

Summary
Faithful DNA replication and accurate chromosome segregation are the key machineries of genetic transmission. Disruption of these processes represents a hallmark of cancer and often results from loss of tumor suppressors. PTEN is an important tumor suppressor frequently mutated or deleted in human cancer. Loss of PTEN has been associated with aneuploidy and poor prognosis in cancer patients. In mice, Pten deletion or mutation drives genomic instability and development of various tumors with high penetrance. PTEN deficiency induces DNA replication stress, confers stress tolerance, and disrupts mitotic spindle architecture, leading to accumulation of structural and numerical chromosome instability. Therefore, PTEN guards the genome by controlling multiple processes of chromosome inheritance. Here we summarize current studies that reveal the PTEN function in promoting high-fidelity transmission of genetic information. We also discuss the PTEN pathways of genome maintenance and highlight potential targets for cancer treatment.
Keywords: checkpoint, chromosome segregation, DNA replication, genome, mitotic spindle, PTEN
Graphical Abstract
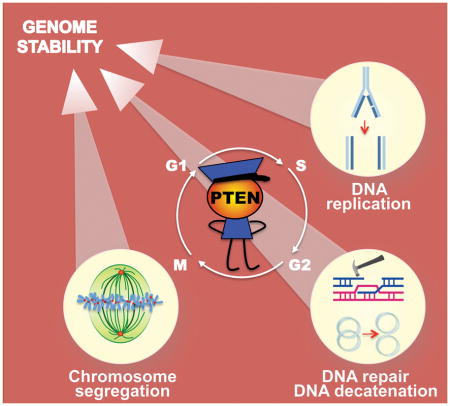
PTEN is a guardian of the genome. However, the role of PTEN in guarding the genome has not been revealed until recently. PTEN controls multiple fundamental processes of genomic transmission. By physically interacting with key molecules in DNA replication, DNA repair/decatenation, and chromosome segregation during cell cycle, PTEN maintains genomic stability.
Introduction
Preservation of genomic integrity requires accurate duplication and transmission of genetic information stored in DNA from one parent cell to two daughter cells during cell division. Execution of this fundamental process is challenging because DNA is highly susceptible to damage by intrinsic and extrinsic genotoxic factors, whereas accumulation of DNA damage leads to malignant transformation. Emerging evidence suggests that cell-intrinsic genotoxic stress, as compared to environmental insult, plays a more compelling role in driving genomic instability and cancer evolution. For example, a world-wide cancer etiological study revealed that random mistakes made during normal DNA replication not only are unavoidable but also play a major role in causing heritable mutations that drive cancer evolution [1]. In addition to genetic errors generated during DNA replication, aberrant chromosome segregation during mitosis is an important cause of chromosome instability (CIN).
CIN may or may not drive malignant transformation, which largely depends on the genetic context. For example, numerical CIN resulting from BubR1 insufficiency predisposes p53+/− mice to thymic lymphomas but had no impact on tumorigenesis in Rb+/− mice. More interestingly, BubR1 insufficiency even reduces the incidence of prostate intraepithelial neoplasia in Pten+/− mice [2]. These observations suggest distinct tumorgenesis outcomes may result from different genetic interactions in a tumor suppressor network. Nevertheless, a recent study demonstrated a direct causal relationship between aberrant chromosome inheritance and tumor development. Mitotic errors manifested by centrosome amplification promote aneuploidy and cause spontaneous tumorigenesis, even in the absence of additional genetic alterations [3].
To cope with the error-prone processes of DNA replication and chromosome segregation, multiple checkpoints have evolved to ensure the proper order, integrity, and fidelity of the major events of the cell cycle. For example, the mitotic spindle assembly checkpoint (SAC) monitors proper microtubule attachment to the kinetochore of each chromosome prior to initiating chromosome segregation. Enhanced mitotic surveillance by SAC reinforcement can protect genomic integrity against aneuploidy and prevent tumorigenesis [4]. Besides SAC, there are additional surveillance mechanisms such as the intra-S and G2 checkpoints that coordinate to monitor DNA replication and repair, restricting cells with damaged DNA from entering mitosis. Checkpoint defects thus allow uncontrolled cell cycle progression and CIN accumulation, which impairs genomic integrity and promotes malignant transformation.
PTEN is a phosphatase encoded by a tensin-homolog gene on chromosome 10 that has been found frequently deleted in human cancer [5, 6]. The primary substrate of PTEN phosphatase is phosphatidylinositol-3, 4, 5-trisphosphate (PIP3), a catalytic product of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) that phosphorylates the 3 position hydroxyl group of the inositol ring of phosphatidylinositol [7]. Due to its frequent loss in human cancer and its catalytic activity of inhibiting the oncogenic PI3K pathway, PTEN has been recognized as a bona fide tumor suppressor. Soon after the identification of PTEN as an antagonist of PI3K signaling, a number of studies revealed its function in regulating cell cycle progression, primarily by controlling the G1-S transition [8–10]. The regulation of G2-M progression by PTEN was reported later [11] and significantly, PTEN plays an essential role in activating the DNA damage checkpoint to prevent genetic instability [12].
Although the tumor suppressor function of PTEN has been mainly attributed to its antagonism of the PI3K pathway, it is increasingly clear that PTEN has additional PI3K-independent functions. In addition to its cytoplasmic localization, PTEN was also found in the nucleus [13] and alongside these observations, nuclear PTEN alone was reported to arrest cell cycle progression independent of the PI3K pathway [14–16]. Importantly, nuclear PTEN plays multiple roles in maintaining the structural integrity of chromosomes and promoting DNA repair in response to genotoxic stress [17, 18]. Moreover, PTEN is directly involved in the core machinery of genetic transmission by regulating both DNA replication [19–21] and chromosome segregation [22–24] (Figure 1). In this review, we will discuss how PTEN controls diverse signaling pathways and molecules to guard the genome.
