammatory mediators promote the sensation of itch remain poorly defined. In AD, the type 2 cytokines IL-4, IL-5, and IL-13 are known to drive skin inflammation (Gittler et al., 2012; Weidinger and Novak, 2016). Further, mice that overexpress IL-4 or IL-13 in the skin develop AD-like disease and robust chronic itch (Chan et al., 2001; Zheng et al., 2009). Notably, dupilumab, a recently approved monoclonal antibody that blocks both IL-4 and IL-13 signaling via their shared receptor subunit IL-4Rα, has demonstrated remarkable efficacy in treating AD (Beck et al., 2014; Simpson et al., 2016; Thaçi et al., 2016). Despite these observations, it remains unclear whether these canonical proinflammatory mediators also function as pruritogens. Further, whether downstream signaling pathways are conserved across the immune and nervous systems and can be targeted to treat chronic itch remains unexplored.
In this study, we demonstrate that sensory neurons are directly activated by the classical immune signaling molecules IL-4 and IL-13 along previously defined itch-sensory pathways in mice. Further, we show that IL-4 can activate human sensory neurons, provoking the hypothesis that neuronal type 2 cytokine signaling contributes to itch in both mice and humans. By employing sensory neuron-specific genetic deletion of IL-4Rα, we found a previously unrecognized mechanism by which neuronal IL-4Rα critically mediates chronic itch. Surprisingly, we found that, rather than triggering acute itch, activation of neuronal IL-4Rα sensitizes sensory neurons to multiple other pruritogens. Based on IL-4Rα signaling biology, we hypothesized that type 2 cytokine-mediated activation of sensory neurons would be dependent on neuronal Janus kinase (JAK) signaling. Consistent with this hypothesis, both pharmacologic JAK inhibition and sensory neuron-specific genetic deletion of JAK1 resulted in abatement of chronic itch in mice. Although JAK inhibitors are well-established anti-inflammatory agents (Schwartz et al., 2016), whether they exhibit additional neuromodulatory properties is not known. In support of this possibility, we observed a dramatic improvement of itch in a small cohort of CIP patients treated off-label with the JAK inhibitor tofacitinib after having failed other anti-inflammatory therapies. Thus, these signaling mechanisms previously ascribed to the immune system may represent new targets within the sensory nervous system for the treatment of chronic itch. Beyond itch, the discovery of these neuroimmunologic pathways may reveal new insights into sensory perception at multiple barrier surfaces and how these pathways modulate behavior.
RESULTS
Sensory neurons are directly activated by type 2 cytokines
Although type 2 cytokines are well-established mediators of skin inflammation in AD (Gittler et al., 2012; Weidinger and Novak, 2016), it is not clear how these cytokines promote chronic itch. When we examined mouse dorsal root ganglia (DRG), which contain sensory neurons that innervate the skin, we found expression of IL-4Rα (Il4ra) and IL-13Rα1 (Il13ra1) but not IL-5Rα (Il5ra) (Figure 1A). As expected, we also observed expression of the receptor for the known cytokine pruritogen IL-31 (Il31ra) (Figure 1A) (Cevikbas et al., 2014; Dillon et al., 2004; Sonkoly et al., 2006). Similarly, human DRG also expressed IL-4Rα (IL4RA), IL-13Rα1 (IL13RA1), and IL-31RA (IL31RA), but not IL-5Rα (IL5RA) (Figure 1B). Further, we found that murine DRG and trigeminal ganglia (TG) express comparable patterns of Il4ra, Il13ra1, and Il31ra (Figure 1C). Taken together, these findings suggest that the type 2 cytokines IL-4 and IL-13 may directly activate sensory neurons.
Figure 1. Type 2 cytokines activate mouse and human sensory neurons.
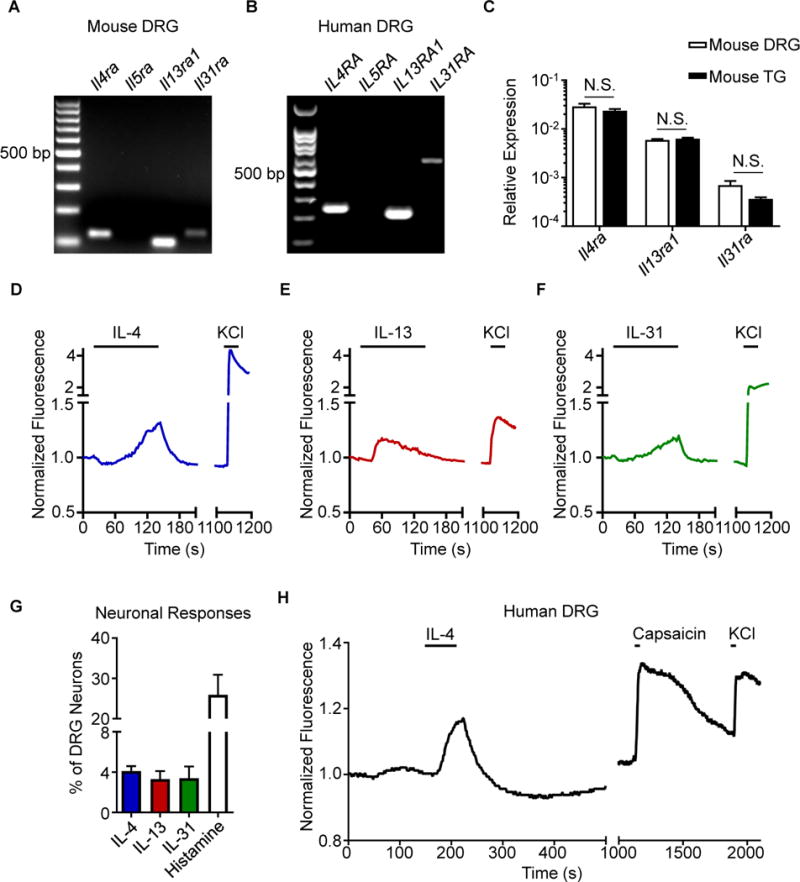
(A) Representative gel of RT-PCR product of whole mouse dorsal root ganglia (DRG), n = 4 wild type (WT) mice.
(B) Representative gel of RT-PCR product of whole human DRG, N = 3 donors.
(C) Comparison of Il4ra, Il13ra1, and Il31ra expression by RT-qPCR in whole mouse DRG and trigeminal ganglia (TG), n = 4 WT mice.
(D–F) Representative calcium imaging trace of mouse DRG neurons in response to (D) recombinant murine (rm)IL-4 (300 nM), (E) rmIL-13 (300 nM), and (F) rmIL-31 (300 nM) and potassium chloride (KCl, 100 mM).
(G) rmIL-4-, rmIL-13-, rmIL-31-, and histamine-responsive DRG neurons as a percentage of total KCl-responsive neurons, n > 500 neurons from 2 WT mice.
(H) Representative calcium imaging trace of human DRG neurons in response to recombinant human IL-4 (300 nM), capsaicin (500 nM), and KCl (50 mM), n > 40 neurons from a male donor. Data are represented as mean ± SEM. Black bars in calcium imaging traces indicate timing of challenges. See also Figure S1.
We next sought to functionally test whether type 2 cytokines can directly stimulate sensory neurons using ratiometric calcium imaging. Stimulation of mouse DRG neurons with IL-4 (Figure 1D) or IL-13 (Figure 1E) resulted in responses similar to IL-31 stimulation (Figure 1F) in a subset of neurons. Overall, IL-4 or IL-13 stimulation led to calcium responses in 4.1% and 3.3% of total DRG neurons, respectively (Figure 1G), and the frequency of sensory neurons responsive to IL-4 and IL-13 increased in a dose-dependent manner (Figures S1A–D). Consistent with previous reports (Cevikbas et al., 2014; Liu et al., 2009), approximately 3.3% and 20% of neurons were responsive to IL-31 and histamine, respectively (Figure 1G), demonstrating that IL-4 and IL-13 stimulation results in responses from a proportion of neurons similar to other canonical pruritogens like IL-31. As expected due to the lack of Il5ra expression (Figure 1A), IL-5 stimulation did not trigger responses from DRG neurons (Figures S1E–F). Collectively, these results demonstrate that type 2 cytokines can directly activate a subset of mouse sensory neurons.
Given the similar expression profile of type 2 cytokine receptors in mouse and human DRG (Figures 1A–B), we next sought to examine whether human DRG neurons would also respond directly to type 2 cytokines. Thus, we obtained viable sensory neurons from human cadaveric donors and stimulated these cells with IL-4 (Valtcheva et al., 2016). Similar to our observations in mice (Figure 1D), we found that a subset of human sensory neurons directly responded to IL-4 (Figure 1H). Together, these findings indicate that sensory neuron responses to type 2 cytokines such as IL-4 are conserved between mice and humans.
Type 2 cytokines activate itch-sensory pathways
To thoroughly characterize neurons that express type 2 cytokine receptors, we reanalyzed two previously published data sets of neuronal transcriptional profiles (Chiu et al., 2014; Usoskin et al., 2015). First, we found that Il4ra expression was favored on sort-purified non-peptidergic and peptidergic pruriceptive and nociceptive sensory neurons compared to proprioceptive neurons (Figure S2A) (Chiu et al., 2014). Second, using a single-cell RNA-seq data set (Usoskin et al., 2015), we found that families of small-diameter neurons predicted to mediate itch (NP1, NP2, and NP3) were enriched in neurons that expressed Il4ra and Il13ra1 compared to other families of nociceptive (PEP1 and PEP2) and mechanoreceptive (TH) neurons (Figure 2A). Although previously defined pruriceptors such as Mas-related G protein-coupled receptor A3 (MrgprA3) and IL-31RA were highly specific to defined itch subsets, Il4ra expression spanned multiple pruriceptive clusters (Figure 2A). Consistent with these findings, we found that IL-4- and IL-13-responsive neurons were predominantly small-diameter neurons, similar to histamine-responsive neurons, using calcium imaging (Figure 2B, Figure S2B). Next, to examine whether IL-4 or IL-13 directly activates established itch-sensory pathways, we exposed DRG neurons to successive challenges with defined pruritogens. When DRG neurons were serially stimulated with IL-4, and then IL-31, followed by histamine, 89% of IL-4-responsive neurons also responded to IL-31, histamine, or both (Figure 2C). Similarly, 75% of IL-13-responsive neurons also responded to IL-31, histamine, or both (Figure S2C). Additionally, 63% of neurons that responded to IL-4 also responded to IL-13 (Figure 2D), while 45% of IL-4-responsive neurons also responded to the previously reported cytokine pruritogen thymic stromal lymphopoietin (TSLP) (Wilson et al., 2013a) (Figure S2D). Collectively, these studies indicate that both IL-4 and IL-13 can activate families of neurons that include previously identified itch-sensory pathways.
Figure 2. Type 2 cytokines activate itch-sensory pathways.
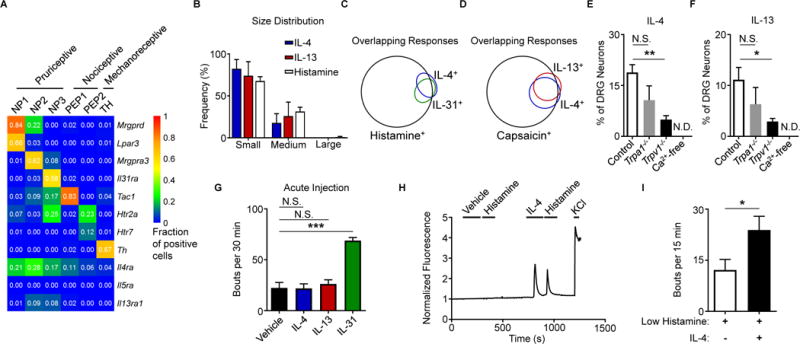
(A) Expression of selected genes in mouse DRG neuron populations clustered into functional subsets based on single-cell RNA-seq data. Numbers indicate fraction of positive cells within individual clusters by thresholding method. Full data set and methods available in Usoskin et al., 2015.
