

Abstract
Abnormal placental function in maternal diabetes affects fetal health and can predispose offspring to metabolic diseases in later life. There are fetal sex-specific differences in placenta structure and gene expression, which may affect placental responses to maternal diabetes. The present study examined the effects of maternal diabetes on indices of mitochondrial biogenesis in placentae from male and female offspring. Mitochondrial DNA (mtDNA) copy number and expression of key regulators of mitochondrial biogenesis were assessed in placentae from 19 diabetic and 23 non-diabetic women. The abundance of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and mitochondria transcription factor A (TFAM) were lower in female placentae compared to males, but not mtDNA content. In male offspring, maternal diabetes was associated with decreased placental PGC-1α and TFAM, and mitochondrial DNA (mtDNA) content. Male placental TFAM levels were highly correlated with PGC-1α and mtDNA content. However, despite decreased PGC-1α, concomitant changes in TFAM and mtDNA content by diabetes were not observed in females. In addition, TFAM abundance in female placentae was not correlated with PGC-1α or mtDNA content. In summary, placental PGC-1α/TFAM/mitochondrial biogenesis pathway is affected by maternal diabetes and offspring sex. Decreased PGC-1α in response to maternal diabetes plausibly contributes to impaired mitochondrial biogenesis in placentae of male offspring, which may affect long-term health and explain some of enhanced risk of future metabolic diseases in males.
Keywords: Placenta, maternal diabetes, fetal sex, PGC-1α, TFAM, mitochondrial DNA
Graphical abstract
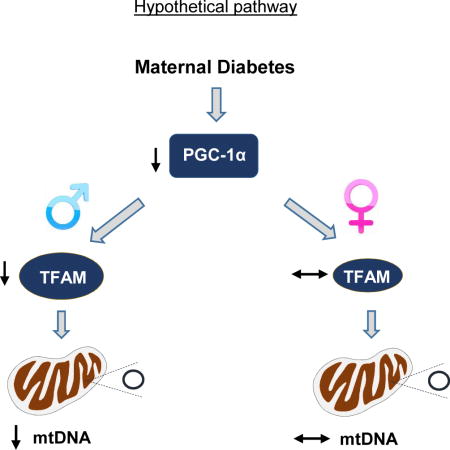
Introduction
Maternal Diabetes during pregnancy not only has short-term impacts on fetal development, but also increases the risk for offspring to develop type 2 diabetes, obesity, and other metabolic disorders later in life [1–6]. A sibling study in Pima Indians demonstrated the important role of the prenatal environment per se in metabolic programming, showing that offspring exposed to diabetes in utero had a 3.7-fold higher risk of diabetes compared to siblings born before their mother developed diabetes [7]. While the exact mechanisms underlying this phenomenon remain unclear, hyperglycemia during pregnancy is considered a major factor [1, 2, 8]. Among offspring, maternal hyperglycemia is associated with increased fetal/neonatal adiposity [9], lower insulin sensitivity, altered β-cell function [8], and higher long-term risks of type 2 diabetes [2, 10, 11] and overweight/obesity [12].
Fetal sex-associated differences have been observed. Pregnant women carrying a male fetus have poorer pancreatic beta cell function and are at increased risk of gestational diabetes [13]. In male infants born to women with gestational diabetes, maternal fasting blood glucose concentration is a major predictor of adiposity, but has little relationship to adiposity in female infants. Conversely, maternal BMI is the primary predictor of adiposity for female infants but has little effect in males [14].
The placenta is the critical organ mediating all communications between mother and fetus, and thus must be involved in the effects of maternal diabetes on offspring [15]. Functional changes occurring in response to maternal diabetes include reduced fatty acid oxidation, impaired mitochondrial function, and increased production of reactive oxygen species [16, 17]. Abnormal placental function could in turn affect nutrient transfer and alter the constituency of bio-active molecules released into the fetal circulation, ultimately affecting fetal growth and predisposing offspring to metabolic disease in later life [18–20]. Furthermore, sexual dimorphism in structure, function [21] and gene expression [22] in human placenta has been shown, suggesting that sex-specific placental responses and adaptation may mediate certain fetal sex-associated differences.
Our previous studies demonstrated a decrease in the catalytic subunits of AMP activated protein kinase (AMPKα1) in placentae of diabetic women [23]. AMPK plays an essential role in maintaining cell energy homeostasis and it regulates the expression and activation of PGC-1α, the master regulator of mitochondrial biogenesis and energy metabolism [24–26]. In the present work, we examined the effects of maternal diabetes and fetal sex on PGC-1α and the mitochondrial biogenesis pathway in human placenta. PGC-1α stimulates mitochondrial biogenesis by activating transcription factors that promote expression of TFAM, which directly regulates mitochondrial DNA transcription and replication. We compared the abundance of PGC-1α and TFAM proteins, and mitochondrial DNA copy number in the placentae of infants born to women with diabetes during pregnancy to controls and demonstrate that maternal diabetes impacts the placental PGC-1α/TFAM/mitochondrial biogenesis pathway in a sex-dependent manner.