Figure 1.

A schematic diagram that summarizes the multifaceted PTEN functions in the nucleus and the CIN consequences of PTEN loss.
PTEN is physically involved in the maintenance of chromosome structural integrity
Inaccurate chromosome transmission results in structural and numerical CIN, which has been manifested in cells lacking PTEN. An earlier report of our work described centromere breakage as a signature of CIN in PTEN-deficient cells, highlighting the importance of PTEN in maintaining the structural integrity of chromosomes [17] (Figure 2A). The study also demonstrated the physical association of PTEN with interphase centromeres and mapped the centromere-binding domain of PTEN to its carboxyl (C) terminus. C-terminal truncation limits the ability of PTEN to interact with the centromere protein CENP-C, however catalytically deficient PTEN remains active in centromere binding [17]. These data suggest a phosphatase-independent function of PTEN in protecting the structure of chromosomes.
Figure 2.

PTEN maintains chromosome integrity (A) and chromatin condensation (B). A: PTEN is associated with centromeric proteins such as CENP-C to protect centromere stability. Loss of PTEN causes centromeric breakage, manifested by centromeric and acentric fragments [17, 25]. B: PTEN interacts with histones and non-histone chromatin architectural proteins to maintain a condensed chromatin structure. Loss of PTEN results in disruption of chromatin anchorage of histone H1 and non-histone heterochromatin protein HP1α and induces histone H4 acetylation. Hyperacetylation of histone H4 impairs the interaction between PTEN and chromatin components and deteriorates chromatin decondensation, which constitutes a positive feedback loop [27].
To further demonstrate the importance of the PTEN C terminus in tumor suppression, we generated a mouse model of C-terminal Pten deletion (Pten+/ΔC) by knock-in of a point mutation originally identified in patients with Cowden syndrome, a disease associated with cancer susceptibility [25]. Despite the lack of the PTEN C-terminal region, the truncated form of PTEN retains its catalytic activity. Nevertheless, the intact phosphatase function of Pten fails to protect the genome against CIN in the absence of a single allele of the C-terminal region and as a result, Pten+/ΔC mice develop spontaneous tumors in multiple organs. Moreover, heterozygous deletion of the Pten C terminus induces accumulation of both structural and numerical CIN including prominent centromere breakage, the structural signature of CIN caused by PTEN deficiency. These results further demonstrate the phosphatase-independent activity of PTEN in maintaining chromosome integrity and reveal haploinsufficiency of Pten C-terminal function in guarding the genome and suppressing tumorigenesis. These findings also explain why nearly 40% of somatic PTEN mutations identified in sporadic cancer patients occur in the C-terminal region. In Cowden syndrome, germline PTEN truncation mutations comprise over 80% of the total mutations in this region [26]. These findings illustrate the importance of the C-terminal region of PTEN in mechanisms relevant to tumor suppression.
PTEN dysfunction induces genomic fragility and disrupts chromatin architecture
In addition to segmental chromosome breakage as a signature CIN resulting from Pten deficiency, C-terminal haploinsufficiency of Pten also causes genome-wide copy number alterations [25] and global chromatin decondensation [27, 28]. In particular, copy number changes found in various tumor-prone tissues from Pten+/ΔC mice mainly affect common fragile sites. For example, Pten+/ΔC tissues exhibit reduced copy numbers and abnormal transcripts of multiple fragile genes including Fhit and Wwox, due to altered splicing and translocation [25]. Both FHIT and WWOX are biomarkers of neoplastic transformation [29], deregulation of which in tissues lacking the Pten C terminus represents overall chromosome fragility. Indeed, Pten C terminal truncation induces transcriptome remodeling manifested by large-scale alterations of the global gene expression profile [27]. Interestingly, over 70% of all differentially expressed genes are upregulated and these include many oncogenes such as Kras and Braf. These data indicate overall activation of gene transcription when PTEN function is compromised, which is related to an aberrant chromatin environment [27].
Consistent with the physical association of PTEN with centromeres [17], PTEN has been found to co-localize with heterochromatin and to maintain chromatin compaction [27, 28] (Figure 2B). In the absence of PTEN or its C terminus, chromatin compaction is disrupted particularly in the heterochromatin regions where centromeres and telomeres are clustered. The maintenance of heterochromatin rigidity by PTEN relies on its functional interaction with histones and non-histone chromatin components such as heterochromatin protein 1 (HP1). PTEN utilizes its C-terminal region to physically associate with linker histone H1 to keep adjacent nucleosomes in close proximity, which is accompanied by recruitment of HP1α and a deacetylated histone H4. The dynamic equilibrium of chromatin condensation can be impaired by either PTEN mutations that affect its interaction with histone H1 or enhanced histone H4 acetylation at lysine 16. In the latter scenario, hyperacetylation of histone H4 at K16 disrupts the interaction between PTEN and histone H1, leading to HP1α dissociation and chromatin decondensation [27, 28] (Figure 2B). On the other hand, loss of PTEN or its C terminus induces histone H4 acetylation, which neutralizes the positive charge of histones and subsequently causes chromatin unfolding. As such, PTEN deficiency triggers a positive feedback loop between histone H4 hyperacetylation and histone H1 unloading to impair chromatin homeostasis leading to aberrant transcriptome reprogramming [27].