(B) Representative size distribution of rmIL-4-, rmIL-13-, and histamine-responsive mouse DRG neurons classified as small-, medium-, and large-diameter neurons (<18 μm, 18–25 μm, and >25 μm), n > 500 neurons from a WT mouse.
(C–D) Representative Venn diagrams of overlapping responses of mouse DRG neurons to stimulation with rmIL-4 with subsequent challenge with (C) rmIL-31 (300 nM) and histamine (50 μM) or with (D) rmIL-13 (4.0 μg/mL) and capsaicin (300 nM), n > 300 neurons from a WT mouse.
(E–F) Responses to (E) rmIL-4 (4 μg/mL) and (F) rmIL-13 (4 μg/mL) of mouse DRG neurons from Trpa1−/− mice, Trpv1−/− mice, and in calcium-free conditions as a percentage of total KCl-responsive neurons compared to WT control mice, n > 300 neurons per group.
(G) Total scratching bouts elicited in response to intradermal (i.d.) cheek injection of vehicle control (0.1% BSA in PBS, 10 μL), rmIL-4, rmIL-13, or rmIL-31 (all 2.5 μg/10 μL), n ≥ 5 mice per group.
(H) Representative calcium imaging trace of mouse DRG neurons responding to low levels of histamine (5 μM) after vehicle control or rmIL-4 (4 μg/mL) challenge, n > 200 neurons from 2 WT mice.
(I) Cheek-directed scratching bouts elicited in response to i.d. cheek injection of a low dose of histamine (4 mM, 10 μL) with or without rmIL-4 (2.5 μg), n ≥ 7 mice per group.
Data are represented as mean ± SEM. Black bars in calcium imaging traces indicate timing of challenges. See also Figure S2.
Earlier studies have shown that pruritogens such as histamine and the MrgprA3 agonist chloroquine are differentially dependent on the transient receptor potential (TRP) channels TRPA1 and TRPV1 for their activity via extracellular calcium influx (Liu et al., 2009; Wilson et al., 2013b). Given the substantial overlap in IL-4- and IL-13-responsive neurons with neurons responsive to the TRPV1 agonist capsaicin (91%, Figure 2D), we sought to test whether neuronal activation by type 2 cytokines is dependent on these TRP channels. Neurons from both Trpa1−/− and Trpv1−/− mice demonstrated reduced responses to both IL-4 (Figure 2E) and IL-13 (Figure 2F), although only significantly reduced in Trpv1−/− mice, and these responses were entirely dependent on extracellular calcium (Figures 2E–F). Together, our findings indicate that type 2 cytokines directly activate sensory neurons via TRP channel-dependent calcium influx.
Based on our findings that type 2 cytokines directly activate itch-sensory neurons, we hypothesized that intradermal (i.d.) administration of IL-4 and IL-13 would evoke acute itch. Surprisingly, in contrast to IL-31, high doses of either IL-4 or IL-13 did not elicit acute itch (Figure 2G). Thus, these findings suggest that type 2 cytokines do not function as potent acute pruritogens despite activating itch-sensory neurons.
Several studies have shown that type 2 inflammation-associated cytokines can modulate the transcriptional activity and responses of multiple immune cell populations to subsequent stimuli (Halim et al., 2016; Martinez-Gonzalez et al., 2016). Thus, we hypothesized that sensory neurons would similarly respond to type 2 cytokines by altering their responsiveness to other pruritogens. To test this, we exposed DRG neurons to multiple pruritogens including histamine (Figure 2H), chloroquine (Figure S2E), TSLP (Figure S2F), and IL-31 (Figure S2G) both before and after exposure to IL-4. We observed that IL-4 sensitized subsets of neurons to previously subthreshold levels of histamine (Figure 2H) as well as other pruritogens (Figures S2E–G). In support of these findings, an i.d. co-injection of a low dose of histamine with IL-4 caused significantly more scratching in mice than histamine alone (Figure 2I). Therefore, our findings demonstrate that although type 2 cytokines are not potent acute pruritogens themselves, they act to sensitize sensory neurons to many different pruritogens. These observations mirror prior human studies that have shown that challenge with pruritogens in lesional skin of AD patients results in amplified itch responses compared to control subjects (Andersen et al., 2017; Ikoma et al., 2003). Thus, we hypothesized that, rather than acute itch, neuronal type 2 cytokine signaling promotes pathologic chronic itch and that interrupting these signals may represent an effective strategy to target chronic itch.
Neuronal type 2 cytokine signaling is necessary for the development of chronic itch
To explore the role of type 2 cytokines in chronic itch, we employed an established model of AD-like skin inflammation in which mice were treated with the topical irritant MC903 (calcipotriol) (Figure S3A) (Kim et al., 2013, 2014; Li et al., 2006). Compared to control mice treated with vehicle only (ethanol, EtOH), MC903-treated mice developed robust skin inflammation as measured by skin thickening (Figure S3B), histologic features of AD (Figures S3C–D), and a characteristic AD-associated RNA-seq profile (Figure S3E), as well as marked chronic itch (Figure S3F). To ensure that MC903 does not directly stimulate sensory neurons to elicit itch, we challenged sensory neurons with MC903 and did not observe significant activation (Figure S3G).
To directly investigate the contributions of sensory neuron-intrinsic type 2 cytokine signaling to chronic itch, we generated mice that lack IL-4Rα specifically on sensory neurons (NaV 1.8-Cre+ Il4rafl/fl, IL-4RαΔneuron). Deletion of Il4ra, but not Il2rg or Il13ra1, was confirmed in sensory ganglia of IL-4RαΔneuron mice (Figure 3A). Further, we confirmed diminished responses to IL-4 and IL-13 in sensory neurons from IL-4RαΔneuron mice (Figures 3B–C). In contrast, sensory neurons from IL-4RαΔneuron mice showed normal responses to other pruritogens including IL-31 (Figure 3D) and histamine (Figure 3E) indicating no gross developmental abnormalities in these mice. Strikingly, upon induction of skin inflammation (Figure 3F), IL-4RαΔneuron mice demonstrated a significant reduction in scratching behavior compared to littermate controls (Figure 3G). This decrease was greater than what we observed in Il31ra−/− mice treated with MC903 (Figures S4A–C) and comparable to decreases in scratching behavior found in other models of itch in which pruriceptors were globally deleted (Liu et al., 2009; Morita et al., 2015). Additionally, skin inflammation in MC903-treated IL-4RαΔneuron mice as reduced as determined by ear thickness measurements (Figure 3H) and histopathologic assessment (Figures 3I–J). IL-4RαΔneuron mice also demonstrated a reduction in type 2 immune cells in the skin (Figures S4D–G) and a distinct skin transcriptional profile (Figure S4H). Thus, deletion of neuronal IL-4Rα signaling results in attenuation of both itch and skin inflammation.
Figure 3. Neuronal IL-4Rα expression is necessary for chronic itch.
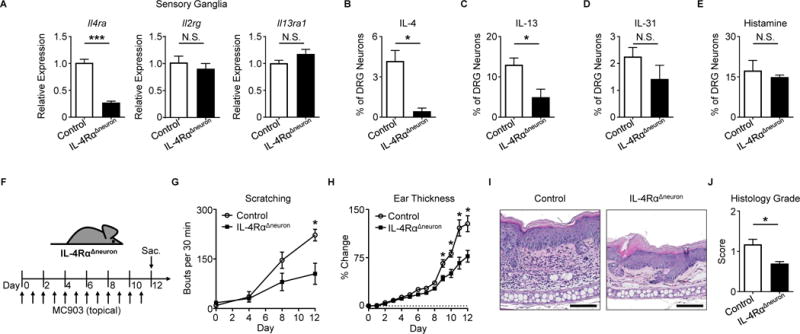
(A) Quantification of Il4ra, Il2rg, and Il13ra1 expression by RT-qPCR in whole sensory trigeminal ganglia (TG) of IL-4RαΔneuron mice, n ≥ 4 mice per group.
(B–E) Representative responses of DRG neurons from an IL-4RαΔneuron mouse to (B) rmIL-4 (1 μg/mL), (C) rmIL-13 (4 μg/mL), (D) rmIL-31 (3 μM), and (E) histamine (50 μM) as a percentage of total KCl-responsive neurons compared to neurons from a littermate control mouse, n > 300 neurons per group.
(F) Experimental schematic indicating daily topical treatment with MC903 to the ears of IL-4RαΔneuron and littermate control mice.
(G) Scratching behavior of IL-4RαΔneuron mice compared to littermate control mice over the course of MC903 treatment, n ≥ 7 mice per group.
(H) Ear thickness measurements, (I) representative H&E histopathology, and (J) histology score of MC903-treated IL-4RαΔneuron mice compared to littermate control mice, n ≥ 4 mice per group. Scale bars indicate 100 μm. Data are represented as mean ± SEM. See also Figures S3–S5.
Given that skin inflammation was reduced, we sought to ensure that the decrease in scratching behavior in these mice was not due to spurious deletion of IL-4Rα in non-neuronal tissues. Although the NaV1.8-Cre system has high specificity for sensory neurons (Agarwal et al., 2004), we elected to investigate the fidelity of this Cre in the setting of skin inflammation. While we observed NaV1.8 expression in cells from the DRG under steady-state (Figures S5A–B) and inflammatory conditions (Figure S5C), no evidence of NaV1.8 expression was found in cells from the skin or skin-draining lymph nodes (Figures S5A–C). Additionally, analysis of the skin and skin-draining lymph nodes after MC903 treatment did not yield differences in expression levels of Il4ra, Il2rg, or Il13ra1 in IL-4RαΔneuron mice (Figures S5D–F). Overall, these findings demonstrate that neuronal IL-4Rα signaling is critical for the elicitation of chronic itch.
Disruption of neuronal JAK1 signaling reduces chronic itch
Given that type 2 cytokines are known to signal through JAK-dependent pathways in immune cells (Kelly-Welch et al., 2003), we chose to examine whether itch-sensory neurons also express JAK signaling components. Again using a previously published single-cell RNA-seq data set (Usoskin et al., 2015), we identified that JAK1 is enriched in pruriceptive neurons (Figure S6A). We then examined whether activation of IL-4Rα on sensory neurons results in increased JAK1 signaling and found that, following stimulation with IL-4, more sensory neurons exhibited JAK1 phosphorylation (Figures S6B–D). To functionally assess whether type 2 cytokines activate sensory neurons in a JAK1-dependent manner, we generated mice in which JAK1 is conditionally deleted in sensory neurons (NaV1.8-Cre+ Jak1fl/fl, JAK1Δneuron) and found that neuronal IL-4 responses were abrogated in the absence of JAK1 using calcium imaging (Figure 4A). Given the importance of JAK1 in many cellular processes, we sought to ensure that neurons from JAK1Δneuron mice were developmentally and functionally intact. We performed a morphological assessment of DRG neurons and found a comparable size distribution between JAK1Δneuron mice and littermate controls (Figure S6E). Additionally, no significant differences were observed in neuronal responses across a number of pruritogens (Figures S6F–I). Rotarod performance was also found to be equivalent between JAK1Δneuron mice and controls (Figures S6J–K). Collectively, these data indicate that neuronal development and function are intact in JAK1Δneuron mice. Thus, given the enrichment of JAK1 expression in pruriceptive neurons (Figure S6A) and our data demonstrating the dependence of chronic itch on neuronal IL-4Rα (Figures 3F–G), we hypothesized that neuronal JAK1 would be required for chronic itch.
Figure 4. Disruption of neuronal JAK1 reduces chronic itch.