Research Design and Methods
Subjects for Placenta Samples
Placental samples were obtained from self-identified Native American and Hispanic women with gestational diabetes (GDM), pregestational type 2 diabetes, or with normoglycemic pregnancies. Mothers and offspring were enrolled into a prospective longitudinal study on the impact of in utero exposure to diabetes, as previously described [27]. Gestational and type 2 diabetes in the mothers was diagnosed according to ADA guidelines [28]. Women were excluded if the infants were small for gestational age, had a major malformation, or chromosome abnormality. They also were excluded if they delivered prior to 37 weeks gestation, had type 1 diabetes, pre-eclampsia, chronic hypertension, renal disorders or a smoking history of more than 5 cigarettes per day during pregnancy. The protocol was approved by the Institutional Review Boards of the University of Oklahoma Health Science Center, the Chickasaw Nation, and the Choctaw Nation of Oklahoma. Maternal fasting blood glucose concentrations (before the fasting oral glucose tolerance tests) were obtained retrospectively from medical records.
Placentae Dissection
Term placentae were dissected as soon as possible after delivery, generally within one hour, and processed as previously described [23]. An approximately three cm diameter core was obtained by cutting from the fetal surface down through the maternal surface. The core was cut into thirds such that one-third was fetal-side tissue, one-third was maternal-side tissue and the other the middle third. Only the fetal-side placenta samples were used in the present study. The fetal membrane was removed and the remaining tissue washed with icecold saline, blotted dry and stored at −80°C.
Western Blot Analysis
Placental samples were lysed and homogenized in protein lysis buffer containing a protease and phosphatase inhibitor cocktail (Pierce Biotechnology, Rockford, IL; part number 78443). Protein concentrations were measured by BCA assay (Pierce, Rockford, IL). Thirty μg of protein lysate was treated with reducing agent (beta-mercaptoethanol), subjected to sodium dodecyl sulfated polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membrane followed by incubation with antibodies specific for PGC-1α (Abcam, Cambridge, MA), TFAM, AMPKα1, or β-actin (Cell Signaling Technology, Danvers, MA). The proteins of interest were detected by enhanced chemiluminescence (Pierce, Rockford, IL) and analyzed by imaging densitometry with Image Lab Software (Bio-Rad, Hercules, CA).
Mitochondrial DNA copy number
DNA was isolated from placental tissue using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma, St. Louis, MO) with proteinase K and RNase treatment, according to the manufacturer’s instructions. Mitochondrial DNA copy number was estimated by comparing the abundance of the mitochondrial tRNALeu(UUR) gene (Determined by quantitative real-time PCR, forward primer: 5′-CACCCAAGAACAGGGTTTGT; reverse: 5′-TGGCCATGGGTATGTTGTTA) and with that of the nuclear β2-macroglobulin gene (forward: 5′-TGCTGTCTCCATGTTTGATGTATCT; reverse: 5′-TCTCTGCTCCCCACCTCTAAGT).
Statistical methods
Group descriptive statistics are presented as mean ± SD and group count (percentage). Differences in characteristics between two groups were assessed using Student’s t-test for continuous measures; chi-squared test for counts. The Generalized Linear Model (GLM) framework was used to determine the significance of the effect of diabetes on PGC-1α and TFAM levels after adjusting for the covariates listed in Table 1. Differences in the effect of diabetes due to sex of infant were assessed by fitting a diabetes × sex interaction. Correlations were calculated as standardized regression coefficients. Data analyses used Excel and IBM SPSS Statistics (IBM Corp. Released 2011. IBM SPSS Statistics for Windows, Version 20.0. Armonk, NY: IBM Corp). P-values <0.05 were treated as statistically significant for the purposes of discussion.
Table 1.
Characteristics of Research Subjects Providing Placenta Samples
| DM | Control | P-value DM vs Control | |
|---|---|---|---|
| n | 19 (16 GDM, 3 Type 2 diabetes) | 23 | |
| Maternal Age, yrs | 31 ± 5 | 27 ± 5 | 0.021 |
| Maternal race | 0.48 | ||
| American Indian | 13 (68%) | 17 (74%) | |
| Hispanic | 6 (32%) | 6 (26%) | |
| Maternal HbA1C, % | 5.6 ± 0.7 | 5.1 ± 0.2 | 0.013 |
| Maternal BMI | 34.7 ± 5.3 | 33.5 ± 4.3 | 0.40 |
| Gestational age, weeks | 38.9 ± 0.7 | 39.4 ± 0.8 | 0.045 |
| Mode of delivery | 0.77 | ||
| vaginal | 14 (74%) | 16 (70%) | |
| cesarean | 5 (26%) | 7 (30%) | |
| Sex of infant | 0.55 | ||
| male | 10 (53%) | 10 (43%) | |
| female | 9 (47%) | 13 (57%) | |
| Birth weight, kg | 3.47 ± 0.60 | 3.36 ± 0.44 | 0.50 |
Values are means ± SD. BMI: Body Mass Index (kg/m2). DM: diabetes
Results
Characteristics of Study Population
Demographics for participants providing placental samples are shown in Table 1. Compared with non-diabetic controls, those with diabetes had higher HbA1c, were older, and delivered slightly earlier (about 4 days) in gestation. Among the diabetic mothers, five were treated with glyburide, seven with insulin, one with metformin, and six with diet alone. The diabetic and control groups were no different with respect to ethnicity, body mass index, infant sex, birth weight, or mode of delivery.
