

SUMMARY
The evolutionary success of parasitoid wasps, a highly diverse group of insects widely used in biocontrol, depends on a variety of life history strategies in conflict with those of their hosts [1]. Drosophila melanogaster is a natural host of parasitic wasps of the genus Leptopilina. Attack by L. boulardi (Lb), a specialist wasp to flies of the melanogaster group, activates NF-κB-mediated humoral and cellular immunity. Inflammatory blood cells mobilize and encapsulate Lb eggs and embryos [2–5]. L. heterotoma (Lh), a generalist wasp, kills larval blood cells and actively suppresses immune responses. Spiked virus-like particles (VLPs) in wasp venom have clearly been linked to its successful parasitism of Drosophila [6], but VLP composition and their biotic nature have remained mysterious. Our proteomics studies reveal that VLPs lack viral coat proteins but possess a pharmacopoeia of (a) eukaryotic vesicular transport system, (b) immunity, and (c) previously unknown proteins. These novel proteins distinguish Lh from Lb VLPs; notably, some proteins specific to Lh VLPs possess sequence similarities with bacterial secretion system proteins. Structure-informed analyses of an abundant Lh VLP surface/spike-tip protein, p40, reveal similarities to the needle-tip invasin proteins SipD/IpaD of Gram negative bacterial type 3 secretion systems that breach immune barriers and deliver virulence factors into mammalian cells. Our studies suggest that Lh VLPs represent a new class of extracellular organelles and share pathways for protein delivery with both eukaryotic microvesicles and bacterial surface secretion systems. Given their mixed prokaryotic/eukaryotic properties, we propose the term Mixed Strategy Extracellular Vesicles (MSEVs) to replace VLP.
Keywords: Drosophila, immune suppression, hemocytes, parasitoid wasps, venom, virus-like particles, secretion systems, T3 secretion system, proteomics, mixed strategy extracellular vesicles
Graphical abstract
RESULTS
Sister Leptopilina species produce different VLPs
Larvae of parasitic wasps of the Leptopilina genus feed on Drosophila larval host tissues, and eclose into free-living adults (Fig 1 A). The VLP-producing generalist/specialist L. heterotoma (Lh)/L. boulardi (Lb) wasps differ greatly in their infection of their natural host D. melanogaster, as seen in the anterior lobes of the fly larval lymph glands. Hematopoietic progenitors are housed in the gland’s medulla (marked by Dome-Meso-GFP) and mature hemocytes, in the cortex (GFP-negative, Fig 1 B). Lb 17 infection induces lamellocyte differentiation (Fig 1 C), while Lh 14 attack causes cell loss in both the medulla and the cortex, and few cells survive (Fig 1 D). The differences in the virulence of Lh versus Lb is attributed to differences in the VLPs produced by these wasps [7] and the mechanism of Lh-induced cell killing are not understood. VLPs from Lb 17 and Lh 14 differ in morphology; Lb 17 VLPs have fewer spikes and are somewhat larger than Lh VLPs (Fig 1 E, F) [8]. A peripheral membrane lipid bilayer (~ 10 nm) surrounds Lh VLPs, which lack the typical coat-like structure found in some viruses (Fig 1 G). Because of their key role in wasp parasitism of Drosophila spp. [4, 5], we hypothesized that differences in the composition of spiked particles produced in the venom of both wasps underlie these contrasting infection strategies.
Fig 1. Effects of wasp attack on host lymph glands and comparison of VLP morphologies.
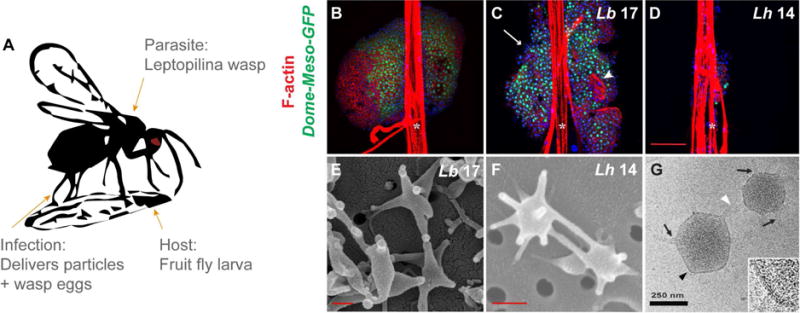
(A) Infection by female Leptopilina spp. parasitic wasps introduces not only wasp eggs into the body cavities and hemolymph of fruit fly larvae, but also venom gland products which includes spiked, 300-nm VLPs. VLP bioactivity is known to be necessary for the infective success of L. heterotoma, rather than other venom constituents [6, 9]. (B) Intact anterior lymph gland lobes from uninfected control Dome-MESO-GFP fly larvae. GFP marks the stem-like progenitors in the medulla. (C) Dome-MESO-GFP glands of Lb 17-infected larvae show lamellocyte differentiation (white arrowhead) and lobe dispersal (white arrow). (D) Progenitors are depleted in Dome-MESO-GFP anterior lobes infected with Lh 14. (B – D) White asterisks mark dorsal vessels. (E) Scanning EMs of Lb 17 and (F) Lh 14 VLPs. (G) CryoEM of Lh 14 VLPs: The external lipid bilayer is contiguous, extending from spike bases (black arrows) at the VLP core to spike tips (white arrowhead). The black arrowhead marks area in zoom, bottom right.
To characterize Lh VLP proteomes and examine differences in VLP protein compositions fundamental to Lh- versus Lb-specific virulence, we identified a high-confidence proteomic dataset common to VLPs from two independently-isolated, isogenized strains (Lh 14 and Lh NY), whose fine structures and activities on host cells are indistinguishable [3, 9]. Peptide sequences from each VLP proteome (Lh 14 and Lh NY wasps) were first aligned against RNA-Seq Lh 14 transcripts [10], translated to open reading frames (ORFs). We thus obtained a common set of Lh VLP proteins (present in both proteomes; Tables S1, S2) and verified these VLP protein sequences at > 90% identity against two Lh expressed sequence tags (ESTs; ~ 30 proteins or 20% [11] and ~ 70 proteins or 45% [12] of the common proteome). To identify candidate pathogenicity effectors, we compared the common Lh VLP dataset to abdominal transcripts of Lb 17 and a distantly-related species Ganaspis sp.1 (G1) that lacks spiked VLPs [10, 11, 13] (Fig 2). A summary of our major findings follows.
Fig 2.
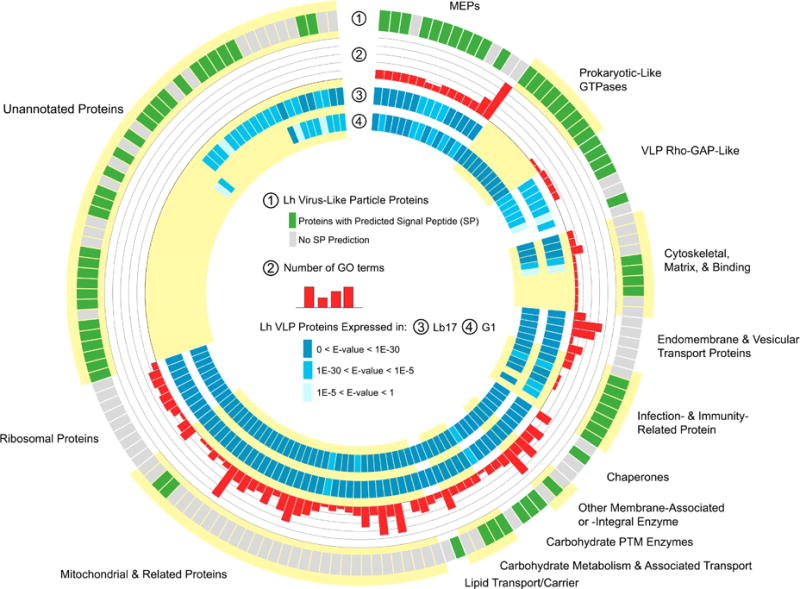
Lh VLP proteome
Lh VLP proteins are arranged by known/predicted functions and annotations. Key provided in center of figure. (Layer 1, outer most layer) Signal peptide predictions are most commonly found in the categories of virulence, immunity, and novel proteins. (Layer 2) GO (gene ontology) terms for conserved cell biology proteins are abundant. (Layers 3, 4) The cytoskeletal/fibronectin proteins and the majority of novel sequences lack similarity to abdominal transcripts from both (Layer 3) Lb 17 or (Layer 4) G1 female wasps. (See also Tables S1, S2; Data S1, S2.)
Lh VLPs are rich in eukaryotic microvesicular proteins
No proteins with significant homology to structural proteins of any known virus, including polydnaviruses (PDVs) associated with ichneumonid and braconid wasps, which prey on Coleoptera, Hymenoptera and Lepidoptera [14], were found in the Lh proteome. The 161 proteins common to Lh 14 and Lh NY VLP proteomes were categorized as (1) conserved eukaryotic proteins with core biological function (42%, Class 1); (2) virulence/immunity-associated proteins (24%, Class 2); or (3) novel sequences (34%, Class 3) (Figs 2, 3 A). Of the ~ 160 VLP proteins, 25% are Lh-specific (i.e., they are not expressed by Lb 17 [10]) and most (27/41, 66%) of these proteins are novel (Class 3) (Fig 2). Class 1 sequences contain orthologs of Drosophila and mammalian extra- and intracellular vesicle (including microvesicle and exosome) components as well as membrane proteins (Fig 3 A – C). The presence of transmembrane (e.g., Na/K pump, SERCA calcium pump) and vesicle transport proteins (e.g., H+-ATPase, heat shock cognate 70, Rab proteins, and soluble NSF attachment protein receptor) (Figs 2, 3 A – C) in the proteome suggest that Lh VLPs are not viral but instead share functional properties with eukaryotic extracellular organelles called microvesicles, produced by animals cells, and specialized to transfer proteins between different cell types [15].