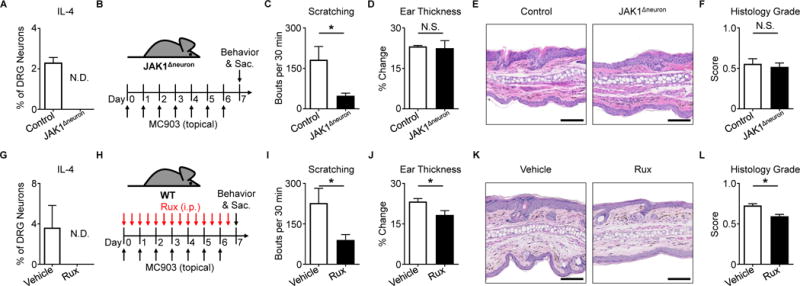
(A) Representative responses to rmIL-4 (1 μg/mL) of DRG neurons from a JAK1Δneuron mouse compared to a littermate control mouse as a percentage of total KCl-responsive neurons, n ≥ 200 neurons per group.
(B) Experimental schematic indicating MC903 treatment of JAK1Δneuron and littermate control mice.
(C) Scratching behavior, (D) ear thickness measurement, (E) representative H&E histopathology, and (F) histology score of JAK1Δneuron mice compared to littermate control mice on Day 7, n ≥ 8 mice per group.
(G) Representative responses to rmIL-4 (1 μg/mL) of DRG neurons from a WT mouse after incubation with ruxolitinib (Rux, 1 μg/mL) compared to vehicle control as a percentage of total KCl-responsive neurons, n ≥ 200 neurons per group.
(H) Experimental schematic indicating twice daily intraperitoneal (i.p.) injection of vehicle control or Rux (100 μg) during MC903 treatment to the ears of WT mice.
(I) Scratching behavior, (J) ear thickness measurement, (K) representative H&E histopathology, and (L) histology score of vehicle control and Rux-treated mice on Day 7, n ≥ 10 mice per group.
Scale bars indicate 100 μm. Data are represented as mean ± SEM. See also Figure S6.
To directly investigate the dependence of chronic itch on neuronal JAK1 signaling, we treated JAK1Δneuron mice with MC903 (Figure 4B). Upon induction of AD-like disease, JAK1Δneuron mice exhibited a marked reduction in scratching behavior (Figure 4C) even in the presence of skin inflammation (Figures 4D–F). To complement this genetic approach, we employed pharmacologic inhibition of JAK signaling using the JAK inhibitor ruxolitinib. Similar to our findings in JAK1Δneuron mice (Figure 4A), we found that application of ruxolitinib to sensory neurons resulted in a reduction in responses to IL-4 (Figure 4G). We then used MC903 treatment to induce AD-associated itch in WT mice while concurrently treating with ruxolitinib (Figure 4H) and observed that systemic intraperitoneal (i.p.) delivery of ruxolitinib significantly reduced scratching behavior (Figure 4I). Surprisingly, skin thickening (Figure 4J) and histologic parameters of AD-like disease (Figures 4K–L) demonstrated only a slight reduction in the setting of systemic ruxolitinib treatment, despite its well-established anti-inflammatory properties. Thus, we speculated that the anti-itch effects of ruxolitinib may be mediated predominantly through direct neuronal JAK inhibition rather than through suppression of skin inflammation. To test whether JAK1 signaling is necessary for itch even in the absence of robust skin inflammation, we employed a model of chronic itch not associated with overt inflammation using twice daily treatment with a mixture of acetone, ether, and water (AEW) (Miyamoto et al., 2002; Valtcheva et al., 2015; Wilson et al., 2013b). Strikingly, we found that JAK1Δneuron mice treated with AEW scratched significantly less than littermate controls (Figures S6L–M). Collectively, these findings demonstrate that neuronal JAK1 is a critical mediator of chronic itch even in the absence of robust skin inflammation. Thus, JAK inhibition may represent a broad therapeutic strategy for chronic itch disorders.
Chronic idiopathic pruritus (CIP) patients exhibit severe itch in the absence of overt skin inflammation
Chronic idiopathic pruritus (CIP) is an itch disorder of unknown etiology and, in contrast to AD, does not present with overt skin inflammation. However, CIP is strongly associated with aging and believed to be a manifestation of systemic immune senescence (Patel and Yosipovitch, 2010; Reich et al., 2011). As a result, it has been proposed that CIP patients exhibit a mild type 2 immune profile due to the loss of type 1 immunity (Berger and Steinhoff, 2011). In support of this, we recently showed that subsets of CIP patients exhibit features of systemic type 2 inflammation associated with low-grade peripheral eosinophilia and elevation of IgE (Xu et al., 2016). Although successful elimination of skin inflammation in AD limits itch symptoms, patients with CIP frequently suffer from severe itch even with potent immunosuppression.
Unlike AD, a characteristic inflammatory skin disease (Figure 5A), CIP presents with grossly normal skin findings (Figure 5B). AD histopathology demonstrates features of skin inflammation including hyperkeratosis, acanthosis, and a mixed dermal inflammatory infiltrate (Figure 5C). In stark contrast, histopathology of CIP exhibits only minimal inflammation upon biopsy from even the most pruritic cutaneous sites (Figure 5D). Indeed, histologic grading of patient biopsies confirmed that CIP patients exhibit markedly lower levels of skin inflammation compared to AD patients (Figure 5E). Remarkably, despite much milder skin inflammation, CIP patients report more itch that AD patients (Figure 5F). Thus, chronic itch can manifest both in the setting of robust skin inflammation as well as in the absence of notable inflammatory processes.
Figure 5. CIP is a distinct chronic itch disorder that exhibits severe itch despite minimal skin inflammation.
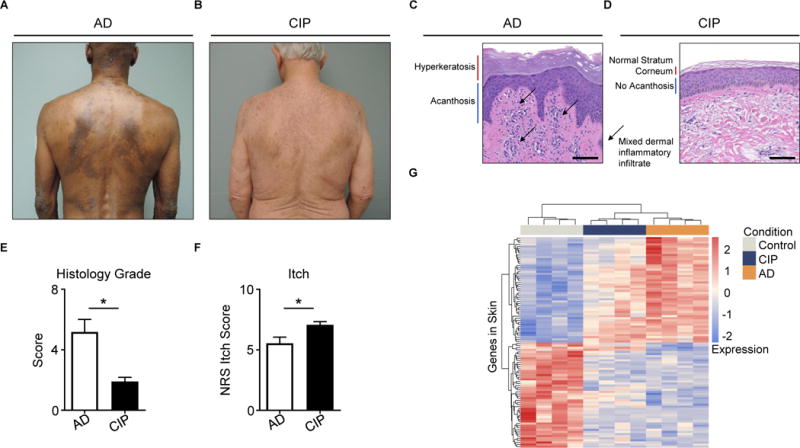
(A–B) Representative clinical pictures of (A) atopic dermatitis (AD) and (B) chronic idiopathic pruritus (CIP).
(C–D) Representative H&E histopathology of pruritic sites from (C) AD and (D) CIP.
(E) Histology score of AD and CIP patient biopsies, N ≥ 4 biopsies per group.
(F) Numerical Rating Scale (NRS) itch scores of AD and CIP patients, N ≥ 22 patients per group.
(G) Clustering of AD, CIP, and control skin samples by row Z-scores of the regularized logarithm of gene expression values of the top 100 differentially expressed genes from RNA-seq of AD versus control skin, N = 4 donors per condition.
Scale bars indicate 100 μm. Data are represented as mean ± SEM. See also Tables S1–S3.
To characterize CIP at the molecular level, we performed RNA-seq on skin from CIP patients, on lesional skin from AD patients, and on skin from control subjects (Table S1). Clustering of samples by expression of the top 100 most differentially expressed genes between control and AD skin revealed that CIP skin is more closely related to AD skin than control skin (Figure 5G). However, gene set enrichment analysis (GSEA) (Subramanian et al., 2005) comparing CIP and AD skin revealed a number of specific transcriptional programs that distinguish CIP from AD. This analysis showed that AD skin is associated with broad activation of immune responses (Table S2) and that CIP skin is enriched with a variety of transcriptional programs including those for cell morphology and calcium transport (Table S3). Similar studies examining the skin of mice with itch in the setting of minimal inflammation have uncovered homologous transcriptional programs (Wilson et al., 2013b). Collectively, these findings demonstrate that CIP is a chronic itch disorder that shares molecular signatures with AD that distinguish it from control skin despite marked differences in inflammation.
JAK inhibition improves pruritus in patients with CIP
Recent studies in AD have demonstrated rapid and significant reduction of itch symptoms in response to treatment with JAK inhibitors (Bissonnette et al., 2016; Levy et al., 2015). While these studies sought to investigate JAK inhibition as an anti-inflammatory treatment, our preclinical finding that disruption of neuronal JAK1 signaling limits both inflammatory (Figures 4B–C) and non-inflammatory itch (Figures S6L–M) suggests that JAK inhibition may represent a novel neuromodulatory approach to target itch. Therefore, despite the absence of overt skin inflammation in CIP, we hypothesized that CIP patients may benefit from JAK blockade.
Due to the lack of approved therapeutics specifically for CIP, we prescribed off-label treatment with the JAK inhibitor tofacitinib to five patients with severe CIP. All of these patients had previously failed multiple other off-label treatments including potent immunosuppression (Table S4). However, one month following treatment with oral tofacitinib, all five patients demonstrated marked improvement in their itch (Figure 6A). Strikingly, patients who closely tracked their itch over time reported rapid onset of itch relief with JAK blockade in contrast to other prior immunosuppressive therapy (Figure 6B). Thus, these findings suggest that JAK inhibition may represent a novel therapeutic strategy for patients with chronic itch disorders that are resistant to conventional anti-inflammatory agents. Taken together, our study reveals that classical cytokine signaling pathways are functional within the sensory nervous system and mediate chronic itch. The discovery of the significance of these pathways may provide new insights into sensory biology and promote the development of new therapies.
Figure 6. Patients with refractory CIP improve when treated with JAK inhibition.
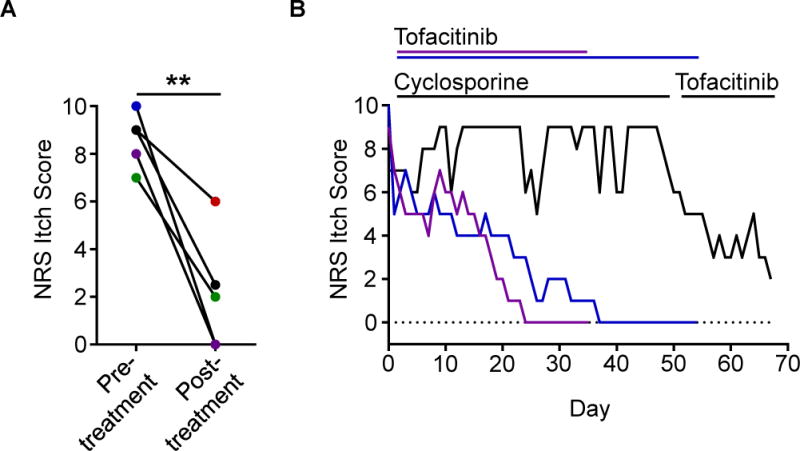
(A) NRS itch scores for a cohort of CIP patients (N = 5) given the JAK inhibitor tofacitinib. Individual patients are shown by unique colors. Significance was calculated using a paired t-test.
(B) Daily NRS itch scores of three CIP patients treated with tofacitinib including one patient treated with cyclosporine immediately preceding tofacitinib treatment (black).
See also Table S4.