Placental PGC-1α and TFAM expression is lower in female offspring
We initially compared placental PGC-1α/TFAM pathway and mitochondrial DNA content between male and female offspring of non-diabetic controls. Both PGC-1α and its downstream target TFAM [29] in female placentae were approximately 60% that in male placentae (Figure 1A and 1B). The level of AMPK phosphorylation (P-T172-AMPKα) was also lower in female placentae compared to males (Figure 1A and 1B). However, protein abundance of AMPKα1 (T-AMPKα1, Figure 1A and 1B) and mtDNA copy number (Figure 1C) were no different between placentae from females and males.
Figure 1.
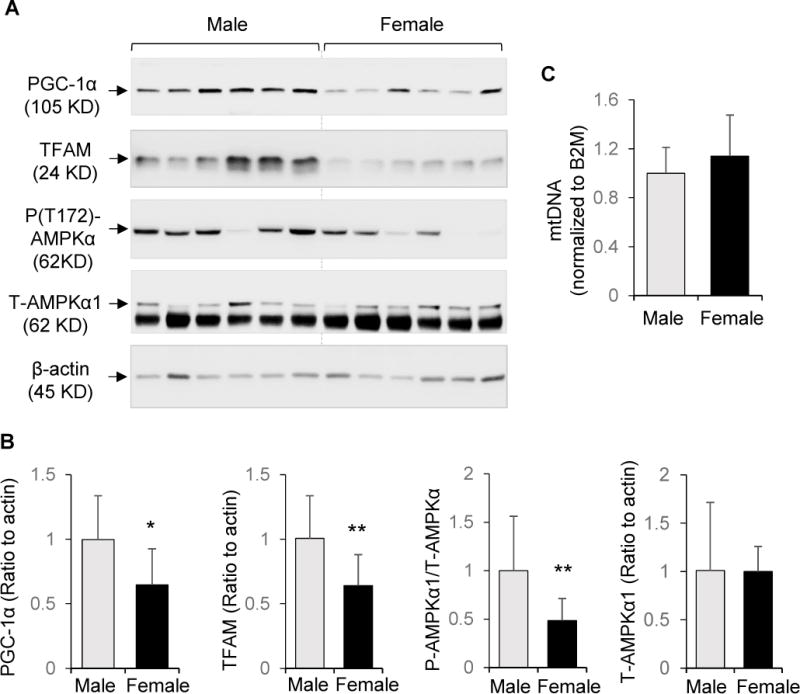
PGC-1α and TFAM protein abundance in placentae from controls. Protein extracts were subjected to Western blot analysis and DNA extracts were analyzed by quantitative RT-PCR. A: Representative blots from male and female offspring. B: Quantitation of PGC-1α, TFAM, P-(T172)-AMPKα, and total AMPKα1 (T-AMPKα1, mean ± SD) from Western blots, using β-actin as reference. C: Fold change of mitochondrial tRNALeu(UUR) gene DNA copy number normalized to nuclear β2 microglobin (B2M). For males, n=10. For females, n = 13. * P<0.05; ** P<0.01.
Maternal DM alters placental PGC-1α/TFAM and mitochondrial DNA content in a sex-dependent manner
We next examined the effects of maternal diabetes on indices of mitochondrial biogenesis. Overall, including values obtained from both male and female offspring, maternal diabetes was associated with a reduction in PGC-1α (by 38%, p<0.01) and TFAM (by 37%, p<0.01) abundance in placentae. After adjusting for the effects of each covariate listed in Table 1, the effect of diabetes on PGC-1α (p for diabetes effect < 0.02) and TFAM (p for diabetes effect < 0.007) remained significant regardless of covariate.
The effects of maternal diabetes were examined separately for each sex. In male offspring, PGC-1α (Figure 2A) and TFAM (Figure 2B) proteins, as well as mtDNA copy number (Figure 2C), were significantly lower in placentae of diabetic women than controls. Placental TFAM in males was highly correlated with PGC-1α (Figure 2D), and there was significant correlation between mitochondrial DNA copy number and TFAM protein abundance (Figure 2E). However, in female offspring, despite reduced placental PGC-1α abundance (Figure 3A), diabetes was not associated with a decrease in TFAM (Figure 3B), nor mtDNA copy number (Figure 3C). In addition, there was no significant association between placental PGC-1α and TFAM in females (Figure 3D), nor between mtDNA copy number and TFAM levels (Figure 3E). Additional analysis on the overall data of both males and females revealed that for TFAM, the diabetes × sex interaction term was significant (p = 0.0021), validating the differential effect of sex on the offsprings’ response to diabetes exposure.