Fig 3. Lh VLPs are enriched in microvesicular/exosomal and membrane-associated proteins.
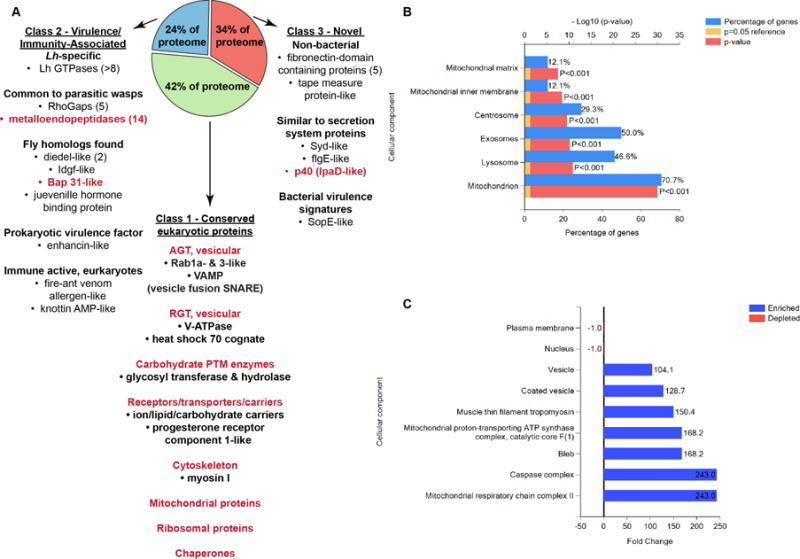
(A) Select example Lh VLP proteins, many of which are expected to be membrane-associated via integral or other biochemical mechanisms, are displayed by their proteomic Classes 1 – 3 (bulleted within descriptive subclasses). Example subclasses and individual proteins found in enrichment analyses (B, C) are shown in red. AGT = anterograde transport; RGT = retrograde transport. (B, C) Enrichments from Vesiclepedia: The organelle character of Lh VLPs based on GO Terms of predicted orthologs is (B) significant and (C) highly enriched. (B) Among VLP proteins with annotated orthologs, 71% are mitochondrial, of which 12% are localized to the mitochondrial inner membrane. Approximately 50% of conserved sequences in the proteome are common to microvesicles/exosomes. (C) Vesicular and mitochondrial, including that of the caspase complex, terms are the most over-represented. Furthermore, genes within the GO Term (GO:0008303) for pro-apoptotic caspase complexes were more than 200 times over-represented. (See also Tables S1, S2.)
Diverse pathogenicity mechanisms are housed in VLPs
Candidate immune-modulating (Class 2) VLP proteins include two diedel-like proteins with high similarities to sequences from insect viruses (60 and 62% similarity to NP_059254.1, Xestia c-nigrum granulovirus; Fig 2; Data S1). Interestingly, a Drosophila diedel modulates the IMD/NF-κB-dependent antimicrobial cascade [16] and the VLP diedel proteins may similarly suppress host signaling. An Lh VLP enhancin-like protein shows similarity to Yersinia spp. enhancins (42% similarity to WP_012413443.1, Yersinia pseudotuberculosis; Fig 2; Data S1), although enhancins are also found in insect viruses [17]. Additional Class 2 immunity and development proteins include: (1) imaginal disc growth factor 4-like sequence (Idgf4) (83% similarity to XP_008560038.1, Microplitis demolitor); (2) fire ant (Solenopsis)-derived venom allergen (62% similarity to XP_008560038.1, Nasonia vitripennis); and (3) B-cell receptor-associated protein 31 (89% similarity to XP_008554920.1, Microplitis demolitor) (Fig 2; Data S1). Two VLP proteins may protect and regulate parasite development: an antimicrobial/antifungal-like knottin protein (46% similarity to XP_014233229.1, Trichogramma pretiosum) and a predicted hemolymph juvenile hormone binding protein (56% similarity to ABV82429.1, Drosophila melanogaster) (Fig 2; Data S1) [11].
In the Class 2 set, we also identified two families of invertebrate immunity proteins (Figs 2, 3 A). At least 6 Lh RhoGAPs were found that, like Lb GAP of Lb [18], may inhibit parasite encapsulation. A group of 14 metalloendopeptidases (MEPs) were also identified in the proteome and, although they are structurally similar to proteins from diverse kingdoms, their virulence functions may be similar to those of MEPs from parasitic wasps Venturia canescens [19] and Nasonia vitripennis [20]. The diversity of predicted activities of Class 2 proteins likely facilitates Lh success across a broad range of Drosophila spp.
The abundance of novel proteins in Class 3 of the proteome was intriguing. Domain identifications predict viral domains (Pox L5 (PF04872), Baculo_PEP_C (PF04513.10), and Baculo_11_kDa (PF06143.9)) in three VLP sequences [21, 22]. One of these, Baculo_11_kDa VLP sequence also shows similarity to phage tail tape measure proteins (data not shown). In addition to the multiple gene families in Lh VLPs that are common to Lb and G1 (e.g., RhoGAPs and MEPs; Figs 2, 3 A), Lh-specific gene families include fibronectin domain-containing sequences and a new family of GTPases (Figs 2, 3 A; Data S2). Multiple members of gene families are expressed in wasp venoms [23–25], and identification of the gene families in Lh VLPs suggests that gene duplications and neofunctionalization underlie the powerful virulence strategy of Lh.
Novel endomembrane-active GTPases
Because the novel GTPase peptides are of high abundance in the Lh VLP proteomes and are absent from the Lb abdominal transcriptome, we investigated their predicted structures and functions in detail. All of the three small (SmGTPase) and five large GTPase (LgGTPase) sequences have N-terminal signals for secretion as well as key residues for GTP hydrolysis (Fig 4 A – C; Data S2). Five of the 8 (small and large) GTPase family members possess prokaryotic domains present in eubacterial and/or archaeal (e.g., PF09488, Fig 4 A, A′) proteins. Beyond a few proteins from parasitic wasps (N. vitripennis, G1, and L. clavipes), the closest putative homologs of these GTPases are prokaryotic (Fig 4 B; Data S2).
Fig 4. Structural characteristics of prokaryote-like Lh VLP GTPases and p40.
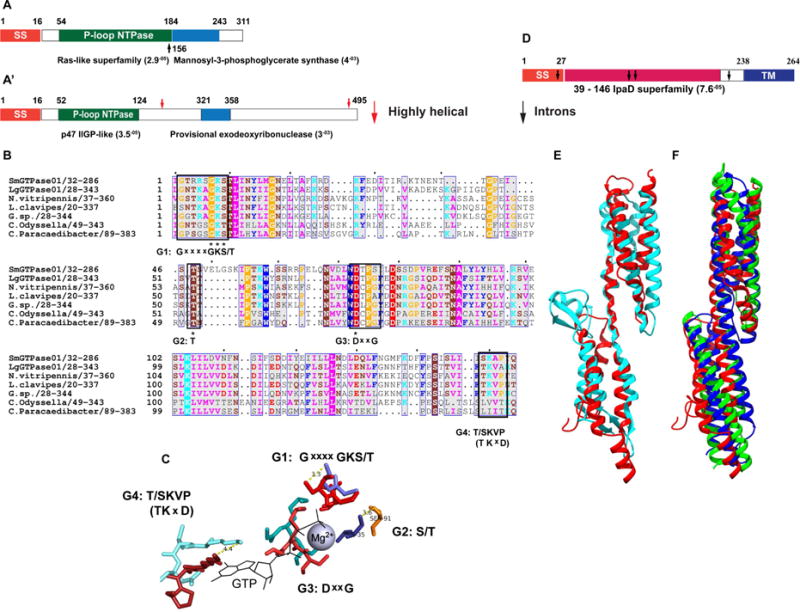
(A, A′) Domain architectures of representative SmGTPase01 (A) and LgGTPase01 (A′) based on Conserved Domains Database (CDD) and PFAM 26.0 (see Methods). (A, A′) SS = signal sequence. Starts/stops are labeled with residue number. The E-values based on CDD domain predictions are listed adjacent to domains. Black and red arrows mark overlapping domain predictions in SmGTPase01 (A) and a highly helical region in LgGTPase0 (A′), respectively. (B) A multi-sequence alignment (MSA) of Sm & LgGTPase01 (SmGTPase01 used as query) reveals that the most significantly similar sequences in the NCBI nr and TSA databases (Lh ESTs excluded) are both prokaryotic and eukaryotic (N = Nasonia; C = Candidatus). Four predicted active site G motifs are labeled below the conserved consensus residues (black boxes) in the MSA. Only the G4 consensus motif ((T/S)KVP) differs from the canonical Ras G4 motif (NKxD) [40]. Asterisks mark 100% conservation in the motifs. The coloring scheme is according to conventional physiochemical properties and sequence conservation. 100% and 99 – 50% conservation levels are indicated by white lettering and blue column boxes, respectively. (C) The predicted geometry of the G motifs in of SmGTPase01 active site (warm, orange tones) superimposed on that of HRas active site (1QRA; cool, blue tones). RMSD = 3.37 Å (calculation is based on the full-length structures and is normalized to 1QRA), TM-score = 0.74 [TM-Score > 0.5 indicates the same fold]. Distances (Å) between functionally critical residues of SmGTPase01 and HRas are indicated by dotted lines. (D) p40 domain architecture. SS = signal sequence; TM = transmembrane domain. Black arrows mark intron insertion sites. Based on CDD prediction, the central domain shares sequence and structural similarity with IpaD superfamily proteins. (E) Structural superposition of IpaD (blue, 2J0; residues 39-284) and p40 model (red, residues 28-187). The N-termini are oriented to the top right corner. The predicted signal sequence and C-terminal transmembrane helix were omitted for modeling. RMSD = 4.73 Å, TM-score = 0.56225. (F) Structural superposition of p40 model (red) to chicken spectrin (green, 1CUN; RMSD = 2.9 Å) and to human plectin (blue, 3PDY; RMSD = 3.0 Å), using the DALI server. (See also Fig S1, Data S2.)
