

Abstract
OleT is a cytochrome P450 enzyme that catalyzes the removal of carbon dioxide from variable chain length fatty acids to form 1-alkenes. In this work, we examine the binding and metabolic profile of OleT with shorter chain length (n ≤ 12) fatty acids that can form liquid transportation fuels. Transient kinetics and product analyses confirm that OleT capably activates hydrogen peroxide with shorter substrates to form the high-valent intermediate Compound I and largely performs C–C bond scission. However, the enzyme also produces fatty alcohol side products using the high-valent iron oxo chemistry commonly associated with insertion of oxygen into hydrocarbons. When presented with a short chain fatty acid that can initiate the formation of Compound I, OleT oxidizes the diagnostic probe molecules norcarane and methylcyclopropane in a manner that is reminiscent of reactions of many CYP hydroxylases with radical clock substrates. These data are consistent with a decarboxylation mechanism in which Compound I abstracts a substrate hydrogen atom in the initial step. Positioning of the incipient substrate radical is a crucial element in controlling the efficiency of activated OH rebound.
Graphical abstract
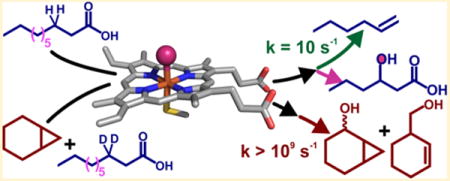
OleT, a cytochrome P450 (CYP) enzyme, has recently attracted a great deal of attention because of the unusual chemistry that it catalyzes. Using hydrogen peroxide as a cosubstrate, OleT removes the carboxylate from Cn chain length fatty acid (FA) substrates to produce Cn−1 terminal alkenes1 and carbon dioxide.2 This decarboxylation reaction is of considerable commercial interest as a potential means of intercepting microbial fatty acid biosynthesis and increasing the heating value of drop-in hydrocarbon biofuels.3,4 The scope of OleT-catalyzed decarboxylations, which has been expanded to include structurally diverse substrates that include a range of benzylic5 and dioic6 carboxylic acids, also suggests that it may serve as a useful biocatalytic platform for the synthesis of important commodity chemicals. The OleT-catalyzed C–Cα scission reaction is a significant departure from the function of most CYPs. This is perhaps best highlighted by comparison to those that are critical for xenobiotic detoxification, where an oxygen atom derived from atmospheric dioxygen is inserted into chemically unreactive substrates, generating a metabolite that is more water-soluble than the parent compound.
Analysis of the hydrocarbon content of Jeotgalicoccus species has demonstrated that long chain fatty acids (Cn, where n ≥ 20) are the native substrates for OleT in vivo.1 In the metabolism of eicosanoic acid (C20 FA or EA), the enzyme displays nearly exclusive (≥95%) chemoselectivity for 1-nonadecene production in vitro.2–4 Isotope tracer experiments have demonstrated that both substrate carboxylate oxygens are retained in the CO2 product.2 The lack of detectable products intermediary with respect to alkene formation from single-turnover reactions eliminates possible mechanisms whereby sequential oxygenations give rise to the atypical metabolite, such as those observed for other C–C bond-cleaving CYPs.7–9 The OleT conversion of a fatty acid to a terminal alkene thus requires, in sum, the removal of a substrate hydride (H−). Although the OleT C–Cα bond scission reaction would then appear to be quite different from mechanisms for archetypal P450 hydroxylations, which proceed via substrate hydrogen atom (H•) abstraction by the iron(IV)–oxo π cation intermediate (Compound I or P450-I) and subsequent rapid •OH radical recombination through a process termed “oxygen rebound,”10–12 several lines of evidence strongly connect the two mechanisms. The primary sequence of OleT and the X-ray crystal structure determined by Munro, Leys, and colleagues13 have revealed that OleT belongs to the P450 fatty acid peroxygenase (CYP152) family.14 CYP152s include the well-characterized CYPs BSβ and SPα that utilize the substrate carboxylate as a general acid to efficiently utilize H2O2 as an oxidant for the hydroxylation of FAs at positions adjacent to the terminal carboxylate, affording alcohol products with variable regioselectivity.14 Although the active intermediate responsible for FA hydroxylation has not been directly observed in BSβ or SPα, a significant steady-state kinetic isotope effect (KIE) for hydroxylation15 strongly suggests a scenario involving H• abstraction by P450-I and ensuing •OH rebound.
Transient kinetic studies by our laboratory have shown that the initial stages of OleT decarboxylation may result from a mechanism similar to those proposed for CYP aliphatic hydroxylations. Single-turnover, rapid mixing studies of OleT bound to perdeuterated EA (EA-d39) with H2O2 result in the generation of a Compound I intermediate that we term Ole-I that accumulates to a high level.2 The optical features of Ole-I suggest that it is electronically similar to the high-valent oxo complexes observed in rapid mixing studies of other CYPs16–18 and the structurally dissimilar thiolate ligated heme enzyme aromatic peroxygenase (APO)19 with peroxyacids, all potent hydroxylation catalysts that are not known to perform desaturative decarboxylations. On this basis, we originally proposed a radical mechanism for 1-alkene production whereby Ole-I abstracts H• from the Cβ position to form a substrate radical, resulting in formation of the iron(IV)–hydroxide, Compound II (Figure 1). Subsequent one-electron oxidation of the substrate by Compound II, coupled to transfer of a proton, would restore the aqua–ferric resting state of the enzyme and generate a substrate carbocation. Such a species could then readily eliminate CO2 and form the Cα=Cβ bond. This type of mechanism is similar to those proposed for desaturations performed by some CYPs,20,21 and O2 activating non-heme mononuclear22 and dinuclear iron enzymes23–25 and synthetic complexes26 that closely structurally resemble monooxygenation catalysts. In support of this scheme, we have recently measured a large (kH/kD > 8) substrate 2H KIE for the decay of Ole-I and have isolated an additional intermediate during EA decarboxylation. Concomitant with the decay of Ole-I, a new intermediate is formed that has optical spectroscopic properties18,27 and a reactivity toward phenols27,28 that are consistent with its assignment as an iron(IV)–hydroxide species (Ole-II).29 In the decarboxylation of EA, Ole-II decays slowly (~10 s−1 at 5 °C), suggesting that the long chain substrate is positioned in such a way that the rapid oxygen rebound characteristic of CYPs is thwarted, allowing alkene formation to occur.
Figure 1.
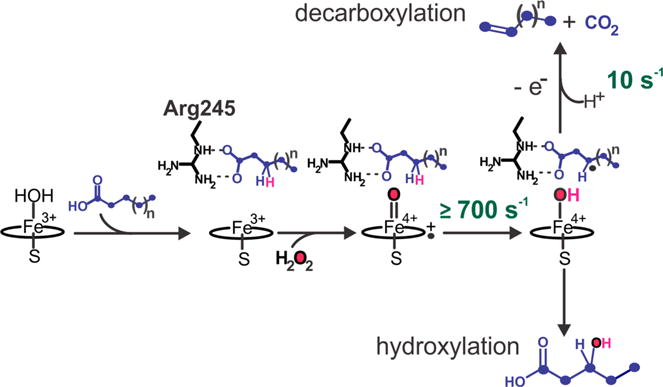
Proposed bifurcative mechanism for the decarboxylation (top) and hydroxylation (bottom) reactions catalyzed by cytochrome P450 OleT with long and short chain (Cn) fatty acid substrates. The rate constants for Compound I and II decay are derived from earlier stopped flow studies with eicosanoic acid (Cn, where n = 20).2,29 For long chain length (CL) substrates (Cn, where n ≥ 20), Cn−1 alkene formation has been shown to be the predominant (≥95%) pathway.2–4,30 Shorter CL substrates have been observed to result in mixed chemoselectivity, producing variable percentages of fatty alcohols and Cn−1 alkenes.1,3,4,30,31
A more precise understanding of the factors that control the branchpoint between monooxygenation and decarboxylation would allow OleT and related enzymes to be rationally engineered for fungible and targeted fuel production. A demonstrated limitation of OleT1,3,4,30,31 and other CYP152s32 is the production of a variable and unwanted proportion of Cn alcohol products, in addition to the Cn−1 alkene, upon reduction of the substrate chain length (CL) below C20. Within the framework of the proposed mechanistic scheme in Figure 1, it is unknown whether the unusually long stability of Ole-II, and the presumptive substrate radical, stems from underappreciated electronic changes in ferryl intermediates that are not readily apparent from the optical data, positional restraints of the substrate imposed by the protein, or structural contributions from the secondary coordination sphere. To better gauge the factors that contribute to this bifurcation, we have examined the binding and reactivity of short chain length FAs (C8–C12), substrates that would be metabolized to the most desirable hydrocarbon “drop-in” biofuels. Although shorter CL substrates bind to the enzyme with a substantially lower affinity, they are efficiently metabolized and support the efficient generation of Ole-I. The production of alkenes with these substrates is accompanied by the formation of alcohol products with complete incorporation of oxygen from H2O2. An analysis of products from the OleT metabolism of norcarane and methylcyclopropane radical clock probes provides a direct measure of the proficiency of radical recombination. The measured radical lifetimes are in sharp contrast with kinetic data for Ole-II decay for both short and long chain FAs and are similar to those measured for many other P450 hydroxylases. These studies provide compelling evidence that OleT alkene formation proceeds by a radical mechanism that bifurcates after hydrogen atom transfer (HAT) and alludes to the importance of substrate active-site chemistry, rather than the electronic structure of iron intermediates, in controlling reaction partitioning in this group of enzymes. The competition of oxygen rebound to form an alcohol and electron transfer to produce a carbocation is guided by exquisite coordination of the FA substrate.
