

SUMMARY
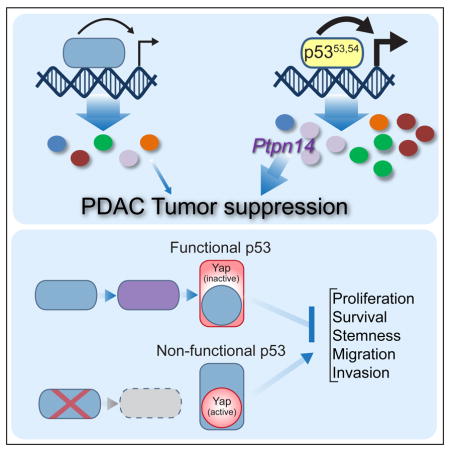
The p53 transcription factor is a critical barrier to pancreatic cancer progression. To unravel mechanisms of p53-mediated tumor suppression, which have remained elusive, we analyzed pancreatic cancer development in mice expressing p53 transcriptional activation domain (TAD) mutants. Surprisingly, the p5353,54 TAD2 mutant behaves as a “super-tumor suppressor”, with an enhanced capacity to both suppress pancreatic cancer and transactivate select p53 target genes, including Ptpn14. Ptpn14 encodes a negative regulator of the Yap oncoprotein and is necessary and sufficient for pancreatic cancer suppression, like p53. We show that p53 deficiency promotes Yap signaling and that PTPN14 and TP53 mutations are mutually exclusive in human cancers. These studies uncover a p53-Ptpn14-Yap pathway that is integral to p53-mediated tumor suppression.
In brief/eTOC blurb
Mello et al. find that a p53 mutant harboring mutations in the second transcriptional activation domain has enhanced tumor suppression capacities due to hyperactivation of the p53 target gene Ptpn14. Ptpn14 suppresses Yap activity and is required for p53 tumor suppressor activity in pancreatic cancer.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is an extremely deadly cancer, with patients displaying a 5-year survival rate of only 8% (Siegel et al., 2016). This high rate of mortality is attributable both to an inability to detect the disease early and the cancer being highly refractory to treatment (Ryan et al., 2014). Molecular genetic characterization of PDACs has identified several common gene alterations, including activating mutations in the KRAS oncogene in 95% of cases and inactivating mutations in the CDKN2A, TP53, and/or SMAD4 tumor suppressor genes (Jones et al., 2008). Studies using genetically engineered mouse models have demonstrated that Kras activation along with mutation of one copy of any of these tumor suppressor genes is sufficient to drive PDAC development (Aguirre et al., 2003; Bardeesy et al., 2006a; Bardeesy et al., 2006b; Hingorani et al., 2005; Izeradjene et al., 2007). Recent studies continue to illuminate the contribution of new proteins to PDAC development, such as the Yap transcriptional coactivator, which is essential for progression of preneoplastic lesions to PDAC in mouse models and can even substitute for Kras activation (Kapoor et al., 2014; Zhang et al., 2014).
The TP53 tumor suppressor gene is mutated in ~75% of human PDACs, underscoring the importance of p53 inactivation for PDAC development (Jones et al., 2008). Mouse models have recapitulated this observation, where Kras activation in combination with point mutation or deletion of one copy of the Trp53 gene is sufficient to induce PDAC with characteristic features of the human disease, including genomic instability and metastatic capacity (Bardeesy et al., 2006a; Hingorani et al., 2005). In particular, progression from preinvasive Pancreatic Intraepithelial Neoplasia lesions (PanINs) to invasive PDACs is associated with loss of heterozygosity (LOH) of Trp53, highlighting p53’s crucial role as a barrier to invasive pancreatic cancer development (Baumgart et al., 2010; Lüttges et al., 2001). Understanding how p53 acts in this context, and which pathways are dysregulated when it is lost, could provide insight into the molecular mechanisms that enable PDAC progression.
p53 is a cellular stress sensor, responding to diverse stress signals such as oncogenic stimuli, DNA damage, and nutrient deprivation by restraining cellular expansion through cell-cycle arrest or apoptosis (Brady and Attardi, 2010; Vousden and Prives, 2009). The best-characterized functions of p53 are in inducing proliferative arrest or apoptosis in response to acute DNA damage, although p53 also regulates various other cellular functions, including metabolism, motility, and differentiation (Bieging et al., 2014). p53 acts primarily as a transcriptional activator that binds defined consensus sites throughout the genome and drives expression of specific genes involved in the different processes regulated by p53. However, despite some understanding of the genes involved in specific cell biological functions of p53, the genes critical for tumor suppression have remained enigmatic. Surprisingly, recent studies have shown that the classical DNA damage-induced p53 responses of apoptosis and cell-cycle arrest and the p53 target genes involved in these responses – Cdkn1a (p21), Pmaip1 (Noxa), and Bbc3 (Puma) – are dispensable for tumor suppression (Brady et al., 2011; Li et al., 2012; Valente et al., 2013). These findings suggest that the key mediators of p53 in tumor suppression remain to be identified.
The p53 protein comprises distinct domains involved in sequence-specific DNA binding, tetramerization and transcriptional activation. p53 harbors two discrete transcriptional activation domains (TADs) identified using in vitro reporter assays but whose in vivo roles have not been fully investigated (Raj and Attardi, 2017). To query the role of each p53 TAD in p53 function in vivo, we generated p53 knock-in mouse strains expressing mutants altered in the first (p5325,26), second (p5353,54) or both TADs (p5325,26,53,54; Brady et al., 2011). Genome-wide transcriptomic analysis demonstrated that p5325,26 is severely compromised for transactivation of most known p53 target genes, including Cdkn1a and Mdm2, but retains the ability to activate a small subset of p53 target genes, including Bax. Furthermore, although unable to trigger cell-cycle arrest or apoptosis in response to acute DNA damage, p5325,26 retains full activity in suppressing diverse cancers in vivo (Brady et al., 2011; Jiang et al., 2011). In contrast, p5325,26,53,54 is completely defective for transactivation and tumor suppression, highlighting the importance of transcriptional activation for p53-mediated tumor suppression. Notably, in these experiments, the p5353,54 mutant appeared uncompromised with respect to wild-type p53 in both transactivation of classical p53 target genes and suppression of lung cancer. Here we leverage these p53 TAD mutant mouse strains to unravel transcriptional programs underlying p53-mediated tumor suppression in a well-established model for PDAC (Hingorani et al., 2003).
RESULTS
The p5353,54 mutant is a hyperactive tumor suppressor
To interrogate the mechanisms by which p53 suppresses pancreatic cancer development, we utilized a well-established model for PDAC in which KrasG12D is expressed in the pancreas by virtue of a Pdx1-Cre transgene (Hingorani et al., 2003). We generated Kras+/LSL-G12D;Pdx1-Cre mice heterozygous for p53 TAD mutant alleles. Given extensive studies on p5325,26 (Brady et al., 2011; Jiang et al., 2011), we focused on the less-characterized p5353,54 and the TAD-dead p5325,26,53,54 mutants (Figure 1A and 1B). In these mice, expression of the mutant KrasG12D and Trp53 TAD mutant alleles is silenced through upstream transcriptional stop elements flanked by loxP recombination sites (Lox-Stop-Lox or LSL). We performed this study in mice heterozygous for the wild-type Trp53 allele, as the wild-type Trp53 allele is typically lost through LOH in pancreatic cancer. We also included cohorts homozygous for wild-type Trp53 (Trp53+/+), heterozygous for a Trp53 null allele (Trp53+/−), and heterozygous for a LSL-WT Trp53 allele (Trp53+/LSL-WT). Cohorts of mice were generated, aged, and examined for pancreas cancer-free survival upon morbidity (Figure 1B).
Figure 1. Analysis of pancreatic cancer suppression potential of p53 TAD mutants.

(A) Schematic of p53 transcriptional activation domain (TAD) mutants analyzed in this study. The p53 DNA binding domain (DBD), tetramerization domain (T) and C-terminal domain (C) are also shown. (B) Schematic for pancreatic cancer study. Kras+/LSL-G12D;Pdx1-Cre;Trp53+/+ (n=16), Kras+/LSL-G12D;Pdx1-Cre;Trp53+/LSL-WT (n=14), Kras+/LSL-G12D;Pdx1-Cre;Trp53+/LSL-53,54 (n=10), Kras+/LSL-G12D;Pdx1-Cre;Trp53+/LSL-25,26,53,54 (n=12), and Kras+/LSL-G12D;Pdx1-Cre;Trp53+/− (n=8) mouse cohorts were analyzed for pancreatic cancer-free survival. (C) Kaplan-Meier analysis of PDAC-free survival of cohorts listed in (B). Labels indicate the p53 status of each cohort. (D) Representative histological images of the most advanced lesions found in each of the cohorts studied. The same field was analyzed in consecutive sections by H&E, Muc5ac, and Ck19 immunostaining. (E) Table summarizing the cohort genotypes and percentage of mice with PanINs (pancreatic intraepithelial neoplasias), IPMNs (intraductal papillary mucinous neoplasms), PDACs (pancreatic ductal adenocarcinomas), and metastatic lesions. Differences in pancreatic cancer incidence in Kras+/LSL-G12D;Pdx1-Cre;Trp53+/LSL-53,54 (p=0.019, n=10), Kras+/LSLG12D;Pdx1-Cre;Trp53+/LSL-25,26,53,54 (p=0.0003, n=12) and Kras+/LSLG12D;Pdx1-Cre;Trp53+/− (p=0.042, n=8) mice are relative to p53 wild-type mice (Kras+/LSLG12D;Pdx1-Cre;Trp53+/+, n=16 and Kras+/LSLG12D;Pdx1-Cre;Trp53+/LSL-WT, n=14), using the two-tailed Fisher’s exact test. Each mouse was scored for the most advanced lesions found. (F) Representative H&E and Ck19 staining on organs with metastases from Kras+/LSLG12D;Pdx1-Cre;Trp53+/LSL-25,26,53,54 and Kras+/LSLG12D;Pdx1-Cre;Trp53+/− mice. The scale bar in each panel applies to all images in that panel.