The predicted active site of a representative SmGTPase (SmGTPase01) coordinates GTP and the NTPase cofactor, Mg2+. Close alignment of the predicted SmGTPase01 active site with the active site structure of HRas, the canonical small GTPase, supports the domain analyses results (Fig 4 A, A′, & C). The large GTPases are predicted to fold into C-terminal coiled-coils (Fig 4 A′). These findings suggest a curious blend of prokaryotic and eukaryotic properties within this new family of Lh VLP proteins, which likely have GTPase enzymatic activity and are likely secreted from wasp cells for incorporation into vesicles.
T3SS-like VLP proteins: Similarities between VLP p40 and bacterial SipD/IpaD
Prokaryotic protein motifs were identified in nearly 10% of novel Class 3 sequences. Overlapping protein motifs, [Bacillus PF05103; fungal PF15577] associated with cell division and microtubule binding, respectively, were identified in a single Class 3 protein. KEGG Mapper BlastKAOLA identified (a) Syd-like (SecY-interacting, Type 2 secretion systems) and (b) flgE-like (bacterial flagellar hook) proteins with low-to-mid scores. A sopE-like (bacterial GEF toxin) protein was also found. The presence of bacterial secretion system and flagellar proteins is especially interesting as these macromolecular assemblies are structurally and functionally related and the Type 3 secretion system (T3SS) of the self-assembling bacterial flagella are thought to be ancestral to the ones found in the needle/injectisome of pathogens [26].
Our previous antibody staining and inhibition studies uncovered an abundant 40 kDa surface protein of Lh VLPs (“p40”) which is necessary for lamellocyte lysis [9]. Early in VLP biogenesis, p40 is associated with the membranes of canals that emanate from the cytoplasm of secretory cells of the venom gland, where it is synthesized. Once in the canal lumen, p40 is then associated with membranous vesicles that are released from secretory cells. The vesicles mature into spiked VLPs which carry p40 both on their surfaces and spikes [9, 27]. The bacterial T3SS domain from IpaD/SipD/BipD (PRK15330, E = 7.60−05) proteins was found in residues 39-146 of p40 (Fig 4 D). This assignment was made by peptide mapping, cloning and expressing polyhistidine-tagged p40 in bacteria. In western blot experiments, anti-p40 antibody recognized this bacterially-expressed protein (Fig S1 A). As expected, p40 is detected in wasp venom extracts (Fig S1 B). We were unable to identify a putative Lb 17 or G1 p40 ortholog (Fig 2) and, to our knowledge, p40 represents the first eukaryotic protein with an IpaD/SipD-like domain.
IpaD-like proteins from Shigella/Salmonella/Burkholderia spp. are tip proteins of T3SS injectisomes, mediating contact and regulated delivery of effectors into the host cytoplasm of non-phagocytic cells [28]. IpaD expression in mammalian macrophages triggers apoptosis [29], reminiscent of the TUNEL-positive death of fly macrophages upon Lh infection [7]. Unlike the bacterial proteins, p40 is predicted to encode a C-terminal transmembrane helix in addition to an N-terminal secretion signal (Figs 4 D; S1 C). p40’s transmembrane domain (this study) and its extracellular localization in venom gland canals [27] suggest that p40 exits venom gland secretory cells in association with microvesicle-like structures. This interpretation is in agreement with the extracellular vesicular proteomic profile of VLPs (Fig 3).
Given the unexpected parallels in their structures and surface/tip localizations, we hypothesized that, like IpaD/SipD on the T3SS injectisome, p40 on VLP spikes facilitates invasive contact with the plasma membrane of non-phagocytic lamellocytes to deliver VLP contents. To test this idea, we carried out ab initio modeling of p40. The knowledge-based energetics of the p40 model are similar to crystal structures of similar length (ProSA Z-Score = −6.23). 82% of model residues are found in expected local environments (3D Verify). Superimposition of the p40 model against IpaD confirmed the T3SS protein-like fold in p40 (Figs 4 E; S1 C) [30].
Surprisingly, high-scoring matches to this fold included the vertebrate actin-binding proteins spectrin and plectin (Fig 4 F, superimposed with p40), further strengthening structural parallels between p40 and IpaD/SipD. Searches for the most similar structures within the N-terminal half of IpaD family proteins also returned actin-binding proteins (talin, vinculin, α-catenin) [31]. These proteins are known to reprogram the actin cytoskeleton leading to the profuse membrane ruffling observed in non-phagocytic mammalian cell invasions by Salmonella and Shigella [31].
DISCUSSION
The composition of Lh VLPs is complex and interesting in multiple respects but the most conspicuous observations are an absence of viral structural proteins and the presence of conserved eukaryotic proteins with microvesicular signature. Abundant Lh-unique proteins, including currently novel proteins, have an unexpected diversity of domains, especially those previously found exclusively in prokaryotic proteins. The mechanisms that contributed to the evolution of VLP proteins (horizontal gene transfer or others [32]) remain unknown.
Lh VLPs lack the defined symmetry and external coat found in many true viruses including PDVs. Reminiscent of eukaryotic organelles, precursors and mature VLPs exhibit heterogeneity in their shapes, sizes, and spike numbers [9, 33]. Moreover, it is noteworthy that, unlike PDVs which are fully formed in the cells of their origin and then released by lysis or budding [14], Leptopilina VLPs assume their final shape outside the cells in which at least some of their proteins and vesicular constituents are synthesized [9, 27]. Also, there is currently no evidence for the presence of nucleic acids in VLPs, which further distinguishes them from DNA-containing PDVs.
VLPs are unlike endosymbiotic bacteria of Leptopilina wasps [34]. Antibiotic-treatment of L. victoriae (sister species of Lh that make VLPs and carry cytoplasmic-incompatibility-inducing Wolbachia) did not affect genomic amplification of p40 or SmGTPase01 genes. Furthermore, VLP gene loci were amplified not only from female Lh 14 genomes but also male wasp genomes, even though VLPs are not produced in males. BrdU incorporation studies did not support the possibility of DNA-based VLP replication in the venom gland (our unpublished results). The non-replicating nature of the particles, genomic encoding of VLP protein genes, and vesicular signature of their proteome strongly suggest that Lh VLPs are neither viruses nor endosymbiotic bacteria but instead represent a new class of genomically-encoded, microvesicle-like organelles. Extracellular vesicles are produced by prokaryotic and eukaryotic cells and VLPs carry a diversity of potential immune-suppressive proteins. We, thus, propose the alternative moniker, MSEV (Mixed Strategy Extracellular Vesicles), to replace the VLP term.
Virulence factors of parasitic wasps have diversified in response to the variety and complexities of their hosts’ immune systems. With a broad host range [3], Lh wasps parasitize many Drosophila spp. whose own distinct immune responses are supported by varying numbers and types of blood cells [35]. This may explain, first, the diversity of putative virulence and cell death proteins with homologs across the biological kingdoms [viral (diedel), bacterial (p40, Syd-like), and eukaryotic (fire ant allergen, B-cell receptor-associated protein 31, etc.)], and second, the presence of MSEV paralogs [Lh-specific GTPase and Lh-/Lb-common Rho GAP, MEPs, and diedel gene families] in the proteome. Multiple members presumably perform redundant or overlapping cell-specific functions for rapid and robust immune suppression, much like the IκB/Cactus-like ankyrin-repeat proteins of distantly-related bracoviral and ichnoviral PDV proteins that block NF-κB signaling [36, 37].
The parallels between the 3D-structure and locations of p40 with the well-characterized T3SS IpaD-like prokaryotic proteins are provocative and suggest that p40 likely contributes to Lh MSEVs’ unique blood cell-killing activities. T3SS assemblies are widespread and are used by bacteria to infect plants and animals [26]. Pseudomonas aeruginosa use their own T3SS to rapidly infect and kill adult flies. While cytotoxic to macrophages, P. aeruginosa infection activates the NF-κB-dependent IMD antimicrobial pathway [38]. It is thus possible that elements of the pathogenic bacterial systems have been co-opted by wasps to attack the fly’s cellular immune system. In this scenario, intracellular protein complexes within lamellocytes would be under direct selective pressure to respond to MSEV-based T3SS-like virulence.
The presence of prokaryotic-like (particularly, T3SS/flagellar-like) proteins, hints at the possibility that either MSEV spikes evolved from primordial flagellar/needle-like structures or they share evolutionary history with such structures. These findings also support a hypothesis proposed by Martin and colleagues [39] that the eukaryotic endomembrane system may have arisen from bacterial outer membrane vesicles. In this regard, characterization of the prokaryotic protein motifs that comprise nearly 10% of the novel proteins outlined above will be especially revealing. The molecular mechanisms by which MSEV proteins deplete and destroy its well-characterized hosts’ immune system will suggest how virulence factors are acquired by insect parasites, how these factors evolve, and how insects might serve as reservoirs of disease. Answering these questions is likely to lead to new cost-effective therapies for treating emerging infections and opportunistic diseases.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shubha Govind (sgovind@ccny.cuny.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly strains
All fly stocks were healthy and free of common laboratory infections. Genetically stable stocks with the desired, uniform genotypes were used for infections and crosses were not necessary. The y w strain of D. melanogaster (used as wild type) and Dome-MESO-GFP (gift from M. Crozatier [41]) (see Key Resource Table (KRT)) were used. Dome-MESO-GFP is a synthetic construct that replaces lacZ with GFP expression in P{dome-lacZ.MESO} (Fly Base FBtp0022619) and produces GFP marked regions of domeless (the fly JAK/STAT receptor) expression [41]. Flies were raised on standard fly food [cornmeal (65 g/L), sucrose (140 g/L), agar (10 g/L), and yeast (35 g/L) with tegosept made in 95% ethanol (22.6 ml/L) and propionic acid (5 ml/L)] (see KRT) medium and maintained long term at 18°C with inspection and flipping to new vials every three to four weeks. Prior to experimental work, fly stocks were moved to fresh food, shifted to 25°C, and maintained for at least two generations. Adults were used for 2 to 3 rounds of egglays to produce larvae for wasp infections. Mid to late second instar larvae (both male and female) were used for infections and sacrificed during dissection.