PTEN controls DNA replication to ensure faithful and efficient genome duplication
Loss of PTEN gives rise to multiple distinct phenotypes of DNA replication defects that include spontaneous elevation of replication stress, enhanced stress tolerance, bypass of the intra-S checkpoint, and failure of stalled fork recovery. Defective DNA replication in PTEN-deficient cells may result from compromised assembly of the replisome and DNA damage response machinery due to chromatin decondensation.
PTEN maintains a baseline level of replication fork progression
PTEN depletion reduces the level of multiple DNA replication and repair proteins on chromatin. These include the DNA sliding clamp in the basal replication machinery PCNA (proliferating cell nuclear antigen) [30], the checkpoint protein CHK1 (checkpoint kinase 1) [31], and the repair protein Rad51 [32]. Although these replication regulators function at different stages and serve for distinct purposes, depletion of PTEN causes a dramatic and consistent reduction of chromatin association of all three proteins [19]. The failure of recruiting important replication proteins to chromatin occurs in the absence of replication perturbation, which reflects a diminished repository of necessary components for the DNA replication machinery. As expected, DNA replication forks in PTEN-null cells progress less efficiently with an increased frequency of fork stalling, even in the absence of exogenous sources of replication stress [19]. These observations suggest that PTEN deficiency confers a genetically intrinsic perturbation of the baseline level of DNA replication, which represents an endogenous source of replication stress.
PTEN maintains the intra-S checkpoint integrity and activity
The intra-S checkpoint is among various DNA damage checkpoints that block cell cycle progression to allow repair of DNA damage before transition to the next phase [33]. Faithful and efficient DNA replication relies on timely assembly of the replisome as well as precise coordination of the helicase-mediated DNA unwinding and the polymerase-mediated DNA synthesis. As every nucleotide in the genome must be screened and duplicated during DNA replication, the replication fork is a sensitive detector of DNA damage. When stalled, the replication fork itself can activate the intra-S checkpoint. This occurs in response to DNA lesions that block DNA polymerases while the helicase continues to proceed with DNA unwinding ahead of the damage site [34]. The uncoupling between the polymerase and the helicase generates single stranded DNA (ssDNA) that is subsequently coated by replication protein A (RPA). Therefore, RPA-coated ssDNA serves as a common intermediate structure necessary for activation of the intra-S checkpoint [35, 36].
Elevated DNA replication stress and stress tolerance in PTEN-deficient cells
Spontaneous chromosome fragility and CIN in PTEN-null cells [17, 25] results in a reduction of baseline DNA replication progression but does not arrest cells in S phase [19]. These data could suggest either insufficient levels of DNA damage or an impaired intra-S checkpoint resulting from PTEN depletion. We found that in the presence of enhanced levels of replication stress induced by DNA polymerase inhibitor aphidicolin (APH), PTEN-null cells remain largely unresponsive while wild-type cells exhibit a prominent S-phase arrest [19]. Consequently, PTEN-null cells prematurely exit S phase and proceed to G2/M despite the presence of unreplicated DNA and elevated CIN. Therefore, loss of PTEN not only confers endogenous replication stress but also causes stress tolerance in response to exogenous replication perturbation, leading to bypass of the intra-S checkpoint and deleterious accumulation of CIN. Stress tolerance and checkpoint bypass manifested in PTEN-null cells appears to be a combined consequence of defective replication licensing and compromised replisome assembly.
Aberrant licensing of replication origins and impaired assembly of the replisome in cells lacking PTEN
During the cellular response to exogenous replication stress, PTEN depletion aggravates uncoupling of the DNA polymerase and helicase, likely by aberrantly promoting the activity of DNA helicases. MCM2 (minichromosome maintenance protein 2) is a key component of the pre-replication complex (pre-RC) that forms during licensing of eukaryotic DNA replication. MCM2 has been identified as a protein substrate of PTEN phosphatase and hyperphosphorylation of MCM2 is associated with aberrant licensing of DNA replication in PTEN-null cells [20]. Hyperactive helicase in PTEN-deficient cells may promote DNA unwinding despite hydroxyurea (HU)-induced depletion of dNTPs, the building blocks for fork progression. HU-mediated inhibition of DNA synthesis in PTEN-deficient cells is accompanied by unrestrained fork progression [20]. This may inevitably result in generation or expansion of ssDNA that requires the protection by RPA. However, the level of RPA associated with chromatin and replication forks is reduced in the absence of PTEN [21]. The reduction of RPA recruitment onto replication forks prevents timely formation of RPA-coated ssDNA, the common mediator for activation of the intra-S checkpoint. This may directly lead to a failure to recruit necessary components of the replisome such as PCNA and checkpoint proteins such as CHK1, demonstrating compromised intra-S checkpoint activity [19]. These findings collectively suggest that PTEN maintains the intra-S checkpoint integrity and activity by temporal and spatial coordination of replication licensing with replisome assembly.