As anticipated, our analyses revealed reduced pancreatic cancer-free survival in Kras+/LSL-G12D;Pdx1-Cre;Trp53+/− mice relative to those with wild-type p53 (Kras+/LSL-G12D;Pdx1-Cre;Trp53+/+ and Kras+/LSL-G12D;Pdx1-Cre;Trp53+/LSL-WT mice; Figure 1C). Moreover, Kras+/LSL-G12D;Pdx1-Cre;Trp53+/LSL-25,26,53,54 mice succumbed to pancreatic cancer with a latency similar to the Kras+/LSL-G12D;Pdx1-Cre;Trp53+/− cohort, indicating that Trp5325,26,53,54 behaves as a null allele in this context and highlighting the importance of p53 transcriptional activity for pancreatic cancer suppression, as seen in other tumor models (Brady et al., 2011; Jiang et al., 2011). The most surprising finding, however, was that Kras+/LSL-G12D;Pdx1-Cre;Trp53+/LSL-53,54 mice exhibited longer pancreatic cancer-free survival than wild-type p53 controls, suggesting that TAD2 mutation protects against pancreatic cancer (Figure 1C). Notably, the most advanced lesions in Kras+/LSL-G12D;Pdx1-Cre;Trp53+/LSL-53,54 mice were PanINs, as determined by histopathological analysis and positivity for the PanIN marker Muc5ac, while all other cohorts presented with more advanced lesions, classified as PDAC by pathology, Cytokeratin 19 (Ck19)-positivity, and absence of Muc5ac staining (Figure 1D, 1E). Pancreatic cancers were of a variety of differentiation states (Figure S1). In addition, metastatic lesions were observed in all cohorts except Kras+/LSL-G12D/+;Pdx1-Cre;Trp53+/LSL-53,54 mice (Figure 1E, F). Liver metastases were the most common among all the cohorts, with rare spleen and lung metastases in some animals. Collectively, these findings demonstrate that transcriptional activation potential is critical for p53 to suppress pancreatic cancer development. Moreover, these experiments reveal a surprising enhanced capacity of the p5353,54 mutant to suppress PDAC development, a property that could provide mechanistic insight into p53 tumor suppressor pathways.
Combined ChIP-seq and transcriptomic analyses reveal p53 target genes hyperactivated by p5353,54
To gain insight into the underlying basis of the enhanced tumor suppressor activity of p5353,54, we leveraged transcriptomics data that we had previously generated to study the p53 mutants in HrasV12-expressing mouse embryonic fibroblasts (MEFs), a model for neoplastic cells (Brady et al., 2011). While classical p53 target genes were not hyperactivated by p5353,54 compared to wild-type p53 (Brady et al. 2011), we sought to determine whether some p53-dependent genes might be induced to a higher level by p5353,54 than by wild-type p53. Indeed, we were able to derive a list of genes induced to a greater extent (≥1.3 fold) by p5353,54 than wild-type p53. Using ChIP-seq data from primary MEFs (Kenzelmann Broz et al., 2013), we identified 103 direct p53 target genes hyperactivated by p5353,54 (Figure 2A). This enhanced activation of select p53 target genes by p5353,54 relative to wild-type p53 is also observed by qRT-PCR analysis in KrasG12D-expressing lung cancer cells that we derived from mice (Figure 2B), suggesting that the enhanced activation of target genes p5353,54 is a broad, cell type-independent effect. The hyperactivation of certain p53 target genes by p5353,54 may in part relate to the fact that p5353,54 is more stable than wild-type p53 (Figure 2C), presumably due to its diminished ability to interact with Mdm2 (Figure 2D). p5353,54 hyperactivity likely results not only from augmented stability but also from enhanced transcriptional activity on specific genes, given its highly selective effects on particular p53 target genes. Indeed, in silico structural analyses show that the TAD2 amphipathic α-helix in both human and mouse p53 allows binding to interacting partners through hydrophobic interactions, and that changing WF in human p53 (or FF in mouse p53) to QS is predicted to decrease the affinity of the interaction (Figure 2E). These observations in turn suggest that perturbed interactions with any negative regulators such as Mdm4 could provide a potential mechanism for the enhanced transcriptional activity of p5353,54.
Figure 2. p53 transcriptomics and ChIP-seq analysis reveal genes hyperactivated by p5353,54.

(A) Genes activated in both HrasV12;Trp5353,54/53,54 and HrasV12;Trp53+/+ MEFs relative to HrasV12;Trp53−/− MEFs were further filtered based on fold change (HrasV12;Trp5353,54/53,54 vs. HrasV12;Trp53+/+ ≥ 1.3 fold) and on direct binding by p53, as established by p53 ChIP-seq data. The heat map shows the expression of 103 genes that satisfy these criteria in MEFs of these genotypes as well as in HrasV12;Trp5325,26,53,54/25,26,53,54 MEFs. (B) qRT-PCR analysis of p53 target gene expression in KrasG12D-expressing lung cancer cells derived from tumors in Kras+/LA2;Trp53LSL-WT/LSL-WT, Kras+/LA2;Trp53LSL-25,26/LSL-25,26, Kras+/LA2;Trp53LSL-53,54/LSL-53,54 or Kras+/LA2;Trp53LSL-25,26,53,54/LSL-25,26,53,54 mice and transduced with Ad-Cre to allow for the recombination of the lox-stop-lox cassette. Expression is relative to Actb (n=1, triplicate). Data are presented as mean ± SD. (C) Western blot analysis of p53 in HrasV12-expressing MEFs homozygous for the different p53 variants. Cells were transduced with Ad-Cre (Adenovirus-Cre) to allow for the recombination of the lox-stop-lox cassette present in the different Trp53 alleles. Gapdh is a loading control. (D) Immunoprecipitation (IP) of p53 in MEFs homozygous for different Trp53 alleles, followed by immunoblotting for Mdm2 and p53. HC denotes the immunoglobulin heavy chain. Input is 2.5% of the total amount immunoprecipitated. Actin serves as a loading control. The fraction of Mdm2 bound to each p53 variant is indicated at the bottom and is relative to wild-type p53. (E) In silico structural modeling showing how the 53,54 residues in human and mouse p53 interact with HMGB1, a representative p53 TAD2-interacting protein, and how this interaction is affected with the QS mutations. (F) Heat map showing the subset of the 103 genes identified in HrasV12-expressing MEFs whose expression is p53-dependent in PDAC, based on the comparison of PDAC cells from Kras+/LSL-G12D;Ptf1a-Cre;Trp53+/fl and Kras+/LSL-G12D;Ptf1a-Cre;Cdkn2a+/fl mice, where tumors undergo LOH for Trp53 or Cdkn2a (Collisson et al., 2011).
The 103 genes hyperactivated by p5353,54 likely underlie its enhanced tumor suppressor function. Gene ontology analysis revealed a significant enrichment of genes in one category – that of “biological adhesion” (FWER p value: 0.043). To pinpoint those most relevant to pancreatic cancer, we identified those genes whose expression is p53-dependent in PDAC by analyzing microarray data from PDACs from Kras+/LSL-G12D;Ptf1a-Cre;Trp53+/fl and Kras+/LSL-G12D;Ptf1a-Cre;Cdkn2a+/fl mice, which display loss of Trp53 or Cdkn2a through LOH, and therefore lack or express p53, respectively (Collisson et al., 2011). In this analysis, we found that 16 genes display clear p53-dependent expression in pancreas cancer (Figure 2F). One gene – Ptpn14 (protein tyrosine phosphatase non-receptor type 14) – was a particularly compelling candidate to pursue given both its function in biological adhesion (Wadham et al., 2003) and its reported mutation in human colorectal cancers, suggesting a role in tumor suppression in humans (Wang et al., 2004). Moreover, Ptpn14 was reported as a critical negative regulator of Yap (Huang et al., 2013; Liu et al., 2013; Michaloglou et al., 2013; Wang et al., 2012), which itself has a central role in driving pancreatic cancer progression (Kapoor et al., 2014; Zhang et al., 2014). We therefore hypothesized that a p53-Ptpn14-Yap axis may be an important component of pancreatic cancer suppression.
Ptpn14 is a p53 target gene, bound and regulated in different contexts
To examine the role of Ptpn14 in the p53 pathway, we first sought to establish whether it is a direct p53 target. Using our MEF p53 ChIP-seq data (Kenzelmann Broz et al., 2013), we found that p53 binds to four intronic regions in the mouse Ptpn14 locus, all of which carry response elements strongly matching the p53 consensus binding sequence (Figure 3A). Importantly, p53 binding is conserved in a p53 ChIP-seq data set we generated from human fibroblasts (Younger et al., 2015), although in this case p53 binds only one intron (Figure 3B).
Figure 3. Ptpn14 is a bona fide p53 target gene.

(A–B) p53 ChIP-seq profiles showing peaks in the Ptpn14 locus in doxorubicin-treated MEFs (A) and the PTPN14 locus in human fetal fibroblasts (B). Exons are represented by gray boxes and introns by dashed lines. The orientation of the vertices indicates whether the gene is on the sense (vertex down) or antisense (vertex up) strand. Transcription start sites (TSS) are marked by arrows. Inverted red triangles mark significant “called” peaks, and numbers denote the distance from the TSS. p53 response elements in each peak are indicated, with red denoting nucleotides in the conserved cores. Spacers between the two half-sites and number of mismatches relative to the consensus are indicated. (C) qRT-PCR for Ptpn14 expression in HrasV12-expressing MEFs homozygous for different Trp53 alleles, relative to cells with wild-type Trp53 (n=1, triplicate). (D) qRT-PCR for Ptpn14 expression in KrasG12D-expressing lung adenocarcinoma cells homozygous for each of the different Trp53 alleles (See Fig. 2B; n=1, triplicate). (E) qRT-PCR for Ptpn14 expression in Eμ-myc-driven B-cell lymphomas with and without p53-ER activation by 4-OHT (n=1, triplicate). (F) qRT-PCR for Ptpn14 expression in MEFs 6 hr after treatment with 8 Gy ionizing radiation, relative to untreated MEFs with wild-type p53 (n=3, triplicate). In all qRT-PCR experiments, Ptpn14 expression is relative to Actb. Data represent mean ± SD; * p ≤ 0.05; ** p ≤ 0.001, two-tailed unpaired Student’s t-test.
To solidify the notion that Ptpn14 is a bona fide p53 target gene, we examined the p53-dependence of Ptpn14 expression in multiple contexts. We first confirmed that Ptpn14 is indeed p53-dependent and is hyperactivated by the p5353,54 mutant relative to wild-type p53 in HrasV12-expressing MEFs (Figure 3C). We next showed that Ptpn14 expression is p53-dependent in multiple cell types, including cells derived from well characterized mouse GEMMs: KrasG12D-driven lung adenocarcinomas and Eμ-myc transgene-triggered B-cell lymphomas (Figure 3D, E). Furthermore, as in oncogene-expressing MEFs, Ptpn14 is hyperactivated by the p5353,54 mutant in KrasG12D-driven lung adenocarcinomas (Figure 3D). In addition, Ptpn14 is induced by ionizing radiation in MEFs in a p53-dependent fashion, indicating that it is also activated by DNA damage signals, not only oncogene expression (Figure 3F). Finally, we observe that PTPN14 expression is p53-dependent in human HCT116 colorectal cancer cells (Figure S2), indicating that the ability of p53 to activate PTPN14 is conserved in humans. Together, these findings demonstrate that Ptpn14/PTPN14 is a p53 target gene in diverse mouse and human cell contexts.