Leptopilina spp. parasitoid wasp strains
All wasp stocks were healthy and free of common laboratory infections. The following wild type parasitoid wasp species and strains were used: Leptopilina heterotoma 14 (Lh 14) [3], L. heterotoma NY (Lh NY) [9], L. victoriae (Lv) [33], and L. boulardi 17 (Lb 17) [3]. (A detailed genotypic notation system has not been formalized for Leptopilina spp. as mutant or transgenic wasps have not been produced to date.) Lh (strains 14 and NY) and L. victoriae are sister species with VLPs that are indistinguishable from each other [9]. Wasp stocks were maintained at 18°C long-term via infestation of healthy and infection-free mid to late second instar y w D. melanogaster larvae raised on the same fly food medium as the “fly-alone” cultures (see Fly Strain details, above, for husbandry and housing of flies). Adult wasps were moved to 25°C prior to experimental infections. Free-living, adult female wasps were collected with males at approximately one week post eclosion (PE). Mated females were used in experimental infections and were used for infections only once. For VLP purification, female wasps were sorted from males for dissection of venom gland apparatus (males do not possess venom glands).
Bacteria
E. coli BL21 cells (see KRT for genotype) were used for expression of the p40 central domain (CD).
METHOD DETAILS
Wasp infections, dissections, and imaging
For experimental infections, cultures containing both male and female mid to late second instar D. melanogaster larvae were exposed to 10 – 12 mated, 1 week PE, Lh 14 or Lb 17 female wasps [3] for 12 hours at 25°C. Wasps were removed and larvae were allowed to recover in the medium for 5 hours at 25°C. Infected larvae were scooped from the medium, scored for age, matched to y w control wasp-infected larvae, washed in deionized water, washed in phosphate buffered saline (1X PBS, pH 7.2, 25°C), and dissected to obtain larval lymph gland tissue. Infection was validated during dissection by the presence of wasp eggs (free floating or attached onto larval gut tissue) and uninfected hosts were discarded.
Dissections and slide preparations were performed at 25°C. Tissue was dissected on untreated glass slides by bleeding larvae and pulling the cuticle bilaterally below the mouth hooks to leave the mouth parts, lymph gland, and dorsal vessel. After air drying, excess tissue was removed. Samples were fixed in 4% paraformaldehyde, washed, permeabilized (1X PBS with 0.3% Triton X-100), and washed again in 1X PBS. Counterstains were applied (Rhodamine-tagged Phalloidin at 0.5 mg/ml and Hoechst 33258 at 0.02 mg/mL) and samples were washed and then mounted in VectaShield.
Samples were imaged with a Zeiss 510 confocal at 40X. Laser settings were based on GFP negative and uninfected control tissue and all images were scanned using the same instrument settings. Images were processed with Zeiss LSM image browser. Figures were compiled in Adobe Photoshop v12.0.4 and CC 2015.5 and Illustrator CC 2015.
Results shown in Fig 1 were validated in dissections of at least twenty animals (n = 20 lymph glands) each, infected by either wasp. Results have also been validated by infections and dissection of at least three other D. melanogaster genotypes with replicates of at least five animals (n = 5 lymph glands). All infection-related changes to lymph gland morphologies were included in our conclusions (no data were excluded). No strategies for randomization and/or stratification, blinding, or sample-size estimation were utilized, or found to be necessary, for this study.
VLP purification
Three hundred each Lh 14 [3] and Lh NY [9] female wasps (see Wasp Strain section, above, for husbandry and housing details) of 1 to 2 weeks PE were sacrificed by submerging in 70% ethanol and rinsed at least three times each in deionized water and 0.1X PBS (pH 7.2) at 4°C. Venom gland complexes were dissected in PBS by grasping the ovipositor and gently pulling. Only intact, dissected venom gland complexes with a VLP long gland, reservoir, and ovipositor were used in the purification protocol; other body parts were discarded. Tissue was collected in 1X PBS (4°C), gently crushed with a sterile pestle in 200μL of 1X PBS (4°C), shook vigorously in 300 μL 1X PBS (4°C), pulse agitated, and added to the top of a 4°C Nycodenz gradient. The gradient of 50 to 10% Nycodenz in 1X PBS (5 bands of 900 μL, each) was prepared in a clean, RNA-free Beckman 9 cm centrifuge tube and pre-chilled. Whole VLPs were separated from other tissues into a single band by ultracentrifugation (20,000 RPM, 7°C, 80 mins). The VLP band was removed from the gradient, washed with 1X PBS (4°C), pelleted (11,700 RPM, 7°C, 35 mins), resuspended in a minimum of 1X PBS (4°C), and stored (4°C). Extraction of VLPs from ~ 300 long gland complexes has consistently yielded upwards of thousands of VLPs.
Electron microscopy
For cryo electron microscopy (EM), purified VLPs were pipetted onto a holey carbon coated grid. Excess fluid was blotted (Whatman #1) and the grid was plunge frozen (liquid ethane) and stored in liquid nitrogen. Samples were visualized with a Technai G2 (200kV) at the New York Structural Biology Center. Membrane surrounding Lh VLPs (observed via cryo EM in Fig 1) was present in all VLPs of more than two sample preparations and this result confirmed previously-published findings from transmission electron microscopy experiments [6, 9].
For scanning EM (SEM), purified VLPs (washed and re-suspended, PBS) were fixed in glutaraldehyde (3% in 0.085M sodium cacodylate buffer, overnight, 4°C), followed by cacodylate buffer (0.085M, 1 hr, 4°C). After washing (glass distilled water) and fixing in osmium tetroxide (1% in 0.085M cacodylate buffer, 1 hr, 4°C), VLPs were filtered onto polycarbonate membranes (0.1 μm pores). Filtered samples were then dehydrated in serial ethanol washes (technical grade, to 70%) and stored overnight. The membranes were washed (amyl acetate), dried, and mounted on pin stubs. Membranes were gold-palladium plated and stored at 60°C until imaged on a Ziess Supra 55 SEM. Parameters for SEM imaging of Lh, Lv, and Lb VLPs were consistent for all samples and technical replicates; data presented are representative of many particles from at least two replicates per species’ VLP.
MS/MS analysis of Lh VLP proteins
Purified VLPs were separated on a 1-D SDS-PAGE gel as per standard protocols. Bands were excised, destained, reduced, alkylated, and trypsin digested. Peptides from combined additional lanes (L457) were also analyzed. Peptides were extracted (Applied Biosystems POROS 20 R2 beads), cleaned-up (C18 ZipTips), dried, and reconstituted in 2% acetonitrile/0.1% formic acid. Lh 14 peptides were trapped (Waters Symmetry® C18 trap column (180 μm × 100 mm, 5 μm particles)), washed, and separated on a Waters BEH130 C18 column (1.7 μm particle size) (Waters NanoAcquity UPLC (Milford, MA)). Lh NY peptides were separated on a Waters BEH130 C18 column (75 μm × 150 mm). The MS analysis was performed on an LTQ-Orbitrap (ThermoFisher, CA).
The instrument RAW files were analyzed using Proteome Discoverer (PD) 1.4.0.288 with a work template that contained a Target Decoy PSM Validator node (peptide spectrum matches) with both Sequest and Mascot algorithms. Mascot searches, independent of PD, were also conducted. Peptide mass tolerance was set to 10 ppm, and the fragment mass tolerance was 0.6 Da. The enzyme was set to “trypsin” with two maximum missed cleavage sites and the search was against VLPSwiss_20140319.fasta (1332969 entries). Carbamidomethylation of cysteine was specified as a fixed modification. Deamidation of asparagine and glutamine and oxidation of histidine, methionine and tryptophan were specified as variable modifications. Methylation at aspartic acid residues was specified only for Mascot analyses conducted without PD. The .msf output files were integrated into Scaffold (version Scaffold_4.7.3, Proteome Software Inc.) which was used to validate MS/MS based peptide and protein identifications.
Identification of Lh VLP proteins
VLP proteins (Tables S1, S2) reported here had at least two proteomic peptides, identified at 99% or greater probability by either PD or solo Mascot searches, that aligned to a target sequence. In addition, four proteins are included in the list where only one peptide aligned at 99% more probability (see below, Protein sequence annotations subsection). Full-length target protein sequences were translated from RNA-Seq Lh 14 (GAJC00000000.1) transcripts translated to ORFs [10].
The number of aligned peptides varied slightly between PD and Mascot analyses conducted without PD. The PD results have been deposited to the ProteomeXchange Consortium via the PRIDE [42] partner repository (see KRT for link).
In this report, VLP composition is based on the subset of proteins common to both the Lh 14 and NY strains. The common, full-length VLP protein ORFs (Tables S1, S2) were annotated via (A) primary sequence analyses, and (B) structure-based analyses and predictions for select proteins (described below). Note that the Lh 14 dataset is larger and select Lh 14-unique sequences were preferentially included.
Additional verification of protein sequences
The VLP peptides were also searched against other proteomes (Uniprot D. melanogaster, A. mellifera, H. sapiens, viral, prokaryotic and archaea databases [43]). The full-length proteins identified from the RNA-Seq ORFs [10] were BLASTed against Lh ESTs (NCBI LIBEST 028179 and 028205, [11, 12]), providing an alternative method of ORF sequence verification. This step also identified the full, or near full, length VLP protein clones in our Lh EST collection (e.g., Lh VLP Sm & LgGTPas01). Roughly 20% and ~ 45% of the common VLP proteins were identified in the Heavner et al. 2013 [11] and the Colinet et al. 2013 [12] studies. (See below, for methods of identification of absence/presence of expression of each Lh VLP protein in Lb 17 and G1 abdominal transcriptomes (Fig 2).)