DISCUSSION
Chronic itch is an example of how sensory responses that are normally employed by mammals to remove noxious stimuli can become dysregulated and pathologic. While itch is a highly debilitating symptom of multiple distinct medical conditions, chronic itch can also present in the absence of a clearly defined disorder as seen in CIP (Berger and Steinhoff, 2011; Mollanazar et al., 2016; Xu et al., 2016). Although itch is an often overlooked symptom in patient care, clinical studies have established that chronic itch has a profoundly negative impact on quality of life (Kini et al., 2011; Yosipovitch and Bernhard, 2013). However, despite its substantial burden on society, there are currently no specific treatments indicated for chronic itch.
In the setting of AD, the type 2 cytokines IL-4 and IL-13 coordinate an inflammatory response that is critical for disease pathogenesis and the eventual development of debilitating chronic itch. However, the mechanisms by which proinflammatory mediators such as the type 2 cytokines elicit itch remain poorly understood. In this study, we provide three conceptual advances that broaden our understanding of chronic itch. First, we demonstrate that activation of the classical immune signaling molecule IL-4Rα directly stimulates both mouse and human sensory neurons. Second, we show that sensory neuron-intrinsic activation of this signaling pathway (IL-4Rα and JAK1) is necessary for chronic itch in inflammatory and non-inflammatory settings. Finally, we show that CIP patients, who are recalcitrant to immunosuppressive therapy, improve when treated with systemic JAK inhibitors. Taken together, the current study identifies novel functions of immune signaling pathways within the sensory nervous system that represent promising targets for the treatment of chronic itch.
Despite the observation that type 2 cytokines can directly activate itch-sensory neurons in vitro, we found that, in contrast to other cytokine pruritogens, both IL-4 and IL-13 do not function as acute pruritogens in vivo. However, in the setting of AD-like skin inflammation where many cytokines are known to be highly upregulated, neuron-specific deletion of IL-4Rα was sufficient to abate chronic itch. To better understand how disruption of neuronal type 2 cytokine signaling could play such a central role in reducing chronic itch, we explored whether neuronal IL-4Rα signaling could alter the responsiveness of sensory neurons to other pruritogens. Indeed, we identified that IL-4 rapidly sensitizes subsets of neurons to a variety of pruritogens. Additionally, we observed that IL-4 significantly amplifies scratching behavior to low doses of known pruritogens like histamine. Thus, we speculate that type 2 cytokines may act as master regulators of chronic itch by intensifying itch responses to multiple pruritogens present in inflamed skin.
In immune cells, cytokine signaling through the JAK-STAT pathway results in transcriptional changes to promote cellular activation. However, although transcriptional alterations remain possible in neurons, our calcium imaging studies indicate that neuronal responses to IL-4Rα stimulation occur within seconds to minutes. Surprisingly, we observed that these rapid responses were dependent on both JAK1 as well as TRP channels. Thus, we predict that alterations in classical STAT-mediated transcriptional changes alone are insufficient to explain how IL-4Rα modulates sensory neurons. Previous studies have shown that TRP channels harbor several phosphorylation sites and can be regulated by multiple kinases (Sun and Dong, 2016; Zheng, 2013). Although JAKs typically phosphorylate STAT proteins within immune cells, our current study provokes the hypothesis that JAK proteins may have novel functions in neurons and may regulate TRP channels by unknown pathways. However, we note that such rapid alteration of sensory neuron function does not exclude the role of type 2 cytokines in modulating transcription or otherwise altering pruriceptors or ion channels within neurons. Future studies will be required to better understand how both rapid sensitization and transcriptional changes induced by type 2 cytokines may impact sensory function.
Classically, the dominant source of type 2 cytokines in AD lesional skin was believed to be the adaptive immune system via T helper type 2 (Th2) cells (Weidinger and Novak, 2016). Further, recent studies have shown that Th2 cell-derived IL-31 can elicit scratching (Cevikbas et al., 2014; Dillon et al., 2004; Sonkoly et al., 2006). Thus, both AD-associated inflammation and itch were largely believed to be mediated by the adaptive immune system. However, we and others have recently shown that innate immune cell populations such as group 2 innate lymphoid cells (ILC2s) and basophils are critical mediators of skin inflammation independent of the adaptive immune system and are essential sources of type 2 cytokines in vivo (Kim et al., 2013, 2014; Roediger et al., 2013; Salimi et al., 2013). Based on our new data demonstrating that neuronal IL-4Rα signaling critically mediates chronic itch, we speculate that innate immune cells such as ILC2s and basophils play important roles in promoting AD-associated itch, in addition to adaptive Th2 cells. Identification of the precise contributions of both adaptive and innate immune cells to the development of chronic itch requires further investigation.
Recently, the epithelial cell-derived cytokines TSLP and IL-33 have been shown to be master initiators of type 2 inflammation via their effects on a variety of cells including Th2 cells, ILC2s, and basophils (Hammad and Lambrecht, 2015; Oetjen et al., 2016). These cytokines are believed rapidly signal barrier breach in order to generate innate immune responses and help prime adaptive immune cells. Strikingly, two recent studies have demonstrated that both TSLP and IL-33 can function as pruritogens (Liu et al., 2016; Wilson et al., 2013a). Additionally, we and others have shown that epithelial cell-derived cytokines are highly expressed in the setting of AD-like disease (Kim et al., 2013; Li et al., 2006; Salimi et al., 2013). Despite high expression of these cytokines and robust activation of numerous other downstream cytokine pathways in AD, our study identifies that sensory neuron-specific deletion of IL-4Rα or JAK1 is sufficient to abate chronic itch. Thus, it appears that both neuronal IL-4Rα and JAK1 signaling are exquisitely robust targets for chronic itch. However, the cascade in which the epithelium simultaneously initiates both immune cell infiltration and itch responses, which are regulated by neuronal IL-4Rα and JAK1 signaling, calls for more exploration.
Clinical trials for dupilumab, an anti-IL-4Rα monoclonal antibody, have repeatedly demonstrated rapid and remarkable improvement of itch symptoms in AD patients (Beck et al., 2014; Simpson et al., 2016; Thaçi et al., 2016). Additionally, significant improvement of itch in AD patients has been observed in early clinical studies employing JAK inhibitors (Bissonnette et al., 2016; Levy et al., 2015). Previously, the changes in itch observed with these therapies have been attributed to the anti-inflammatory properties of either IL-4Rα or JAK blockade. However, our study demonstrates that sensory neurons co-opt classical immune signaling pathways to mediate chronic itch in multiple settings. Thus, we speculate that the rapid improvement of pruritus observed in patients treated with IL-4Rα or JAK blockade may be mediated, at least in part, by disruption of these signals in sensory neurons and that these therapies may abate chronic itch in diseases beyond overt inflammatory skin disorders. Strikingly, a recently published clinical trial for nemolizumab, an anti-IL-31RA monoclonal antibody, also described itch improvement in patients with moderate-to-severe AD (Ruzicka et al., 2017). Given our current study demonstrating a direct role of neuronal IL-4Rα and JAK1 in chronic itch, whether combinations of IL-4Rα-, JAK1-, and IL-31RA-blockade can lead to synergistic therapeutic improvements of itch in patients demands deeper investigation. However, larger prospective, randomized-controlled trials will be required to fully determine the efficacy of such treatments.
Immunity at barrier surfaces is believed to have evolved to mediate rapid expulsion of pathogens and potential toxins by employing the specialized functions of the epithelial, immune, and nervous systems. Whereas the epithelium was previously thought to be solely a physical barrier, recent advances have highlighted its direct role in a variety of immune functions, demonstrating that crosstalk between multiple cellular systems is essential for proper host defense (Artis and Spits, 2015; Hammad and Lambrecht, 2015). In addition to the immune system and the epithelial barrier itself, the mammalian host also employs behavioral responses initiated by the sensory nervous system such as coughing and scratching to mechanically remove noxious stimuli. Our current study demonstrates that classical immune mediators are an essential part of sensory circuits that modulate host behavior by activating and sensitizing the sensory nervous system. Given that the innate immune system employs type 2 cytokines to amplify immune responses at barrier surfaces, we speculate that the sensory nervous system may have evolved the ability to use these signals to similarly intensify sensory responses. Thus, direct neuronal priming by type 2 cytokines may be a key first step in the host response to promote protective behavioral responses like scratching. By identifying previously unrecognized neuroimmunologic pathways, we have uncovered novel therapeutic approaches to combat conditions in which these sensory processes become pathologic as in chronic itch.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian S. Kim (briankim@wustl.edu).
Experimental Model and Subject Details
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). The protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University School of Medicine. Human samples were collected from fully informed and consented individuals, and human studies were approved by the Washington University in St. Louis Institutional Review Board (IRB).
Animal studies
All mice were housed in standard group housing and husbandry conditions (social housing, 12 hour light-dark cycle, 23°C, food and water ad libi tum) at Washington University School of Medicine. C57Bl/6, Rosa26-LSL- tdTomato, and Rosa26-LSL-GFP mice were purchased from The Jackson Laboratory. NaV1.8-Cre mice were provided by Dr. Rohini Kuner (Heidelberg University). Il4raflox mice were generated by Dr. Frank Brombacher (ICGEB, Cape Town). Jak1flox mice were purchased from Nanjing Biomedical Research Institute of Nanjing University. NaV 1.8 reporter mice were generated by crossing NaV 1.8-Cre+ mice with Rosa26-LSL-tdTomato+ or Rosa26-LSL-GFP+ mice. IL-4RαΔneuron (NaV 1.8-Cre+ Il4rafl/fl) mice were generated by crossing the NaV 1.8-Cre+ and Il4raflox mice. JAK1Δneuron (NaV 1.8-Cre+ Jak1fl/fl) mice were generated by crossing the NaV 1.8-Cre+ and Jak1flox mice. Trpv1−/− mice were kindly provided by Dr. Durga Mohapatra at Washington University and also purchased from The Jackson Laboratory. Trpa1−/− mice were provided by Dr. Hongzhen Hu at Washington University. Il31ra−/− mice were provided by Bristol-Myers Squibb. Genotyping of mice was performed using standard PCR. Naïve adult (8–12 week) mice of both sexes were used in this study. Mice were either randomly assigned to treatment groups or assigned to groups based on genotype. No differences between sexes were observed, and no analyses of the influence of sex were performed.
Human studies
Diagnoses of AD and CIP were made by a board-certified dermatologist based on clinical and laboratory assessment (Eichenfield et al., 2014; Xu et al., 2016). Demographics of tissue donors and patients are included in Table S1 and Table S4. Given the mixture of sexes, no analyses of the influence of sex were performed. Human dorsal root ganglia (hDRG) were acquired from de-identified US transplant donors under an IRB-exempt protocol.
Primary neuron cultures
Matched donors of both sexes were used for primary culture of mouse neurons. Primary human neuron cultures were derived from a male donor. All cultures were kept under a humidified atmosphere of 5% CO2 at 37°C.
Method Details
RNA isolation from mouse DRG and TG and RT-qPCR
For RNA isolation, mouse dorsal root ganglia (DRG) or trigeminal ganglia (TG) were harvested, cleaned of connective tissues, and homogenized with a bead homogenizer (BioSpec) in 1 mL Trizol Reagent (Life Technologies). Total RNA was extracted with the RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. Following extraction, samples were treated with DNase (Turbo DNA-Free Kit, Invitrogen) following the manufacturer’s instructions. Next, cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression levels were determined by RT-qPCR (StepOnePlus, Applied Biosystems) using the TaqMan Gene Expression qPCR Master Mix (Applied Biosystems) following the manufacturer’s instructions. Gene expression was normalized to Gapdh and relative expression was calculated using the ΔΔCt method. For gel electrophoresis, products from RT-qPCR reactions were loaded onto a 2% agarose gel with ethidium bromide (1 μg/mL) and run at 140 V for 25 minutes. Primer and probe sequences used for each gene were selected from pre-validated PrimeTime qPCR Assays (Integrated DNA Technologies) as listed in the Key Reagents table.