Ptpn14 suppresses pancreatic cell proliferation and transformation
To ascertain whether p53 acts through Ptpn14 to suppress pancreatic cancer, we investigated whether Ptpn14 itself has growth suppressive activity in pancreatic cancer cells. We first utilized overexpression approaches to determine whether Ptpn14 is sufficient to induce growth arrest. Indeed, introduction of HA-Ptpn14 into mouse p53-deficient pancreatic cancer cells (Kras+/LSL-G12D;Pdx1-Cre;Trp53fl/fl or KPC) is sufficient to inhibit cell cycle progression, as assessed by BrdU incorporation (Figure 4A), commensurate with that seen upon HA-p53 overexpression. Moreover, this growth arrest correlates with suppressed clonogenic potential in a low density plating assay and with decreased anchorage-independent growth in a soft agar assay (Figure 4B). Similarly, HA-Ptpn14 overexpression inhibits proliferation, clonogenic potential, and anchorage-independent growth of MIA PaCa-2 and PANC-1 human p53 mutant pancreatic cancer cells (Figure 4C and 4D, Figure S3A). Interestingly, the HA-p53-mediated arrest is impaired in Ptpn14-knockdown cells, indicating that p53 relies at least in part on Ptpn14 to drive cell cycle arrest (Figure 4E, Figure S3B). Together, these findings show that Ptpn14 can impede cell cycle progression and limits both clonogenic potential and anchorage-independent growth in both mouse and human cells.
Figure 4. Ptpn14 overexpression drives growth arrest.

(A) The effect of HA-GFP, HA-Ptpn14 or HA-p53 expression on cell cycle progression in Kras+/G12D;Pdx1-Cre;Trp53fl/fl mouse PDAC cells was examined by BrdU immunostaining cells expressing each antigen (detected by GFP or HA immunofluorescence). (Left) The average BrdU incorporation ± SD from (n=3), with 100 cells counted per experiment, is shown. (Right) Representative images are shown; arrows point to BrdU+ cells, while arrowheads point to BrdU− cells. (B) (Left) Average colony number ± SD of KPC cells infected with an empty vector or a HA-Ptpn14 vector for low plating and soft agar assays (n=2, triplicate). (Right) Representative images of crystal violet-stained low plating (2 weeks after seeding), Giemsa-stained soft agar (4 weeks after seeding) and Ptpn14 localization by immunofluorescence. (C) The effect of HA-GFP, HA-Ptpn14 or HA-p53 expression on cell cycle progression in PANC-1 and MIA PaCa-2 human PDAC cells was examined by BrdU immunostaining. The average BrdU incorporation ± SD, with 100 cells counted per experiment, is shown (n=3). (D) (Left) Average colony number ± SD of MIA PaCa-2 cells infected with an empty vector or a HA-Ptpn14 vector for both low plating and soft agar assays (n=2, triplicate). (Right) Representative images of crystal violet-stained low plating (2 weeks after seeding), Giemsa-stained soft agar (4 weeks after seeding) and Ptpn14 localization by immunofluorescence. (E) The effect of HA-p53 expression on cell cycle progression in KPC PDAC cells expressing luciferase control shRNA (shCont) or shPtpn14-2 was examined by BrdU immunostaining. The average BrdU incorporation ± SD was assessed, with 100 cells counted per experiment (n=3). * p ≤ 0.05; ** p ≤ 0.001, two-tailed unpaired Student’s t-test. The scale bar in each panel applies to all images in that panel.
Knockdown of a key tumor suppressor in the p53 pathway should promote transformation in wild-type p53-expressing cells but not p53 null cells. We tested this hypothesis by knocking down Ptpn14 expression in two different p53-proficient (Kras+/LSL-G12D;Pdx1-Cre;Cdkn2afl/fl or KIC) PDAC cell lines and examining the ability of cells to grow in clonogenic and anchorage-independent growth assays. Ptpn14 knockdown resulted in increased colonies in both assays, using either of two different Ptpn14 shRNAs, indicating that Ptpn14 indeed displays tumor suppressor activity (Figures 5A–C, Figures S4A–B). Interestingly, the extent of enhanced growth was comparable to that achieved by p53 knockdown, highlighting the potency of this target gene in transformation suppression. Rescue experiments verified that the shRNA specifically targeted Ptpn14 (Figure S4C). Importantly, the effects of Ptpn14 knockdown were dependent on p53, as Ptpn14 knockdown in p53-deficient KPC cells did not enhance colony growth in clonogenic growth assays (Figure 5B). To assess whether Ptpn14 is also a tumor suppressor in vivo, we knocked down Ptpn14 in two different p53-proficient KIC PDAC cell lines and injected these cells subcutaneously into immunocompromised mice. Ptpn14 knockdown increased tumor size, with a growth enhancement comparable to that achieved by p53 knockdown (Figure 5D), indicating that Ptpn14 also behaves as a tumor suppressor in vivo. Collectively, these findings demonstrate that the p53 target gene Ptpn14 has potent p53-dependent tumor suppression activity.
Figure 5. Ptpn14 is a pancreatic cancer suppressor.

(A) Western blot for Ptpn14 and p53 in PDAC cells from Kras+/LSL-G12D;Pdx1-Cre;Cdkn2afl/fl (KIC) and KPC mice after introducing a luciferase control shRNA (shCont), either of two Ptpn14 shRNAs (shPtpn14-1 and shPtpn14-2), or p53 shRNA (shp53). Ptpn14 quantification relative to Gapdh loading control is shown below the blot. (B) (Left) Low plating experiment quantification, showing the average colony number ± SD after introduction of the indicated shRNA into two lines of KIC cells (KIC1 and KIC2) and one line of KPC cells (n=3, triplicate). (Right) Representative images of crystal violet-stained low plating assays 2 weeks after seeding. (C) (Left) Average soft agar colony number ± SD after introduction of the indicated shRNA into KIC1 and KIC2 cells (n=2, triplicate). (Right) Representative images of soft agar experiment 4 weeks after seeding. (D) (Left) Tumor volume (average ±SD): KIC cells transduced with luciferase control shRNA, p53 shRNA, Ptpn14 shRNA-1 or Ptpn14 shRNA-2 were injected subcutaneously into ICR/Scid mice and tumor volume was measured as a function of time (n=3). (Right) Representative tumor images at the end of the experiment (22 days), with quantification of average tumor weight ± SD below. Each individual point represents a mouse. * p ≤ 0.05; ** p ≤ 0.001, two-tailed unpaired Student’s t-test.
Ptpn14 restrains pancreatic cancer cell proliferation through effects on Yap
Ptpn14 interacts with multiple proteins, such as β-Catenin (Wadham et al., 2003) and p130Cas (Zhang et al., 2013), but its best-characterized binding partner is Yap, a transcriptional coactivator and component of the Hippo growth regulatory pathway (Huang et al., 2013; Liu et al., 2013; Michaloglou et al., 2013; Wang et al., 2012; Wilson et al., 2014). Ptpn14 also interacts with Kibra and Lats1, other Hippo pathway components that negatively regulate Yap (Poernbacher et al., 2012; Wilson et al., 2014). Given that Yap is critical for progression of PanINs to PDACs in mice (Zhang et al., 2014), we sought to assess whether the growth inhibitory effects of Ptpn14 in pancreatic cancer cells are mediated via effects on Yap. We first delineated domains important for Ptpn14 function in inhibiting cell cycle progression. Ptpn14 is characterized by several functional domains, including N-terminal FERM (F for 4.1 protein, E for ezrin, R for radixin and M for moesin) domains that allow bridging between the cytoskeleton and the plasma membrane (Tsukita et al., 1997), central PPxY motifs that mediate interaction with WW domain-containing proteins such as Yap to drive cytoplasmic sequestration, and a C-terminal protein tyrosine phosphatase domain. We introduced a panel of V5-tagged Ptpn14 mutants altered in these key domains into p53 null KPC PDAC cells and assessed their ability to inhibit proliferation (Figure 6A, B; Figure S5A). We found that the PPxY motifs, which are required for Yap binding, are critical for Ptpn14-mediated growth arrest. Furthermore, the C-terminal domain, which complexes with Kibra to activate the Lats1 kinase and thereby indirectly block Yap activity, is also important for efficient proliferative arrest (Poernbacher et al., 2012; Wilson et al., 2014). In contrast, the amino-terminal FERM domains were dispensable for proliferative arrest. All mutants showed similar sub-cellular localization patterns, indicating that mislocalization did not account for lack of activity (Figure 6B). Taken together, the requirement for the PPxY motif and C-terminus for inhibiting proliferation in pancreatic cancer cells is consistent with an important role for Ptpn14 in regulating Yap in these cells. Indeed, HA-Ptpn14 overexpression promoted Yap cytoplasmic localization in pancreatic cancer cells (Figure 6C). The phosphatase activity of Ptpn14 was dispensable for its ability to relocalize Yap and to suppress colony formation (Figure S5B and S5C), suggesting that biological activity in this context does not require catalytic activity, as reported previously in some contexts (Wang et al., 2012). Importantly, the enhanced colony growth observed upon Ptpn14 knockdown in p53-proficient KIC PDAC cells was attenuated upon treatment of these cells with verteporfin, a Yap inhibitor (Liu-Chittenden et al., 2012), implicating Yap in the enhanced transformation triggered by Ptpn14 knockdown (Figure 6D). Together, these findings demonstrate that Ptpn14 negatively regulates Yap to induce growth arrest in pancreatic cancer cells.
Figure 6. Ptpn14 restrains Yap activity.

(A) Schematic representation of wild-type Ptpn14 and a panel of mutants altered in key domains, including the FERM, PPxY and protein tyrosine phosphatase domains. PPxA1/PPxA2 denotes alanine mutations in both PPxY domains (Wilson et al., 2014). (B) HA-GFP or V5-tagged Ptpn14 variants were expressed in KPC mouse PDAC cells and BrdU-positivity of cells expressing each antigen (detected by GFP, HA or V5 immunofluorescence) was assessed. (Left) The average BrdU incorporation from 3 experiments ± SD, with 100 cells counted per experiment, is shown. (Right) Representative immunofluorescence images showing the localization pattern for each Ptpn14 variant. (C) Effects of HA-Ptpn14 on YAP localization in PANC-1 human PDAC cells, compared to HA-GFP. (Left) Graph showing the average percentage of cells with YAP nuclear exclusion from 3 experiments ± SD. (Right) Representative images with arrows pointing to HA-GFP-positive cells with nuclear YAP and arrowheads pointing to Ptpn14-expressing cells without nuclear Yap. (D) Colony forming ability of KIC cells treated with the Yap inhibitor verteporfin (10 μM) after introduction of control, p53, Ptpn14-1 or Ptpn14-2 shRNAs. (Left) Low plating experiment quantification, showing the average colony number ± SD of 3 experiments. (Right) Representative images show the crystal violet-stained colonies 2 weeks after seeding. (E) Sorting scheme to isolate PanIN cells from Kras+/G12D;Pdx1-Cre;Trp53+/+ and Kras+/G12D;Pdx1-Cre;Trp53−/− mice, based on CD133 positivity. Side-scatter area (SSC-A) and CD133 area (CD133-A) are used to define the PanIN population. (F) Yap GSEA signature (YAP1_UP) found enriched in Kras+/G12D;Pdx1-Cre;Trp53−/− cells relative to Kras+/G12D;Pdx1-Cre;Trp53+/+ cells. Nominal p value is indicated. The heat map represents the expression of the genes that contributed to the enrichment score. (G) Heat map showing the expression of an expanded list of 114 Yap-activated target genes based on 3 different signatures in Kras+/G12D;Pdx1-Cre;Trp53+/+ and Kras+/G12D;Pdx1-Cre;Trp53−/− PanIN cells. (H) (Left) Representative histological images of PanIN lesions from Kras+/G12D;Ptf1a-Cre;Trp53+/+ (n=4) and Kras+/G12D;Ptf1a-Cre;Trp53fl/fl (n=3) mice. The same field was analyzed by H&E and Yap staining, at 2 different magnifications, as indicated by the scale bars. Box indicates region of magnification. (Right) Average percentage of cells in PanINs with nuclear Yap ± SD. At least 300 nuclei were analyzed per mouse. * p ≤ 0.05; ** p ≤ 0.001, two-tailed unpaired Student’s t-test.