MATERIALS AND METHODS
Chemicals and Reagents
Chemicals, unless otherwise indicated, were purchased from Sigma-Aldrich. Isopropyl β-D-thiogalactopyranoside and all buffers used in this study were purchased from Research Products International (Mt. Prospect, IL). Imidazole and Luria broth were purchased from bio-WORLD (Dublin, OH). Isotopically labeled H218O2 (90% 18O) was purchased from ICON Isotopes (Summit, NJ). Perdeuterated fatty acids were purchased from CDN Isotopes (Pointe-Claire, QC). Protiated fatty acids, N,O-bis(trimethylsilyl)-trifluoroacetamide (BSTFA), and trimethylchlorosilane (TMCS) (99:1) were purchased from Supelco (Bellefonte, PA). Radical clock substrates were synthesized according to published procedures.33,34
Heterologous Expression and Purification of P450 OleT
Wild-type cytochrome P450 OleT from Jeotgalicoccus sp. ATCC 8456 was overexpressed in Escherichia coli and purified as previously described.2,29 Adventitiously bound fatty acids were removed in a procedure similar to that previously reported that includes treatment with a 5–10-fold molar excess of H2O2 and desalting on a PD10 column. Here, we have utilized a subsequent step involving incubation of the protein for 15 h at 4 °C with BioBeads SM2 resin (Bio-Rad). The protein was separated from the resin with a syringe filter and verified for complete conversion to the low-spin substrate-free form by optical spectroscopy. Protein concentrations were determined using an extinction coefficient (ε417 = 109 mM−1 cm−1) determined by the pyridine hemochromagen method.35
Guaiacol Oxidation Studies
The oxidation of guaiacol to the polymerized tetraguaiacol product was used as a probe to determine the optimal chain length and isotopic composition of fatty acid substrates that efficiently activate OleT yet still permit sufficient entry of small molecules into the active site. An 800 μL reaction mixture contained 200 mM KPi (pH 7.5), 200 mM NaCl, 2 μM OleT, 2 mM guaiacol, and 800 μM fatty acid and was allowed to incubate for 1 h while being stirred at 4 °C. The reaction was initiated with 1 mM H2O2, and tetraguaicol formation was monitored at 470 nm using a Hewlett-Packard 8453 spectrophotometer. Initial rates were determined using a tetraguaiacol extinction coefficient of 26.6 mM−1 cm−1.
Determination of Substrate Dissociation Constants
A 500 μL reaction mixture containing 5 μM OleT in 200 mM KPi (pH 7.5) and 200 mM NaCl was titrated with sequential additions of a 10 mM fatty acid stock dissolved in ethanol with a Hamilton gastight syringe. The amount of ethanol never exceeded 5% (v/v). In contrast to similar plots for the binding of EA,2,13 the relatively high dissociation constants of C8 and C6 fatty acids did not necessitate fitting to a quadratic Morrison expression.36 In these cases, the additive absorbance changes at 392 and 417 nm were fit as a function of substrate concentration [S] using Origin software with the following hyperbolic expression to determine dissociation constants (Kd).
In the case of C10 and C12 FAs, which have KD values within an order of magnitude of the enzyme concentration utilized, the following Morrison expression was utilized for KD determination.
Using spectral deconvolution procedures to determine the fraction HS as a function of Stot resulted in similar KD values. The addition of small molecules (for example, guaiacol, benzene, and norcarane) to FA-bound OleT did not induce any additional spectroscopic changes in the enzyme.
Product Analysis of OleT with Short Chain Fatty Acid Substrates
A sealed vial contained a 2 mL reaction mixture of 5 μM OleT and 500 μM fatty acid (prepared from a 20 mM stock in ethanol) in 200 mM KPi (pH 7.5). Each reaction was terminated by the addition of 1 mL of 12 M HCl and each mixture extracted with 4 mL of chloroform using a gastight Hamilton syringe. Dual internal standards consisted of an alkene (125 nmol of hexadecene) and a FA (500 nmol). C12 FA was used as the standard for reactions of C8, C14 FA for reactions of C10, and C10 FA for reactions of C8. Vials were centrifuged, and the organic phase was dried using N2 and subsequently derivatized with 200 μL of BSTFA and TMCS (99:1) at 68 °C for 30 min. The quantity of fatty acid metabolized and fatty alcohol products, both nonvolatile, can be determined empirically for all CL substrates by comparing the peak areas of monotrimethylsilylated (TMS) fatty acids and di-TMS product alcohols to internal standards with response factors that have been determined in previous studies.32 For reactions using C12 FA as a substrate, all of the products are nonvolatile, and we have found that the sum of all products accurately accounts for ≥98% of the substrate metabolized. This is not the case for reactions of OleT with C10 FA and C8 FA because of the volatility of the Cn−1 alkene products, nonene and heptene, respectively, that are produced. For these substrates, the presence of alkenes was verified by headspace gas chromatography (GC) analysis, but products were quantified by subtraction of the product alcohols from the total amount of substrate metabolized. Peaks were identified on the basis of their retention time, mass spectral fragmentation patterns, and comparison to authentic standards. To minimize the level of overly oxidized products (e.g., ketones and di-OH FAs), low concentrations of H2O2 relative to FA were used (2:1 on a molar basis). Gas chromatography–mass spectrometry (GC–MS) analysis of C10 and C8 reactions showed no evidence of these products within detection limits. Headspace GC analysis employed similar reaction conditions in a septum-sealed vial. A reaction mixture from which H2O2 was omitted was used as a control. Nonane (112 nmol) was added as an internal standard, and 250 μL of the reaction headspace was injected onto a GC Hewlett-Packard 5890 GC system using a Hamilton syringe needle. The gas chromatograph was equipped with a J&W Scientific DB-5MS column (30 m × 0.25 mm, 0.5 μm). The following oven temperature program was used: 300 °C injector temperature, held at 50 °C for 3 min, heated at a rate of 5 °C min−1 to 100 °C, held at 100 °C for 3 min.
Insertion of Oxygen into Fatty Alcohol Products
A 2 mL reaction mixture containing 200 mM KPi (pH 7.5), 1 mM FA, and 5 μM OleT was initiated by the addition of hydrogen peroxide [2 mL, 500 μM H216O2 or H218O2 (90% 18O)] over the course of 1 h. Upon completion, the mixture was extracted with an equal volume of chloroform and 100 nmol of dodecanoic acid was added as an internal standard. The combined mixture was vortexed for 15 min and centrifuged at 4 °C for 20 min. The organic phase was removed and derivatized as described above. Samples were analyzed on a Hewlett-Packard 5890 gas chromatograph with an Rtx-5 fused silica column (30 m × 0.25 mm inside diameter, film thickness of 0.25 μm) using a temperature gradient from 70 to 300 °C at a rate of 10 °C/min. The column fed directly into a Waters VG 705 magnetic sector mass spectrometer and was ionized using a 70 eV electron impact energy.
Stopped Flow Spectroscopy
Transient kinetic experiments were conducted using methods we have previously described2,29 with minor modifications. Because of the lower affinity of C12 and C10 FA for OleT, it was necessary to add these substrates in a large molar excess to the enzyme to generate a fully saturated enzyme:substrate (E:S) complex. The OleT:DA and OleT:LA complexes were generated by addition of 500 μM FA to 20 μM OleT in 200 mM KPi (pH 7.5) and allowed to incubate for 2 h at 4 °C. The measured Compound I decay rate constants exceed those for substrate association steps by at least an order of magnitude. As a result, the spectral changes monitored in short time regimes approximate single-turnover conditions. For stopped flow experiments that included a saturating norcarane concentration, 5 μL of neat norcarane was added to 2 mL of the OleT:FA reaction mixture. Samples were centrifuged to remove any remaining nonsolubilized fatty acid and subsequently loaded into one syringe of an Applied Photophysics SX20 stopped flow spectrophotometer. E:S complexes were rapidly mixed with H2O2 in 200 mM KPi, and the reaction was monitored by a photodiode array (PDA) or at 370 or 440 nm with a photomultiplier tube (PMT). Ole-I decay rates were obtained by fitting the PMT time courses at 370 or 440 nm to the following summed two-exponential expression in Pro-Data software.
where At,obs is the observed absorbance, 1/ti is the reciprocal relaxation time (RRT) (inverse seconds), ai is the amplitude of phase i, t is the time (seconds), and A∞ is the final absorbance. The observed RRTs were analyzed as a function of H2O2 using the following kinetic scheme where Fe3+(HS) and Fe3+(LS) are the high-spin and low-spin forms of the enzyme, respectively, and Ole-I and Ole-II have the usual designations:
We have previously found in reactions of EA that only the fast, larger amplitude phases (RRT1) at 370 and 440 nm demonstrate appreciable isotopic sensitivity. This is also the case for DA and LA. The RRT1 at 370 nm, which corresponds to Ole-I decay, is identical in value to the fast RRT at 440 nm, or Ole-II formation. The RRT1 (1/τobs) versus [H2O2] plot for deuterated substrates was fit using the following hyperbolic expression
where the apparent is provided by k−1/k1 and the rate constant for C–D bond cleavage by Ole-I is provided by k2 (the asymptote of the hyperbolic plot). The y-intercept (k−2) is zero because of the irreversibility of C–D bond cleavage. The slower and smaller amplitude RRT at 370 or 440 nm (RRT2), which is insensitive to H2O2, provides the decay rate constant of Ole-II (k3).