PTEN promotes recovery of stalled replication forks
Uncoupling of MCM2-mediated origin licensing and RPA-mediated checkpoint priming not only impairs the intra-S checkpoint but may also prevent repair of stalled replication forks. Indeed, PTEN-deficient cells fail to resume DNA replication in response to exogenous perturbation, leading to detrimental accumulation of stalled forks [19]. The failure of stalled fork restart in PTEN-null cells is associated with compromised recruitment of Rad51 following acute inhibition of DNA synthesis by nucleotide depletion [19]. Rad51 is well known to function in homologous recombination (HR)-mediated double strand break (DSB) repair [37], which has been shown to contribute to PTEN-mediated genomic stability [17, 38, 39]. Nevertheless, HR-dependent DNA repair is unlikely responsible for Rad51-mediated PTEN function in promoting replication fork restart. Instead, Rad51 is recruited on stalled forks without forming discrete filamentous-like foci in a PTEN-dependent manner following acute replication stress. Hence, the PTEN-Rad51 pathway of promoting reactivation of stalled forks is consistent with a HR-independent stress response that requires formation of a Rad51-ssDNA complex [40]. Rad51 loading on ssDNA relies on the preceding RPA recruitment and RPA-mediated stabilization of ssDNA [41]. Depletion of PTEN results in impairment of RPA1 recruitment on DNA replication forks [21], which may occur more specifically on ssDNA at stalled forks. As such, even severe fork stalling in PTEN-deficient cells may not be able to trigger Rad51 loading without preceding ssDNA stabilization by RPA. Therefore, PTEN facilitates consecutive recruitment of stress-responsive proteins with diverse functions to promote recovery of stalled DNA replication forks in response to acute DNA replication stress.
PTEN ensures faithful duplication of the genome by controlling multiple fundamental mechanisms of DNA replication: 1) PTEN maintains normal unperturbed fork progression to avoid endogenous source of replication stress; 2) PTEN warrants functional coupling between helicase-mediated unwinding of DNA strands and polymerase-mediated DNA synthesis to reduce the risk of helicase-polymerase uncoupling and ssDNA exposure; 3) PTEN promotes timely recruitment of RPA1 for ssDNA protection; 4) PTEN interacts with Rad51 on nascent forks to promote stalled fork recovery (Figure 3). Thus, loss of PTEN results in spatial and temporal dissociation of replication fork behavior at various levels. For example, slower fork progression due to frequent fork stalling appears to conflict with an overall shorter S phase due to checkpoint bypass. A similar conflict is also evident between faster DNA unwinding and slower fork elongation, which is likely due to helicase-polymerase uncoupling. Therefore, PTEN deficiency confers a basal level of endogenous replication stress and impairment of stress-responsive checkpoint activity, resulting in premature S phase exit in the presence of unreplicated DNA.
Figure 3.

PTEN controls the DNA replication machinery. PTEN functions at the DNA replication fork to maintain a baseline level of fork progression in the absence of acute exogenous perturbation [19]. In response to replication stress, PTEN interacts with single-strand DNA (ssDNA) binding protein RPA1 to stabilize DNA replication forks [21] and recruits Rad51 to promote reactivation of stalled forks [19]. In addition, PTEN is required for the functionality of the intra-S checkpoint [19] and the MCM2-mediated coordination of DNA helicase with polymerase [20] to prevent uncontrolled DNA replication licensing and unscheduled G2/M transition.
PTEN controls the G2-M transition and mitotic fidelity to prevent CIN
Premature S phase exit inevitably rushes PTEN-deficient cells to enter G2/M despite incomplete DNA replication. This is an example of uncontrolled cell cycle progression, which is a hallmark of cancer. Unscheduled phase transition usually results from impairment of checkpoint function, and PTEN deficiency notably causes checkpoint dysfunction throughout all phases of the cell cycle. In G2 phase, Pten deficient cells exhibit a defective DNA damage checkpoint in response to radiation treatment [12]. In addition to regulating the G2-M transition, PTEN also directly controls mitotic surveillance mechanisms to ensure proper spindle assembly and high-fidelity chromosome segregation [22–24].
PTEN regulates multiple checkpoints during the G2-M transition
Acute replication stress fails to arrest PTEN-deficient cells in S phase and as a consequence, a significantly greater G2/M population is found in cells lacking PTEN as compared to wild-type cells [19]. As stalled forks may collapse and consequently result in DSB formation, these data seem to suggest that PTEN-deficient cells may retain a proficient DNA damage checkpoint to arrest cells in G2 and permit DSB repair. However, studies have indicated this is an incorrect prediction. Instead, a defective DNA damage checkpoint manifests in Pten-null embryonic stem cells in response to ionizing radiation (IR) [12]. The failure of checkpoint response to IR-induced DNA damage has been attributed to aberrant phosphorylation of CHK1 and resultant cytoplasmic sequestration and degradation. Impairment of the DNA damage checkpoint results in abnormal progression into mitosis with increased breaks and gaps in metaphase chromosomes, which is also associated with an elevated frequency of aneuploidy in PTEN-deficient breast carcinoma [12]. These observations demonstrate the critical role of PTEN in activation of the DNA damage checkpoint and the importance of PTEN-mediated G2 checkpoint activity for preventing CIN and aneuploidy.