p53 Acts as a Tumor Suppressor through the Ptpn14-Yap axis
Our finding that p53-mediated transformation suppression in pancreatic cancer depends on Ptpn14 suggests that p53 acts at least in part by modulating Yap signaling. To directly test this idea, we took a multipronged approach. First, we interrogated whether p53 loss exerts effects through Yap by attenuating p53 expression through RNA interference in KIC PDAC cells and treating these cells with verteporfin. As with shPtpn14-expressing cells, verteporfin treatment attenuated colony formation in p53 shRNA-expressing PDAC cells significantly more than in luciferase control shRNA-expressing cells, suggesting that Yap contributes to the enhanced transformation triggered by p53 knockdown (Figure 6D). Similarly, Yap knockdown inhibited enhanced colony formation in p53-deficient KPC cells (Figures S5D–E). Next, we examined the effects of p53 deficiency on Yap target gene expression in pre-malignant mouse PanIN lesions to determine whether p53 loss promotes Yap signaling during mouse pancreatic cancer development. Toward this end, we collected pancreata from Kras+/LSL-G12D;Pdx1-Cre;Trp53+/+ and Kras+/LSL-G12D;Pdx1-Cre;Trp53−/− mice, at 6–8 and 4–6 weeks, respectively, ages at which we verified that only PanINs are present. We dissociated the pancreas, sorted the CD133+ ductal epithelial cells that make up the PanINs (Lee et al., 2013; Sugiyama et al., 2007), and used these cells for RNA-seq analysis (Figure 6E). Gene Set Enrichment Analysis (GSEA) revealed a Yap signature induced upon p53 deficiency (Figure 6F). Moreover, expanding this analysis to examine the expression of a gene set comprising YAP target genes from 2 different MSigDB signatures and genes previously described as Yap targets (Dupont et al., 2011) showed that ~40% of Yap-activated genes are induced in the p53-deficient PanINs, further supporting the idea that p53 loss promotes the induction of a Yap transcriptional program (Figure 6G, Table S1). Finally, we tested whether PanINs from Kras+/LSL-G12D;Ptf1a-Cre;Trp53−/− mice displayed more Yap nuclear localization than those from Kras+/LSL-G12D;Ptf1a-Cre;Trp53+/+ mice. Indeed, the percentage of cells with Yap nuclear staining was significantly higher in p53-deficient PanINs than in p53-expressing ones, supporting the notion that p53 restrains Yap signaling in incipient pancreatic cancers (Figure 6H). These studies thus collectively illuminate a heretofore unknown connection between p53 deficiency and Yap activation in pancreatic cancer development.
The p53-Ptpn14-Yap axis is altered in human cancers
To examine the significance of the p53-Ptpn14-Yap axis in human pancreatic cancer, we first examined Ptpn14 expression during human pancreatic cancer development. Immunostaining for Ptpn14 on tissue microarrays comprising samples representing different stages of pancreatic cancer development revealed that Ptpn14, while robustly expressed at the plasma membrane in PanINs, is greatly reduced during the progression to PDAC, consistent with it having tumor suppressor activity (Figure 7A). Moreover, p53 deficiency was particularly associated with reduced Ptpn14 membrane localization in PDACs, as tumors harboring TP53 mutations display a significant loss of Ptpn14 staining, while tumors with wild-type TP53 predominantly exhibit Ptpn14 plasma membrane localization (Figure 7B). We also similarly analyzed these samples for Yap localization and found that tumors carrying TP53 mutations predominantly display Yap nuclear localization, while tumors with wild-type TP53 frequently show lack of Yap nuclear localization (Figure 7B). Moreover, by leveraging TCGA data to compare the expression of Yap-inducible genes in tumors that preserve wild-type p53 function with those harboring TP53 mutations, we found that p53 deficiency stimulates Yap signaling in human pancreatic cancer as well as in a variety of other human cancer types, including tumors driven by oncogenic Kras and those with wild-type Kras expression (Figure 7C, Table S2, Table S3, Figure S6A). Consistent with this idea, verteporfin selectively inhibits the growth of p53-deficient human cancer cells of a variety of types but not p53-proficient counterparts (Figure S6B). Moreover, Ptpn14 overexpression inhibits the clonogenic potential of p53-deficient human cancer cells but not that of wild-type counterparts (Figures S6C–D). Collectively, these findings demonstrate that p53 deficiency generally promotes Yap activity in a host of cancers, which can enable transformation.
Figure 7. The p53-Ptpn14-Yap tumor suppressive axis is important in human cancers.

(A) (Left) Representative images of Ptpn14 immunohistochemistry on PanIN (n=53) and PDAC (n=109) samples. (Right) Percentage of samples with Ptpn14 plasma membrane staining. (B) (Left) Representative images of Ptpn14 and Yap immunohistochemistry in PDAC samples with known p53 status (n=32 and n=64, for TP53 wild-type and mutant respectively; defined by sequencing of the TP53 gene). (Right) Percentage of samples with Ptpn14 plasma membrane staining and with Yap nuclear localization. (C) Heat map showing the fold-change in the expression of Yap-activated genes in tumor samples harboring TP53 mutations relative to tumors with wild-type p53. Only genes where p value for summary statistics across different tumors was ≤ 9.8E-17 are shown. Asterisks denote significant changes (p ≤ 0.05) within each gene/tumor type, based on a linear regression model. The black box highlights the results obtained in pancreatic cancer (PAAD). Ovarian cancer (OV), lung squamous cell carcinoma (LUSC), lung adenocarcinoma (LUAD), breast cancer (BRCA), bladder cancer (BLCA), kidney cancer (KIRC), glioblastoma (GMB), uterine cancer (UCEC), acute leukemia (LAML), head and neck carcinoma (HNSC), colon (COAD) and rectal adenocarcinomas (READ). (D) Map depicting TP53 and PTPN14 mutations in gastrointestinal cancers. Mutual exclusivity was evaluated using the DISCOVER algorithm. (E) Proposed model incorporating Ptpn14 and Yap into the p53 tumor suppression program. For the TMA analysis, * p ≤ 0.05; ** p ≤ 0.001, Fisher test. For the heat map of the fold-change, * p ≤ 0.05, based on a linear model built for each Yap target gene.
To provide more direct evidence of the p53-Ptpn14 transcriptional program in pancreatic cancer suppression, we assessed whether PTPN14 is mutated in human cancers and whether its mutation is mutually exclusive with TP53. Because of the limited number of pancreatic cancer samples, we chose to examine PTPN14 status in an expanded set of samples of gastrointestinal cancers, including pancreas, colon, esophageal, gastric and rectal cancers. Interestingly, we found that PTPN14 is mutated in 5% of these cancers and that these mutations are typically mutually exclusive with TP53 (Figure 7D), consistent with p53 and Ptpn14 being in the same pathway. Additionally, we found that higher levels of PTPN14 expression predict increased overall survival in PDAC patients (Figure S6E), consistent with tumor suppressor activity. Collectively, these findings support the idea that Ptpn14 lies in the same pathway as p53 and underscore the importance of the p53-Ptpn14-Yap axis in cancer suppression in both mice and humans (Figure 7E).
DISCUSSION
Despite the critical role of p53 in tumor suppression, the transcriptional programs downstream of p53 in tumor suppression have remained elusive. Recent studies have challenged the significance of the classical p53 functions of cell cycle arrest and apoptosis, as well as key effectors of these pathways, for tumor suppression (Brady et al., 2011; Li et al., 2012; Valente et al., 2013). Here, we strive to shed light on the pathways of p53-mediated tumor suppression by studying a panel of p53 TAD mutant mouse strains in a pancreatic cancer model.
Although hyperactive p53 mutants have been described previously (reviewed in Van Nostrand and Attardi, 2014), the p5353,54 mutant is unique in displaying enhanced tumor suppressor capability. Mice expressing either of two N-terminal deletion variants – p53Δ40 or p53m – display premature aging, which is at least in part attributable to effects on gene expression (Maier et al., 2004; Tyner et al., 2002). Moreover, expression of the C-terminal deletion mutants p53ΔN31 or p53ΔCTD in mice provokes early adult lethality accompanied by phenotypes of dyskeratosis congenita and hematopoietic failure/impaired cerebellar development, respectively, phenotypes correlated with hyperactivation of p53 target genes. The p5353,54 mutant is distinct in that it enhances tumor suppression without provoking any additional detrimental effects and is reminiscent of super-p53 mice with multiple copies of p53, which are tumor resistant but fail to age prematurely (Garcia-Cao et al., 2002). Embryos and mice expressing the p5353,54 mutant are viable and healthy, unlike those expressing these other hyperactive mutants (Van Nostrand et al., 2014). The exact mechanism by which p5353,54 acts is not well understood, but its hyperactivity is likely due at least in part to mildly enhanced stability resulting from a compromised ability to bind the Mdm2 ubiquitin ligase, as predicted by our molecular modeling and previous structural studies (Chi et al., 2005; Shan et al., 2012) and supported by our co-immunoprecipitation experiments. However, slightly increased stability is not likely to fully account for the enhanced tumor suppressor activity of the p5353,54 mutant, as it has very selective effects on the p53-dependent transcriptome. The precise basis for the enhanced transcriptional activity remains unclear, but disruption of interactions with any partner that negatively regulates p53 transcriptional activity, such as Mdm4, would explain transcriptional hyperactivity. Our in silico structural analyses of the p53-Hmgb1 interaction suggest that W53Q;F54S mutations would perturb any interaction mediated by an amphipathic α-helix structure in TAD2. Which TAD2 binding partner is most relevant in this scenario, however, remains unclear and will be interesting to deduce in future studies.