Radical Clock Studies
A 2 mL reaction mixture containing 200 mM KPi (pH 7.5), 200 mM NaCl, 1 mM perdeuterated decanoic acid (DA-d19) prepared in ethanol, 25 μM OleT, and 2 μL of neat radical clock substrate (norcarane or methylphenylcyclopropane) was allowed to stir and incubate at room temperature for 15 min prior to the addition of hydrogen peroxide (2 mL, 5 mM) via a syringe pump over the course of 1 h. After completion of the reaction, 400 μL of CDCl3 (stabilized with Ag) was added to each sample for product extraction. The mixture was vortexed vigorously for 1 min and centrifuged at 16200 × g. The organic layer was removed from the bottom of the conical centrifuge tube and placed in a GC–MS vial for immediate analysis. Samples that were not immediately analyzed were stored at −80 °C. Products were analyzed on an Agilent 7890A GC system (Rtx-5 G27 30 m column with fused silica) with an Agilent 5975C mass spectrometer with EI/CI capabilities (Princeton), or on an Agilent 5977E GC/MSD bundled system with an HP-5MS column (Barnard). For methylphenylcyclopropane products, the sample was injected at an oven temperature of 35 °C, held for 2 min, and heated at a rate of 10 °C/min to a final temperature of 225 °C. For norcarane-derived products, two slightly different methods were employed. For method A (Princeton), the initial oven temperature was 30 °C, which was held for 6 min and subsequently increased at a rate of 10 °C/min to 225 °C. For method B (Barnard), the initial oven temperature was 35 °C, with a 5 min hold time, followed by a 10 °C/min gradient to a final oven temperature of 225 °C. The rate constants for the oxygen rebound reactions were determined by multiplying the ratio of ring-closed to ring-opened products by the intramolecular radical rearrangement rate for each probe: krearr = 2 × 108 s−120 and 3 × 1011 s−137 for norcarane and methylcyclopropane, respectively. The radical lifetime is the reciprocal of this value.
RESULTS
The Metabolism of Short Chain Length FAs Results in Alcohol Products from Radical Recombination
The chemical outcome of OleT has been shown to vary significantly with the fatty acid CL. Published studies that have probed the OleT metabolism of longer CL (Cn, where n ≥ 16) fatty acids are largely consistent and generally show a high degree of chemoselectivity of the enzyme.1–3,13 However, there is significant ambiguity in the metabolic profiles of short to mid CL substrates (C8–C12) despite their importance for the production of the most desired biofuels. Ultraviolet–visible (UV–vis) titration studies (Figure S1) examined the binding and efficiency of low- to high-spin-state conversion for this set of substrates. The data, summarized in Table 1, show a systematic decrease in both the affinity and propensity for high-spin conversion as the chain length of the FA is shortened. This is consistent with the general trend previously reported for C12–C22 saturated substrates by Belcher et al.13 The measured dissociation constants are several orders of magnitude lower than those reported for EA (<1 μM), most likely because of the extensive loss of hydrophobic contacts with the enzyme upon reduction of the acyl chain.
Table 1.
Binding Parameters of Short Chain Fatty Acids and OleT
| fatty acid | KD (μM) | maximal high-spin conversion (%) |
|---|---|---|
| C12:0 | 2.8 ± 0.4 | 61 |
| C10:0 | 25 ± 8 | 36 |
| C8:0 | 134 ± 11 | 25 |
| C6:0 | 164 ± 15 | 19 |
Reported turnover data for OleT and shorter chain FAs have shown significant variability, which may be attributed to differences in the turnover conditions utilized, the overall efficiency of substrate conversion, and, possibly, the analytical methods employed.3,4,31 For example, turnover studies of C12 FA (or lauric acid, LA), using either a surprisingly efficient NAD(P)H/O2/redox partner (CamAB) system and headspace GC detection3 or a photochemical turnover system and liquid:liquid extraction,4 have found that alkenes are exclusively generated or not produced in detectable levels, respectively. Here, we have utilized turnover conditions in which H2O2 is slowly introduced into the reaction mixture. We have shown that both OleT and another CYP152 enzyme that we have termed CYP-MP retained catalytic activity with these methods and produced high levels of products, facilitating accurate product quantitation.32 Using the H2O2 perfusion approach, the shorter CL FAs were nearly completely metabolized within 2 h. A representative chromatogram of the reaction headspace of the C10 FA reaction is shown in Figure S2 and revealed the presence of a significant amount of alkene, consistent with previous results using the CamAB/O2 turnover system. GC–MS analysis of quenched samples was utilized to detect and quantify any possible nonvolatile products. Representative chromatograms of the Cn TMS-derivatized alcohol products and Cn−1 alkenes are shown in Figure S3. Product distributions are summarized in Table 2. For all substrates, 1-alkene was the dominant reaction product formed. However, analysis of the GC–MS chromatograms following derivatization also revealed the presence of a significant fraction of fatty alcohols for some substrates. This was most apparent for the reaction of C12, which showed that alcohols, mostly localized to Cβ but also present at other positions, comprised nearly 30% of the total product formed. Although the distribution of products (alkenes vs alcohols) varied among the CLs tested here, the regiospecificity of hydroxylation showed a strong bias (>70%) toward the Cβ position for all substrates.
Table 2.
Metabolic Profile of Short Chain Fatty Acids with OleT and Slow H2O2 Addition
| fatty acid | % metabolized | product distribution (%)
|
||
|---|---|---|---|---|
| 1-alkene | alcohols | alcohol regiospecificity | ||
| C12:0 | 98.3 ± 0.2 | 61.4 ± 7.6a | 38.6 ± 7.6 | 7.0 ± 1.3% α |
| 71.5 ± 15.5% β | ||||
| 21.5 ± 4.4% γ | ||||
| C10:0 | 100 | 90.0 ± 0.8b | 10.0 ± 0.8 | 100% β |
| C8:0 | 100 | 88.4 ± 0.4b | 11.6 ± 0.8 | 89.0 ± 5.3% β |
| 11.3 ± 2.3% α | ||||
Determined from direct quantitation.
Determined from subtraction of product alcohols from the total FA metabolized.
Given the unusual stability of Ole-II in the reaction of EA and, by extension, a presumptive substrate Cβ radical, oxygen isotope labeling studies probed whether the observed product alcohols specifically resulted from an authentic oxygen rebound event or from a process involving diffusive escape of these more weakly bound substrates after the radical is formed.38,39 Reactions using C12 FA, which formed the most alcohol products, were performed with H216O2 or H218O2 as an oxidant in sealed vials, quenched, and analyzed by MS following trimethylsilylation (TMS). A comparison of the demethylated [m – 15] fragment ions from the TMS-derivatized alcohols is shown in Figure 2. When the reaction was performed with H218O2, a diagnostic shift of the fragment ion from m/z 345 to 347 was observed, consistent with complete (≥95%) integration of 18O from the oxidant. No appreciable exchange with solvent was observed within the available limits of detection. This indicates that the substrate radical, once formed, is retained within the OleT active site and that oxygenation via radical recombination occurs in a canonical fashion.
Figure 2.
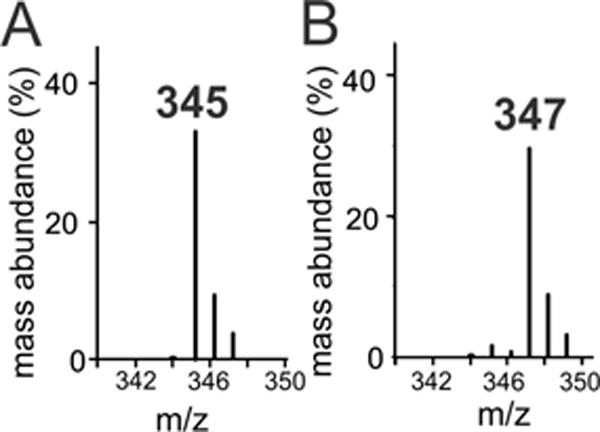
Gas chromatography mass spectra of the trimethylsilylated Cβ alcohols produced by OleT using lauric acid as a substrate and (A) H216O2 or (B) H218O2 as an oxidant.