In addition to regulating the DNA damage checkpoint, PTEN is involved in another G2 checkpoint named “decatenation checkpoint” to resolve DNA entanglements before initiating mitosis [42]. In comparison with the DNA damage checkpoint, the decatenation checkpoint has a closer intrinsic connection with replication stress because DNA decatenation aims to separate intertwined DNA strands that are formed mainly during DNA replication [43]. PTEN-deficient cells or tissues harbor increased common fragile sites [25] due to elevated DNA replication stress [19–21], which is detectable as ultrafine anaphase bridges (UFBs) in mitotic cells by immunofluorescence analysis with the polo-like kinase-interacting checkpoint helicase (PICH) [42]. UFBs are the characteristic structure arising from DNA replication stress and resultant chromosome fragility and DNA entanglements [44], representing a defective DNA decatenation checkpoint. UFBs occur spontaneously in a significantly greater number of PTEN-null cells as compared to wild-type cells, indicating an increase of endogenous DNA catenation stress [42]. Inhibition of topoisomerase II α (TOP2A), the major enzyme involved in DNA decatenation [43], further exacerbates PICH bridges in anaphase cells null for PTEN [42]. Mechanistic investigation reveals that PTEN promotes TOP2A stability to activate the DNA decatenation checkpoint and to allow elimination of entangled DNA prior to initiation of chromosome segregation. Dysfunction of this process leads to accumulation of entangled DNA due to topological constraints and decatenation checkpoint defects.
During the transition from DNA replication and chromosome segregation, PTEN controls multiple checkpoints to avoid unscheduled cell cycle progression with entangled DNA and unrepaired DNA damage. PTEN depletion confers defective surveillance of DNA lesions generated during both DNA replication and the G2-M transition, which poises an accumulative challenge to the substantial task of segregating the genome in mitosis.
PTEN maintains proper spindle architecture and mitotic fidelity
Impairment of multiple pre-mitotic checkpoints creates an unsecured fast track for cells with numerous DNA lesions to enter mitosis, vulnerable to mitotic stress and resultant segregation errors. In agreement with this prediction, emerging studies have reported various mitotic defects in PTEN-null cells: aberrant spindle assembly in prometaphase, chromosome misalignment and missegregation in metaphase, anaphase bridges and lagging chromosomes in anaphase, chromosome non-disjunction in telophase, and mitotic catastrophe [22–24, 45, 46]. At least some of these mitotic defects may stem from abnormal pre-mitotic processes such as incomplete or defective DNA replication, repair, and decatenation. Nevertheless, recent studies uncovered specific impairment of the core machinery that controls mitotic spindle assembly and chromosome segregation following PTEN depletion [22–24]. These findings suggest that PTEN directly controls the assembly and functionality of mitotic spindles to ensure timely and accurate alignment and segregation of mitotic chromosomes.
PI3K/AKT-dependent γ-tubulin/PLK1/PTEN feedback loop on the mitotic spindle
The mitotic spindle is a self-organized macro-molecular structure comprised of microtubules (MTs), MT-associated proteins, and motor proteins. Nucleation of MTs is required for mitotic spindle assembly, and centrosomes are the major site of MT nucleation. MTs nucleate to form mitotic spindle poles and generate antiparallel MT arrays that interact with motor proteins to support the bipolar configuration of mitotic spindles [47]. PTEN was first reported to localize at mitotic centrosomes in neonatal human epidermal keratinocytes, and its centrosome localization was shown to rely on AKT-mediated recruitment of γ-tubulin and polo-like kinase 1 (PLK1) [46]. Interestingly, knockdown of PTEN results in elevated whole-cell levels of γ-tubulin and PLK1, which leads to centrosome amplification. Moreover, AKT inhibition abrogates the increase of γ-tubulin and PLK1 in PTEN knockdown cells and reduces the number of cells with supernumerary centrosomes [46]. These data suggest that PTEN restrains an AKT-mediated cellular depository of γ-tubulin and PLK1 to prevent centrosome amplification, and on the other hand, the centrosome deposit of γ-tubulin and PLK1 is essential for recruiting PTEN to mitotic spindle poles. It is therefore possible that PTEN, γ-tubulin, and PLK1 constitute a signaling feedback loop at centrosomes to maintain the stability of mitotic spindle poles.
PI3K-independent formation of the centrosmal PLK1/PTEN/DLG1/EG5 complex
The function of PTEN at mitotic centrosomes has been recently elucidated in a greater detail [22]. PLK1-dependent PTEN association with centrosomes has been confirmed and this process requires the PLK1 catalytic activity that had been shown to phosphorylate the PTEN multisite phosphorylation region near its C-terminal tail [48, 49]. PLK1-dependent recruitment of PTEN to centrosomes creates a docking site for subsequent loading of additional proteins including DLG1 (discs large MAGUK scaffold protein 1) and EG5 (also known as KIF11, kinesin family member 11). DLG1 is a PDZ-domain protein that interacts with the PDZ-binding domain of the PTEN C-terminal tail, which is a prerequisite for loading of EG5, a motor protein essential for centrosome movement and bipolar spindle formation. Moreover, PTEN-mediated recruitment of the DLG1/EG5 complex also requires EG5 phosphorylation. The phosphorylation-dependent and PDZ-mediated formation of PLK1/PTEN/DLG1/EG5 complex on mitotic centrosomes plays a critical role in regulating spindle pole movement to establish symmetrical bipolar spindles. Importantly, the regulation of spindle pole motility by PTEN relies on its C-terminal PDZ-binding domain in a PI3K-independent manner. Deletion of the PDZ-binding domain from the Pten gene locus in mice results in a reduced interaction between Pten and Dlg1, as well as increased aneuploidy and tumor susceptibility [22]. These data demonstrate the importance of the PTEN PDZ-binding domain in promoting mitotic spindle motility and suppressing aneuploidy and cancer susceptibility.