Protein sequence annotations
The automated, pipe-line annotation algorithms of (1) BLAST2GO, E <= 10−5 (v2.7.0) [44], (2) InterProScan (v5.3–46) [45], and (3) FastAnnotator [46] were used to characterize all (common set) VLP proteins identified from VLP peptide alignments to Lh transcriptome ORFs (See above). Multiple bioinformatic methods were used to avoid algorithm bias and increase accuracy. Two related criteria were used as measures of reliability of annotations: (a) E values (E <= 10−5); and (b) percent identity and percent similarity (applied only when at least 75% of the query aligned to the hit).
For manual annotations, BLASTs were conducted via NCBI [47] (nr and TSA databases, default parameters [48]). To identify potential Lh VLP protein homologs in microbiota, the unannotated full-length VLP proteins were pBLAST searched against a subset of all nr archaea, viruses, and prokaryotic genomes at higher sensitivity (GenBank, E <= 10). To identify Lh VLP proteins expressed by Lb 17 and G1, all VLP sequences were tBLASTn searched with default parameters against GAJA00000000.1 (Lb 17) and GAIW00000000.1 transcriptomes (Ganaspis sp1 (G1)) [10]. The Conserved Domains Database (CDD) [49, 50] and PFAM [51] were used for domain identifications and architectures. Manual annotation results are reported in main text only if they were confirmed by a second method. Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) numbers were assigned using GhostKOALA [52]. KO numbers were then used to obtain model ortholog annotations.
For pairwise and multiple-sequence alignments, T-Coffee [53], Needleman [54], ClustalOmega [55], and MUSCLE [56, 57] (EBI webserver, default parameters) were used and ESPript 3.0 [58] was used for visualizations. FunRich [59] was used for functional enrichment and over-representation analyses against two extracellular vesicle databases, Exocarta [60, 61] and Vesiclepedia [62]. Circos (v0.69) was used to visualize the proteome [63].
The four proteins with only on a single aligned peptide (99% or greater peptide probability) (Fig 3; Tables S1, S2) passed the (1) manual inspections of their MS/MS spectra and fragments, (2) solo Mascot searches, and (3) Exocarta/Vesiclepedia enrichment analyses. Of these four proteins, one is classified as novel (Class 3; Figs 2, 3 A), while the other three have sequence similarities with eukaryotic proteins (Class 1; Fig 2, 3) of extracellular vesicles (Fig 3 B, C).
In silico structural predictions of SmGTPase01
A high-quality structural model was created for SmGTPase01 using a hybrid approach. A MODELLER [64] model was created using three templates: GIMAP crystal structure 3V70 and two ab initio models [65]. 3V70 was chosen using threading metaservers (LOMETS [66] and PHYRE2 [67]) and was evaluated in single-template MODELLER trials. Loop modeling and side-chain optimization were done using Loopy [68] and SCWRL4 [69], respectively. The active site of this full-length model is presented in Fig 4 C; the remaining details of the model will be published elsewhere.
The SmGTPase01 co-factor, Mg2+, was placed in the active site based on COFACTOR [70] and was checked for positioning using WHAT IF [71]. GTP was placed and checked for energy minimization in the SmGTPase01 active site with Autodock Vina [72]. The cofactor and substrate placements were confirmed with predictions from COACH [73], BSP-SLIM [74], and 3D Ligand [75].
The qualities and knowledge-based energy values of our models were assessed using ProSA-web [76] and Verify3D [77]. TM-Align was used to compare crystal structures and our in silico models [30]. STRIDE [78] was used to define secondary structures from molecular coordinates. Crystal structure molecular files came from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do).
Cloning and structural predictions of p40
For p40-specific proteomics, peptides isolated and digested from an anti-p40 positive SDS-PAGE gel band were sequenced at the Harvard Microchemistry Facility by HPLC-MS/MS (Finnigan LCQ quadrupole ion trap) and then BLAST queried against NCBI GAJC00000000.1 RNA-Seq Lh 14 database.
For p40 cloning and expression, the p40 IpaD-like central domain (CD) was amplified from venom gland cDNA and cloned into pCR2.1-TOPO. Primer sequences are listed in Key Resource Table. The p40 central domain (amino acids 26 – 240, Fig 4 D) was expressed from a pTrcHisA subclone by addition of IPTG (1 mM, ThermoFisher Scientific). E. coli BL21cells were lysed by freeze-thaw in lysis buffer (10% triton X, 150 mM NaCl, 1 mM EDTA and 50 mM Tris HCl, pH 6.8) with protease inhibitors (AEBSF-HCl 100 mM, aprotinin 80 μM, bestatin 5 mM, E-64 1.5 mM, leupeptin 2 mM, pepstatin A 1 mM, Sigma or Fermentas). Protein concentrations in all assays were determined with Bradford reagent [79].
For the verification of p40 identity via western analyses, bacterial proteins were separated (SDS-PAGE, 5% stacking and 12% resolving), transferred to membrane (nitrocellulose, HyBond, Amersham Life Science), and blocked (PBS, pH 7.4, 0.1 % Tween 20, 5% nonfat dry milk, 3% bovine serum albumin (1 hr, 25°C)). Primary antibodies used were anti-p40 (1:1000) or anti-6X-His (1:1000; 12 hr, 4°C). Alkaline-phosphatase-linked anti-mouse secondary antibody (1:2,500; 1 hour, 25°C), 5-bromo-4-chloro-3′-indolyphosphate (BCIP, Amresco), and nitro-blue tetrazolium (NBT, Biotium) solutions in NaCl-Tris-MgCl2 buffer (NTM, pH 9.5) were used for detection.
For p40 structural predictions, ab initio and template fragment assembly methods [80] [65] were used along with N-terminal predictions from MODELLER’s loop methods [81–83]. Structural optimizations were generated using ModRefiner [84]. The most N- and C-terminal residues of p40 are predicted to be a signal peptide and transmembrane helix and were not modeled (Fig S1 C). Crystal structures (http://www.rcsb.org/pdb/home/home.do) similar to our in silico p40 model were identified using DaliLITE [85].
Antibiotic treatment of Lv
To cure Lv adults of the strain of Gram negative Wolbachia endosymbionts [34], three successive generations of Lv females were cultivated from infections of y w D. melanogaster egglays on fly food medium containing rifampicin at 0.75 mg/g (see Fly and Wasp Strain sections, above, for husbandry and housing details). The next six generations of Lv wasps were cultivated on larvae raised on standard, antibiotic-free medium, to assure no direct drug effects. Isogenized control wasps did not receive antibiotic treatment and were otherwise reared the same way as experimental wasps. For PCR amplification of wasp genomic DNA, antibiotic-treated and control male and female wasps, no more than 1 week PE, were sorted into microfuge tubes and frozen at −20°C. Genomic DNA template was amplified for the absence or presence of (a) Lv-specific gatB and coxA genes of Wolbachia, and (b) VLP protein genes (p40 and Sm/LgGTPase01) (see Analyses of genomic sequences, below, and KRT). Results from all PCR amplification experiments supported the conclusions in this study.
Analyses of genomic sequences
Total genomic DNA from Lh, antibiotic-treated Lv, and control wasps was obtained using a Qiagen DNA Blood and Tissue kit. Primer sequences are listed in KRT; the following notation is used:
y = pyrimidines; r = purines; and k = T and G.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details regarding experimental replicates are provided with each Method. As indicated in the Wasp infections, dissections, and imaging subsection, n in this study refers to the whole lymph gland of an animal (i.e., two lobes). More than 35 lymph glands from larvae of different genotypes yielded consistent results after wasp infection. Similarly, examination of numerous VLPs in each sample, as well as repetition of different EM protocols provided supporting information regarding VLP morphologies. Confidence in proteomics results is based on peptide characterizations of purified VLP preparations from two independent strains of Lh. The criteria of agreement between multiple, independent methods for identification and annotation of VLP proteins (both novel and previously characterized) were met. Confidence in quantitative bioinformatics results (e.g., E values for BLAST results and domain predictions, RMSDs for protein model analyses, p-values for enrichments) is based on the algorithms intrinsic to these methods and is described in the corresponding primary references. Where possible, more than one computational approach (supported by different algorithms and metaservers) was used to strengthen interpretation by avoiding biases arising from a single computational methodology. No additional statistical analyses or methods to determine assumption credibility and/or sample size were conducted for this study.
DATA AND SOFTWARE AVAILABILITY
.mzid files for all VLP protein gel bands for both L. heterotoma 14 and NY (see MS/MS analysis of Lh VLP proteins methodology, above) were exported from Scaffold and submitted to PRIDE, a ProteomeXchange repository member (see KRT for links). .mzid files provide the data for VLP peptide identification in the mzIdentML proteomics format (Tables S1, S2), a HUPO Proteomics Standards Initiative (Proteomics Informatics Standards group) standard.
Supplementary Material
(A) Anti-p40 antibody reacts at the expected molecular weight (top bands) for the His-tagged p40 central domain (CD, residues 26-240) from bacterial extracts, and (B) with venom extracts (VGE) from L. heterotoma (Lh) and L. victoriae (Lv) female wasps. In panel A, S=supernatant; P=pellet. Uninduced and induced refer to bacterial extracts prepared without or with IPTG induction for p40 CD expression. p40's identity was also confirmed with anti-His antibody (not shown). The p40-positive bands at higher molecular weights (B) suggest higher-order protein associations and/or post-translational modifications. (C) Comparison of the Shigella flexneri IpaD (2J0O) structure and L. heterotoma p40 model (Figure 4 E): The residue numbers for the p40 model do not include the predicted signal peptide. The first and last model residues are 26 and 213 of the predicted full length protein, respectively. The p40 model lacks the short α-helix and β-hairpin at residues 208-251 in IpaD and the model's local quality drops in this region. 310 helices are found in the p40 model, in addition to α-helices. 310 and α-helices psi/phi angles are similar. These 310 helices could render as α-helices, given slight conformational and energetic shifts in the model. Experimental methods are necessary to validate these p40 model predictions.