RNA isolation from human DRG and RT-PCR
Human dorsal root ganglia (hDRG) were acquired from de-identified US transplant donors under an IRB-exempt protocol. After hDRG extraction, fat, dura, and connective tissues were removed as previously described (Valtcheva et al., 2016). The hDRG were subsequently stored in RNAlater (Sigma-Aldrich) at −80°C until RNA isolati on was performed. For RNA isolation, tissue from one half of one hDRG was homogenized in 1 mL Trizol Reagent following the manufacturer’s instructions. Following total RNA extraction, genomic DNA was eliminated and cDNA synthesized using the Maxima H Minus First Strand cDNA Synthesis Kit with dsDNase (Thermo Scientific) following the manufacturer’s instructions. RT-PCR product was loaded onto a 2% agarose gel with ethidium bromide (1 μg/mL) and run at 100 V for 45 minutes. RT-PCR was performed using primer sets listed in the Key Reagents table.
Overnight mouse DRG neuron culture
Mouse DRG neurons from both sexes were isolated and cultured using a previously published protocol with slight modification (Malin et al., 2007). Laminectomies were performed on mice and bilateral DRG were removed. After removal of connective tissues, DRG were transferred to 1 mL Ca2+/Mg2+-free Hank’s Balanced Salt Solution (HBSS) containing 1 L saturated NaHCO3, 0.35 mg L-cysteine (Sigma-Aldrich), and 20 U papain (Worthington) and incubated at 37°C for 20 minutes. The suspension was then centrifuged, and the supernatant was removed and replaced with 1 mL Ca2+/Mg2+-free HBSS containing 3.75 mg collagenase type II (Worthington) and 7.5 mg dispase (Gibco) and incubated at 37°C for 20 minutes. After digestion, neurons were gently triturated, pelleted, and then resuspended in Neurobasal-A culture medium (Gibco) containing 2% B-27 supplement (Gibco), 100 U/mL penicillin plus 100 μg/mL streptomycin (Sigma-Aldrich), 100 ng/mL nerve growth factor (NGF, Sigma-Aldrich), 20 μg/mL glial cell-derived neurotrophic factor (GDNF, R&D Systems) and 10% heat-inactivated FBS (Sigma-Aldrich). Neurons were then plated on glass coverslips pre-coated with poly-L-lysine (Sigma-Aldrich) and laminin (Sigma-Aldrich) and cultured under a humidified atmosphere of 5% CO2 at 37°C for 18–24 hours before use.
Calcium imaging of mouse DRG neurons
Cultured mouse DRG neurons were loaded with 4 μM Fura-2 AM (Invitrogen) in DRG culture medium at 37°C for 45 minutes. Cells were then washed three times and incubated in calcium imaging buffer (130 mM NaCl, 3 mM KCl, 2.5 mM CaCl2, 0.6 mM MgCl2, 10 mM HEPES, 10 mM glucose, 1.2 mM NaHCO3, pH 7.45) at room temperature for 30 minutes before use. Calcium-free imaging buffer was prepared by the omission of CaCl2 and the addition of 10 mM EDTA (Sigma-Aldrich). Murine IL-4, IL-5, IL-13, and TSLP (Peprotech and/or R&D Systems) and IL-31 (Bristol-Myers Squibb) were used at 300 nM, while BAM8-22 (Genescript), histamine (Sigma-Aldrich), chloroquine (Sigma-Aldrich), capsaicin (Sigma-Aldrich) were used at 10 μM, 50 μM, 1 mM, and 300 nM, respectively, unless otherwise indicated. Where indicated, sensory neurons were also challenged with corresponding dilutions of control vehicle (0.1% BSA in PBS). Only sensory neurons that responded to a final challenge of 100 mM KCl were used in analyses. Fluorescence was recorded at 340 nm and 380 nm excitation wavelengths (F340, F380) using an inverted Nikon Ti-S microscope with NIS-Elements imaging software (Nikon Instruments). Fluorescence ratios (F340/F380) were normalized to baseline and used to reflect changes in intracellular Ca2+ and neuronal activation upon stimulation. Data was then analyzed in R where cells were considered responsive if they demonstrated a change in fluorescence ratio >10% of baseline. Cell diameter was calculated as the diameter of a circle of an equivalent area equal to the cross-sectional area of an imaged neuron. Neurons were classified as a small-, medium-, or large-diameter neuron if the diameter was <18 μm, 18–25 μm, or >25 μm, respectively.
Human DRG neuron culture and calcium imaging
Human DRG were recovered in the operating room and transported in N-methyl-D-glucamine (NMDG) artificial cerebrospinal fluid (aCSF) solution where they were dissociated and plated on glass coverslips following the procedures described in Valtcheva et al., 2016. Neurons were incubated for 3 days in media containing Neurobasal-A supplemented with B-27, 100 U/mL penicillin plus streptomycin, 2 mM Glutamax (Gibco), and 5% FBS at 37°C and 5% CO2. Fura-2 AM (3 μg in 3 μL DMSO) was administered to coverslips in 1 mL of media and after 45 minutes, coverslips were moved to external solution containing (in mM): 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 30 glucose, 10 HEPES for calcium imaging following the protocols in Valtcheva et al., 2016. Calcium imaging was performed on an Olympus BX51 microscope with Rolera Bolt camera (Q-Imaging) and a CoolLED pE-4000 (365/385) illumination system controlled via MetaFluor software (Molecular Devices). Recombinant human IL-4 (300 nM, R&D Systems), capsaicin (500 nM) and KCl (50 mM) were administered to the bath.
Scratching behavior assessment
All behavioral tests were performed on sex- and age-matched adult mice (8–12 weeks old). For behavioral assessments, mice were first acclimated to the recording room and behavior chambers at least one day prior to testing. On the day of testing, mice were again acclimated to the recording room and behavior chambers and then video recorded. The video recordings were then manually scored for number of scratching bouts per 30 minute block by an observer blind to treatment and genotype. One scratching bout was defined as one instance of lifting the hind paw from the floor, scratching, and returning the paw to the floor or placing the paw in the animal’s mouth. For chronic itch models, mice were recorded before daily treatments. For acute injection of pruritic compounds, the cheeks of mice were shaved at least 5 days prior to behavioral testing. On testing days, acclimated mice were injected intradermally into the cheek with 10 μL of vehicle control (0.1% BSA in PBS), 10 μL containing 2.5 μg of IL-4, IL-13, or IL-31, or 10 μL of a 4 mM histamine solution unless otherwise indicated. Scoring of scratching behavior commenced immediately after the injected mouse was returned to the recording chamber.
MC903 treatment and pathology assessment
For induction of AD-like inflammation and itch, mice were topically treated once daily with 2 nmol of MC903 (calcipotriol, Tocris Bioscience) for 7 days or 1 nmol of MC903 for 12 days on both ears in 20 μL of ethanol (vehicle) as previously described (Kim et al., 2013, 2014). Ear thickness measurements were performed daily with dial calipers as previously described (Kim et al., 2013, 2014). At the end of the MC903 treatment, murine ear skin tissues were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin before sectioning and staining with Hematoxylin & Eosin (H&E). Histology score was determined by the following formula using ImageJ analysis software (NIH) as previously described: (total number of lymphocytes per high power field (HPF) + thickness of the epidermis measured in microns from the basement membrane to the top of the stratum corneum) divided by 100 (Kim et al., 2014). All images were captured with a NanoZoomer 2.0-HT System (Hamamatsu).
Mouse and human skin RNA extraction
Samples of murine skin were obtained and stored in RNAlater at −80°C before processing following the manufacturer’s instructions. Human skin tissues were obtained as 4-mm punch biopsies or de-identified surgical tissue and stored in RNAlater at −80°C before processing. To extract whole tissue RNA, samples were homogenized using a bead homogenizer and processed using the Qiagen RNeasy Kit following the manufacturer’s instructions. Following total RNA extraction, samples were treated with DNase (Turbo DNA-Free Kit, Invitrogen) following the manufacturer’s instructions before sequencing library preparation.
Library preparation and RNA-sequencing
Library preparation, sequence alignment, and determination of transcript abundance were carried out by the Genome Technology Access Center (GTAC) at Washington University School of Medicine. Briefly, library preparation was performed with 1 μg of total RNA, and RNA integrity was determined using an Agilent Bioanalyzer. Ribosomal RNA was removed by a hybridization method using Ribo-ZERO kits (Illumina). mRNA was then fragmented in buffer containing 40 mM Tris acetate (pH 8.2), 100 mM potassium acetate, and 30 mM magnesium acetate and heated to 94°C for 150 seconds. mRNA was reverse tr anscribed to yield cDNA using SuperScript III RT enzyme (Invitrogen, per manufacturer’s instructions) and random hexamers. A second strand reaction was performed to yield ds-cDNA. cDNA was blunt ended, had an A base added to the 3′ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 12 cycles using primers incorporating unique index tags. Fragments were sequenced on an Illumina HiSeq-3000 using single reads extending 50 bases.
RNA-sequencing reads were aligned to the Ensembl release 76 assembly with STAR v2.0.4b (Dobin et al., 2013). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount v1.4.5 (Liao et al., 2014). Transcript counts were produced by Sailfish v0.6.3 (Patro et al., 2014). Sequencing performance was assessed for total number of aligned reads, total number of uniquely aligned reads, genes and transcripts detected, ribosomal fraction, known junction saturation, and read distribution over known gene models with RSeQC v2.3 (Wang et al., 2012).
Differential gene expression and gene set enrichment analysis
Differential gene expression analysis and sample clustering from RNA-sequencing data were performed with the DESeq2 package available in R using default configurations (Love et al., 2014). Genes with a two-fold or greater change and an adjusted p-value < 0.1 were considered significant. Dendrograms in heat maps were formed by hierarchical clustering on the Euclidean distances between samples. Gene set enrichment analysis (GSEA) was carried out using the preranked gene list feature available in the javaGSEA application (Broad Institute). Genes were preranked by the product of the fold change and the inverse of the adjusted p-value of differential expression as determined by DESeq2. Gene sets were derived from the Biological Process Ontology (Molecular Signatures Database).
Flow cytometry
Ear skin and skin-draining lymph nodes (sdLN) were harvested from NaV1.8 reporter mice (NaV1.8-GFP or NaV1.8-tdTomato) as previously described (Kim et al., 2013). Ears were cut and separated using forceps and digested in 0.25 mg/ml Liberase TL (Roche) in DMEM media for 90 minutes at 37°C and 5% CO2. Resulting digested skin and sdLN were mashed through 70 μm cell strainers and washed with DMEM media supplemented with 5% FBS to yield single cell suspensions. Suspensions of DRG neurons were prepared using the same procedure used for culturing. Skin samples were assessed as previously described for flow cytometry (Kim et al., 2013, 2014). Basophils were defined as Live CD49b+ FcεRIα+ cells negative for expression lineage (Lin) markers CD3, CD5, CD11c, CD19, NK1.1, and CD117 (c-Kit). ILC2s were defined as Live CD45+ CD25+ IL-33R+ cells negative for expression of lineage (CD3, CD5, CD11b, CD11c, CD19, NK1.1, and FcεRIα) markers. Th2 cells were defined as Live CD45+ CD3+ CD4+ IL-33R+ cells. Flow cytometry data was acquired using an LSRFortessa X20 (BD Biosciences) and analyzed using FlowJo 10 (Tree Star).