PTEN in maintaining mitotic phosphorylation equilibrium and mitotic spindle architecture
In addition to utilizing the non-catalytic PDZ-binding domain to regulate the motility of mitotic spindles, PTEN also uses its phosphatase function to control the spindle architecture. Based on an immunofluorescence tracing of PTEN throughout the cell cycle, a dynamic localization profile of PTEN during mitosis has been uncovered. PTEN begins to appear around centrosomes in prophase, followed by enrichment in the separating centrosomes at prometaphase and spreading in the whole spindle region during metaphase. Subsequently, PTEN accumulates at the spindle crest in anaphase 1 and is found on both the spindle poles and the midbody in anaphase 2, followed by gathering in the cleavage furrow in telophase [23]. This PTEN localization profile during cell division largely overlaps with that of EG5, the MT plus end-directed MT motor. Mitotic spindle architectural analysis reveals a common phenotype of spindle shortening in cells expressing a catalytic deficient form of PTEN or a hyperphosphorylated form of EG5. Further mechanistic study demonstrates that PTEN maintains a proper length of mitotic spindles by physically interacting with EG5 and suppressing its phosphorylation [23]. EG5 phosphorylation is required for the formation of the PTEN-DLG1-EG5 complex on centrosomes [22], while an abnormal level of EG5 phosphorylation impairs PTEN-mediated maintenance of the mitotic spindle size [23]. These findings suggest that phosphorylation of important motor proteins such as EG5 must be precisely controlled to satisfy a dynamic equilibrium for proper spindle assembly, which is ensured by an optimal balance between mitotic kinases and phosphatases.
Spindle pole integrity controlled by multiple PTEN pathways
Besides the reduced size of mitotic spindles, fragmentation of spindle poles is an additional common phenotype of both PTEN deficiency and EG5 hyperphosphorylation [23]. A non-phosphorylatable form of EG5 can rescue pole fragmentation in PTEN-deficient cells, demonstrating that an aberrant elevation of EG5 phosphorylation is responsible for the impairment of pole integrity. Of note, the mobility of mitotic spindle poles requires EG5 phosphorylation [22], but an excessive level of EG5 phosphorylation disrupts the pole integrity. These seemingly conflicting observations highlight the importance of a precise control and optimal equilibrium of EG5 phosphorylation for maintaining spindle pole stability and promoting pole movement during chromosome segregation. Although PTEN deficiency or EG5 hyperphosphorylation can both lead to spindle pole fragmentation, this aberrant phenotype may not be attributed solely to the PTEN phosphatase function as catalytically deficient PTEN can also partially restore the stability of mitotic spindle poles [23]. It is possible that the maintenance of mitotic spindle pole integrity and stability requires multiple PTEN pathways including the AKT-γ-tubulin/PLK1-PTEN pathway [46] and the PTEN-EG5 or PLK1-PTEN-DLG1-EG5 pathway [22].
Proper chromosome disjunction and cytokinesis to avoid polyploidy: limiting PLK1 activity by PTEN
PLK1 is a versatile kinase that controls various mitotic functions during cell division, including spindle bipolarity, mitotic entry and exit, as well as cytokinesis [50]. In addition to the involvement in the AKT-γ-tubulin/PLK1-PTEN [46] and PLK1-PTEN-DLG1-EG5 [22] pathways of spindle pole regulation, PLK1 also serves as a PTEN target to control mitotic exit and cytokinesis [24]. Loss of PTEN results in elevated expression and phosphorylation levels of PLK1, with concomitant phenotypes of chromosome non-disjunction, cytokinesis failure, and polyploidy. Moreover, PTEN-null cells or cells with a phospho-mimicking form of PLK1 exhibit resistance to mitotic spindle poisons [24]. These observations support a hypothesis that PTEN may suppress PLK1-mediated defects of cell division and acquisition of a survival advantage. As expected, PTEN reduces PLK1 phosphorylation and phosphorylation-dependent protein stability to prevent chromosome non-disjunction and polyploidy. Additionally, chemical inhibition of PLK1 or ectopic expression of non-phosphorylatable PLK1 rescues PTEN-null cells from polyploidization and improves their sensitivity to anti-cancer drugs that target mitotic spindles [24]. These data establish a PTEN-PLK1 pathway in controlling mitotic exit to prevent polyploidy, which is distinct from the spindle pole regulatory pathways mediated by the AKT-γ-tubulin/PLK1-PTEN cascade or the PLK1-PTEN-DLG1-EG5 complex (Figure 4). Moreover, the identification of PLK1 and EG5 as signalling mediators of the PTEN mitotic pathway has profound implications for therapeutic targeting of deregulated PTEN signalling that occurs in many cancer patients.
Figure 4.

PTEN controls the mitotic machinery by maintaining normal architecture of the mitotic spindle and promoting high-fidelity chromosome alignment and segregation. In the absence of PTEN, aberrant phosphorylation of AKT mediates increased cellular repository of both γ-Tubulin and PLK1, leading to centrosome amplification [46]. Loss of PTEN phosphatase function results in hyperphosphorylation of PLK1 and EG5, which causes spindles shortening, spindle pole fragmentation, chromosome misalignment and non-disjunction, polyploidy, as well as mitotic catastrophe [23, 24]. Depletion of the PTEN C-terminal PDZ binding domain impairs the formation of a PLK1-PTEN-DLG1-EG5 complex on prometaphase centrosomes, leading to a reduction of centrosome movement [22]. Therefore, PTEN promotes mitotic spindle assembly and stability by facilitating the formation of a protein complex associated with centrosome maturation [22] and by suppressing phosphorylation of multiple proteins responsible for mitotic defects associated with PTEN loss [23, 24, 46].