A summary of proteomic data for proteins common to Lh 14 (Gel01) and Lh NY (Gel02) VLPs purified on Nycodenz gradients is presented here. The data are organized by our in-house VLP_Swiss-Prot identifiers. The wasp strain (column 6) from which the greatest number of VLP protein peptides were detected is provided first (columns 2 – 5). Data shown include protein identification probability, peptide to protein alignment coverage, exclusive unique spectra, and exclusive unique peptides. The number of unique peptides detected from the second Lh strain’s VLPs (column 8) for each protein is given in column 7.
This table provides additional information not presented in the peptide-to-ORF table (Table S1). The peptide sequences detected for each protein from both Lh 14 (Gel01) and Lh NY (Gel02) VLP preparations are provided along with post-translational modifications (columns 1, 2, and 11, respectively). The SDS-PAGE gel band of origin for each peptide can be found in column 23 (i.e., spectrum file ID). Any redundancies in protein identifications per peptide are provided in columns 21 and 22.
Data S1: Select infection- and immunity-related Lh VLP protein alignments (Related to Figs 2, 3A)
Seven (A–G) Class 2 Lh VLP proteins aligned with their most similar putative homologs from prokaryotic, viral, and eukaryotic species (emphasis on Hymenotpera and Diptera). If sequences were trimmed, the absolute residue range is given following the species of origin. The coloring scheme is based on physiochemical properties. ORF = open reading frame
(A) Two diedel-like Lh VLP sequences with pfam13164 domains identified (E = 2.29e−12 and 1.17e−07, diedel-like 1 and 2, respectively) are aligned with nine similar sequences (BLASTp nr, 25 – 53% identity; 7e−08 <= E <= 3e−01). Five of the putative homologs are from Drosophila spp. and are four from dsDNA insect viruses (granulovirus, ascovirus, and entomopoxvirus). Both sequences contain secretion signal motifs, and multiple predicted disulfide bridges. The D. melanogaster diedel, a putative homolog, is a negative regulator of the JAK-STAT pathway.
(B) A Lh VLP enhancin-like protein is aligned with similar prokaryotic sequences (BLAST2GO and Delta BLAST nr, 20 – 42% identity; 5.33e−04 <= E <= 9e−03). Multiple sequences from Yersinia, Listeria, and other pathogenic bacterial species were found in our BLASTs and, in a few cases, these sequences were annotated as M60 peptidases. The VLP sequence contains a putative secretion signal motif, but no known domains were identified. It is notable that enhancin homologs encoded in viral genomes were not uncovered in our searches.
(C) A Lh VLP GH18 chitinase-like superfamily (CDD cd02873 domain, E = 0) protein is aligned with five similar sequences (BLASTp nr, 50 – 75% identity; 0 <= E <= 2 e−143) from other insects, including the yellow-fever mosquito, the Tobacco horn worm, and three parasitoid wasps (the Jewel and two Braconid wasps). The VLP protein sequence encodes a predicted secretion signal and is predicted to be an Imaginal disc growth factor (Idgf)-like protein, a superfamily that diverged from chitinase-like proteins.
(D) The Lh VLP venom allergen-like (CDD domain cd05380, E = 1.95 e−42) protein is aligned with five similar sequences (BLASTp nr, 37 – 42% identity; 6 e−41<= E <= 6 e−34) from other insects, including the red imported fire ant, the Panamanian leafcutter ant, and three parasitoid wasps (Jewel, a Braconid wasp, and a Chalcidoid egg parasite). A eukaryotic-specific SCP domain, best characterized in plant pathogenesis defense proteins, has been identified, as well as a putative secretion signal.
(E) The Lh VLP Bap31-like (CDD domain cd05380, E = 1.95 e−42) protein is aligned with five similar sequences (BLASTp nr, 59 – 76% identity; 1 e−126 <= E <= 4 e−83) from other insects, including two species of ant (Florida carpenter and red imported fire ant), wasp (a Braconid and the Jewel parasitoid), and Drosophila mojavensis. Canonical Bap31 proteins regulate ER-stress-mediated apoptosis. Similar to these proteins, the Lh VLP protein is predicted to encode three transmembrane helices.
(F) The Lh VLP knottin-like (pfam11410 domain, E = 5.19 e−03) protein is aligned with five similar sequences (BLASTp nr, 33 – 52% identity; 3 e−08 <= E <= 2 e−02), four from insect species and one from the eudicotyledon, Mesembryanthemum crystallinum. Among the most similar putative homologs are sequences from diverse insects: Hymenoptera (sawfly and wasp), Diptera, and Hemipteran species. The sequence contains a putative secretion signal motif and three predicted disulfide bridges. 40% of the VLP sequence shows notable conservation (54% identity) when compared to a secreted ion channel toxin from the spider Chilobrachys guangxiensis (Arachnida: Theraphosidae). Knottins are classified as a cystine-rich plant antimicrobial peptide family.
(G) The Lh VLP hemolymph juvenile hormone binding protein (JHBP)-like (pfam06585 domain, E = 6.10 e−17) protein is aligned with four similar sequences (BLASTp nr, 24 – 30% identity; 1 e−11 <= E <= 6 e−03) from a wheat stem sawfly, the Panamanian leafcutter ant, the diamondback moth, and Drosophila melanogaster. No high identity homologs were found, but moderately similar homologs were found encoded by wasps, ants, and arthropods, in general. Homologs in Drosophila spp. were found, but are not functionally well characterized. The regions of similarities are within 80 to 90% of the length of the VLP protein sequence, but largely only in comparisons to putative homologs from other hymenoptera.
Data S2: Lh VLP GTPase alignments (Related to Figs 2, 3A, & 4A – C)
This document provides alignments of select small and large VLP GTPases (including SmGTPases01/LgGTPases01) with putative homologs/paralogs from (A) Lh; (B) prokaryotic and eukaryotic species; (C) Ganaspis sp.1 (G1); and (D, E) Leptopilina clavipes (Lc). The far N-and C-termini show the most variation among the sequences and were trimmed. The residue range displayed is indicated after the species of origin. The coloring scheme is according to physiochemical properties. ORF = open reading frame.
(A) Eight Lh VLP GTPases are presented here. Additional putative GTPase family members exist, but are not presented here. The first three sequences of ~ 300 residues, are SmGTPases, while the following five sequences of ~ 500 residues are LgGTPases. Among the eight family members shown here, the percent similarities within the N-terminal regions range from ~ 25 to 60% and average pairwise similarity over their full lengths is 45%. Only seven insertion/deletion sites exist in this 222 amino acid alignment and large blocks of identity (> 80% of the alignment length) are present. Highly similar secretion signal peptides are predicted at the N-termini of all GTPases (not shown).
(B) The five most-similar ORFs found in the NCBI TSA transcriptome for G1 (GAIW00000000.1) are shown aligned with Lh VLP Sm/LgGTPase01s. Like the Lh VLP GTPases, several putative G1 GTPases have predicted secretion signal peptides (not shown). Potential G1 homologs demonstrate >= 31% and 35% identity (E <= 3e−03 and 4e−59) to Lh VLP SmGTPase01 and LgGTPase01, respectively (given 75% alignment coverage).
(C) The five most-similar ORFs found in the NCBI TSA transcriptome for Lc (GAXY00000000.2) are shown aligned with Lh VLP Sm/LgGTPase01s. Similar to Lh and G1 GTPases, multiple Lc GTPases have predicted signal sequence peptides. The Lc transcripts identified show similarity in the N-terminal G domain, but are generally much shorter than the Lh VLP GTPases. Lh VLP SmGTPase01 shows <= 34% identity to Lc transcripts (>= 75% alignment criterion, E >= 2e−34) and LgGTPase01 shows <= 44% identity (E >= 3e−99), respectively. Lc also parasitizes Drosophila spp., but its comparative virulence and phylogenetic distance are poorly characterized in comparison to Lh and Lb.
(D) The most similar Lh and Lc sequences have been extracted from (C) and are shown here.
Acknowledgments
We would like to thank E. Miller, J. Crissman, & J. Berriman, along with Govind and Singh lab members (M. Badri, H. Chiu, A. Hudgins, J. Morales, I. Paddibhatala, R. Rajwani, S. Shamburger, and B. Wey) for discussions, experimental assistance, and reagents. This work was supported by grants from NASA (NNX15AB42G), NSF (IOS-1121817), NIH (1F31GM111052-01A1 and 5G12MD007603-30).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, M.H., J.R., S.S., S.G.; Validation, M.H., J.R., S.S., S.G.; Investigation (bench), M.H., J.R., G.G., G.D., R.W., S.G.; Investigation (bioinformatics), M.H., G.G., G.R., M.S., J.K., S.S., S.G.; Data Curation, M.H., G.D., G.R., J.K., S.G., S.S., R.W.; Writing – original draft, M.H., J.R., G.G., G.R., G.D., M.S., R.W., S.S., S.G.; Writing – review & editing, M.H., J.R., G.D., S.B., R.W., S.S., S.G.; Visualization, M.H., J.R., G.R., S.S., S.G.; Supervision, S.G., S.S., R.W.; Funding Acquisition, M.H., S.G., S.B.