Short Chain Substrates Efficiently Generate Ole-I
The binding of fatty acid is thought to be obligatory for the efficient activation of H2O2 by CYP152 enzymes whereby the substrate carboxylate serves as a general acid to facilitate proton rearrangement for the generation of P450-I.40,41 In transient kinetic studies of OleT, we have found that Ole-I is detected in reactions with H2O2 only when the enzyme is bound to FA.2 The efficient turnover number, high chemoselectivity, and Cβ selectivity of shorter CL FAs with OleT suggest that these substrates should occupy a position very similar to that of EA within the active site. Stopped flow studies directly interrogated the reaction of C10 FA [decanoic acid (DA)] with OleT. Rapid mixing of the ternary OleT:DA-d19 complex with H2O2 resulted in the formation of readily detectable levels of Ole-I within the dead time of the instrument (Figure 3A). Over the course of 300 ms, Ole-I decayed to the low-spin Fe3+–OH2 enzyme. Although the spectroscopic features of Ole-I generated with DA-d19 are identical to those we have observed with EA, the level of accumulation is markedly lower for the shorter substrate (<20% for DA vs >60% for EA). In principle, this could arise from an enhanced reactivity (C–D abstraction rate) of Ole-I with DA-d19, compromised H2O2 activation, or a more complex kinetic behavior. To address these possibilities, the absorbance changes of Ole-I were monitored at 370 nm using a photomultiplier tube (PMT). When using high H2O2 concentrations, the time course revealed a biphasic behavior that could be fit to a two-summed exponential expression with well-resolved reciprocal relaxation times (RRT1 ~ 60 s−1, and RRT2 ~ 8 s−1) (Figure S4). The RRTs are similar to the decay rate constants of Ole-I and Ole-II in the reaction of EA-d39 (80 and 10 s−1, respectively). This suggests that the poor accumulation of Ole-I did not result from a faster reaction of Ole-I with DA. The H2O2 dependence of both RRTs was examined, allowing for a full dissection of the kinetic rate constants for the DA reaction and efficiency of H2O2 activation. RRT1, which corresponds to the rate of Ole-I decay, revealed a hyperbolic dependence on H2O2 concentration. RRT2, the decay rate constant for Ole-II, was independent of H2O2 (Figure 3B). Using nonlinear kinetic fitting procedures, we measured an Ole-I decay rate constant (k1 = 62 ± 4 s−1), the asymptote of the plot, and an apparent KD for H2O2 . The Ole-II decay rate constant (k2) was 9 ± 1 s−1. Thus, the activation of OleT and the kinetic rate constants for Ole-I and Ole-II decay are similar to those of short (DA) and long CL (EA) substrates. This is consistent with the similar chemical nature [i.e., C–D bond dissociation energy (BDE)] of both classes of substrates and the largely similar reaction outcome.
Figure 3.
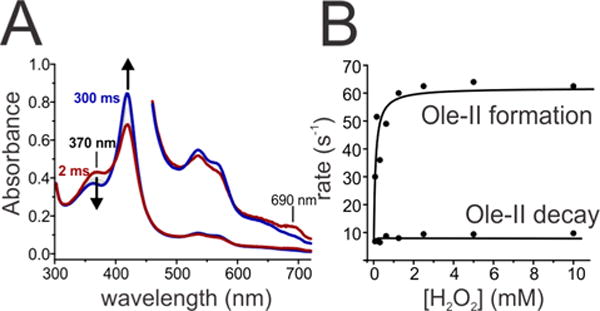
Transient kinetic studies of the reaction of OleT bound to perdeuterated decanoic acid (DA-d19) and H2O2 at 4 °C. (A) Photodiode array spectra show that Compound I decays over the course of 300 ms to the ferric low-spin enzyme. (B) The decay of OleT Compound I (Ole-I) was fit to a biexponential expression and plotted vs H2O2 concentration, revealing the rate constants of Ole-I and Ole-II decay (~60 and 10 s−1, respectively).
OleT Compound I-Catalyzed Oxidation of Alternative Substrates
To expand the inventory of substrates that could be probed in reactivity studies of Ole-I, particularly for the radical clock measurements described below, we have employed the “decoy” strategy from the pioneering work of Watanabe, Shoji, and co-workers.41–43 This has proven to be a highly adaptable approach for many CYP152 enzymes and permits BSβ and SPα to oxidize substrates that do not contain a carboxylate. We directly tested whether the binding of a short CL FA would support the formation of Ole-I yet still permit sufficient access of a secondary molecule for oxidation. Enzyme–substrate (E–S) complexes were prepared with EA-d39 or deuterated C12 FA (lauric acid, LA-d23) and rapidly mixed with 10 mM H2O2 in the presence or absence of the radical clock norcarane. When using EA-d39, the initial 1 ms spectra recorded after mixing revealed the presence of an admixture of species. These included Ole-I, with a Soret maximum at 370 nm and an additional band at 690 nm, Ole-II, and the Fe3+–OH2 product state (Figure 4A, red spectrum). The inclusion of norcarane did not significantly alter the relative composition of these species (Figure 4A, black spectrum). The formation and decay of Ole-II were followed at 440 nm using PMT detection under both conditions. The time courses are nearly superimposable (Figure 4B). In the presence of norcarane, the measured rate constants for Ole-II formation (Ole-I decay) and Ole-II decay were 83 ± 3 and 10 ± 2 s−1, respectively. These values are identical to those we have measured for the reaction of EA-d39 in the absence of a secondary molecule.2,29 This demonstrates that in the presence of a long chain FA, norcarane cannot efficiently access the heme iron to react with Ole-I.
Figure 4.
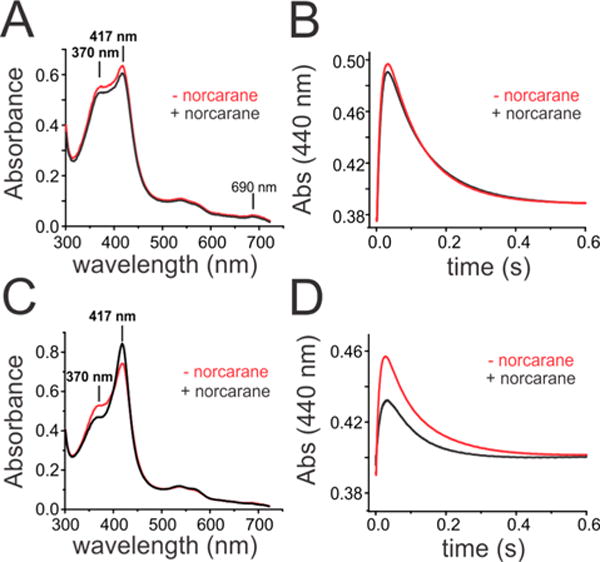
Transient kinetic studies of the reaction of OleT bound to perdeuterated eicosanoic acid (EA-d39) or lauric acid (LA-d23) with H2O2 at 4 °C in the presence (black) or absence (red) of the radical clock norcarane. (A and C) Photodiode array spectra taken at 1 ms after mixing. (B and D) Time course of OleT Compound II formation and decay monitored at 440 nm.
Rapid mixing of the LA-d23-bound enzyme with H2O2 also revealed the presence of Ole-I at levels of accumulation that were similar to those of reactions of DA-d19 (Figure 4C, red trace). In the presence of norcarane, however, the 1 ms spectra revealed a significantly different distribution of species (Figure 4C, black trace). The absorption maximum corresponding to Ole-I at 370 nm was significantly lower and accompanied by the presence of higher levels of the Fe3+–OH2 product form of the enzyme with a Soret peak at 417 nm. When the time course at 440 nm was examined, it was also apparent that Ole-II also failed to accumulate when norcarane was included in the reaction mixture (Figure 4D). However, an examination of the RRTs for Ole-II formation and decay indicated that the kinetic parameters were largely insensitive to the presence of the small molecule. The observed RRT values for Ole-II formation (RRT1) were 105 ± 10 and 66 ± 3 s−1 and the decay rate constants of Ole-II (RRT2) 10.4 ± 0.5 and 12.5 ± 0.6 s−1 for reactions in the absence and presence of norcarane, respectively. Provided that the activation of H2O2 is insensitive to the presence of small molecules such as norcarane, a decrease in the level of accumulation of both OleT ferryl intermediates suggests a branched oxidation mechanism from Ole-I. This implies that the products of radical clock oxidation characterized below derive from the same Ole-I oxidant that initiates the decarboxylation reaction sequence. Additionally, the lower level of accumulation of Ole-II with norcarane suggests that reactions with the radical clock substrate are substantially different from those with FAs and are likely accompanied by a much faster rebound step.