The identification of PTPN14 as a p53 target gene implicates the Hippo pathway, which regulates organ size and tissue regeneration (Yu et al., 2015), as a central component of the p53 tumor suppression pathway. In the Hippo pathway, a cascade of kinases ultimately impinges upon Yap and the related transcriptional coactivator Taz to block their nuclear localization and associated transcriptional activity. Pathological Yap activation is involved in the initiation and progression of various cancers, including pancreatic cancer, by promoting programs essential for tumorigenesis, such as proliferation, survival, stemness, epithelial-to-mesenchymal transition and metastasis programs (Yu et al., 2015; Zanconato et al., 2016b). However, while dysregulation of the Hippo pathway is a key event in many tumor types, mutations in Hippo pathway components themselves are rare, and the mechanisms that liberate Yap during tumorigenesis remain incompletely understood, underscoring the critical need to identify upstream regulators of this pathway (Plouffe et al., 2016; Zanconato et al., 2016b). Our results show that mouse tumors harboring complete p53 inactivation display significantly enhanced active nuclear Yap, suggesting that the wild-type p53 is key for negatively regulating Yap activity in PDAC and in other cancers. Ptpn14 binding and sequestering Yap in the cytoplasm (Wang et al., 2012) provides a reasonable rationale for how p53 might restrict Yap signaling and consequent tumorigenic phenotypes. In addition, Ptpn14, via interactions with Kibra, activates Lats1 to inhibit Yap (Wilson et al., 2014). Activation of Lats1/2 by PtpnN14 may also have the additional effect of stimulating p53 activity by binding Mdm2, driving a positive feedback loop for p53 activity (Aylon et al., 2006; Matallanas et al., 2011). Our data highlighting the requirement of Ptpn14 in tumor suppression has been bolstered by recent human cancer genome studies in which PTPN14 was shown to be mutated in both kidney cancer and basal cell carcinoma, leading to Yap nuclear localization (Bonilla et al., 2016; Mehra et al., 2016). These observations are in keeping with studies in immortalized mammary cell lines, in which Ptpn14 knockdown enhances colony growth (Liu et al., 2013; Michaloglou et al., 2013; Wang et al., 2012). Collectively, our findings highlighting p53-Ptpn14 as an axis upstream of Yap suggests an important pathway through which Yap can be regulated during cancer development.
Our study has potential therapeutic implications. First, our findings with the p5353,54-expressing mice suggest the possibility of mild hyperactivation of the p53 tumor suppressor response without provoking detrimental effects in normal tissues. It may be possible to develop either a chemopreventive agent or cancer therapeutic for wild-type p53-expressing cancers that would perturb the interaction of the TAD2 domain with interacting partners and mimic the p5353,54 mutant to enhance tumor suppression in vivo. Second, our discovery that Yap is activated in p53-deficient tumors suggests it as a potential therapeutic target in the many cancers that carry p53 mutations. As our experiments suggest, inhibiting Yap activity in p53-deficient cells dampens transformation potential. Many therapeutic strategies aimed at targeting Yap are being explored (Moroishi et al., 2015; Zanconato et al., 2016a), and verteporfin is currently being tested in a clinical trial for the treatment of locally advanced pancreatic cancer (Huggett et al., 2014). It will be worthwhile to evaluate the particular efficacy of such therapies in p53-deficient cancers. Through such strategies, future studies will leverage our enhanced understanding of p53 tumor suppression pathways to develop improved approaches to cancer therapy.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact Laura D. Attardi at attardi@stanford.edu
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse analysis
All animal experiments were in accordance with the Stanford University APLAC (Administrative Panel on Laboratory Animal Care). p53 TAD mutant mice (Brady et al., 2011; Johnson et al., 2005) were bred to mice carrying KrasLSL-G12D (Jackson et al., 2001) and Pdx1-Cre alleles (Hingorani et al., 2003) to generate cohorts for pancreatic cancer experiments. Mice were aged and humanely sacrificed upon signs of morbidity. Kras+/G12D;Pdx1-Cre;Trp53+/+ mice (6–8 weeks old) and Kras+/G12D;Pdx1-Cre;Trp53−/− mice (4–6 weeks old) were used for the sorting of PanIN cells and RNA-seq. We verified that only PanINs are present in those animals, at the specified ages. Kras+/LSL-G12D;Ptf1a-Cre;Trp53+/+ and Kras+/LSL-G12D;Ptf1a-Cre;Trp53fl/fl mice were used for the Yap localization analysis (Jonkers et al., 2001; Kawaguchi et al., 2002). 4–6 week-old animals were humanely sacrificed and pancreatic tissue was processed and sectioned for analysis.
Subcutaneous Tumor Model
1 × 106 cells Kras+/G12D;Pdx1-Cre;Cdkn2afl/fl (KIC) cells transduced with shRen (LPE-Ren_713, Mirimus, Inc.), shp53 (LPE-Trp53_1224, Mirimus, Inc.), or shPtpn14 hairpins (LPE-Ptpn14_8252 and LPE-Ptpn14_10201 or shPtpn14-1 and shPtpn14-2 respectively, Mirimus, Inc.) were injected subcutaneously into the posterior flanks of 5-week-old male ICR/Scid mice (Taconic Biosciences). 4–6 tumors were injected per hairpin. Tumor volume was measured using electronic calipers. At the final time point, tumors were resected and weighed.
Cell lines
KPC and KIC cells were derived from tumors of Kras+/LSL-G12D;Pdx1-Cre;Trp53fl/fl and Kras+/LSL-G12D;Pdx1-Cre;Cdkn2afl/fl mice respectively and were a kind gift from Dr. Nabeel Bardeesy (Bardeesy et al., 2006a). Mouse NSCLC cell lines were generated from lung tumors dissected from KrasLA2/+;Trp53LSL-WT/LSL-WT, KrasLA2/+;Trp53LSL-25,26/LSL-25,26, KrasLA2/+;Trp53LSL-53,54/LSL-53,54, or KrasLA2/+;Trp53LSL-25,26,53,54/LSL-25,26,53,54 mice. The KrasLA2/+ allele undergoes spontaneous recombination to express oncogenic KrasG12D (Johnson et al., 2001). Cells were transduced with Ad-Cre (Adenovirus-Cre) previous to the experiment, to allow for the recombination of the lox-stop-lox cassette present in the different Trp53 alleles. Eμ-Myc B cell lymphoma cells were derived from Eμ-myc;Trp53ER/− mice and were a kind gift of Dr. Lin He. The cells were then treated with 1 μM of 4-hydroxytamoxifen (4-OHT) or with vehicle (EtOH), to generate p53 proficient and deficient cell lines. Pancreatic (CAPAN2-p53 WT, Panc-1-p53 MUT and MIA PaCa-2-p53 MUT) cancer cells were procured from ATCC. Lung (A549-p53 WT and H1299-p53 MUT) and breast (MCF7-p53 WT and MDA-MB-231-p53 MUT) cancer cells were obtained from laboratories at Stanford University School of Medicine. Colorectal (HCT116 p53 WT and HCT116 p53 KO) cells were originally obtained from Dr. Bert Vogelstein. All cells were grown in DMEM containing 10% FBS and penicillin/streptomycin and incubated at 37°C in an incubator with 5% CO2, with the exception of Capan 2 cells, which were grown in McCoy’s 5A medium containing 10% FBS and penicillin/streptomycin.
Tissue microarrays
Tissue microarrays were made from pancreatic cancer specimens from 126 patients from Stanford Hospital diagnosed with pancreatic cancer. Specifically, tumor samples from patients who had undergone resection of pancreatic tumors at Stanford Hospital between 1995 and 2013 were requested from the Department of Pathology. A variety of tumors were chosen for the array, including mucinous cystic neoplasms, intraductal papillary mucinous neoplasms, serous cystadenomas, and adenocarcinoma. The median age of our patient population was 66 (range: 15 – 91), with a roughly even distribution of gender. This retrospective study was approved by Stanford University Institutional Review Board, with a waiver of informed consent to use patient tissue and specimens.
METHOD DETAILS
Tissue staining and immunohistochemistry
Hematoxylin and eosin (H&E) staining and immunohistochemistry was performed on paraffin-embedded tissues using standard protocols. Imunohistochemistry and tissue microarray (TMA staining) was performed using antibodies directed against Ck19 (TROMA-III; 1:200, DSHB-University of Iowa), Muc5ac (145-P1; 1:500, Thermo Scientific), Yap (#14074; 1:100, Cell Signaling), and Ptpn14 (sc-68384; 1:500, Santa Cruz). In brief, immunohistochemistry staining was performed as follows: 4 μm thick sections from paraffin blocks were re-hydrated, unmasked with citrate buffer (0.1 %) in a pressure cooker for 13 minutes, peroxidase-quenched for 20 minutes (H2O2, 3 %), blocked for 30 minutes in PBS supplemented with 10% goat serum and incubated overnight at 4° C with primary antibody. On the next day, the sections were incubated with biotinylated antibody compatible with the primary antibody used (1:1000, for details, see the Key Resources Table) for 30 minutes and subsequently incubated with VECTASTAIN Elite ABC HRP Kit (Vector Laboratories), according to manufacturer’s instructions. The sections were washed with PBS in between different incubations. Chromogenic staining was performed using the DAB peroxidase kit (Vector Laboratories) and the sections were counter-stained using hematoxylin (H-3401, Vector Laboratories). Comparisons of H&E and immunohistochemistry from the same field were performed using consecutive sections. Pictures were taken using a Leica DM6000B microscope (Leica Microsystems) and/or with a NanoZoomer 2.0-RS slide scanner (Hamamatsu), and composite images were assembled using Photoshop’s auto-align layers function. TMAs were made of triplicate 1 mm tissue cores from Whipple specimens from 126 patients from Stanford Hospital using a tissue arrayer (Beecher Instruments, Silver Spring, MD). Representative areas from each case were reviewed and selected by a board-certified surgical pathologist based on H&E-stained sections. TP53 status was defined by targeted sequencing. TMA analysis of Ptpn14 in PanINs and PDACs was performed “per core”, on 53 core biopsies with lesions histologically identified as PanINs and 109 core biopsies with lesions identified as PDAC. TMA analysis of how Ptpn14 localization is affected by p53 status in PDACs was performed in a subset of patients with known p53 status (32 harboring intact TP53 and 64 harboring TP53 mutations). TMA analysis of Yap localization in PDACs was performed similarly.
qRT-PCR
RNA collection was performed using Trizol reagent (Invitrogen) and reverse transcription was performed with MMLV reverse transcriptase (Invitrogen). Quantitative PCR was performed in triplicate using SYBR green (Life Technologies) and a 7900HT Fast Real-Time PCR machine (Applied Biosystems). Expression analysis in different cell types was performed using specific primers for each gene (Table S4).