Immunofluorescence of DRG neurons
Mouse DRG neurons were extracted and fixed in 4% paraformaldehyde (PFA) for 5 minutes on ice. The fixed cells were then stained overnight with rabbit anti-mouse/human phospho-JAK1 (Tyr1022, Tyr1023) primary antibody (59H4L5, Invitrogen) at 4°C. Secondary goat anti-rabbit antibody conjugated to Alexa Fluor 555 (Invitrogen) was used to stain for 1 hour at 4°C. VECTASHIELD mounting medium with DAPI was used for mounting and staining of nuclei (Vector Laboratories). All imaging and analysis was performed on a Nikon C2+ Confocal Microscope System with NIS-Elements imaging software (Nikon Instruments).
Rotarod testing
A rotarod system of accelerating treadmills (Ugo Basile) was used to assess coordinated motor activity and general motor disability. Acceleration from 5 rpm to 40 rpm over 5 minutes was used. The time to failure was defined as falling from the rod or one complete passive rotation. Mice were trained over 3 days with 3 trials per day with an intratrial interval of 15 minutes. On testing day, mice we first acclimated to the recording room. Time to failure was recorded over 3 trails with an intratrial interval of 15 minutes. Presented data represents 3 averaged trials.
Pharmacologic JAK inhibition
Stock ruxolitinib (Selleck Chemicals) solutions were prepared in DMSO (Sigma-Aldrich) per the manufacturer’s instructions. For in vitro inhibition, cells were incubated with 1 μg/mL ruxolitinib during Fura-2 dye loading and imaged in standard calcium imaging buffer that also contained 1 μg/mL ruxolitinib. Control neurons were incubated with a corresponding amount of DMSO and imaged in stardard calcium imaging buffer with a corresponding dilution of DMSO. For systemic treatments, mice treated twice daily with 100 μg ruxolitinib diluted in 100 μl PBS by intraperitoneal (i.p.) injection. Control mice were injected with corresponding vehicle control (DMSO in PBS) of equivalent volumes.
Mouse model of chronic itch without robust inflammation
The acetone-ether-water (AEW) model of non-inflammatory itch was used as previously described (Miyamoto et al., 2002; Zhao et al., 2013). Briefly, the nape of the neck of mice was shaved prior to the start of treatment. On treatment days, a cotton square soaked with a 1:1 mixture of acetone (Sigma-Aldrich) and ether (Sigma-Aldrich) was applied to the nape of the neck for 15 seconds. Immediately after this first application, a second cotton square soaked with distilled water was applied to the neck for 30 seconds. Treatments were applied twice daily for 5 days.
Human histopathology
Human skin tissues were obtained as 4-mm punch biopsy specimens then fixed in 10% paraformaldehyde (PFA) and embedded in paraffin. Sections were then stained with H&E. Histology score was determined by the following formula using ImageJ analysis software (NIH) as previously described: (total number of lymphocytes per high power field (HPF) + thickness of the epidermis measured in microns from the basement membrane to the top of the stratum corneum) divided by 100 (Kim et al., 2014).
Numerical Rating Scale (NRS) itch scoring
Patients in the specialty Itch Clinic at the Center for Advanced Medicine at Washington University School of Medicine are routinely assessed prospectively for their NRS itch score. Briefly, patients were asked to rate their itch intensity over the past 24 hours from 0 to 10 where 0 represents “no itch” and 10 represents the “worst itch imaginable”. For AD and CIP patient comparisons, NRS itch scores were obtained by retrospective analysis of charts from patients seen between August 2014 and February 2016. For patients who received treatment with tofacitinib (5 mg by mouth twice daily), NRS itch scores were obtained at the beginning of treatment and after one month of treatment. Recordings of daily NRS itch scores of three patients are reported in Figure 6B during tofacitinib treatment.
Quantification and Statistical Analyses
Statistical analysis for RNA-seq data is described above. Data in figures represent mean ± standard error of the mean (SEM) unless otherwise indicated. Data from independent experiments were pooled when possible or represent at least two independent replicates. All significance tests were chosen considering the design of experiments, and we assumed normal distribution and variance of data. Sample sizes were chosen based upon pilot experiments in order to accurately detect statistical significance as well as considering technical feasibility, resource availability, and ethical animal and sample use. No data were excluded from statistical analyses, unless due to technical errors. Statistical significance was determined by unpaired Student’s t-test with Welch’s correction unless noted otherwise. Further statistical details of experiments can be found in the figure legends. Statistical analyses were performed using Prism 7 (GraphPad Software). Significance is labeled as: ***p < 0.001, **p < 0.01, *p < 0.05, N.D., Not Detected, N.S., Not Significant.
Supplementary Material
Figure S1. DRG neurons respond to type 2 cytokines in a dose-dependent manner. Relates to Figure 1.
(A) Average calcium imaging trace of IL-4-responsive mouse dorsal root ganglia (DRG) neurons challenged with vehicle and increasing doses of recombinant murine (rm)IL-4. Gray traces represent 24 IL-4-responsive neurons (from 256 KCl-responsive neurons) with average response shown in black.
(B) Quantification of DRG neuron responses to increasing doses of rmIL-4 as a percentage of total KCl-responsive neurons, n > 400 neurons from 2 WT mice.
(C) Average calcium imaging trace of IL-13-responsive mouse DRG neurons challenged with vehicle control and increasing doses of rmIL-13. Gray traces represent 15 IL-13-responsive neurons (from 248 KCl-responsive neurons) with average response shown in black.
(D) Quantification of DRG neuron responses to increasing doses of rmIL-13 as a percentage of total KCl-responsive neurons, n > 400 neurons from 2 WT mice.
(E) Average calcium imaging trace of mouse DRG neurons challenged with rmIL-5 (4 μg/mL), histamine (Hist, 50 μM), capsaicin (Cap, 300 nM), and KCl (100 mM). Gray traces represent individual KCl-responsive neurons with average response shown in black.
(F) Quantification of DRG neuron responses to rmIL-5, histamine, and capsaicin as a percentage of total KCl-responsive neurons, n > 300 neurons from a WT mouse.
Black bars in calcium imaging traces indicate timing of challenges. Data are represented as mean ± SEM.
Figure S2. Type 2 cytokine receptors are enriched on NaV1.8+ sensory neurons. Relates to Figure 2.
(A) Expression of selected genes in mouse DRG neuron populations generated from sort-purified sensory neurons shown as row Z-scores of microarray data. Neurons were sorted by Isolectin B4 (IB4), NaV1.8, and/or parvalbumin (Parv) expression. Full data set and methods available in Chiu et al., 2014.
(B) Total number of rmIL-4-, rmIL-13-, histamine-, and KCl-responsive mouse DRG neurons classified as small-, medium-, and large-diameter neurons (<18 μm, 18–25 μm, and >25 μm), n > 500 neurons from a WT mouse.
(C) Representative Venn diagram of overlapping responses of mouse DRG neurons to stimulation with rmIL-13 (300 nM), rmIL-31 (300 nM), and histamine (50 μM), n > 300 neurons from a WT mouse.
(D) Representative Venn diagram of overlapping responses of mouse DRG neurons to stimulation with rmIL-4 (4 μg/mL) and rmTSLP (4 μg/mL), n > 100 neurons from a WT mouse.
(E–G) Representative calcium imaging trace of mouse DRG neurons responding to (E) chloroquine (100 μM, CQ), (F) rmTSLP (0.25 μg/mL), and (G) rmIL-31 (3 μM) after vehicle control or rmIL-4 (4 μg/mL) challenge, n > 200 neurons per group.
Black bars in calcium imaging traces indicate timing of challenges.
Figure S3. MC903 treatment results in AD-like disease and robust chronic itch. Relates to Figure 3.
(A) Experimental schematic indicating daily topical treatment with vehicle control (ethanol, EtOH) or MC903 to the ears of WT mice.
(B) Ear thickness measurements as percent change from baseline over the course of control or MC903 treatment, n ≥ 4 mice per group.
(C) Representative H&E histopathology and (D) histology score of control and MC903-treated mice, n ≥ 4 mice per group.
(E) Comparison of row Z-scores of the regularized logarithm of gene expression values of selected AD-associated genes from RNA-seq of the skin of control and MC903-treated mice, n = 4 mice per group.
(F) Scratching behavior of control and MC903-treated mice on Day 0, 4, 8, and 12, n ≥ 4 mice per group.
(G) Representative responses of DRG neurons to increasing concentrations of MC903 as a percentage of total KCl-responsive neurons, n > 300 neurons from a WT mouse.
Scale bars indicate 100 μm. Data are represented as mean ± SEM.
Figure S4. Il31ra−/− mice are not protected from AD-like itch, and IL-4RαΔneuron mice have reduced infiltration of type 2 immune cells into the skin with a distinct skin transcriptional profile. Relates to Figure 3.
(A) Experimental schematic indicating daily topical treatment with MC903 to the ears of Il31ra−/− mice and WT control mice.
(B) Scratching behavior and (C) ear thickness measurement of Il31ra−/− mice compared to control mice on Day 7, n ≥ 9 mice per group.
(D) Experimental schematic indicating daily topical treatment with MC903 to the ears of IL-4RαΔneuron and littermate control mice.
(E) Basophil, (F) group 2 innate lymphoid cell (ILC2), and (G) T helper type 2 (Th2) cell frequencies as measured by flow cytometry from the ear skin of IL-4RαΔneuron mice at Day 12, n ≥ 3 mice per group.
(H) Comparison of row Z-scores of the regularized logarithm of gene expression values of the top 100 differentially expressed genes from RNA-seq of the skin of MC903-treated IL-4RαΔneuron mice compared to littermate control mice as determined by p-value. Genes are listed based on increasing adjusted p-value, n ≥ 4 mice per group.
Data are represented as mean ± SEM.
Figure S5. NaV1.8 expression is restricted to sensory neurons under both steady-state and inflammatory conditions. Relates to Figure 3.
(A) Experimental schematic indicating treatment of NaV1.8 reporter mice with MC903.
(B) Representative flow cytometry plots of the bulk DRG, skin, and skin-draining lymph node (sdLN) of naïve NaV1.8 reporter mice compared to non-reporter control mice.
(C) Representative flow cytometry plots of the bulk DRG, skin, and sdLN of MC903-treated NaV1.8 reporter mice compared to non-reporter control mice at Day 12.
(D) Experimental schematic indicating daily topical treatment with MC903 to the ears of IL-4RαΔneuron and littermate control mice.
(E–F) Quantification of Il4ra, Il2rg, and Il13ra1 by RT-qPCR in the (E) skin and (F) sdLN of IL-4RαΔneuron mice treated with MC903, n ≥ 3 mice per group.
Data are represented as mean ± SEM.
Figure S6. JAK1Δneuron mice are developmentally normal and are protected from chronic itch even in the absence of robust skin inflammation. Relates to Figure 4.
(A) Expression of the JAK family of genes in mouse DRG neuron populations clustered into functional subsets based on single-cell RNA-seq data. Numbers in heat map indicate fraction of positive cells within individual clusters by thresholding method. Full data set and methods available in Usoskin et al., 2015.
(B–C) Representative immunofluorescent images of dissociated DRG neurons at (B) baseline and (C) after 5 minutes of stimulation with rmIL-4 (1 μg/mL). Red staining indicates phosphorylated JAK1 (pJAK1). Blue staining indicates DAPI, n = 3 WT mice.
(D) Frequency of pJAK1+ cells as a percentage of all DAPI+ cells at baseline and after stimulation with rmIL-4, n ≥ 70 cells per high power field.
(E) Representative distribution of diameters of dissociated DRG neurons from a JAK1Δneuron and a littermate control mouse, n ≥ 200 neurons per group.
(F–I) Representative responses to (F) histamine (50 μM), (G) BAM8-22 (10 μM), (H) rmTSLP (4 μg/mL), and (I) rmIL-31 (300 nM) of DRG neurons from a JAK1Δneuron mouse compared to a littermate control mouse as a percentage of total KCl-responsive neurons, n ≥ 300 neurons per group.