The “decoy” approach was further optimized with a series of short CL FAs using the oxidation of guaiacol to the polymerized tetraguaiacol product as a facile colorimetric probe. C8 and C10 FAs were the most proficient for producing tetraguaiacol (Figure S5). We next tested whether changing the isotopic composition of DA would further facilitate the metabolism of a secondary probe substrate. The guaiacol oxidation rate improved approximately 5-fold when using the more inert DA-d19. This is consistent with the transient kinetic data for norcarane, whereby both the FA and secondary substrate, guaiacol in this case, are competitively oxidized by Ole-I. Enhanced guaiacol oxidation with short CL FAs may result from direct entry of the small molecule into the active site, as with norcarane, or by opening a more distal site in the enzyme pocket for guaiacol to bind for ensuing long-range electron transfer. Intriguingly, perfluorinated DA failed to promote guaiacol oxidation, most likely because of an unfavorable binding configuration or alteration of the carboxylate pKa.
Considering the efficient incorporation of oxygen into product alcohols, we sought to time the radical recombination reaction. Radical clock substrates have been widely used for many decades to probe the reaction mechanisms of C–H oxidizing enzymes and biomimetic model systems.20,33,44–60 However, these probes have not been examined with H2O2-utilizing CYPs. The efficient oxidation of guaiacol using DA-d19 served as a template for testing whether a similar approach could facilitate the oxidation of norcarane (bicyclo[4.1.0]heptane) and methylphenylcyclopropane (MCP). The observed product yields for radical clock substrates were highest when using DA-d19. A representative GC chromatogram of the chloroform-extracted major reaction products of norcarane in the presence of DA-d19 is shown in Figure 5. The peaks were well-resolved and easy to analyze on the basis of MS data and our earlier assignments. The distribution of major species is shown in the inset of Figure 5 and summarized in Table 3.
Figure 5.
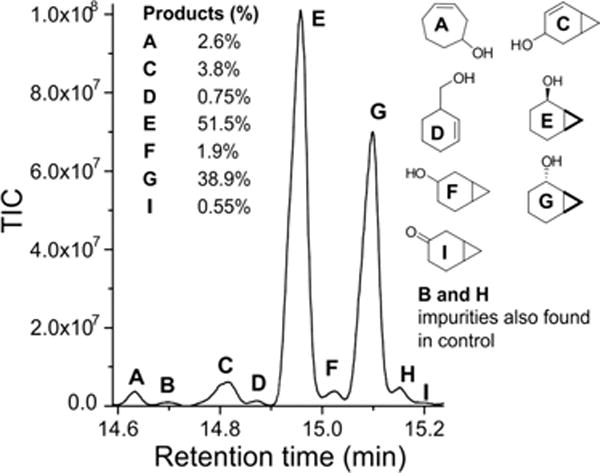
Total ion chromatogram of the products from the oxidation of the radical clock norcarane using perdeuterated decanoic acid (DA-d19) to trigger the formation of OleT Compound I using H2O2. The product ratios of major products are shown in the inset.
Table 3.
Product Distribution Profiles of Mechanistically Significant Products from the Oxidation of Norcarane and Methylphenylcyclopropane in the Presence of DA-d19
| norcarane | product yield (%)
|
radical lifetime (ps) | ||||||
|---|---|---|---|---|---|---|---|---|
| cation | radical | E | G | F | I | C | ||
| trial 1 | 2.6 | 0.75 | 51.5 | 38.9 | 1.9 | 0.55 | 3.8 | 42 |
| trial 2 | 1.4 | 0.47 | 47.6 | 33.4 | 29 | |||
| methylcyclopropane | product yield (%)
|
radical lifetime (ps) | |
|---|---|---|---|
| ring-opened | ring-closed | ||
| trial 1 | 59.9 | 40.1 | 2.2 |
| trial 2 | 58.1 | 41.9 | 2.4 |
The oxidation of norcarane by OleT produced an informative array of hydroxylation and desaturation products. The major products were exo- (G) and endo-2-norcaranol (E), as has been generally observed for other P450 hydroxylases. Significant amounts of the desaturation product, 2-norcarene, and its hydroxylated derivative, 4-hydroxy-2-norcarene (C), were also identified. These products are thought to derive either from a substrate cation through deprotonation or from a second C–H abstraction adjacent to the incipient radical.53 However, no 3-norcarene was detected. In addition, both the radical-rearranged product, 3-hydroxymethylcyclohexene (D), and the cation-rearranged product, 3-cycloheptenol (A), were also observed in readily detectable amounts. Notably, very similar amounts of the cation-rearranged product were observed in studies of the fungal heme-thiolate hydroxylase APO.61 On the basis of the averaged ratio of rearranged products to norcaranol (G + E), a radical lifetime of ~35 ps was calculated. Using the same approach with DA-d19 as a decoy, the oxidation of MCP also afforded both unrearranged and rearranged alcohol products (Figure S6). While mechanistically less informative, the product ratio is also consistent with the presence of a short-lived (picosecond) substrate radical.
DISCUSSION
We have examined the ability of OleT to convert short chain fatty acids (C6–C12), those most valuable for the development of biosynthetic strategies for fuel production, into alkenes. These compounds were evaluated as substrates for OleT using equilibrium binding, steady-state, and transient kinetic methods. The binding was markedly weaker for this set of substrates and was accompanied by a lower degree of high-spin conversion of the heme iron. Nonetheless, these shorter chain substrates efficiently activate H2O2 to generate Ole-I, and OleT can capably produce 1-alkenes with this panel of substrates. This is in contrast to some of the other recently identified enzymes that catalyze hydrocarbon synthesis that may have a more narrowed substrate scope.22
Importantly, however, various amounts of Cβ hydroxylation (10–28%) accompanied the desaturative decarboxylation of shorter chain length fatty acids. For LA, hydroxylation was also observed at Cα (7%) and Cγ (22%) positions. Although they are minor products, the incorporation of labeled oxygen from H218O2 into the alcohol of the hydroxyacid demonstrates that OleT is capable of prototypical aliphatic oxidations. The radical clock substrates norcarane and methylphenylcyclopropane (MPC) were also hydroxylated in the presence of deuterated FAs. This result indicates that the same Ole-I high-valent intermediate can catalyze two very different reactions, decarboxylation and hydroxylation.
A key question this work set out to answer is what controls the bifurcation between hydroxylation and decarboxylation in OleT. The Compound I intermediate generated by CYPs and porphyrin models is a very versatile reagent for chemical transformations. Depending on the balance between the nature of the axial ligand, steric hindrance near the active site, and the electronic structure of the substrate, this ferryl porphyin π-cation radical is capable of initial hydrogen atom abstraction that can be followed by either •OH rebound, heteroatom rebound, abstraction of a second hydrogen atom to form an internal double bond, or transfer of an electron from the substrate to form a substrate cation from which C–C bond cleavage reactions such as decarboxylation can occur.12,62 A number of studies have sought to clarify the active-site requirements that allow OleT to steer the reaction coordinate toward decarboxylation. Particular focus has centered on potential proton donation pathways (e.g., His85)1,30,63 and hydrogen bonding interactions with solvent64 for tuning the barrier for oxygen rebound. Although we cannot exclude a possible contribution of these interactions for tuning iron–oxo reactivity, on the basis of the results presented herein, we propose that the position of the fatty acid substrate, bound to key residues in the active site, is critical for steering the reaction at several stages of catalysis. First, substrate binding allows proton rearrangement of the bound peroxide and facilitates the formation of Ole-I. Without the substrate carboxylic acid, the peroxide bond is not cleaved and Ole-I cannot efficiently form. Binding is also accompanied by a conversion to the high-spin state. Thus, this switch also provides a vacant coordination site for peroxide to bind. As a consequence of incomplete spin-state conversion upon binding shorter FAs, the relevant ferryl intermediates accumulate to lower levels but have very similar reactivities. The importance of the substrate for the generation of Ole-I is also supported by the data from the oxidation of radical clocks. Norcarane is a substrate for OleT only when a carboxylic acid is introduced.
Once Ole-I is generated, it abstracts a hydrogen atom from the FA β-carbon. However, after the formation of Ole-II, we propose that the substrate is held too firmly in place by the substrate binding pocket to move the additional angstrom or two required to allow the enzyme to donate •OH back to the substrate. This scenario is reminiscent of Fe2+- and α-ketoglutarate-dependent halogenases such as SyrB2, which can transfer either a •Cl or •OH to substrates based on exquisite (subangstrom) positioning of the carbon radical.65 For SyrB2, orientation of the amino acid substrate relative to the rebound species is reinforced by its attachment to a phosphopantetheine arm. The substrate binding pocket of OleT must be exquisitely arranged to orient the Cβ to frustrate the rebound step and allow time for oxidation of the substrate radical. The crystal structure of EA-bound OleT13 and the salient protein–ligand interactions generated in Ligplot66 are shown in Figure 6. The FA orientation is maintained through electrostatic interactions with Arg245 and an extensive series of hydrophobic contacts with the enzyme at C8 and below. An ~45° bend in the acyl chain originates at C12. Molecular dynamics studies have suggested that interactions with several of these residues, including Leu78, Phe173, Pro246, and Val292, significantly contribute to the substrate binding free energy and may serve to maintain the Cβ hydrogen at a relatively long distance (as much as 5.2 Å for C8) from the oxoferryl species.67 Although some anchoring at the bend of the fatty acid is lost with short CL substrates, our data show that they can slide into the correct orientation to ensure Cβ abstraction and are sufficiently immobile to enforce decarboxylation. The importance of substrate positioning has been reiterated in recent mutagenesis studies, which have demonstrated that substitution of Arg245 results in compromised activity, likely because of poor activation of H2O2, and switches the enzyme to the hydroxylation of shorter CL (Cn, where n ≤ 16) FAs.30 Thus, an attractive means of engineering OleT for the decarboxylation of shorter substrates may be to further constrict the active-site channel and reinforce contacts at various positions along the acyl chain.