Cell culture experiments
Overexpression experiments for BrdU incorporation analysis were performed using constructs in which GFP, p53 and Ptpn14 cDNAs were cloned into a pCDNA vector carrying an HA-tag. GFP and p53 constructs were described (Brady et al., 2011). The pCDNA3.1-3XHA-Ptpn14 construct was generated by PCR amplification and insertion of the cDNA into AscI and PacI restriction sites in pCDNA3.1-3XHA plasmid (gift of S. Artandi). pCDNA3-V5-Ptpn14-del-N, pCDNA3-V5-Ptpn14-del-C, and pCDNA3-V5-Ptpn14-PPxY plasmids were a kind gift from Jianmin Zhang (procured from Addgene, plasmids # 61004, 61005 and 61006, respectively) (Wilson et al., 2014). Vectors were transfected into cells using Lipofectamine 2000 (Invitrogen), according to manufacturer’s instructions. 24 hr after transfection the cells were pulsed for 4 hr with BrdU and then immunostained for HA or V5 tags and BrdU. Immunofluorescence was performed using these antibodies: anti-HA (rabbit; 1:200, Invitrogen), anti-V5 (rabbit; 1:100, Novus Biological), and anti-BrdU (mouse; 1:50, BD Biosciences). Briefly, the cells were fixed with 4% paraformaldehyde for 15 minutes, washed in PBS, permeabilized in PBS + 0.25 % Triton X-100 for 15 minutes, washed in PBS, incubated with anti-HA or V5 antibody for 30 minutes, washed in PBS, and subsequently incubated with anti-rabbit fluorescein-labeled secondary antibody (goat; 1:200, Vector Laboratories) for 30 minutes. The cells were then treated with hydrochloric acid (1.5 N) for 10 minutes, washed in PBS, incubated with anti-BrdU antibody, washed in PBS, and subsequently with anti-mouse alexa 546-labeled secondary antibody (goat; 1:200, Invitrogen).
Western blotting on cell extracts mouse was performed using these antibodies: p53 (rabbit CM5; 1:150, Vector Labs), Ptpn14 (sc-373766; 1:100, Santa Cruz and rabbit-anti-Ptpn14; 1:500, a generous gift from Dr. Khew Goodall). Co-immunoprecipitation experiments were performed in MEFs homozygous for different Trp53 alleles. The MEFs were infected with Ad-Cre virus and grown for 3 days. Cells were harvested by trypsinization, washed with 1X PBS and lysed using ice cold NP-40 lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 0.5 mM EDTA, 10% Glycerol) containing protease inhibitors (Roche cOmpleteTM, Cat # 11 697 498 001). 1 mg total protein was used for each immunoprecipitation reaction (IP) reaction in 500 μl final volume. To setup the IP reactions, lysates were first pre-cleared using 50% slurry of BSA-blocked Protein A sepharose beads (GE Cat # 17-0780-01) by incubating for 30 minutes at 4°C. Each pre-cleared lysate was incubated with 2.5 μg p53 antibody overnight at 4°C with gentle agitation to allow p53 immune complexes to form. The immune complexes were retrieved by incubating with 50 μl of 50% slurry of BSA blocked Protein A sepharose beads for 2 hours at 4°C. Following this incubation, the supernatant was discarded and the beads were washed 3 times using wash buffer (0.5% NP-40 containing lysis buffer). The immobilized immunoprecipitated complexes were eluted by boiling the beads in 2X SDS sample buffer. The eluates were resolved on a 10% SDS-PAGE gel and the proteins were electro-blotted onto PVDF membranes (Millipore, Immobilon-P, Cat # IPVH20200) for probing with anti-Mdm2 antibody. To confirm the p53 IP, blots were stripped (Thermo Fisher Restore stripping buffer, Cat # 21059) and re-probed using p53 antibody. Inputs represent 2.5% of the lysate subjected to immunoprecipitation. Anti-p53 (NCL-L-p53-CM5p, Leica Biosystems) and anti-Mdm2 (Abcam ab16895, 1:500 dilution) antibodies were used.
Clonogenic assays were performed by infecting cells with an empty pMGIB retroviral vector (Venteicher et al., 2008) or with pMGIB-3xHA-Ptpn14. The Ptpn14 cDNA was cloned into pMGIB using AscI and PacI restriction sites (Venteicher et al., 2008). The cells were selected in blasticidin, and 150 cells were plated per well in triplicate in 6 well plates. The cells were grown for 8 to 14 days, depending on the cell lines used. At the end of the experiment, the cells were fixed with formalin and stained using crystal violet (0.1 %). Anchorage-independent growth assays were performed by plating 6,000 cells per well in triplicate in 6 well plates. Cells were plated in DMEM containing 10% FBS, penicillin/streptomycin and 0.4 % agarose, and were grown for 4 weeks. At the end of the experiment, cells were stained using a Giemsa solution (0.02 % in PBS). Clonogenic assays were scored using ImageJ (Schneider et al., 2012) and anchorage-independent growth assays were scored using OpenCFU (Geissmann, 2013). For knockdown experiments, Kras+/G12D;Pdx1-Cre;Cdkn2afl/fl (KIC) or Kras+/G12D;Pdx1-Cre;Trp53fl/fl (KPC) PDAC cells from mice were retrovirally infected using virus made with LPE-miR-E constructs (Mirimus Inc.) harboring either control luciferase or p53 shRNAs (LPE-Ren_713 and LPE-Trp53_1224) or shRNAs against Ptpn14 (LPE-Ptpn14_8252 and LPE-Ptpn14_10201, or shPtpn14-1 and shPtpn14-2 respectively) and then subjected to puromycin selection. For the colony assay experiments with verteporfin, the cells were plated as previously described and verteporfin (10 μM) was added 24 hr after plating.
Cell viability assays in verteporfin-treated cell lines were performed using CellTiter-Blue (Promega). 10,000 or 25,000 cells were plated in triplicate into 96 well plates in 200 μL media. Four hours prior to drug addition, CellTiter-Blue reagent (40 μL, 1:2 dilution) was added to the cells. DMSO or 10 μM verteporfin (dissolved in DMSO) was then added to the cells, and the plates were then immediately read on a fluorescence plate reader (0 hr). At later time points (24 hr and 48 hr), CellTiter-Blue reagent was added to the plates 4 hr prior to the scanning. Values for CellTiter-Blue fluorescence were averaged across triplicate wells, then background fluorescence subtracted (determined from empty wells containing CellTiter-Blue, media and DMSO/verteporfin). Fold change was then calculated, based on fluorescence values at 0 hr.
Data analysis and mining
The Significant Analysis of Microarrays (SAM) method (Tusher et al., 2001) was used to compare the expression profiles of p53 wild-type and p5353,54 cells with those of p53−/− cells. By comparing genes differentially expressed in p53 wild-type and p5353,54 we were able to find 317 probes (~240 genes) that were present in both lists and that were at least 1.3 fold more expressed in the p5353,54 cells. From those 240 genes, 103 were found to be within 10 kb of a p53 ChIP-seq peak found in a ChIP-seq experiment performed in doxorubicin-treated MEFs (Kenzelmann Broz et al., 2013; GSE46240). p53 ChIP-seq data in human cells was previously published (Younger et al., 2015; GSE55727). Kras+/G12D;Ptf1a-Cre;Cdkn2a+/fl and Kras+/G12D;Ptf1a-Cre;Trp53+/fl PDAC gene expression profiles were published (Collisson et al., 2011; GSE17891).
Mutual exclusivity between TP53 and PTPN14 mutations was evaluated in gastrointestinal cancers, including pancreatic (PAAD), esophageal (ESCA), gastric (STAD), colon (COAD) and rectal (READ) cancers using data from TCGA available via the Genomic Data Portal (gdc.cancer.gov). The DISCOVER package, which employs proper false positive rate controls in comparison to the Fisher test (Canisius et al., 2016), was used to test for mutual exclusivity. Mutual exclusivity plots were generated with Bioconductor package ComplexHeatmap (Gu et al., 2016). To evaluate the effect of p53 on Yap downstream genes, we used TCGA PANCAN12 data from Synapse (https://www.synapse.org/#!Synapse:syn300013/wiki/70804). A linear model was built for each Yap target gene to measure the influence of TP53 mutations in each tumor type. Summary statistics were calculated using the function “combine.estimate” and “combine.test” (with z-transform method) from the package survcomp (Schroder et al., 2011).
Cell sorting and RNA-seq expression analysis
Total RNA was extracted from primary PanIN cells sorted from dissociated mouse pancreas, using CD133 as a marker for PanIN cells, as described (Lee et al., 2013; Sugiyama et al., 2007). In brief, we collected pancreata from Kras+/LSL-G12D;Pdx1-Cre;Trp53+/+ and Kras+/LSL-G12D;Pdx1-Cre;Trp53−/− mice, at 6–8 and 4–6 weeks, respectively, ages at which we verified that only PanINs are present. We then dissociated the pancreas through consecutive steps, starting with collagenase (2.5 mg/mL of collagenase D + 0.1 mg/mL DNAse I in HBSS) for 10 minutes at 37° C. After this period, the digested pancreas was passed through a 40 μm strainer and the retained cell clumps were washed in PBS. After centrifugation, the supernatant was removed and the cells were incubated with a warm Trypsin-EDTA solution (0.05 %) for 5 minutes at 37° C. The trypsin reaction was inhibited by adding FACS buffer (10 mM EGTA, 2 % FBS in Ca++, Mg++-free PBS) to the cells and mixing. After centrifugation, the supernatant was removed and the cells were treated with DNAse I (0.1 mg/mL in PBS). After centrifugation, the supernatant was removed and the cells were incubated with a warm dispase solution (2 U/mL of dispase + 0.1 mg/mL DNAse I in PBS) on a rocker for 30 minutes at 37° C. After centrifugation, the dispase solution was removed, and the cells were resuspended in FACS buffer and filtered through a 40 μm strainer. The cells passing through the filter were resuspended at a concentration of 1×106 cells/mL and used for staining and sorting, starting with a blocking step with goat IgG (005-000-003; 1:100, Jackson Immunoresearch Laboratories) and followed by incubation with fluorescently-labeled antibodies directed against CD133 (17-1331-81; 1:100, Ebioscience), CD45 (48-0451-82; 1:100, Ebioscience) and Ter-119 (48-5921-82; 1:100, Ebioscience) for 15 minutes. The cells were washed with FACS buffer and subsequently incubated with the live/dead Aqua Dead Cell Stain Kit (L-34965; Invitrogen), according to the manufacturer’s instructions. Live CD133+CD45−Ter-119− ductal epithelial PanIN cells were sorted using a FACSAria II cell sorter (BD Biosciences). Cells from four mice of each genotype (Kras+/G12D;Pdx1-Cre;Trp53+/+ and Kras+/G12D;Pdx1-Cre;Trp53−/−) were sorted. To achieve a minimum of 1 μg of total RNA for each library, RNA samples from 2 mice of the same genotype were combined, totaling 2 libraries per genotype in this study. RNA-seq libraries were prepared using the Illumina TruSeq Kit (v.2), according to the manufacturer’s instructions. RNA-seq reads were aligned to the mouse genome (mm10) and analyzed using Basespace’s RNA Express pipeline (RNA Express Legacy; version: 1.0.0), which employs the STAR aligner (Dobin et al., 2013) and DESEQ2 (Love et al., 2014) for differential expression analysis. The normalized read matrix was then used as input for Gene Set Enrichment Analysis (GSEA, Subramanian et al., 2005), to search for signature enrichments. The matrix of reads was also used to produce a heat-map for YAP-induced target genes based on a compilation of two MSigDB signatures (CORDENONSI_YAP_CONSERVED_SIGNATURE and YAP1_UP) and a previously published Yap signature (Dupont et al., 2011).