References
- 1.Pennacchio F, Strand MR. Evolution of developmental strategies in parasitic Hymenoptera. Annu Rev Entomol. 2006;51:233–258. doi: 10.1146/annurev.ento.51.110104.151029. [DOI] [PubMed] [Google Scholar]
- 2.Sorrentino RP, Carton Y, Govind S. Cellular immune response to parasite infection in the Drosophila lymph gland is developmentally regulated. Dev Biol. 2002;243:65–80. doi: 10.1006/dbio.2001.0542. [DOI] [PubMed] [Google Scholar]
- 3.Schlenke TA, Morales J, Govind S, Clark AG. Contrasting infection strategies in generalist and specialist wasp parasitoids of Drosophila melanogaster. PLoS Pathog. 2007;3:1486–1501. doi: 10.1371/journal.ppat.0030158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heavner ME, Hudgins AD, Rajwani R, Morales J, Govind S. Harnessing the natural -parasitoid model for integrating insect immunity with functional venomics. Curr Opin Insect Sci. 2014;6:61–67. doi: 10.1016/j.cois.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MJ, Kalamarz ME, Paddibhatla I, Small C, Rajwani R, Govind S. Virulence factors and strategies of Leptopilina spp.: selective responses in Drosophila hosts. Adv Parasitol. 2009;70:123–145. doi: 10.1016/S0065-308X(09)70005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rizki RM, Rizki TM. Parasitoid virus-like particles destroy Drosophila cellular immunity. Proc Natl Acad Sci U S A. 1990;87:8388–8392. doi: 10.1073/pnas.87.21.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu H, Govind S. Natural infection of D. melanogaster by virulent parasitic wasps induces apoptotic depletion of hematopoietic precursors. Cell Death Differ. 2002;9:1379–1381. doi: 10.1038/sj.cdd.4401134. [DOI] [PubMed] [Google Scholar]
- 8.Gueguen G, Rajwani R, Paddibhatla I, Morales J, Govind S. VLPs of Leptopilina boulardi share biogenesis and overall stellate morphology with VLPs of the heterotoma clade. Virus Res. 2011;160:159–165. doi: 10.1016/j.virusres.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu H, Morales J, Govind S. Identification and immuno-electron microscopy localization of p40, a protein component of immunosuppressive virus-like particles from Leptopilina heterotoma, a virulent parasitoid wasp of Drosophila. J Gen Virol. 2006;87:461–470. doi: 10.1099/vir.0.81474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goecks J, Mortimer NT, Mobley JA, Bowersock GJ, Taylor J, Schlenke TA. Integrative approach reveals composition of endoparasitoid wasp venoms. PLoS One. 2013;8:e64125. doi: 10.1371/journal.pone.0064125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heavner ME, Gueguen G, Rajwani R, Pagan PE, Small C, Govind S. Partial venom gland transcriptome of a Drosophila parasitoid wasp, Leptopilina heterotoma, reveals novel and shared bioactive profiles with stinging Hymenoptera. Gene. 2013;526:195–204. doi: 10.1016/j.gene.2013.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colinet D, Deleury E, Anselme C, Cazes D, Poulain J, Azema-Dossat C, Belghazi M, Gatti JL, Poirie M. Extensive inter- and intraspecific vemon variation in closely related parasites targeting the same host: The case of Leptopilina parasitoids of Drosophila. Insect Biochem and Mol Biol. 2013;43:601–611. doi: 10.1016/j.ibmb.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Mortimer NT, Goecks J, Kacsoh BZ, Mobley JA, Bowersock GJ, Taylor J, Schlenke TA. Parasitoid wasp venom SERCA regulates Drosophila calcium levels and inhibits cellular immunity. PNAS. 2013 doi: 10.1073/pnas.1222351110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strand M, Burke G. Polydnaviruses as symbionts and gene delivery systems. PLoS Pathog. 2012;8:e1002757. doi: 10.1371/journal.ppat.1002757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silverman JM, Reiner NE. Exosomes and other microvesicles in infection biology: organelles with unanticipated phenotypes. Cell Microbiol. 2011;13:1–9. doi: 10.1111/j.1462-5822.2010.01537.x. [DOI] [PubMed] [Google Scholar]
- 16.Lamiable O, Kellenberger C, Kemp C, Troxler L, Pelte N, Boutros M, Marques JT, Daeffler L, Hoffmann JA, Roussel A, et al. Cytokine Diedel and a viral homologue suppress the IMD pathway in Drosophila. Proc Natl Acad Sci U S A. 2016;113:698–703. doi: 10.1073/pnas.1516122113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang S, Wang L, Guo W, Zhang X, Peng D, Luo C, Yu Z, Sun M. Bacillus thuringiensis bel protein enhances the toxicity of Cry1Ac protein to Helicoverpa armigera larvae by degrading insect intestinal mucin. Appl Environ Microbiol. 2009;75:5237–5243. doi: 10.1128/AEM.00532-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colinet D, Schmitz A, Depoix D, Crochard D, Poirie M. Convergent use of RhoGAP toxins by eukaryotic parasites and bacterial pathogens. PLoS Pathog. 2007;3:e203. doi: 10.1371/journal.ppat.0030203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asgari S, Reineke A, Beck M, Schmidt O. Isolation and characterization of a neprilysin-like protein from Venturia canescens virus-like particles. Insect Mol Biol. 2002;11:477–485. doi: 10.1046/j.1365-2583.2002.00356.x. [DOI] [PubMed] [Google Scholar]
- 20.Formesyn EM, Heyninck K, de Graaf DC. The role of serine- and metalloproteases in Nasonia vitripennis venom in cell death related processes towards a Spodoptera frugiperda Sf21 cell line. J Insect Physiol. 2013;59:795–803. doi: 10.1016/j.jinsphys.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Shchelkunov SN, Blinov VM, Totmenin AV, Marennikova SS, Kolykhalov AA, Frolov IV, Chizhikov VE, Gytorov VV, Gashikov PV, Belanov EF, et al. Nucleotide sequence analysis of variola virus HindIII M, L, I genome fragments. Virus Res. 1993;27:25–35. doi: 10.1016/0168-1702(93)90110-9. [DOI] [PubMed] [Google Scholar]
- 22.Gross CH, Russell RL, Rohrmann GF. Orgyia pseudotsugata baculovirus p10 and polyhedron envelope protein genes: analysis of their relative expression levels and role in polyhedron structure. J Gen Virol. 1994;75(Pt 5):1115–1123. doi: 10.1099/0022-1317-75-5-1115. [DOI] [PubMed] [Google Scholar]
- 23.Asgari S, Rivers DB. Venom proteins from endoparasitoid wasps and their role in host-parasite interactions. Annu Rev Entomol. 2011;56:313–335. doi: 10.1146/annurev-ento-120709-144849. [DOI] [PubMed] [Google Scholar]
- 24.Strand MR, Burke GR. Polydnaviruses: Nature’s Genetic Engineers. Annu Rev Virol. 2014;1:333–354. doi: 10.1146/annurev-virology-031413-085451. [DOI] [PubMed] [Google Scholar]
- 25.Herniou EA, Huguet E, Theze J, Bezier A, Periquet G, Drezen JM. When parasitic wasps hijacked viruses: genomic and functional evolution of polydnaviruses. Philos Trans R Soc Lond B Biol Sci. 2013;368:20130051. doi: 10.1098/rstb.2013.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hueck C. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrarese R, Morales J, Fimiarz D, Webb BA, Govind S. A supracellular system of actin-lined canals controls biogenesis and release of virulence factors in parasitoid venom glands. J Exp Biol. 2009;212:2261–2268. doi: 10.1242/jeb.025718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiavolin L, Meghraoui A, Cherradi Y, Biskri L, Botteaux A, Allaoui A. Functional insights into the Shigella type III needle tip IpaD in secretion control and cell contact. Mol Microbiol. 2013;88:268–282. doi: 10.1111/mmi.12185. [DOI] [PubMed] [Google Scholar]
- 29.Arizmendi O, Picking WD, Picking WL. Macrophage Apoptosis Triggered by IpaD from Shigella flexneri. Infection and Immunity. 2016;84:1857–1865. doi: 10.1128/IAI.01483-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson S, Roversi P, Espina M, Olive A, Deane JE, Birket S, Field T, Picking WD, Blocker AJ, Galyov EE, et al. Self-chaperoning of the type III secretion system needle tip proteins IpaD and BipD. J Biol Chem. 2007;282:4035–4044. doi: 10.1074/jbc.M607945200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinson EO, Martinson VG, Edwards R, Mrinalini, Werren JH. Laterally Transferred Gene Recruited as a Venom in Parasitoid Wasps. Mol Biol Evol. 2016;33:1042–1052. doi: 10.1093/molbev/msv348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morales J, Chiu H, Oo T, Plaza R, Hoskins S, Govind S. Biogenesis, structure, and immune-suppressive effects of virus-like particles of a Drosophila parasitoid, Leptopilina victoriae. J Insect Physiol. 2005;51:181–195. doi: 10.1016/j.jinsphys.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 34.Gueguen G, Onemola B, Govind S. Association of a new Wolbachia strain with, and its effects on, Leptopilina victoriae, a virulent wasp parasitic to Drosophila spp. Appl Environ Microbiol. 2012;78:5962–5966. doi: 10.1128/AEM.01058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salazar-Jaramillo L, Paspati A, van de Zande L, Vermeulen CJ, Schwander T, Wertheim B. Evolution of a Cellular Immune Response in Drosophila: A Phenotypic and Genomic Comparative Analysis. Genome Biol Evol. 2014;6:273–289. doi: 10.1093/gbe/evu012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kroemer JA, Webb BA. Divergences in protein activity and cellular localization within the Campoletis sonorensis Ichnovirus Vankyrin family. J Virol. 2006;80:12219–12228. doi: 10.1128/JVI.01187-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gueguen G, Kalamarz ME, Ramroop J, Uribe J, Govind S. Polydnaviral ankyrin proteins aid parasitic wasp survival by coordinate and selective inhibition of hematopoietic and immune NF-kappa B signaling in insect hosts. PLoS Pathog. 2013;9:e1003580. doi: 10.1371/journal.ppat.1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fauvarque MO, Bergeret E, Chabert J, Dacheux D, Satre M, Attree I. Role and activation of type III secretion system genes in Pseudomonas aeruginosa-induced Drosophila killing. Microbial Pathogenesis. 2002;32:287–295. doi: 10.1006/mpat.2002.0504. [DOI] [PubMed] [Google Scholar]