Figure 6.
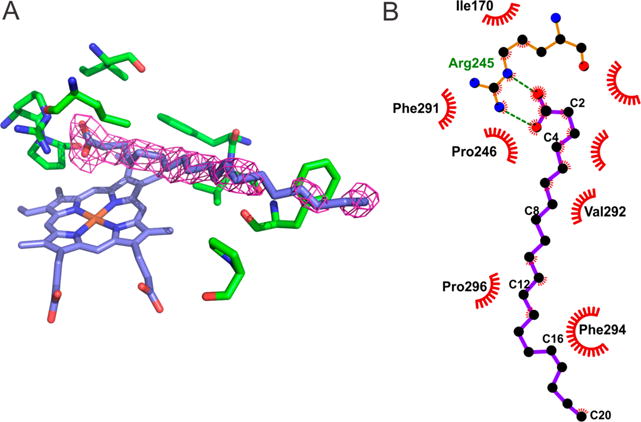
(A) Crystal structure of eicosanoic acid-bound OleT (Protein Data Bank entry 4L40).13 (B) LigPlot66 diagram that shows the residues involved in stabilizing the bound fatty acid in the active-site pocket.
As we have shown in reactions of Ole-II with phenols,29 and in reactions with Aae-APO-II and a wide range of substrates,27 iron(IV)–hydroxides are competent oxidants that can abstract an additional electron from the substrate, forming a cation at the Cβ position. From there, elimination of CO2 and formation of an alkene are highly favorable. An example of this chemistry occurs in isopentenyl pyrophosphate biosynthesis.68 An alternative proposal has suggested that once Ole-II is formed from Cβ-H abstraction, it could abstract an electron from the substrate carboxylate to afford a diradical, which could then rearrange to form an alkene and CO2. While this chemistry is plausible, it would require a tremendous amount of flexibility of the substrate in the pocket during catalysis to ensure that both abstraction steps were favorable. The substrate movements that would be required are inconsistent with product profiles for FAs, which show that the enzyme is highly regioselective.
CONCLUSIONS
In summary, we have confirmed that OleT can catalyze both decarboxylase and monooxygenase reactions. If an FA is bound and an alternative substrate is offered, OleT can behave like a typical CYP monooxygenase and hydroxylate aliphatic substrates that do not contain a carboxylate. A similar result is observed when an active-site carboxylate is introduced into the protein framework69 to mimic the strategem used by APO.70 The radical lifetimes measured for OleT with norcarane, which is a probe sensitive enough to be used as a diagnostic tool for classifying monooxygenases,71 and methylphenylcyclopropane, are indistinguishable from those that have been measured with other CYPs. After initial hydrogen atom abstraction, a caged substrate radical is formed that undergoes fast (picosecond) oxygen rebound.72 Slightly larger than average amounts of cationic-derived product, relative to the amounts seen with other CYPs, were observed with norcarane. This is consistent with a model in which Ole-II is an effective one-electron oxidizing agent. The amount of desaturated products from norcarane is comparable to what has been seen with other enzymes and, as we have argued in other places, a reflection, in part, of the chemistry of norcarane.53 Collectively, these results point toward substrate identity and coordination, rather than electronic structure, as a key element in dictating the reaction outcome.
It is useful to reflect on the available (bio)chemical strategies for the synthesis of deoxygenated biofuels. Many mechanisms for generating biofuels rely on expensive reducing equivalents to release water or catalyze similar types of C–C bond cleavage. In contrast, the approach illustrated by OleT, and shared with the non-heme iron enzyme UndA,22 is an oxidative approach that consumes a toxic reactive oxygen species (H2O2) and releases CO2. For commercialization, the OleT chemistry (or inorganic chemistry modeled on it) would ideally be coupled to CO2 capture or utilization. OleT also offers inspiration for synthetic model chemistry: to design and synthesize a model compound or protein that generates a high-valent intermediate capable of C–H abstraction but then holds the substrate at bay and forces it to decarboxylate.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Science Foundation CAREER Grant 1555066 and an ASPIRE grant from the University of South Carolina Vice President of Research (T.M.M.), National Institutes of Health (NIH) Grant 2R37 GM036298 (J.T.G.), and NIH Grant GM072506 (R.N.A.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00338.
Additional results, including UV–vis binding titrations of C6, C8, C10, and C12 FAs (Figure S1), gas chromatogram of the reaction headspace from OleT:DA reactions (Figure S2), gas chromatograms of the products of OleT reactions of C8, C10, and C12 FAs (Figure S3), time course and fitting for Ole-I decay with DA-d19 (Figure S4), decoy CL screening using guaiacol oxidation as a probe (Figure S5), gas chromatograms and reaction products from MCP oxidations (Figure S6), and MS of ring-opened norcarane reaction products (Figure S7) (PDF)
ORCID
John T. Groves: 0000-0002-9944-5899
Thomas M. Makris: 0000-0001-7927-620X
Notes
The authors declare no competing financial interest.
References
- 1.Rude MA, Baron TS, Brubaker S, Alibhai M, Del Cardayre SB, Schirmer A. Terminal Olefin (1-Alkene) Biosynthesis by a Novel P450 Fatty Acid Decarboxylase from Jeotgalicoccus Species. Appl Environ Microbiol. 2011;77:1718–1727. doi: 10.1128/AEM.02580-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grant JL, Hsieh CH, Makris TM. Decarboxylation of fatty acids to terminal alkenes by cytochrome P450 compound I. J Am Chem Soc. 2015;137:4940–4943. doi: 10.1021/jacs.5b01965. [DOI] [PubMed] [Google Scholar]
- 3.Dennig A, Kuhn M, Tassoti S, Thiessenhusen A, Gilch S, Bulter T, Haas T, Hall M, Faber K. Oxidative decarboxylation of short-chain fatty acids to 1-alkenes. Angew Chem, Int Ed. 2015;54:8819–8822. doi: 10.1002/anie.201502925. [DOI] [PubMed] [Google Scholar]
- 4.Zachos I, Gassmeyer SK, Bauer D, Sieber V, Hollmann F, Kourist R. Photobiocatalytic decarboxylation for olefin synthesis. Chem Commun (Cambridge, U K) 2015;51:1918–1921. doi: 10.1039/c4cc07276f. [DOI] [PubMed] [Google Scholar]
- 5.Wang JB, Lonsdale R, Reetz MT. Exploring substrate scope and stereoselectivity of P450 peroxygenase OleTJE in olefin-forming oxidative decarboxylation. Chem Commun (Cambridge, U K) 2016;52:8131–8133. doi: 10.1039/c6cc04345c. [DOI] [PubMed] [Google Scholar]
- 6.Dennig A, Kurakin S, Kuhn M, Dordic A, Hall M, Faber K. Enzymatic oxidative tandem decarboxylation of dioic Acids to terminal dienes. Eur J Org Chem. 2016;2016:3473–3477. [Google Scholar]
- 7.Cryle MJ, De Voss JJ. Carbon-carbon bond cleavage by cytochrome P450(BioI) (CYP107H1) Chem Commun. 2004:86–87. doi: 10.1039/b311652b. [DOI] [PubMed] [Google Scholar]
- 8.Davydov R, Strushkevich N, Smil D, Yantsevich A, Gilep A, Usanov S, Hoffman BM. Evidence that compound I is the active species in both the hydroxylase and lyase steps by which P450scc converts cholesterol to pregnenolone: EPR/ENDOR/cryoreduction/annealing studies. Biochemistry. 2015;54:7089–7097. doi: 10.1021/acs.biochem.5b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khatri Y, Gregory MC, Grinkova YV, Denisov IG, Sligar SG. Active site proton delivery and the lyase activity of human CYP17A1. Biochem Biophys Res Commun. 2014;443:179–184. doi: 10.1016/j.bbrc.2013.11.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groves JT, McClusky GA. Aliphatic hydroxylation via oxygen rebound - Oxygen-transfer catalyzed by iron. J Am Chem Soc. 1976;98:859–861. [Google Scholar]
- 11.Groves JT, McClusky GA, White RE, Coon MJ. Aliphatic hydroxylation by highly purified liver microsomal cytochrome P450 - Evidence for a carbon radical intermediate. Biochem Biophys Res Commun. 1978;81:154–160. doi: 10.1016/0006-291x(78)91643-1. [DOI] [PubMed] [Google Scholar]