QUANTIFICATION AND STATISTICAL ANALYSIS
Fisher’s exact test was used to calculate tumor incidence in the tumor study performed in the mouse cohorts and to assess differences in Ptpn14 and Yap localization in the tissue microarrays. The unpaired two-tailed Student’s t-test was used for all the other statistical analyses. Error bars represent standard deviation of the mean. Significance was defined as a p value≤0.05, unless otherwise stated. Details and significance values can be found in the figure legends.
DATA AND SOFTWARE AVAILABILITY
Gene expression profiles of HrasV12-expressing MEFs homozygous for different p53 alleles as well as Kras+/G12D;Ptf1a-Cre;Cdkn2a+/fl and in Kras+/G12D;Ptf1a-Cre;Trp53+/fl PDAC gene expression profiles were published (Brady et al., 2011; Collisson et al., 2011; GSE27901 and GSE17891 respectively). The p53 ChIP-seq results used to define p53 target genes in mouse and human cells were also previously published (Kenzelmann Broz et al., 2013; Younger et al., 2015; GSE46240 and GSE55727 respectively). The RNA-seq data from sorted primary PanIN cells were generated by this study and are deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE94566. Software availability is detailed in the Key Resources Table.
KEY RESORUCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PTPN14/Pez (F-12) antibody | Santa Cruz | sc-373766 |
| PTPN14/Pez (H-67) antibody | Santa Cruz | sc-68384 |
| PTPN14/Pez antibody | Laboratory of Dr. Yeesim Khew-Goodall | N/A |
| YAP (D8H1X) XP® rabbit mAb | Cell signaling | 14074 |
| p53 antibody (CM5) | Leica (Novocastra) | P53-CM5P |
| BrdU antibody | Becton Dickinson | 555627 |
| CD133-APC antibody | eBioscience | 17-1331-81 |
| CD45-eFluor 450 antibody | eBioscience | 48-0451-82 |
| Ter-119-eFluor 450 antibody | eBioscience | 48-5921-82 |
| Ck19 antibody | University of Iowa (DSHB) | TROMAIII |
| Gapdh antibody | Fitzgerald | 10R-G109a |
| Actb antibody | Santa Cruz | sc-1615 |
| HA tag monoclonal antibody (2–2.2.14) | ThermoFisher Scientific | 26183 |
| Mdm2 antibody (#2A10) | Abcam | ab16895 |
| Muc5ac antibody | ThermoFisher Scientific | 145-P1 |
| V5 Epitope tag antibody | Novus Biological | NB600-381 |
| Fluorescein goat anti-rabbit IgG | Vector Laboratories | FI-1000 |
| Alexa Fluor 546 goat anti-mouse IgG | Invitrogen | A-11003 |
| Biotinylated goat anti-rat | Vector Laboratories | BA-9401 |
| Biotinylated goat anti-mouse | Vector Laboratories | BA-9200 |
| Biotinylated goat anti-rabbit | Vector Laboratories | BA-1000 |
| Bacterial and Virus Strains | ||
| Ad-Cre or Ad5CMVCre | University of Iowa | VVC-U of Iowa-5 |
| Ad-empty or Ad5CMVempty | University of Iowa | VVC-U of Iowa-272 |
| Biological Samples | ||
| Tissue microarrays | Laboratory of Dr. Albert Koong | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Verteporfin | Fisher Scientific | 530150 |
| Doxorubicin hydrochloride | Sigma | D1515 |
| Critical Commercial Assays | ||
| Illumina TruSeq Kit (v.2) | Illumina | RS-122-2001 |
| VECTASTAIN Elite ABC HRP Kit (Peroxidase, Standard) | Vector Laboratories | PK-6100 |
| DAB Peroxidase (HRP) Substrate Kit (with Nickel), 3,3′-diaminobenzidine | Vector Laboratories | SK-4100 |
| ChromPure Goat IgG, whole molecule | Jackson Immunoresearch Laboratories | 005-000-003 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Invitrogen | L-34965 |
| Deposited Data | ||
| Mouse primary PanINs | This paper | GSE94566 |
| HrasV12-expressing MEFs homozygous for different Trp53 alleles | Brady et al., 2011 | GSE27901 |
| Mouse PDAC gene expression | Collisson et al., 2011 | GSE17891 |
| Mouse p53 CHIP-seq | Kenzelmann-Broz et al., 2013 | GSE46240 |
| Human p53 CHIP-seq | Younger et al., 2015 | GSE55727 |
| TCGA PANCAN12 | Synapse | https://www.synapse.org/#!Synapse:syn300013/wiki/70804 |
| Experimental Models: Cell Lines | ||
| Mouse: KPC pancreatic cancer cell line: Kras+/LSL- G12D;Pdx1-Cre;Trp53fl/fl | Laboratory of Dr. Nabeel Bardeesy | NB490 |
| Mouse: KIC pancreatic cancer cell line: Kras+/LSL- G12D;Pdx1-Cre;Cdkn2afl/fl | Laboratory of Dr. Nabeel Bardeesy | 32a and 33 |
| Mouse: p53WT non-small cell lung cancer cell line: KrasLA2/+;Trp53LSL-WT/LSL-WT | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: p5325,26 non-small cell lung cancer cell line: KrasLA2/+;Trp53LSL-25,26/LSL-25,26 | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: p5353,54 non-small cell lung cancer cell line: KrasLA2/+;Trp53LSL-53,54/LSL-53,54 | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: p5325,26,53,54 non-small cell lung cancer cell line: KrasLA2/+;Trp53LSL-25,26,53,54/LSL-25,26,53,54 | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: Eμ-Myc B cell lymphoma cells: Eμ-myc;Trp53ER/− | Laboratory of Dr. Lin He | N/A |
| Human: Panc-1 pancreatic cancer cell line: TP53 MUT | ATCC | CRL-1469 |
| Human: MIA PaCa-2 pancreatic cancer cell line: TP53 WT | ATCC | CRL-1420 |
| Human: Capan 2 pancreatic cancer cell line: TP53 MUT | ATCC | HTB-80 |
| Human: HCT116 p53 WT colorectal cancer cell line: TP53 WT | Laboratory of Dr. Bert Vogelstein | N/A |
| Human: HCT116 p53 MUT colorectal cancer cell line: TP53−/− | Laboratory of Dr. Bert Vogelstein | N/A |
| Human: A549 lung cancer cell line: TP53 WT | Stanford University School of Medicine laboratories | N/A |
| Human: H1299 lung cancer cell line: TP53 MUT | Stanford University School of Medicine laboratories | N/A |
| Human: MCF7 breast cancer cell line: TP53 WT | Stanford University School of Medicine laboratories | N/A |
| Human: MDA-MB-231 breast cancer cell line: TP53 MUT | Stanford University School of Medicine laboratories | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Trp53flox mice: B6.129P2-Trp53tm1Brn/J | The Jackson Laboratory | 008462 |
| Mouse: Trp53 null− mice: B6.FVB-Tg(Pdx1-cre)6Tuv/Nci | Laboratory of Dr. Tyler Jacks | N/A |
| Mouse: Trp53LSL-53,54 mice: 129-Trp53tm3Att/J | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: Trp53LSL-25,26,53,54 mice: 129-Trp53tm4Att/J | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: Trp53LSL-wt mice: B6.129S4-Trp53tm5Tyj/J | Laboratory of Dr. Tyler Jacks | N/A |
| Mouse: KrasLSL-G12D mice: B6.129S4-Krastm4Tyj/J | Laboratory of Dr. Tyler Jacks | N/A |
| Mouse: Pdx1-Cre mice: B6.FVB-Tg(Pdx1-cre)6Tuv/J | NCI Mouse Repository | 01XL5 |
| Mouse: Ptf1a-Cre mice: 129-Ptf1atm1.1(cre)Cvw/J | Laboratory of Dr. Christopher Wright | N/A |
| Mouse: ICR/Scid mice: IcrTac:ICR-Prkdcscid | Taconic Biosciences | ICRSC-M |
| Oligonucleotides | ||
| See table S4 for primers | ||
| Recombinant DNA | ||
| pMGIB-3xHA- | Laboratory of Dr. Steven Artandi | N/A |
| pMGIB-3xHA-p53 | Laboratory of Dr. Laura Attardi | N/A |
| pMGIB-3xHA-Ptpn14 | This paper | N/A |
| pCDNA3.1-3XHA-Ptpn14 | This paper | N/A |
| pCDNA3.1-3XHA-Ptpn14-C1121S | This paper | N/A |
| pCDNA3-V5-Ptpn14-del-N | Addgene | 61004 |
| pCDNA3-V5-Ptpn14-del-C | Addgene | 61005 |
| pCDNA3-V5-Ptpn14-PPxY | Addgene | 61006 |
| LPE-Ren_713 | Mirimus | 713 |
| LPE-Trp53_1224 | Mirimus | 1224 |
| LPE-Ptpn14_8252 (shPtpn14-1) | Mirimus | 19250 |
| LPE-Ptpn14_10201 (shPtpn14-2) | Mirimus | 19250 |
| pLKO-shYap1 | Addgene | 42540 |
| pLKO-shLacZ | Addgene | 42559 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej-nih-gov.laneproxy.stanford.edu/ij/ |
| GSEA | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| DISCOVER package | Canisius et al., 2016 | http://ccb.nki.nl/software/discover/ |
| ComplexHeatmap | Gu et al., 2016 | http://www.bioconductor.org/packages/devel/bioc/html/ComplexHeatmap.html |
| Survcomp | Schroder et al., 2011 | http://bioconductor.org/packages/release/bioc/html/survcomp.html |
| OpenCFU | Geissmann, 2013 | http://opencfu.sourceforge.net/ |
| RNA Express Legacy; version: 1.0.0 (Basespace) | Illumina, Inc. | https://basespace.illumina.com |
| Adobe Photoshop CS6 | Adobe | Version 13.0.1 x64 |
| Other | ||
Supplementary Material
SIGNIFICANCE.
Since the discovery that classical p53 DNA damage responses are dispensable for p53-mediated tumor suppression, the transcriptional programs underlying p53 tumor suppressor function have remained enigmatic. Here, by identifying a p53 “super-tumor suppressor” that hyperactivates select p53 target genes, we unveil Ptpn14, a negative regulator of Yap, as a central component of p53-mediated tumor suppression. We show that TP53 status dictates the subcellular localization and activity of Yap, establishing p53 as an upstream regulator of the Hippo pathway, and that TP53 and PTPN14 mutations are mutually exclusive in human cancers. Together, these findings illuminate a p53-Ptpn14-Yap axis in tumor suppression, giving critical insight into both mechanisms of Yap activation during tumorigenesis and programs of p53-mediated tumor suppression.
HIGHLIGHTS.
The p5353,54 TAD2 mutant is a super-tumor suppressor in pancreatic cancer.
p5353,54 hyperactivates Ptpn14, a p53 target gene involved in tumor suppression.
p53 negatively regulates Yap through Ptpn14 activation.
p53-Ptpn14-Yap is a key tumor suppressive axis in mice and humans.