Figure 2.
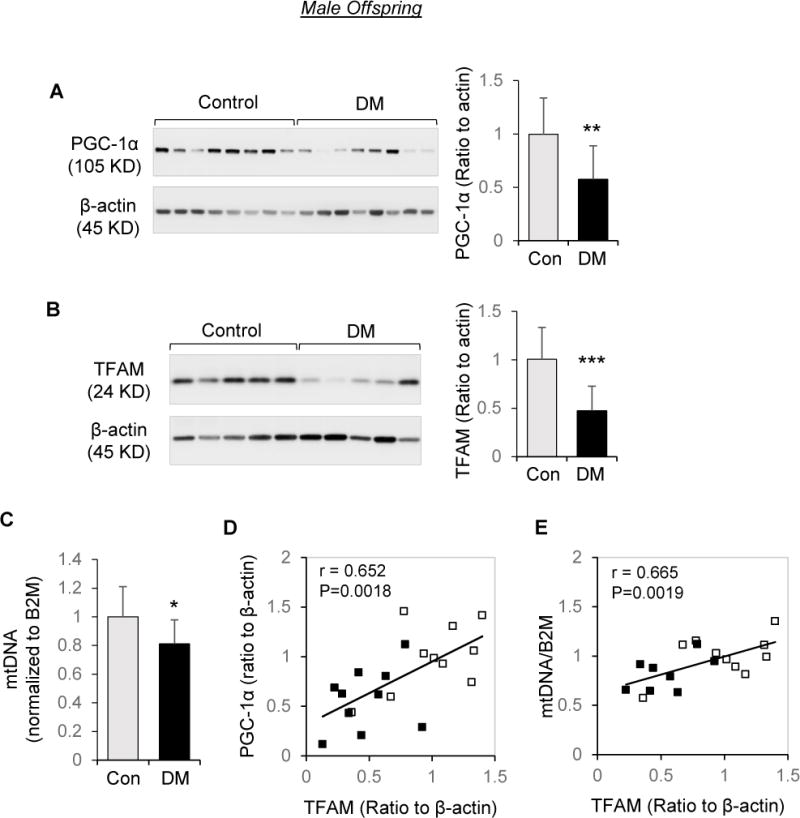
Effects of maternal diabetes on PGC-1α/TFAM/Mitochondrial DNA abundance in placentae of male offspring. Protein extracts subjected to Western blot analysis and DNA extracts were analyzed by quantitative RT-PCR. A and B: Representative Western blots and quantitation of PGC-1α (A) and TFAM (B) in male placentae of diabetic women (DM, n=10) and non-diabetic controls (n=10). C: Fold change of mitochondrial tRNALeu(UUR) gene DNA copy number normalized to β2-microglobin (B2M); n=9 for DM, n=10 for control. D: Correlation between PGC-1α and TFAM protein levels. E: Correlation between mtDNA copy number to TFAM protein levels. Open square: control group; Solid square: DM. Bar graphs are presented as mean ± SD, * P<0.05; ** P<0.01; *** P<0.001.
Figure 3.
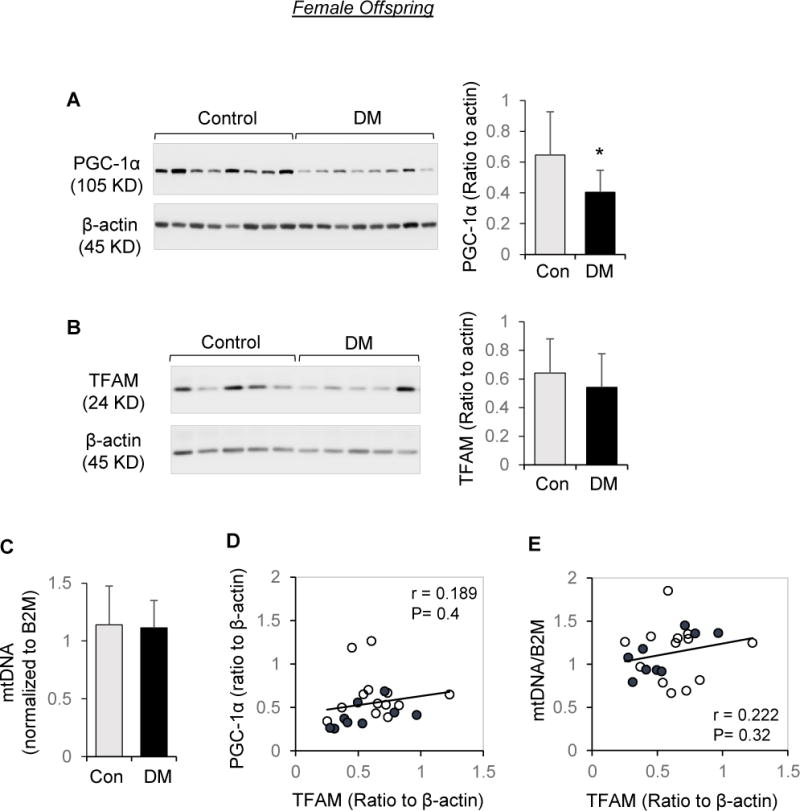
Effects of maternal diabetes on PGC-1α/TFAM/Mitochondrial DNA abundance in placentae of female offspring. Protein extracts subjected to Western blot analysis and DNA extracts were analyzed by quantitative RT-PCR. A, B, and C: Representative Western blots and quantitation of PGC-1α (A) and TFAM (B), and fold change of mitochondrial tRNALeu(UUR) gene DNA copy number normalized to β2-microglobin (C) in female placentae of diabetic pregnancies (DM, n=9) and non-diabetic controls (n=13). Bar graphs are presented as mean ± SD, * P<0.05. D: Correlation between PGC-1α and TFAM protein levels. E: Correlation between mtDNA copy number to TFAM protein levels. Open circle: control group; Solid circle: DM.