- 12.Huang X, Groves JT. Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C-H activation. JBIC, J Biol Inorg Chem. 2017;22:185–207. doi: 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belcher J, McLean KJ, Matthews S, Woodward LS, Fisher K, Rigby SEJ, Nelson DR, Potts D, Baynham MT, Parker DA, Leys D, Munro AW. Structure and biochemical properties of the alkene producing cytochrome P450 OleTJE (CYP152L1) from the Jeotgalicoccus sp. 8456 bacterium. J Biol Chem. 2014;289:6535–6550. doi: 10.1074/jbc.M113.527325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girvan HM, Munro AW. Applications of microbial cytochrome P450 enzymes in biotechnology and synthetic biology. Curr Opin Chem Biol. 2016;31:136–145. doi: 10.1016/j.cbpa.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 15.Matsunaga I, Yamada A, Lee DS, Obayashi E, Fujiwara N, Kobayashi K, Ogura H, Shiro Y. Enzymatic reaction of hydrogen peroxide-dependent peroxygenase cytochrome P450s: kinetic deuterium isotope effects and analyses by resonance Raman spectroscopy. Biochemistry. 2002;41:1886–1892. doi: 10.1021/bi011883p. [DOI] [PubMed] [Google Scholar]
- 16.Kellner DG, Hung SC, Weiss KE, Sligar SG. Kinetic characterization of Compound I formation in the thermostable cytochrome P450 CYP119. J Biol Chem. 2002;277:9641–9644. doi: 10.1074/jbc.C100745200. [DOI] [PubMed] [Google Scholar]
- 17.Rittle J, Green MT. Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 18.Yosca TH, Rittle J, Krest CM, Onderko EL, Silakov A, Calixto JC, Behan RK, Green MT. Iron(IV)hydroxide pK(a) and the role of thiolate ligation in C-H bond activation by cytochrome P450. Science. 2013;342:825–829. doi: 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Peter S, Kinne M, Hofrichter M, Groves JT. Detection and kinetic characterization of a highly reactive hemethiolate peroxygenase compound I. J Am Chem Soc. 2012;134:12897–12900. doi: 10.1021/ja3049223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Auclair K, Hu Z, Little DM, Ortiz de Montellano PR, Groves JT. Revisiting the mechanism of P450 Enzymes with the radical clocks norcarane and spiro[2,5]octane. J Am Chem Soc. 2002;124:6020–6027. doi: 10.1021/ja025608h. [DOI] [PubMed] [Google Scholar]
- 21.Rettie AE, Boberg M, Rettenmeier AW, Baillie TA. Cytochrome P450-catalyzed desaturation of valproic acid in vitro. Species differences, induction effects, and mechanistic studies. J Biol Chem. 1988;263:13733–13738. [PubMed] [Google Scholar]
- 22.Rui Z, Li X, Zhu X, Liu J, Domigan B, Barr I, Cate JHD, Zhang W. Microbial biosynthesis of medium-chain 1-alkenes by a nonheme iron oxidase. Proc Natl Acad Sci U S A. 2014;111:18237–18242. doi: 10.1073/pnas.1419701112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fox BG, Shanklin J, Somerville C, Munck E. Stearoyl-acyl carrier protein delta-9 desaturase from Ricinus communis is a diiron-oxo protein. Proc Natl Acad Sci U S A. 1993;90:2486–2490. doi: 10.1073/pnas.90.6.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guy JE, Abreu IA, Moche M, Lindqvist Y, Whittle E, Shanklin J. A single mutation in the castor Delta(9)-18:0-desaturase changes reaction partitioning from desaturation to oxidase chemistry. Proc Natl Acad Sci U S A. 2006;103:17220–17224. doi: 10.1073/pnas.0607165103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shanklin J, Guy JE, Mishra G, Lindqvist Y. Desaturases: emerging models for understanding functional diversification of diiron-containing enzymes. J Biol Chem. 2009;284:18559–18563. doi: 10.1074/jbc.R900009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bigi MA, Reed SA, White MC. Diverting non-haem iron catalysed aliphatic C-H hydroxylations towards desaturations. Nat Chem. 2011;3:218–224. doi: 10.1038/nchem.967. [DOI] [PubMed] [Google Scholar]
- 27.Wang XS, Ullrich R, Hofrichter M, Groves JT. Heme-thiolate ferryl of aromatic peroxygenase is basic and reactive. Proc Natl Acad Sci U S A. 2015;112:3686–3691. doi: 10.1073/pnas.1503340112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lambeir AM, Dunford HB, Pickard MA. Kinetics of the oxidation of ascorbic acid, ferrocyanide and p-phenolsulfonic acid by chloroperoxidase compounds I and II. Eur J Biochem. 1987;163:123–127. doi: 10.1111/j.1432-1033.1987.tb10744.x. [DOI] [PubMed] [Google Scholar]
- 29.Grant JL, Mitchell ME, Makris TM. Catalytic strategy for carbon-carbon bond scission by the cytochrome P450 OleT. Proc Natl Acad Sci U S A. 2016;113:10049–10054. doi: 10.1073/pnas.1606294113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthews S, Belcher JD, Tee KL, Girvan HM, McLean KJ, Rigby SEJ, Levy CW, Leys D, Parker DA, Blankley RT, Munro AW. Catalytic determinants of alkene production by the cytochrome P450 Peroxygenase OleT(JE) J Biol Chem. 2017;292:5128–5143. doi: 10.1074/jbc.M116.762336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Wang C, Yan J, Zhang W, Guan W, Lu X, Li S. Hydrogen peroxide-independent production of alpha-alkenes by OleTJE P450 fatty acid decarboxylase. Biotechnol Biofuels. 2014;7:28. doi: 10.1186/1754-6834-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amaya JA, Rutland CD, Makris TM. Mixed regiospecificity compromises alkene synthesis by a cytochrome P450 peroxygenase from Methylobacterium populi. J Inorg Biochem. 2016;158:11–16. doi: 10.1016/j.jinorgbio.2016.02.031. [DOI] [PubMed] [Google Scholar]
- 33.Austin RN, Chang HK, Zylstra GJ, Groves JT. The non-heme diiron alkane monooxygenase of Pseudomonas oleovorans (AlkB) hydroxylates via a substrate radical intermediate. J Am Chem Soc. 2000;122:11747–11748. [Google Scholar]
- 34.Smith RD, Simmons HE. Norcarane [Bicyclo[4.1.0]heptane] Org Synth. 1961;41:72–75. [Google Scholar]
- 35.Berry EA, Trumpower BL. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal Biochem. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 36.Morrison JF. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim Biophys Acta. 1969;185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- 37.Fu H, Newcomb M, Wong CH. Pseudomonas-Oleovorans Monooxygenase Catalyzed Asymmetric Epoxidation of Allyl Alcohol Derivatives and Hydroxylation of a Hypersensitive Radical Probe with the Radical Ring-Opening Rate Exceeding the Oxygen Rebound Rate. J Am Chem Soc. 1991;113:5878–5880. [Google Scholar]
- 38.Austin RN, Luddy K, Erickson K, Pender-Cudlip M, Bertrand E, Deng D, Buzdygon RS, van Beilen JB, Groves JT. Cage escape competes with geminate recombination during alkane hydroxylation by the diiron oxygenase AlkB. Angew Chem, Int Ed. 2008;47:5232–5234. doi: 10.1002/anie.200801184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajakovich LJ, Norgaard H, Warui DM, Chang WC, Li N, Booker SJ, Krebs C, Bollinger JM, Pandelia ME. Rapid reduction of the diferric-peroxyhemiacetal intermediate in aldehyde-deformylating oxygenase by a cyanobacterial ferredoxin: evidence for a free-radical mechanism. J Am Chem Soc. 2015;137:11695–11709. doi: 10.1021/jacs.5b06345. [DOI] [PubMed] [Google Scholar]
- 40.Matsunaga I, Sumimoto T, Ueda A, Kusunose E, Ichihara K. Fatty acid-specific, regiospecific, and stereospecific hydroxylation by cytochrome P450 (CYP152B1) from Sphingomonas paucimobilis: substrate structure required for alpha-hydroxylation. Lipids. 2000;35:365–371. doi: 10.1007/s11745-000-533-y. [DOI] [PubMed] [Google Scholar]
- 41.Shoji O, Wiese C, Fujishiro T, Shirataki C, Wunsch B, Watanabe Y. Aromatic C-H bond hydroxylation by P450 peroxygenases: a facile colorimetric assay for monooxygenation activities of enzymes based on Russig’s blue formation. JBIC, J Biol Inorg Chem. 2010;15:1109–1115. doi: 10.1007/s00775-010-0671-9. [DOI] [PubMed] [Google Scholar]
- 42.Shoji O, Fujishiro T, Nagano S, Tanaka S, Hirose T, Shiro Y, Watanabe Y. Understanding substrate misrecognition of hydrogen peroxide dependent cytochrome P450 from Bacillus subtilis. JBIC, J Biol Inorg Chem. 2010;15:1331–1339. doi: 10.1007/s00775-010-0692-4. [DOI] [PubMed] [Google Scholar]
- 43.Shoji O, Fujishiro T, Nakajima H, Kim M, Nagano S, Shiro Y, Watanabe Y. Hydrogen peroxide dependent monooxygenations by tricking the substrate recognition of cytochrome P450BSbeta. Angew Chem, Int Ed. 2007;46:3656–3659. doi: 10.1002/anie.200700068. [DOI] [PubMed] [Google Scholar]