Conclusions and outlook
Our understanding of PTEN as a guardian of the genome has grown significantly since its nuclear function in protecting chromosome integrity was uncovered 10 years ago [17, 51]. The recent discovery of multiple PTEN pathways that contribute to faithful DNA replication and chromosome segregation has provided profound insight into how cells rely on PTEN signaling to transmit their genetic material with fidelity and efficiency. Although distinct PTEN pathways exhibit functional preference, these pathways often converge and overlap in regulating critical processes of genetic transmission. For example, PTEN may integrate its functions in regulating both global chromatin structure and local fork activities to ensure faithful DNA replication. Similarly, proper spindle assembly and chromosome segregation may require both catalytic and non-catalytic activities of PTEN. Advances in imaging and big data technologies now allow us to track the chromosome behavior with high resolution and to systematically explore the protein interactome in the microenvironment of genetic transmission. Using such approaches should allow tracing each stage of phase transition and identifying new PTEN pathways and targets involved in controlling each transition stage. Structural CIN results primarily from non-mitotic defects whereas numerical CIN is generated mainly during mitosis. Nevertheless, both CIN types are tightly interconnected as one can lead to the other (Figure 1). Further efforts are demanded to decipher the relationship between structural and numerical CIN related to PTEN deficiency. Although we are starting to understand the multiple pathways and multifaceted roles of PTEN in maintaining genomic stability, the relative contribution of each pathway to the tumor suppressive function of PTEN remains to be defined and elucidated. Combined employment of mouse models and patient specimens should help interrogate how distinct PTEN pathways work in concert to ensure the fidelity and timely completion of genome transmission.
Acknowledgments
The authors would like to thank the National Institutes of Health grant (R01GM100478) and the Irma T. Hirschl/Monique Weill-Caulier Trust for funding the research in the Shen laboratory.
Abbreviations
- APH
aphidicolin
- CHK1
checkpoint kinase 1
- CIN
chromosome instability
- DLG1
discs large MAGUK scaffold protein 1
- DSB
double strand break
- HP1
heterochromatin protein 1
- HR
homologous recombination
- HU
hydroxyurea
- IR
ionizing radiation
- MCM2
minichromosome maintenance protein 2
- MT
microtubule
- PCNA
proliferating cell nuclear antigen
- PICH
polo-like kinase-interacting checkpoint helicase
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PIP3
phosphatidylinositol-3, 4, 5-trisphosphate
- PLK1
polo-like kinase 1
- Pre-RC
pre-replication complex
- RPA
replication protein A
- SAC
spindle assembly checkpoint
- ssDNA
single stranded DNA
- TOP2A
topoisomerase II α
- UFB
ultrafine anaphase bridge
Footnotes
The authors declare no conflicts of interest.
References
- 1.Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355:1330–4. doi: 10.1126/science.aaf9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell. 2009;16:475–86. doi: 10.1016/j.ccr.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine MS, Bakker B, Boeckx B, Moyett J, et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev Cell. 2017;40:313–22. e5. doi: 10.1016/j.devcel.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weaver RL, Limzerwala JF, Naylor RM, Jeganathan KB, et al. BubR1 alterations that reinforce mitotic surveillance act against aneuploidy and cancer. eLife. 2016;5 doi: 10.7554/eLife.16620. pii: e16620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Yen C, Liaw D, Podsypanina K, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 6.Steck PA, Pershouse MA, Jasser SA, Yung WK, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 7.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 8.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1999;96:2110–5. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheney IW, Neuteboom ST, Vaillancourt MT, Ramachandra M, et al. Adenovirus-mediated gene transfer of MMAC1/PTEN to glioblastoma cells inhibits S phase entry by the recruitment of p27Kip1 into cyclin E/CDK2 complexes. Cancer Res. 1999;59:2318–23. [PubMed] [Google Scholar]
- 10.Sun H, Lesche R, Li DM, Liliental J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci USA. 1999;96:6199–204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kandel ES, Skeen J, Majewski N, Di Cristofano A, et al. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol. 2002;22:7831–41. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puc J, Keniry M, Li HS, Pandita TK, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Ginn-Pease ME, Eng C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Res. 2003;63:282–6. [PubMed] [Google Scholar]
- 14.Chung JH, Ginn-Pease ME, Eng C. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res. 2005;65:4108–16. doi: 10.1158/0008-5472.CAN-05-0124. [DOI] [PubMed] [Google Scholar]
- 15.Liu JL, Sheng X, Hortobagyi ZK, Mao Z, et al. Nuclear PTEN-mediated growth suppression is independent of Akt down-regulation. Mol Cell Biol. 2005;25:6211–24. doi: 10.1128/MCB.25.14.6211-6224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung JH, Eng C. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res. 2005;65:8096–100. doi: 10.1158/0008-5472.CAN-05-1888. [DOI] [PubMed] [Google Scholar]