Negative correlation of maternal blood glucose concentrations with placental PGC-1α/TFAM and mitochondrial DNA content
The results of correlation analysis show that in pregnant women with male offspring, maternal fasting blood glucose concentration was inversely associated with placental PGC-1α (P<0.05) and TFAM (P<0.05) with statistical significance (Figure 4). There was a trend for negative relationship of maternal fasting blood glucose with placental mtDNA copy number (P=0.067).
Figure 4.
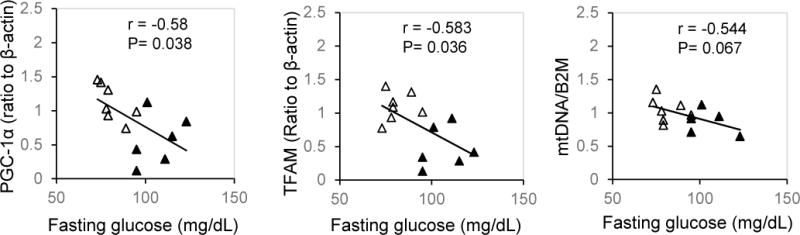
Relation between maternal fasting blood glucose concentrations with PGC-1α/TFAM/Mitochondrial DNA abundance in placentae of male offspring. Graphs show correlation of maternal fasting blood glucose concentrations with placental PGC-1α, TFAM protein levels, and mtDNA copy numbers. Open triangle: control group; Solid triangle: DM.
Discussion
The placenta requires sufficient mitochondrial metabolic activity to meet the high energy demands of gestation required for fetal growth and development [15, 20, 30]. As a key regulator of mitochondrial biogenesis and energy metabolism, PGC-1α is positioned to play a central role in optimizing placental function. In this study, we report that the abundance of PGC-1α in the placentae is reduced in offspring of diabetic women, but the effects on the downstream targets (TFAM and mitochondrial DNA content) are varied depending on offspring sex.
The critical roles of PGC-1α as a determinant of mitochondrial function and energy metabolism have been extensively demonstrated in adipose tissue, skeletal muscle, heart, neurons, and pancreatic β-cells [31, 32]. Much less is known about the regulation and function of PGC-1α in the placenta, especially in the context of maternal diabetes. The placenta is not only the conduit of oxygen to the fetal circulation but is also a significant consumer of oxygen [33]. With the decreased placental blood flow in maternal diabetes [34, 35] the placenta undergoes metabolic remodeling to reduce mitochondrial oxygen consumption in order to preserve oxygen delivery to the fetus [33]. In this regard, the reduction in placental PGC-1α abundance in response to diabetes we report is a plausible mechanism for such placental remodeling. Interestingly. Kelstrup et al [36] recently reported PGC-1α gene expression was 40% lower in skeletal muscle of adults born to mothers with GDM, supporting the ideas that the changes we find in placentae also occur in other offspring organs, likely as an important component of the long-term effects of prenatal exposure to diabetes.
PGC-1α stimulates mitochondrial biogenesis by increasing transcriptional activity of nuclear respiratory factor 1 which, in turn, increases expression of TFAM [29], which directly regulates mitochondrial DNA transcription and replication [37]. We found that maternal diabetes was associated with a significant decrease in TFAM protein levels in placentae of male offspring and was tightly correlated with PGC-1α expression and mitochondrial DNA copy number, consistent with the regulatory role of PGC-1α in TFAM expression and mitochondrial biogenesis. In contrast, although female offspring from diabetic women showed reduced placental PGC-1α, abundance of TFAM and mitochondrial DNA copy number were unaffected, revealing sexual dimorphism in the PGC-1α/TFAM/mitochondrial biogenesis system. This influence of offspring sex is additionally supported by our observation that PGC-1α and TFAM protein abundance, as well as AMPK activation status, were generally lower in female compared to male placentae.