(J) Experimental schematic indicating rotarod training and testing days.
(K) Results of rotarod testing of JAK1Δneuron mice compared to littermate control mice, n ≥ 4 mice per group.
(L) Experimental schematic indicating twice daily acetone-ether-water (AEW) treatment to the nape of the neck of JAK1Δneuron and littermate control mice.
(M) Scratching behavior of JAK1Δneuron and littermate control mice on Day 5 of AEW treatment, n ≥ 8 mice per group.
Scale bars indicate 50 μm. Data are represented as mean ± SEM.
HIGHLIGHTS.
Type 2 cytokines directly activate both mouse and human sensory neurons.
IL-4 enhances neuronal responsiveness to multiple pruritogens.
Sensory neuron-specific deletion of IL-4Rα or JAK1 reduces chronic itch.
Clinical studies demonstrate that JAK inhibitors relieve chronic itch.
Acknowledgments
We thank all our collaborators at Washington University School of Medicine (WUSM) for their advice and discussion. We also thank members of the Division of Dermatology at WUSM for their assistance with patient sample collection. Specifically, we thank Ms. Mary Tabacchi, Ms. Nancy Bodet, and Dr. Lynn Cornelius for their support. This publication was supported, in part, by a grant from LEO Pharma and by the WUSM Institute of Clinical and Translational Sciences (ICTS) which is supported by the NIH National Center for Advancing Translational Sciences (NCATS) Clinical and Translational Science Award (CTSA) Grant UL1TR000448. We also thank the Genome Technology Access Center (GTAC) at WUSM for assistance with RNA-sequencing. GTAC is partially supported by NCI P30CA91842 and by ICTS CTSA UL1TR000448 from the National Center for Research Resources (NCRR). We also thank the WUSM Alafi Neuroimaging Laboratory, supported by NCRR Shared Instrumentation Grant 1S10RR027552. We thank the WUSM Digestive Disease Research Core Center for histology support, supported by NIDDK P30DK052574. We also thank the Immunomonitoring Laboratory (IML) at the Bursky Center for Human Immunology & Immunotherapy Programs (ChiiPs) for flow cytometry support. Research using human DRG in the Gereau Lab is supported by NINDS R01NS042595. The Davidson Lab is supported by NIAMS R21AR068012. The Hu Lab is supported by NIGMS R01GM101218 and NIDDK R01DK103901. The Kim Lab is supported by NIAMS K08AR065577 and R01AR070116, the American Skin Association, and the Doris Duke Charitable Foundation. L.K.O. is supported by NHLBI T32HL007317. A.Z.X. is supported by the Howard Hughes Medical Institute (HHMI) Medical Fellows Program. Il4raflox mice were kindly provided by Dr. Herbert (Skip) Virgin at WUSM and Dr. Ajay Chawla at University of California, San Francisco. We also thank Dr. Steven Saenz for critical discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
M.R.M., J.F., T.M.W., H.N., C.J.G., S.C., A.M.T., A.Z.X, S.V.T., J.L., X.G., L.Y., S.L.H., P.L.W., M.L.C, R.B., A.S., and M.J.M performed experiments and analyzed data. R.W.G and S.D. provided data from human cadaveric dorsal root ganglia. J.R.B., C.S.H., Z.F.C., H.H., and Q.L. helped design the study and interpret results. F.B. generated the Il4raflox mice. L.K.O and B.S.K. performed experiments, designed the study, interpreted the results, and wrote the manuscript.
The authors have no conflicts of interest to declare.
Data and Software Availability
The accession numbers for the RNA-seq data generated from animal studies are GEO: GSE90883 and GEO: GSE100397. Other data are available upon request to the lead contact author.
Additional Resources
Retrospective human studies were approved by that Washington University IRB #201410014, #201412117, #201507042, and #201604088.
References
- Agarwal N, Offermanns S, Kuner R. Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis. 2004;38:122–129. doi: 10.1002/gene.20010. [DOI] [PubMed] [Google Scholar]
- Andersen HH, Elberling J, Sølvsten H, Yosipovitch G, Arendt-Nielsen L. Non-histaminergic and mechanical itch sensitization in atopic dermatitis. Pain. 2017 doi: 10.1097/j.pain.0000000000000980. [DOI] [PubMed] [Google Scholar]
- Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Wilson SR, Hoon MA. Why we scratch an itch: the molecules, cells and circuits of itch. Nat Neurosci. 2014;17:175–182. doi: 10.1038/nn.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, Ming JE, Ren H, Kao R, Simpson E, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130–139. doi: 10.1056/NEJMoa1314768. [DOI] [PubMed] [Google Scholar]
- Berger TG, Steinhoff M. Pruritus in elderly patients—Eruptions of senescence. Semin Cutan Med Surg. 2011;30:113–117. doi: 10.1016/j.sder.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, Wang C, Purohit V, Mamolo C, Papacharalambous J, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. 2016;175:902–911. doi: 10.1111/bjd.14871. [DOI] [PubMed] [Google Scholar]
- Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, Kukova G, Buhl T, Ikoma A, Buddenkotte J, et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: Involvement of TRPV1 and TRPA1. J Allergy Clin Immunol. 2014;133:448–460. doi: 10.1016/j.jaci.2013.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LS, Robinson N, Xu L. Expression of interleukin-4 in the epidermis of transgenic mice results in a pruritic inflammatory skin disease: an experimental animal model to study atopic dermatitis. J Invest Dermatol. 2001;117:977–983. doi: 10.1046/j.0022-202x.2001.01484.x. [DOI] [PubMed] [Google Scholar]
- Chiu IM, Barrett LB, Williams EK, Strochlic DE, Lee S, Weyer AD, Lou S, Bryman GS, Roberson DP, Ghasemlou N, et al. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. ELife. 2014;3 doi: 10.7554/eLife.04660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, Haugen HS, Maurer M, Harder B, Johnston J, et al. Interleukin-31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5:752–760. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenfield LF, Tom WL, Chamlin SL, Feldman SR, Hanifin JM, Simpson EL, Berger TG, Bergman JN, Cohen DE, Cooper KD, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338–351. doi: 10.1016/j.jaad.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. 2016;164:378–391. doi: 10.1016/j.cell.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittler JK, Shemer A, Suárez-Fariñas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQF, Mitsui H, Cardinale I, de Guzman Strong C, Krueger JG, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130:1344–1354. doi: 10.1016/j.jaci.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim TYF, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, McKenzie ANJ. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat Immunol. 2016;17:57–64. doi: 10.1038/ni.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. 2015;43:29–40. doi: 10.1016/j.immuni.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Ikoma A, Rukwied R, Ständer S, Steinhoff M, Miyachi Y, Schmelz M. Neuronal sensitization for histamine-induced itch in lesional skin of patients with atopic dermatitis. Arch Dermatol. 2003;139:1455–1458. doi: 10.1001/archderm.139.11.1455. [DOI] [PubMed] [Google Scholar]
- Kaplan AP. Chronic urticaria: Pathogenesis and treatment. J Allergy Clin Immunol. 2004;114:465–474. doi: 10.1016/j.jaci.2004.02.049. [DOI] [PubMed] [Google Scholar]
- Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. Nociceptive sensory fibers drive Interleukin-23 production from CD301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity. 2015;43:515–526. doi: 10.1016/j.immuni.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and Interleukin-13 signaling connections maps. Science. 2003;300:1527–1528. doi: 10.1126/science.1085458. [DOI] [PubMed] [Google Scholar]
- Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Voorhees ASV, Comeau MR, Artis D. TSLP elicits IL-33–independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5:170ra16–170ra16. doi: 10.1126/scitranslmed.3005374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BS, Wang K, Siracusa MC, Saenz SA, Brestoff JR, Monticelli LA, Noti M, Tait Wojno ED, Fung TC, Kubo M, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol. 2014;193:3717–3725. doi: 10.4049/jimmunol.1401307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kini S, DeLong LK, Veledar E, McKenzie-Brown A, Schaufele M, Chen SC. The impact of pruritus on quality of life: The skin equivalent of pain. Arch Dermatol. 2011;147:1153–1156. doi: 10.1001/archdermatol.2011.178. [DOI] [PubMed] [Google Scholar]
- Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73:395–399. doi: 10.1016/j.jaad.2015.06.045. [DOI] [PubMed] [Google Scholar]
- Li M, Hener P, Zhang Z, Kato S, Metzger D, Chambon P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci. 2006;103:11736–11741. doi: 10.1073/pnas.0604575103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu B, Tai Y, Achanta S, Kaelberer MM, Caceres AI, Shao X, Fang J, Jordt SE. IL-33/ST2 signaling excites sensory neurons and mediates itch response in a mouse model of poison ivy contact allergy. Proc Natl Acad Sci. 2016;113:E7572–E7579. doi: 10.1073/pnas.1606608113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng HJ, Geng Y, et al. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell. 2009;139:1353–1365. doi: 10.1016/j.cell.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc. 2007;2:152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- Martinez-Gonzalez I, Mathä L, Steer CA, Ghaedi M, Poon GFT, Takei F. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity. 2016;45:198–208. doi: 10.1016/j.immuni.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Matterne U, Apfelbacher CJ, Loerbroks A, Schwarzer T, Büttner M, Ofenloch R, Diepgen TL, Weisshaar E. Prevalence, correlates and characteristics of chronic pruritus: a population-based cross-sectional study. Acta Derm Venereol. 2011;91:674–679. doi: 10.2340/00015555-1159. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Nojima H, Shinkado T, Nakahashi T, Kuraishi Y. Itch-associated response induced by experimental dry skin in mice. Jpn J Pharmacol. 2002;88:285–292. doi: 10.1254/jjp.88.285. [DOI] [PubMed] [Google Scholar]
- Mollanazar NK, Sethi M, Rodriguez RV, Nattkemper LA, Ramsey FV, Zhao H, Yosipovitch G. Retrospective analysis of data from an itch center: Integrating validated tools in the electronic health record. J Am Acad Dermatol. 2016;75:842–844. doi: 10.1016/j.jaad.2016.05.035. [DOI] [PubMed] [Google Scholar]
- Morita T, McClain SP, Batia LM, Pellegrino M, Wilson SR, Kienzler MA, Lyman K, Olsen ASB, Wong JF, Stucky CL, et al. HTR7 mediates serotonergic acute and chronic itch. Neuron. 2015;87:124–138. doi: 10.1016/j.neuron.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oetjen LK, Noti M, Kim BS. New insights into basophil heterogeneity. Semin Immunopathol. 2016;38:549–561. doi: 10.1007/s00281-016-0567-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordovas-Montanes J, Rakoff-Nahoum S, Huang S, Riol-Blanco L, Barreiro O, von Andrian UH. The regulation of immunological processes by peripheral neurons in homeostasis and disease. Trends Immunol. 2015;36:578–604. doi: 10.1016/j.it.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel T, Yosipovitch G. The management of chronic pruritus in the elderly. Skin Ther Lett. 2010;15:5–9. [PubMed] [Google Scholar]
- Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich A, Ständer S, Szepietowski JC. Pruritus in the elderly. Clin Dermatol. 2011;29:15–23. doi: 10.1016/j.clindermatol.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, Wood JN, von Andrian UH. Nociceptive sensory neurons drive Interleukin-23 mediated psoriasiform skin inflammation. Nature. 2014;510:157–161. doi: 10.1038/nature13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, Mitchell AJ, Tay SS, Jain R, Forbes-Blom E, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol. 2013;14:564–573. doi: 10.1038/ni.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka T, Hanifin JM, Furue M, Pulka G, Mlynarczyk I, Wollenberg A, Galus R, Etoh T, Mihara R, Yoshida H, et al. Anti-Interleukin-31 Receptor A Antibody for Atopic Dermatitis. N Engl J Med. 2017;376:826–835. doi: 10.1056/NEJMoa1606490. [DOI] [PubMed] [Google Scholar]
- Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST, McKenzie ANJ, et al. A role for IL-25 and IL-33–driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210:2939–2950. doi: 10.1084/jem.20130351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. 2016;12:25–36. doi: 10.1038/nrrheum.2015.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, Silverberg JI, Deleuran M, Kataoka Y, Lacour JP, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335–2348. doi: 10.1056/NEJMoa1610020. [DOI] [PubMed] [Google Scholar]
- Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, Alenius H, Dieu-Nosjean MC, Meller S, Rieker J, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. 2006;117:411–417. doi: 10.1016/j.jaci.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Dong X. Trp channels and itch. Semin Immunopathol. 2016;38:293–307. doi: 10.1007/s00281-015-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot S, Abdulnour REE, Burkett PR, Lee S, Cronin SJF, Pascal MA, Laedermann C, Foster SL, Tran JV, Lai N, et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron. 2015;87:341–354. doi: 10.1016/j.neuron.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaçi D, Simpson EL, Beck LA, Bieber T, Blauvelt A, Papp K, Soong W, Worm M, Szepietowski JC, Sofen H, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387:40–52. doi: 10.1016/S0140-6736(15)00388-8. [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci. 2015;18:145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- Valtcheva MV, Samineni VK, Golden JP, Gereau RW, Davidson S. Enhanced nonpeptidergic intraepidermal fiber density and an expanded subset of chloroquine-responsive trigeminal neurons in a mouse model of dry skin itch. J Pain. 2015;16:346–356. doi: 10.1016/j.jpain.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtcheva MV, Copits BA, Davidson S, Sheahan TD, Pullen MY, McCall JG, Dikranian K, Gereau RW. Surgical extraction of human dorsal root ganglia from organ donors and preparation of primary sensory neuron cultures. Nat Protoc. 2016;11:1877–1888. doi: 10.1038/nprot.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Fernandes H, Mucida D. Neuro-immune interactions at barrier surfaces. Cell. 2016;165:801–811. doi: 10.1016/j.cell.2016.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387:1109–1122. doi: 10.1016/S0140-6736(15)00149-X. [DOI] [PubMed] [Google Scholar]
- Wilson SR, Thé L, Batia LM, Beattie K, Katibah GE, McClain SP, Pellegrino M, Estandian DM, Bautista DM. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell. 2013a;155:285–295. doi: 10.1016/j.cell.2013.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SR, Nelson AM, Batia L, Morita T, Estandian D, Owens DM, Lumpkin EA, Bautista DM. The ion channel TRPA1 is required for chronic itch. J Neurosci. 2013b;33:9283–9294. doi: 10.1523/JNEUROSCI.5318-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AZ, Tripathi SV, Kau AL, Schaffer A, Kim BS. Immune dysregulation underlies a subset of patients with chronic idiopathic pruritus. J Am Acad Dermatol. 2016;74:1017–1020. doi: 10.1016/j.jaad.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosipovitch G, Bernhard JD. Chronic Pruritus. N Engl J Med. 2013;368:1625–1634. doi: 10.1056/NEJMcp1208814. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Huo FQ, Jeffry J, Hampton L, Demehri S, Kim S, Liu XY, Barry DM, Wan L, Liu ZC, et al. Chronic itch development in sensory neurons requires BRAF signaling pathways. J Clin Invest. 2013;123:4769–4780. doi: 10.1172/JCI70528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. Molecular mechanism of TRP channels. Compr Physiol. 2013;3:221–242. doi: 10.1002/cphy.c120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng T, Oh MH, Oh SY, Schroeder JT, Glick AB, Zhu Z. Transgenic expression of Interleukin-13 in the skin induces a pruritic dermatitis and skin remodeling. J Invest Dermatol. 2009;129:742–751. doi: 10.1038/jid.2008.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. DRG neurons respond to type 2 cytokines in a dose-dependent manner. Relates to Figure 1.
(A) Average calcium imaging trace of IL-4-responsive mouse dorsal root ganglia (DRG) neurons challenged with vehicle and increasing doses of recombinant murine (rm)IL-4. Gray traces represent 24 IL-4-responsive neurons (from 256 KCl-responsive neurons) with average response shown in black.
(B) Quantification of DRG neuron responses to increasing doses of rmIL-4 as a percentage of total KCl-responsive neurons, n > 400 neurons from 2 WT mice.
(C) Average calcium imaging trace of IL-13-responsive mouse DRG neurons challenged with vehicle control and increasing doses of rmIL-13. Gray traces represent 15 IL-13-responsive neurons (from 248 KCl-responsive neurons) with average response shown in black.
(D) Quantification of DRG neuron responses to increasing doses of rmIL-13 as a percentage of total KCl-responsive neurons, n > 400 neurons from 2 WT mice.
(E) Average calcium imaging trace of mouse DRG neurons challenged with rmIL-5 (4 μg/mL), histamine (Hist, 50 μM), capsaicin (Cap, 300 nM), and KCl (100 mM). Gray traces represent individual KCl-responsive neurons with average response shown in black.
(F) Quantification of DRG neuron responses to rmIL-5, histamine, and capsaicin as a percentage of total KCl-responsive neurons, n > 300 neurons from a WT mouse.
Black bars in calcium imaging traces indicate timing of challenges. Data are represented as mean ± SEM.
Figure S2. Type 2 cytokine receptors are enriched on NaV1.8+ sensory neurons. Relates to Figure 2.
(A) Expression of selected genes in mouse DRG neuron populations generated from sort-purified sensory neurons shown as row Z-scores of microarray data. Neurons were sorted by Isolectin B4 (IB4), NaV1.8, and/or parvalbumin (Parv) expression. Full data set and methods available in Chiu et al., 2014.
(B) Total number of rmIL-4-, rmIL-13-, histamine-, and KCl-responsive mouse DRG neurons classified as small-, medium-, and large-diameter neurons (<18 μm, 18–25 μm, and >25 μm), n > 500 neurons from a WT mouse.
(C) Representative Venn diagram of overlapping responses of mouse DRG neurons to stimulation with rmIL-13 (300 nM), rmIL-31 (300 nM), and histamine (50 μM), n > 300 neurons from a WT mouse.
(D) Representative Venn diagram of overlapping responses of mouse DRG neurons to stimulation with rmIL-4 (4 μg/mL) and rmTSLP (4 μg/mL), n > 100 neurons from a WT mouse.
(E–G) Representative calcium imaging trace of mouse DRG neurons responding to (E) chloroquine (100 μM, CQ), (F) rmTSLP (0.25 μg/mL), and (G) rmIL-31 (3 μM) after vehicle control or rmIL-4 (4 μg/mL) challenge, n > 200 neurons per group.
Black bars in calcium imaging traces indicate timing of challenges.
Figure S3. MC903 treatment results in AD-like disease and robust chronic itch. Relates to Figure 3.
(A) Experimental schematic indicating daily topical treatment with vehicle control (ethanol, EtOH) or MC903 to the ears of WT mice.
(B) Ear thickness measurements as percent change from baseline over the course of control or MC903 treatment, n ≥ 4 mice per group.
(C) Representative H&E histopathology and (D) histology score of control and MC903-treated mice, n ≥ 4 mice per group.
(E) Comparison of row Z-scores of the regularized logarithm of gene expression values of selected AD-associated genes from RNA-seq of the skin of control and MC903-treated mice, n = 4 mice per group.
(F) Scratching behavior of control and MC903-treated mice on Day 0, 4, 8, and 12, n ≥ 4 mice per group.
(G) Representative responses of DRG neurons to increasing concentrations of MC903 as a percentage of total KCl-responsive neurons, n > 300 neurons from a WT mouse.
Scale bars indicate 100 μm. Data are represented as mean ± SEM.
Figure S4. Il31ra−/− mice are not protected from AD-like itch, and IL-4RαΔneuron mice have reduced infiltration of type 2 immune cells into the skin with a distinct skin transcriptional profile. Relates to Figure 3.
(A) Experimental schematic indicating daily topical treatment with MC903 to the ears of Il31ra−/− mice and WT control mice.
(B) Scratching behavior and (C) ear thickness measurement of Il31ra−/− mice compared to control mice on Day 7, n ≥ 9 mice per group.
(D) Experimental schematic indicating daily topical treatment with MC903 to the ears of IL-4RαΔneuron and littermate control mice.
(E) Basophil, (F) group 2 innate lymphoid cell (ILC2), and (G) T helper type 2 (Th2) cell frequencies as measured by flow cytometry from the ear skin of IL-4RαΔneuron mice at Day 12, n ≥ 3 mice per group.
(H) Comparison of row Z-scores of the regularized logarithm of gene expression values of the top 100 differentially expressed genes from RNA-seq of the skin of MC903-treated IL-4RαΔneuron mice compared to littermate control mice as determined by p-value. Genes are listed based on increasing adjusted p-value, n ≥ 4 mice per group.
Data are represented as mean ± SEM.
Figure S5. NaV1.8 expression is restricted to sensory neurons under both steady-state and inflammatory conditions. Relates to Figure 3.
(A) Experimental schematic indicating treatment of NaV1.8 reporter mice with MC903.
(B) Representative flow cytometry plots of the bulk DRG, skin, and skin-draining lymph node (sdLN) of naïve NaV1.8 reporter mice compared to non-reporter control mice.
(C) Representative flow cytometry plots of the bulk DRG, skin, and sdLN of MC903-treated NaV1.8 reporter mice compared to non-reporter control mice at Day 12.
(D) Experimental schematic indicating daily topical treatment with MC903 to the ears of IL-4RαΔneuron and littermate control mice.
(E–F) Quantification of Il4ra, Il2rg, and Il13ra1 by RT-qPCR in the (E) skin and (F) sdLN of IL-4RαΔneuron mice treated with MC903, n ≥ 3 mice per group.
Data are represented as mean ± SEM.
Figure S6. JAK1Δneuron mice are developmentally normal and are protected from chronic itch even in the absence of robust skin inflammation. Relates to Figure 4.
(A) Expression of the JAK family of genes in mouse DRG neuron populations clustered into functional subsets based on single-cell RNA-seq data. Numbers in heat map indicate fraction of positive cells within individual clusters by thresholding method. Full data set and methods available in Usoskin et al., 2015.
(B–C) Representative immunofluorescent images of dissociated DRG neurons at (B) baseline and (C) after 5 minutes of stimulation with rmIL-4 (1 μg/mL). Red staining indicates phosphorylated JAK1 (pJAK1). Blue staining indicates DAPI, n = 3 WT mice.
(D) Frequency of pJAK1+ cells as a percentage of all DAPI+ cells at baseline and after stimulation with rmIL-4, n ≥ 70 cells per high power field.
(E) Representative distribution of diameters of dissociated DRG neurons from a JAK1Δneuron and a littermate control mouse, n ≥ 200 neurons per group.
(F–I) Representative responses to (F) histamine (50 μM), (G) BAM8-22 (10 μM), (H) rmTSLP (4 μg/mL), and (I) rmIL-31 (300 nM) of DRG neurons from a JAK1Δneuron mouse compared to a littermate control mouse as a percentage of total KCl-responsive neurons, n ≥ 300 neurons per group.
(J) Experimental schematic indicating rotarod training and testing days.
(K) Results of rotarod testing of JAK1Δneuron mice compared to littermate control mice, n ≥ 4 mice per group.
(L) Experimental schematic indicating twice daily acetone-ether-water (AEW) treatment to the nape of the neck of JAK1Δneuron and littermate control mice.
(M) Scratching behavior of JAK1Δneuron and littermate control mice on Day 5 of AEW treatment, n ≥ 8 mice per group.
Scale bars indicate 50 μm. Data are represented as mean ± SEM.