- 17.Shen WH, Balajee AS, Wang J, Wu H, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 18.Bassi C, Ho J, Srikumar T, Dowling RJ, et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341:395–9. doi: 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He J, Kang X, Yin Y, Chao KS, et al. PTEN regulates DNA replication progression and stalled fork recovery. Nat Commun. 2015;6:7620. doi: 10.1038/ncomms8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng J, Liang J, Li J, Li Y, et al. PTEN Controls the DNA Replication Process through MCM2 in Response to Replicative Stress. Cell Rep. 2015;13:1295–303. doi: 10.1016/j.celrep.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Wang G, Li Y, Wang P, Liang H, et al. PTEN regulates RPA1 and protects DNA replication forks. Cell Res. 2015;25:1189–204. doi: 10.1038/cr.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Ree JH, Nam HJ, Jeganathan KB, Kanakkanthara A, et al. Pten regulates spindle pole movement through Dlg1-mediated recruitment of Eg5 to centrosomes. Nat Cell Biol. 2016;18:814–21. doi: 10.1038/ncb3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He J, Zhang Z, Ouyang M, Yang F, et al. PTEN regulates EG5 to control spindle architecture and chromosome congression during mitosis. Nat Commun. 2016;7:12355. doi: 10.1038/ncomms12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Hou SQ, He J, Gu T, et al. PTEN regulates PLK1 and controls chromosomal stability during cell division. Cell Cycle. 2016;15:2476–85. doi: 10.1080/15384101.2016.1203493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Z, Huang C, He J, Lamb KL, et al. PTEN C-terminal deletion causes genomic instability and tumor development. Cell Rep. 2014;6:844–54. doi: 10.1016/j.celrep.2014.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bubien V, Bonnet F, Brouste V, Hoppe S, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. 2013;50:255–63. doi: 10.1136/jmedgenet-2012-101339. [DOI] [PubMed] [Google Scholar]
- 27.Chen ZH, Zhu M, Yang J, Liang H, et al. PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep. 2014;8:2003–14. doi: 10.1016/j.celrep.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong L, Govan JM, Evans EB, Dai H, et al. Nuclear PTEN tumor-suppressor functions through maintaining heterochromatin structure. Cell Cycle. 2015;14:2323–32. doi: 10.1080/15384101.2015.1044174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iliopoulos D, Guler G, Han SY, Druck T, et al. Roles of FHIT and WWOX fragile genes in cancer. Cancer Lett. 2006;232:27–36. doi: 10.1016/j.canlet.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 30.Indiani C, O’Donnell M. The replication clamp-loading machine at work in the three domains of life. Nat Rev Mol Cell Biol. 2006;7:751–61. doi: 10.1038/nrm2022. [DOI] [PubMed] [Google Scholar]
- 31.Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40:477–86. doi: 10.1093/nar/gkr697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Godin SK, Sullivan MR, Bernstein KA. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem Cell Biol. 2016;94:407–18. doi: 10.1139/bcb-2016-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iyer DR, Rhind N. The Intra-S Checkpoint Responses to DNA Damage. Genes. 2017;8:74. doi: 10.3390/genes8020074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cortez D. Unwind and slow down: checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 2005;19:1007–12. doi: 10.1101/gad.1316905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 36.Byun TS, Pacek M, Yee MC, Walter JC, et al. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–52. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol. 2011;22:898–905. doi: 10.1016/j.semcdb.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 38.McEllin B, Camacho CV, Mukherjee B, Hahm B, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70:5457–64. doi: 10.1158/0008-5472.CAN-09-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mukherjee A, Karmakar P. Attenuation of PTEN perturbs genomic stability via activation of Akt and down-regulation of Rad51 in human embryonic kidney cells. Mol Carcinog. 2013;52:611–8. doi: 10.1002/mc.21903. [DOI] [PubMed] [Google Scholar]
- 40.Petermann E, Orta ML, Issaeva N, Schultz N, et al. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruff P, Donnianni RA, Glancy E, Oh J, et al. RPA Stabilization of Single-Stranded DNA Is Critical for Break-Induced Replication. Cell Rep. 2016;17:3359–68. doi: 10.1016/j.celrep.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang X, Song C, Du X, Zhang C, et al. PTEN stabilizes TOP2A and regulates the DNA decatenation. Sci Rep. 2015;5:17873. doi: 10.1038/srep17873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broderick R, Niedzwiedz W. Sister chromatid decatenation: bridging the gaps in our knowledge. Cell Cycle. 2015;14:3040–4. doi: 10.1080/15384101.2015.1078039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Nielsen CF, Yao Q, Hickson ID. The origins and processing of ultra fine anaphase DNA bridges. Curr Opin Genet Dev. 2014;26:1–5. doi: 10.1016/j.gde.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Liu XS, Song B, Elzey BD, Ratliff TL, et al. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem. 2011;286:35795–800. doi: 10.1074/jbc.C111.269050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leonard MK, Hill NT, Bubulya PA, Kadakia MP. The PTEN-Akt pathway impacts the integrity and composition of mitotic centrosomes. Cell Cycle. 2013;12:1406–15. doi: 10.4161/cc.24516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prosser SL, Pelletier L. Mitotic spindle assembly in animal cells: a fine balancing act. Nat Rev Mol Cell Biol. 2017;18:187–201. doi: 10.1038/nrm.2016.162. [DOI] [PubMed] [Google Scholar]
- 48.Choi BH, Pagano M, Dai W. Plk1 protein phosphorylates phosphatase and tensin homolog (PTEN) and regulates its mitotic activity during the cell cycle. J Biol Chem. 2014;289:14066–74. doi: 10.1074/jbc.M114.558155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Li J, Bi P, Lu Y, et al. Plk1 phosphorylation of PTEN causes a tumor-promoting metabolic state. Mol Cell Biol. 2014;34:3642–61. doi: 10.1128/MCB.00814-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Combes G, Alharbi I, Braga LG, Elowe S. Playing polo during mitosis: PLK1 takes the lead. Oncogene. 2017 doi: 10.1038/onc.2017.113. in press. [DOI] [PubMed] [Google Scholar]
- 51.Kritikou E. PTEN - a new guardian of the genome. Nat Rev Mol Cell Biol. 2007;8:179. [Google Scholar]