The precise significance of the sex difference we report is unclear, however it is consistent with emerging evidence that the response of the fetus to environmental influence differs by sex [38]. Males born to mothers with GDM have higher cord blood levels of leptin and c-peptide [39] and increased adiposity both at birth [14] and at school age [40] compared to similarly exposed females. Sexual dimorphism of mitochondrial biogenesis has been demonstrated in the brain, cardiac muscle, liver, and brown adipose tissue of rodents [41, 42], but not previously reported in placenta in the context of maternal diabetes to our knowledge. Although speculative, the sex dependent effects on factors regulating mitochondrial abundance or function, if persistent, offer a potential mechanism that could explain subsequent changes in body composition or metabolic state.
Our findings are consistent with a recent study by Muralimanoharan S. et al [43], which also reported a reduction in placental PGC-1α content in the context of GDM. However, the number of subjects they examined was likely too small to detect any sex differences. These authors also reported reduced oxygen consumption by cultured trophoblast from mothers with GDM, which would be anticipated given our observations of reduced PGC-1α and TFAM. Our study also demonstrated a negative association of maternal blood glucose levels with PGC-1α and TFAM content in placentae of males, supporting the important role of intrauterine hyperglycemia in affecting placenta mitochondrial biogenesis pathway.
The strengths of our study are the examination of relevant human tissue susceptible to the influence of maternal diabetes in a population at increased risk for type 2 diabetes, the determination of correlation between protein levels of key factors of mitochondrial biogenesis and mitochondrial DNA content, and the detection of fetal sex differences. The limitations are the intrinsic variability in expression of placental proteins and the somewhat limited sample size that could obscure other important relationships, such as the effects of medication during pregnancy. Further studies about the potential effects of various aspects of maternal status (e.g. degree of dysglycemia, form of treatment) are warranted and would require studies of larger scope and different design.
In summary, we found that the placental PGC-1α/TFAM/mitochondrial biogenesis pathway is affected by maternal diabetes and offspring sex. The effect on PGC-1α plausibly contributes to impaired placental mitochondrial biogenesis that may ultimately affect long-term health and explain some of enhanced risk of future metabolic disease in males.
Highlights.
Placental PGC-1α and TFAM proteins are lower in female offspring than males.
Placental PGC-1α is reduced in both male and female offspring of diabetic women.
The effects of maternal diabetes on TFAM/mtDNA content are fetal-sex dependent.
Acknowledgments
We thank study coordinators Mary Ayn Tullier, Justin Fowler, Olufolake Olufowote and Shelly Hopper; the Choctaw Nation of Oklahoma and the Chickasaw Nation; and our study participants and families. We thank Drs. Kevin Short and David P Sparling for help and comments. Some data from this manuscript were presented at the American Diabetes Association 76th Scientific Sessions (2016).
Funding: This study was supported by NIH Grants R01 DK089034-03 (S. Chernausek, PI) and P20 MD000528-05 (T. Lyons, Project PI); American Diabetes Association Grant 1-10-CT-09 (S. Chernausek, PI); the CMRI Metabolic Research Program; and Harold Hamm Diabetes Center training grant (S. Jiang).
Abbreviations
- mtDNA
Mitochondrial DNA
- PGC-1α
Peroxisome proliferator-activated receptor-γ coactivator-1α
- TFAM
Mitochondria transcription factor A
- AMPK
AMP activated protein kinase
- GDM
Gestational diabetes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions:
All authors contribute to the conception, design and interpretation of the data. SJ, AMT, and JBT performed the experiments. SJ and SDC wrote the manuscript. CEA did the statistical analysis. All authors revised the manuscript and approved this version to be published.
Duality of interest:
There is no conflict of interest associated with this manuscript.