Acknowledgments
We thank Nabeel Bardeesy for NB490 KPC and 32a and 33 KIC cells, Lin He for Eμ-myc lymphoma cells, Mirimus for shRNAs, Steven Artandi for pCDNA constructs, and Yeesim Khew-Goodall for the Ptpn14 antibody. We thank Alyssa Kaiser, Monte Winslow, and Julien Sage for critical reading of the manuscript. This work was supported by the Canadian Natural Sciences and Engineering Research Council to D.I. and C.H.A. and R21 CA169673, R01 CA140875, and R35 CA197591 grants to L.D.A. The results published here are in part based upon data generated by The Cancer Genome Atlas (TCGA) project established by the NCI and NHGRI.
Footnotes
AUTHOR CONTRIBUTIONS
S.M. and L.D.A designed the study. L.V. performed some BrdU and Yap IF experiments and some verteporfin experiments. N.R. and K.B. performed gene expression analysis by qRT-PCR. N.R. and J.M. performed protein blots and immunoprecipitation studies. B.F. constructed plasmids and performed immunohistochemistry. J.S. and C.C. designed and performed the analysis involving human tumor data. J.L. and S. K. advised and gave support on the sorting of PanIN cells. D.I. and C.A. designed and performed the in silico analysis of the human p53 TAD2 mutant protein. M.K., D.C., T.L., and A.K. generated the tissue microarrays and M.K. performed the Ptpn14 staining. H.V., L.W., and R.H. performed histopathological analysis of the tumor study tissues. S.M. performed all other experiments. S.M. and L.D.A. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes & Development. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes & Development. 2006;20:2687–2700. doi: 10.1101/gad.1447006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng K-h, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16Ink4a and the p19Arf-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proceedings of the National Academy of Sciences. 2006a;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Cheng K-h, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, DePinho RA. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes & Development. 2006b;20:3130–3146. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart M, Werther M, Bockholt A, Scheurer M, Ruschoff J, Dietmaier W, Ghadimi BM, Heinmoller E. Genomic instability at both the base pair level and the chromosomal level is detectable in earliest PanIN lesions in tissues of chronic pancreatitis. Pancreas. 2010;39:1093–1103. doi: 10.1097/MPA.0b013e3181dc62f6. [DOI] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, Seplyarskiy VB, Sharpe HJ, McKee T, Letourneau A, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. 2016;48:398–406. doi: 10.1038/ng.3525. [DOI] [PubMed] [Google Scholar]
- Brady CA, Attardi LD. p53 at a glance. Journal of Cell Science. 2010;123:2527–2532. doi: 10.1242/jcs.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Broz DK, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canisius S, Martens JW, Wessels LF. A novel independence test for somatic alterations in cancer shows that biology drives mutual exclusivity but chance explains most co-occurrence. Genome Biol. 2016;17:261. doi: 10.1186/s13059-016-1114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Lee SH, Kim DH, Ahn MJ, Kim JS, Woo JY, Torizawa T, Kainosho M, Han KH. Structural Details on mdm2-p53 Interaction. Journal of Biological Chemistry. 2005;280:38795–38802. doi: 10.1074/jbc.M508578200. [DOI] [PubMed] [Google Scholar]
- Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA, Serrano M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. Embo j. 2002;21:6225–6235. doi: 10.1093/emboj/cdf595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann Q. OpenCFU, a New Free and Open-Source Software to Count Cell Colonies and Other Circular Objects. PLoS ONE. 2013;8:e54072. doi: 10.1371/journal.pone.0054072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, III, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Huang JM, Nagatomo I, Suzuki E, Mizuno T, Kumagai T, Berezov A, Zhang H, Karlan B, Greene MI, Wang Q. YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene. 2013;32:2220–2229. doi: 10.1038/onc.2012.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M, Kent E, Bown SG, Hasan T, Pogue BW, Pereira SP. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer. 2014;110:1698–1704. doi: 10.1038/bjc.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA, Hingorani SR. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer cell. 2007;11:229–243. doi: 10.1016/j.ccr.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & Development. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Brady CA, Johnson TM, Lee EY, Park EJ, Scott MP, Attardi LD. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proceedings of the National Academy of Sciences. 2011;108:17123–17128. doi: 10.1073/pnas.1111245108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TM, Hammond EM, Giaccia A, Attardi LD. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet. 2005;37:145–152. doi: 10.1038/ng1498. [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell. 2014;158:185–197. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CVE. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Mello SS, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes & Development. 2013;27:1016–1031. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sugiyama T, Liu Y, Wang J, Gu X, Lei J, Markmann JF, Miyazaki S, Miyazaki J, Szot GL, et al. Expansion and conversion of human pancreatic ductal cells into insulin-secreting endocrine cells. eLife. 2013;2:e00940. doi: 10.7554/eLife.00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor Suppression in the Absence of p53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD–YAP complex suppresses the oncogenic activity of YAP. Genes & Development. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yang N, Figel SA, Wilson KE, Morrison CD, Gelman IH, Zhang J. PTPN14 interacts with and negatively regulates the oncogenic function of YAP. Oncogene. 2013;32:1266–1273. doi: 10.1038/onc.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüttges J, Galehdari H, Bröcker V, Schwarte-Waldhoff I, Henne-Bruns D, Klöppel G, Schmiegel W, Hahn SA. Allelic Loss Is Often the First Hit in the Biallelic Inactivation of the p53 and DPC4 Genes During Pancreatic Carcinogenesis. The American Journal of Pathology. 2001;158:1677–1683. doi: 10.1016/S0002-9440(10)64123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes & Development. 2004;18:306–319. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matallanas D, Romano D, Al-Mulla F, O’Neill E, Al-Ali W, Crespo P, Doyle B, Nixon C, Sansom O, Drosten M, et al. Mutant K-Ras Activation of the Proapoptotic MST2 Pathway Is Antagonized by Wild-Type K-Ras. Molecular Cell. 2011;44:893–906. doi: 10.1016/j.molcel.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Mehra R, Vats P, Cieslik M, Cao X, Su F, Shukla S, Udager AM, Wang R, Pan J, Kasaian K, et al. Biallelic Alteration and Dysregulation of the Hippo Pathway in Mucinous Tubular and Spindle Cell Carcinoma of the Kidney. Cancer Discovery. 2016;6:1258–1266. doi: 10.1158/2159-8290.CD-16-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Lehmann W, Martin T, Delaunay C, Hueber A, Barys L, Niu H, Billy E, Wartmann M, Ito M, et al. The Tyrosine Phosphatase PTPN14 Is a Negative Regulator of YAP Activity. PLoS ONE. 2013;8:e61916. doi: 10.1371/journal.pone.0061916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15:73–79. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe SW, Meng Z, Lin KC, Lin B, Hong AW, Chun JV, Guan KL. Characterization of Hippo Pathway Components by Gene Inactivation. Molecular Cell. 2016;64:993–1008. doi: 10.1016/j.molcel.2016.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poernbacher I, Baumgartner R, Marada Suresh K, Edwards K, Stocker H. Drosophila Pez Acts in Hippo Signaling to Restrict Intestinal Stem Cell Proliferation. Current Biology. 2012;22:389–396. doi: 10.1016/j.cub.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Raj N, Attardi LD. The Transactivation Domains of the p53 Protein. Cold Spring Harbor perspectives in medicine. 2017;7 doi: 10.1101/cshperspect.a026047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DP, Hong TS, Bardeesy N. Pancreatic Adenocarcinoma. New England Journal of Medicine. 2014;371:1039–1049. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder MS, Culhane AC, Quackenbush J, Haibe-Kains B. survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics. 2011;27:3206–3208. doi: 10.1093/bioinformatics/btr511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Li DW, Brüschweiler-Li L, Brüschweiler R. Competitive binding between dynamic p53 transactivation subdomains to human MDM2 protein: implications for regulating the p53·MDM2/MDMX interaction. Journal of Biological Chemistry. 2012;287:30376–30384. doi: 10.1074/jbc.M112.369793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: A Cancer Journal for Clinicians. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Rodriguez RT, McLean GW, Kim SK. Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proceedings of the National Academy of Sciences. 2007;104:175–180. doi: 10.1073/pnas.0609490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Yonemura S, Tsukita S. ERM proteins: head-to-tail regulation of actin-plasma membrane interaction. Trends in Biochemical Sciences. 1997;22:53–58. doi: 10.1016/s0968-0004(96)10071-2. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- Valente Liz J, Gray Daniel HD, Michalak Ewa M, Pinon-Hofbauer J, Egle A, Scott Clare L, Janic A, Strasser A. p53 Efficiently Suppresses Tumor Development in the Complete Absence of Its Cell-Cycle Inhibitory and Proapoptotic Effectors p21, Puma, and Noxa. Cell Reports. 2013;3:1339–1345. doi: 10.1016/j.celrep.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Van Nostrand JL, Attardi LD. Guilty as CHARGED: p53’s expanding role in disease. Cell Cycle. 2014;13:3798–3807. doi: 10.4161/15384101.2014.987627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, Lin CY, Lin CJ, Swiderski DL, Vogel H, et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature. 2014;514:228–232. doi: 10.1038/nature13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Meng Z, Mason PJ, Veenstra TD, Artandi SE. Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell. 2008;132:945–957. doi: 10.1016/j.cell.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wadham C, Gamble JR, Vadas MA, Khew-Goodall Y. The Protein Tyrosine Phosphatase Pez Is a Major Phosphatase of Adherens Junctions and Dephosphorylates β-Catenin. Molecular Biology of the Cell. 2003;14:2520–2529. doi: 10.1091/mbc.E02-09-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Huang J, Wang X, Yuan J, Li X, Feng L, Park JI, Chen J. PTPN14 is required for the density-dependent control of YAP1. Genes & Development. 2012;26:1959–1971. doi: 10.1101/gad.192955.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, et al. Mutational Analysis of the Tyrosine Phosphatome in Colorectal Cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- Wilson KE, Li YW, Yang N, Shen H, Orillion AR, Zhang J. PTPN14 Forms a Complex with Kibra and LATS1 Proteins and Negatively Regulates the YAP Oncogenic Function. Journal of Biological Chemistry. 2014;289:23693–23700. doi: 10.1074/jbc.M113.534701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger ST, Kenzelmann-Broz D, Jung H, Attardi LD, Rinn JL. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Research. 2015;43:4447–4462. doi: 10.1093/nar/gkv284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Battilana G, Cordenonsi M, Piccolo S. YAP/TAZ as therapeutic targets in cancer. Current Opinion in Pharmacology. 2016a;29:26–33. doi: 10.1016/j.coph.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016b;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Guo A, Possemato A, Wang C, Beard L, Carlin C, Markowitz SD, Polakiewicz RD, Wang Z. Identification and functional characterization of p130Cas as a substrate of protein tyrosine phosphatase nonreceptor 14. Oncogene. 2013;32:2087–2095. doi: 10.1038/onc.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, Gupta S, Vietsch EE, Laughlin SZ, Wadhwa M, et al. Downstream of Mutant KRAS, the Transcription Regulator YAP Is Essential for Neoplastic Progression to Pancreatic Ductal Adenocarcinoma. Science Signaling. 2014;7:ra42–ra42. doi: 10.1126/scisignal.2005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.