- 44.Atkinson JK, Hollenberg PF, Ingold KU, Johnson CC, LeTadic MH, Newcomb M, Putt DA. Cytochrome P450-catalyzed hydroxylation of hydrocarbons: kinetic deuterium isotope effects for the hydroxylation of an ultrafast radical clock. Biochemistry. 1994;33:10630–10637. doi: 10.1021/bi00201a009. [DOI] [PubMed] [Google Scholar]
- 45.Atkinson JK, Ingold KU. Cytochrome-P450 Hydroxylation of Hydrocarbons - Variation in the Rate of Oxygen Rebound Using Cyclopropyl Radical Clocks Including 2 New Ultrafast Probes. Biochemistry. 1993;32:9209–9214. doi: 10.1021/bi00086a028. [DOI] [PubMed] [Google Scholar]
- 46.Austin RN, Deng D, Jiang Y, Luddy K, van Beilen JB, Ortiz de Montellano P, Groves JT. The diagnostic substrate bicyclohexane reveals a radical mechanism for bacterial cytochrome P450 in whole cells. Angew Chem, Int Ed. 2006;45:8192–8194. doi: 10.1002/anie.200603282. [DOI] [PubMed] [Google Scholar]
- 47.Brazeau BJ, Austin RN, Tarr C, Groves JT, Lipscomb JD. Intermediate Q from soluble methane monooxygenase hydroxylates the mechanistic substrate probe norcarane: evidence for a stepwise reaction. J Am Chem Soc. 2001;123:11831–11837. doi: 10.1021/ja016376+. [DOI] [PubMed] [Google Scholar]
- 48.Chakrabarty S, Austin RN, Deng DY, Groves JT, Lipscomb JD. Radical intermediates in monooxygenase reactions of Rieske dioxygenases. J Am Chem Soc. 2007;129:3514–3515. doi: 10.1021/ja068188v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi SY, Eaton PE, Hollenberg PF, Liu KE, Lippard SJ, Newcomb M, Putt DA, Upadhyaya SP, Xiong YS. Regiochemical variations in reactions of methylcubane with tert-butoxyl radical, cytochrome P-450 enzymes, and a methane monooxygenase system. J Am Chem Soc. 1996;118:6547–6555. [Google Scholar]
- 50.Choi SY, Eaton PE, Kopp DA, Lippard SJ, Newcomb M, Shen RN. Cationic species can be produced in soluble methane monooxygenase-catalyzed hydroxylation reactions; radical intermediates are not formed. J Am Chem Soc. 1999;121:12198–12199. [Google Scholar]
- 51.Choi SY, Shen R, Kopp DA, Eaton PE, Hollenberg PF, Vaz ADA, Coon MJ, Lippard SJ, Newcomb ME. Abstracts of Papers of the American Chemical Society. Vol. 219. American Chemical Society; Washington, DC: 2000. Oxidation of methylcubane with cytochrome P-450 enzymes and methane monooxygenase; p. U142. [Google Scholar]
- 52.Cooper HLR, Groves JT. Molecular probes of the mechanism of cytochrome P450. Oxygen traps a substrate radical intermediate. Arch Biochem Biophys. 2011;507:111–118. doi: 10.1016/j.abb.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cooper HLR, Mishra G, Huang XY, Pender-Cudlip M, Austin RN, Shanklin J, Groves JT. Parallel and competitive pathways for substrate desaturation, hydroxylation, and radical rearrangement by the non-heme diiron hydroxylase AlkB. J Am Chem Soc. 2012;134:20365–20375. doi: 10.1021/ja3059149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Groves JT, Austin RN, Chang HK, Zylstra GJ. The non-heme diiron alkane monooxygenase of Pseudomonas oleovorans (AlkB) hydroxylates via a substrate radical intermediate. J Inorg Biochem. 2001;86:54. [Google Scholar]
- 55.Groves JT, Kruper WJ, Haushalter RC. Hydrocarbon oxidations with oxometalloporphinates. Isolation and reactions of a (porphinato)manganese(V) complex. J Am Chem Soc. 1980;102:6375–6377. [Google Scholar]
- 56.Liu KE, Johnson CC, Newcomb M, Lippard SJ. Radical clock substrate probes and kinetic isotope effect studies of the hydroxylation of hydrocarbons by methane monooxygenase. J Am Chem Soc. 1993;115:939–947. [Google Scholar]
- 57.Newcomb M, Chandrasena REP, Lansakara-P DSP, Kim HY, Lippard SJ, Beauvais LG, Murray LJ, Izzo V, Hollenberg PF, Coon MJ. Desaturase reactions complicate the use of norcarane as a mechanistic probe. Unraveling the mixture of twenty-plus products formed in enzyme-catalyzed oxidations of norcarane. J Org Chem. 2007;72:1121–1127. doi: 10.1021/jo061864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newcomb M, Shen RN, Lu Y, Coon MJ, Hollenberg PF, Kopp DA, Lippard SJ. Evaluation of norcarane as a probe for radicals in cytochome P450-and soluble methane monooxygenase-catalyzed hydroxylation reactions. J Am Chem Soc. 2002;124:6879–6886. doi: 10.1021/ja017858o. [DOI] [PubMed] [Google Scholar]
- 59.Ortiz de Montellano PR, Nelson SD. Rearrangement reactions catalyzed by cytochrome P450s. Arch Biochem Biophys. 2011;507:95–110. doi: 10.1016/j.abb.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valentine AM, LeTadic-Biadatti MH, Toy PH, Newcomb M, Lippard SJ. Oxidation of ultrafast radical clock substrate probes by the soluble methane monooxygenase from Methylococcus capsulatus (Bath) J Biol Chem. 1999;274:10771–10776. doi: 10.1074/jbc.274.16.10771. [DOI] [PubMed] [Google Scholar]
- 61.Peter S, Kinne M, Wang XS, Ullrich R, Kayser G, Groves JT, Hofrichter M. Selective hydroxylation of alkanes by an extracellular fungal peroxygenase. FEBS J. 2011;278:3667–3675. doi: 10.1111/j.1742-4658.2011.08285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Groves JT. Models and Mechanisms of Cytochrome P-450 Action. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 1–44. [Google Scholar]
- 63.Fang B, Xu H, Liu Y, Qi F, Zhang W, Chen H, Wang C, Wang Y, Yang W, Li S. Mutagenesis and redox partners analysis of the P450 fatty acid decarboxylase OleTJE. Sci Rep. 2017;7:44258. doi: 10.1038/srep44258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Faponle AS, Quesne MG, de Visser SP. Origin of the regioselective fatty acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: what drives the reaction to biofuel production? Chem - Eur J. 2016;22:5478–5483. doi: 10.1002/chem.201600739. [DOI] [PubMed] [Google Scholar]
- 65.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc Natl Acad Sci U S A. 2009;106:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laskowski RA, Swindells MB. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 67.Du J, Liu L, Guo LZ, Yao XJ, Yang JM. Molecular basis of P450 OleTJE: an investigation of substrate binding mechanism and major pathways. J Comput-Aided Mol Des. 2017;31:483–495. doi: 10.1007/s10822-017-0013-x. [DOI] [PubMed] [Google Scholar]
- 68.Duan L, Jogl G, Cane DE. The cytochrome P450-catalyzed oxidative rearrangement in the final step of pentalenolactone biosynthesis: substrate structure determines mechanism. J Am Chem Soc. 2016;138:12678–12689. doi: 10.1021/jacs.6b08610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsieh CH, Makris TM. Expanding the substrate scope and reactivity of cytochrome P450 OleT. Biochem Biophys Res Commun. 2016;476:462–466. doi: 10.1016/j.bbrc.2016.05.145. [DOI] [PubMed] [Google Scholar]
- 70.Piontek K, Strittmatter E, Ullrich R, Grobe G, Pecyna MJ, Kluge M, Scheibner K, Hofrichter M, Plattner DA. Structural basis of substrate conversion in a new aromatic peroxygenase: cytochrome P450 functionality with benefits. J Biol Chem. 2013;288:34767–34776. doi: 10.1074/jbc.M113.514521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rozhkova-Novosad EA, Chae JC, Zylstra GJ, Bertrand EM, Alexander-Ozinskas M, Deng DY, Moe LA, van Beilen JB, Danahy M, Groves JT, Austin RN. Profiling mechanisms of alkane hydroxylase activity in vivo using the diagnostic substrate norcarane. Chem Biol. 2007;14:165–172. doi: 10.1016/j.chembiol.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 72.Groves JT. The bioinorganic chemistry of iron in oxygenases and supramolecular assemblies. Proc Natl Acad Sci U S A. 2003;100:3569–3574. doi: 10.1073/pnas.0830019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.