References
- 1.Fetita LS, Sobngwi E, Serradas P, Calvo F, Gautier JF. Consequences of fetal exposure to maternal diabetes in offspring. J Clin Endocrinol Metab. 2006;91(10):3718–24. doi: 10.1210/jc.2006-0624. [DOI] [PubMed] [Google Scholar]
- 2.Dabelea D, Crume T. Maternal environment and the transgenerational cycle of obesity and diabetes. Diabetes. 2011;60(7):1849–55. doi: 10.2337/db11-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Schmidt L, Damm P. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J Clin Endocrinol Metab. 2009;94(7):2464–70. doi: 10.1210/jc.2009-0305. [DOI] [PubMed] [Google Scholar]
- 4.Dabelea D, Mayer-Davis EJ, Lamichhane AP, D’Agostino RB, Jr, Liese AD, Vehik KS, Narayan KM, Zeitler P, Hamman RF. Association of intrauterine exposure to maternal diabetes and obesity with type 2 diabetes in youth: the SEARCH Case-Control Study. Diabetes Care. 2008;31(7):1422–6. doi: 10.2337/dc07-2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dabelea D, Hanson RL, Bennett PH, Roumain J, Knowler WC, Pettitt DJ. Increasing prevalence of Type II diabetes in American Indian children. Diabetologia. 1998;41(8):904–10. doi: 10.1007/s001250051006. [DOI] [PubMed] [Google Scholar]
- 6.Damm P, Houshmand-Oeregaard A, Kelstrup L, Lauenborg J, Mathiesen ER, Clausen TD. Gestational diabetes mellitus and long-term consequences for mother and offspring: a view from Denmark. Diabetologia. 2016;59(7):1396–9. doi: 10.1007/s00125-016-3985-5. [DOI] [PubMed] [Google Scholar]
- 7.Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, Roumain J, Bennett PH, Knowler WC. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000;49(12):2208–11. doi: 10.2337/diabetes.49.12.2208. [DOI] [PubMed] [Google Scholar]
- 8.Bush NC, Chandler-Laney PC, Rouse DJ, Granger WM, Oster RA, Gower BA. Higher maternal gestational glucose concentration is associated with lower offspring insulin sensitivity and altered beta-cell function. J Clin Endocrinol Metab. 2011;96(5):E803–9. doi: 10.1210/jc.2010-2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.H.S.C.R Group. Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study: associations with neonatal anthropometrics. Diabetes. 2009;58(2):453–9. doi: 10.2337/db08-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mele J, Muralimanoharan S, Maloyan A, Myatt L. Impaired mitochondrial function in human placenta with increased maternal adiposity, American journal of physiology. Endocrinology and metabolism. 2014;307(5):E419–25. doi: 10.1152/ajpendo.00025.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Damm P. High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes: the role of intrauterine hyperglycemia. Diabetes Care. 2008;31(2):340–6. doi: 10.2337/dc07-1596. [DOI] [PubMed] [Google Scholar]
- 12.Zhu Y, Olsen SF, Mendola P, Yeung EH, Vaag A, Bowers K, Liu A, Bao W, Li S, Madsen C, Grunnet LG, Granstrom C, Hansen S, Martin K, Chavarro JE, Hu FB, Langhoff-Roos J, Damm P, Zhang C. Growth and obesity through the first 7 y of life in association with levels of maternal glycemia during pregnancy: a prospective cohort study. Am J Clin Nutr. 2016;103(3):794–800. doi: 10.3945/ajcn.115.121780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Retnakaran R, Kramer CK, Ye C, Kew S, Hanley AJ, Connelly PW, Sermer M, Zinman B. Fetal sex and maternal risk of gestational diabetes mellitus: the impact of having a boy. Diabetes Care. 2015;38(5):844–51. doi: 10.2337/dc14-2551. [DOI] [PubMed] [Google Scholar]
- 14.Lingwood BE, Henry AM, d’Emden MC, Fullerton AM, Mortimer RH, Colditz PB, Le Cao KA, LK Callaway. Determinants of body fat in infants of women with gestational diabetes mellitus differ with fetal sex. Diabetes Care. 2011;34(12):2581–5. doi: 10.2337/dc11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gude NM, Roberts CT, Kalionis B, King RG. Growth and function of the normal human placenta. Thromb Res. 2004;114(5–6):397–407. doi: 10.1016/j.thromres.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 16.Visiedo F, Bugatto F, Sanchez V, Cozar-Castellano I, Bartha JL, Perdomo G. High glucose levels reduce fatty acid oxidation and increase triglyceride accumulation in human placenta, American journal of physiology. Endocrinology and metabolism. 2013;305(2):E205–12. doi: 10.1152/ajpendo.00032.2013. [DOI] [PubMed] [Google Scholar]
- 17.Meng Q, Shao L, Luo X, Mu Y, Xu W, Gao C, Gao L, Liu J, Cui Y. Ultrastructure of Placenta of Gravidas with Gestational Diabetes Mellitus. Obstetrics and gynecology international. 2015;2015:283124. doi: 10.1155/2015/283124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desoye G, Hauguel-de Mouzon S. The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care. 2007;30(Suppl 2):S120–6. doi: 10.2337/dc07-s203. [DOI] [PubMed] [Google Scholar]
- 19.Myatt L. Placental adaptive responses and fetal programming. The Journal of physiology. 2006;572(Pt 1):25–30. doi: 10.1113/jphysiol.2006.104968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jansson T, Powell TL. Role of placental nutrient sensing in developmental programming. Clin Obstet Gynecol. 2013;56(3):591–601. doi: 10.1097/GRF.0b013e3182993a2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenfeld CS. Sex-Specific Placental Responses in Fetal Development. Endocrinology. 2015;156(10):3422–34. doi: 10.1210/en.2015-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sood R, Zehnder JL, Druzin ML, Brown PO. Gene expression patterns in human placenta. Proc Natl Acad Sci U S A. 2006;103(14):5478–83. doi: 10.1073/pnas.0508035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tryggestad JB, Vishwanath A, Jiang S, Mallappa A, Teague AM, Takahashi Y, Thompson DM, Chernausek SD. Influence of Gestational Diabetes Mellitus on Human Umbilical Vein Endothelial Cell microRNA. Clin Sci (Lond) 2016 doi: 10.1042/CS20160305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang H, Ward WF. PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30(4):145–51. doi: 10.1152/advan.00052.2006. [DOI] [PubMed] [Google Scholar]
- 25.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98(1):115–24. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 26.Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teague AM, Fields DA, Aston CE, Short KR, Lyons TJ, Chernausek SD. Cord blood adipokines, neonatal anthropometrics and postnatal growth in offspring of Hispanic and Native American women with diabetes mellitus. Reprod Biol Endocrinol. 2015;13:68. doi: 10.1186/s12958-015-0061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.A American Diabetes, Gestational diabetes mellitus, Diabetes Care. 2003;26(Suppl 1):S103–5. doi: 10.2337/diacare.26.2007.s103. [DOI] [PubMed] [Google Scholar]
- 29.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79(2):208–17. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 30.Godfrey KM. The role of the placenta in fetal programming-a review. Placenta. 2002;23(Suppl A):S20–7. doi: 10.1053/plac.2002.0773. [DOI] [PubMed] [Google Scholar]
- 31.Chan MC, Arany Z. The many roles of PGC-1alpha in muscle–recent developments. Metabolism. 2014;63(4):441–51. doi: 10.1016/j.metabol.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–39. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 33.Murray AJ. Oxygen delivery and fetal-placental growth: beyond a question of supply and demand? Placenta. 2012;33(Suppl 2):e16–22. doi: 10.1016/j.placenta.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Eriksson UJ, Jansson L. Diabetes in pregnancy: decreased placental blood flow and disturbed fetal development in the rat. Pediatr Res. 1984;18(8):735–8. doi: 10.1203/00006450-198408000-00012. [DOI] [PubMed] [Google Scholar]
- 35.Vambergue A, Fajardy I. Consequences of gestational and pregestational diabetes on placental function and birth weight. World J Diabetes. 2011;2(11):196–203. doi: 10.4239/wjd.v2.i11.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelstrup L, Hjort L, Houshmand-Oeregaard A, Clausen TD, Hansen NS, Broholm C, Borch-Johnsen L, Mathiesen ER, Vaag AA, Damm P. Gene Expression and DNA Methylation of PPARGC1A in Muscle and Adipose Tissue From Adult Offspring of Women With Diabetes in Pregnancy. Diabetes. 2016;65(10):2900–10. doi: 10.2337/db16-0227. [DOI] [PubMed] [Google Scholar]
- 37.Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, Takio K, Hamasaki N, Kang D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003;31(6):1640–5. doi: 10.1093/nar/gkg251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clifton VL. Review: Sex and the human placenta: mediating differential strategies of fetal growth and survival, Placenta. 2010;31(Suppl):S33–9. doi: 10.1016/j.placenta.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 39.Oken E, Morton-Eggleston E, Rifas-Shiman SL, Switkowski KM, Hivert MF, Fleisch AF, Mantzoros C, Gillman MW. Sex-Specific Associations of Maternal Gestational Glycemia with Hormones in Umbilical Cord Blood at Delivery. Am J Perinatol. 2016;33(13):1273–1281. doi: 10.1055/s-0036-1586509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Regnault N, Gillman MW, Rifas-Shiman SL, Eggleston E, Oken E. Sex-specific associations of gestational glucose tolerance with childhood body composition. Diabetes Care. 2013;36(10):3045–53. doi: 10.2337/dc13-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colom B, Oliver J, Garcia-Palmer FJ. Sexual Dimorphism in the Alterations of Cardiac Muscle Mitochondrial Bioenergetics Associated to the Ageing Process. J Gerontol A Biol Sci Med Sci. 2015;70(11):1360–9. doi: 10.1093/gerona/glu014. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez-Cuenca S, Monjo M, Gianotti M, Proenza AM, Roca P. Expression of mitochondrial biogenesis-signaling factors in brown adipocytes is influenced specifically by 17beta-estradiol, testosterone, and progesterone. Am J Physiol Endocrinol Metab. 2007;292(1):E340–6. doi: 10.1152/ajpendo.00175.2006. [DOI] [PubMed] [Google Scholar]
- 43.Muralimanoharan S, Maloyan A, Myatt L. Mitochondrial function and glucose metabolism in the placenta with gestational diabetes mellitus: role of miR-143. Clin Sci (Lond) 2016;130(11):931–41. doi: 10.1042/CS20160076. [DOI] [PMC free article] [PubMed] [Google Scholar]