- 39.Gould SB, Garg SG, Martin WF. Bacterial Vesicle Secretion and the Evolutionary Origin of the Eukaryotic Endomembrane System. Trends Microbiol. 2016;24:525–534. doi: 10.1016/j.tim.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 40.Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- 41.Oyallon J, Vanzo N, Krzemien J, Morin-Poulard I, Vincent A, Crozatier M. Two Independent Functions of Collier/Early B Cell Factor in the Control of Drosophila Blood Cell Homeostasis. PLoS One. 2016;11:e0148978. doi: 10.1371/journal.pone.0148978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:11033. doi: 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Consortium, U. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 45.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen TW, Gan RC, Wu TH, Huang PJ, Lee CY, Chen YY, Chen CC, Tang P. FastAnnotator–an efficient transcript annotation web tool. BMC Genomics. 2012;13(Suppl 7):S9. doi: 10.1186/1471-2164-13-S7-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pruitt K, Tatusova T, Maglott D. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2004;33:D5010–D5504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2010;39:D225–229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finn RD, Mistry J, Tate P, Coggill P, Heger A, Polington JE, Gavin OL, Gunesekaran G, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J Mol Biol. 2016;428:726–731. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 53.Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011;39:W13–17. doi: 10.1093/nar/gkr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Needleman S, Wunsch C. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 55.Sievers F, Wilm A, Dineen D, Gibson T, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. Fast, scalable generation of high-quality protien multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edgar R. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edgar R. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim A, et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15:2597–2601. doi: 10.1002/pmic.201400515. [DOI] [PubMed] [Google Scholar]
- 60.Keerthikumar S, Chisanga D, Ariyaratne D, Al Saffar H, Anand S, Zhao K, Samuel M, Pathan M, Jois M, Chilamkurti N, et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J Mol Biol. 2016;428:688–692. doi: 10.1016/j.jmb.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simpson RJ, Kalra H, Mathivanan S. ExoCarta as a resource for exosomal research. J Extracell Vesicles. 2012;1 doi: 10.3402/jev.v1i0.18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, Bond VC, Borras FE, Breakefield X, Budnik V, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012;10:e1001450. doi: 10.1371/journal.pbio.1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu S, Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35:3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 68.Xiang Z, Soto CS, Honig B. Evaluating conformational free energies: the colony energy and its application to the problem of loop prediction. Proc Natl Acad Sci U S A. 2002;99:7432–7437. doi: 10.1073/pnas.102179699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roy A, Yang J, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40:W471–477. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hekkelman ML, Te Beek TA, Pettifer SR, Thorne D, Attwood TK, Vriend G. WIWS: a protein structure bioinformatics Web service collection. Nucleic Acids Res. 2010;38:W719–723. doi: 10.1093/nar/gkq453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang J, Roy A, Zhang Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29:2588–2595. doi: 10.1093/bioinformatics/btt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee HS, Zhang Y. BSP-SLIM: a blind low-resolution ligand-protein docking approach using predicted protein structures. Proteins. 2012;80:93–110. doi: 10.1002/prot.23165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wass MN, Kelley LA, Sternberg MJ. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010;38:W469–473. doi: 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 78.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 79.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 80.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 2012;80:1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.John B, Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eswar N, John B, Mirkovic N, Fiser A, Ilyin VA, Pieper U, Stuart AC, Marti-Renom MA, Madhusudhan MS, Yerkovich B, et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003;31:3375–3380. doi: 10.1093/nar/gkg543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J. 2011;101:2525–2534. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Anti-p40 antibody reacts at the expected molecular weight (top bands) for the His-tagged p40 central domain (CD, residues 26-240) from bacterial extracts, and (B) with venom extracts (VGE) from L. heterotoma (Lh) and L. victoriae (Lv) female wasps. In panel A, S=supernatant; P=pellet. Uninduced and induced refer to bacterial extracts prepared without or with IPTG induction for p40 CD expression. p40's identity was also confirmed with anti-His antibody (not shown). The p40-positive bands at higher molecular weights (B) suggest higher-order protein associations and/or post-translational modifications. (C) Comparison of the Shigella flexneri IpaD (2J0O) structure and L. heterotoma p40 model (Figure 4 E): The residue numbers for the p40 model do not include the predicted signal peptide. The first and last model residues are 26 and 213 of the predicted full length protein, respectively. The p40 model lacks the short α-helix and β-hairpin at residues 208-251 in IpaD and the model's local quality drops in this region. 310 helices are found in the p40 model, in addition to α-helices. 310 and α-helices psi/phi angles are similar. These 310 helices could render as α-helices, given slight conformational and energetic shifts in the model. Experimental methods are necessary to validate these p40 model predictions.
A summary of proteomic data for proteins common to Lh 14 (Gel01) and Lh NY (Gel02) VLPs purified on Nycodenz gradients is presented here. The data are organized by our in-house VLP_Swiss-Prot identifiers. The wasp strain (column 6) from which the greatest number of VLP protein peptides were detected is provided first (columns 2 – 5). Data shown include protein identification probability, peptide to protein alignment coverage, exclusive unique spectra, and exclusive unique peptides. The number of unique peptides detected from the second Lh strain’s VLPs (column 8) for each protein is given in column 7.
This table provides additional information not presented in the peptide-to-ORF table (Table S1). The peptide sequences detected for each protein from both Lh 14 (Gel01) and Lh NY (Gel02) VLP preparations are provided along with post-translational modifications (columns 1, 2, and 11, respectively). The SDS-PAGE gel band of origin for each peptide can be found in column 23 (i.e., spectrum file ID). Any redundancies in protein identifications per peptide are provided in columns 21 and 22.
Data S1: Select infection- and immunity-related Lh VLP protein alignments (Related to Figs 2, 3A)
Seven (A–G) Class 2 Lh VLP proteins aligned with their most similar putative homologs from prokaryotic, viral, and eukaryotic species (emphasis on Hymenotpera and Diptera). If sequences were trimmed, the absolute residue range is given following the species of origin. The coloring scheme is based on physiochemical properties. ORF = open reading frame
(A) Two diedel-like Lh VLP sequences with pfam13164 domains identified (E = 2.29e−12 and 1.17e−07, diedel-like 1 and 2, respectively) are aligned with nine similar sequences (BLASTp nr, 25 – 53% identity; 7e−08 <= E <= 3e−01). Five of the putative homologs are from Drosophila spp. and are four from dsDNA insect viruses (granulovirus, ascovirus, and entomopoxvirus). Both sequences contain secretion signal motifs, and multiple predicted disulfide bridges. The D. melanogaster diedel, a putative homolog, is a negative regulator of the JAK-STAT pathway.
(B) A Lh VLP enhancin-like protein is aligned with similar prokaryotic sequences (BLAST2GO and Delta BLAST nr, 20 – 42% identity; 5.33e−04 <= E <= 9e−03). Multiple sequences from Yersinia, Listeria, and other pathogenic bacterial species were found in our BLASTs and, in a few cases, these sequences were annotated as M60 peptidases. The VLP sequence contains a putative secretion signal motif, but no known domains were identified. It is notable that enhancin homologs encoded in viral genomes were not uncovered in our searches.
(C) A Lh VLP GH18 chitinase-like superfamily (CDD cd02873 domain, E = 0) protein is aligned with five similar sequences (BLASTp nr, 50 – 75% identity; 0 <= E <= 2 e−143) from other insects, including the yellow-fever mosquito, the Tobacco horn worm, and three parasitoid wasps (the Jewel and two Braconid wasps). The VLP protein sequence encodes a predicted secretion signal and is predicted to be an Imaginal disc growth factor (Idgf)-like protein, a superfamily that diverged from chitinase-like proteins.
(D) The Lh VLP venom allergen-like (CDD domain cd05380, E = 1.95 e−42) protein is aligned with five similar sequences (BLASTp nr, 37 – 42% identity; 6 e−41<= E <= 6 e−34) from other insects, including the red imported fire ant, the Panamanian leafcutter ant, and three parasitoid wasps (Jewel, a Braconid wasp, and a Chalcidoid egg parasite). A eukaryotic-specific SCP domain, best characterized in plant pathogenesis defense proteins, has been identified, as well as a putative secretion signal.
(E) The Lh VLP Bap31-like (CDD domain cd05380, E = 1.95 e−42) protein is aligned with five similar sequences (BLASTp nr, 59 – 76% identity; 1 e−126 <= E <= 4 e−83) from other insects, including two species of ant (Florida carpenter and red imported fire ant), wasp (a Braconid and the Jewel parasitoid), and Drosophila mojavensis. Canonical Bap31 proteins regulate ER-stress-mediated apoptosis. Similar to these proteins, the Lh VLP protein is predicted to encode three transmembrane helices.
(F) The Lh VLP knottin-like (pfam11410 domain, E = 5.19 e−03) protein is aligned with five similar sequences (BLASTp nr, 33 – 52% identity; 3 e−08 <= E <= 2 e−02), four from insect species and one from the eudicotyledon, Mesembryanthemum crystallinum. Among the most similar putative homologs are sequences from diverse insects: Hymenoptera (sawfly and wasp), Diptera, and Hemipteran species. The sequence contains a putative secretion signal motif and three predicted disulfide bridges. 40% of the VLP sequence shows notable conservation (54% identity) when compared to a secreted ion channel toxin from the spider Chilobrachys guangxiensis (Arachnida: Theraphosidae). Knottins are classified as a cystine-rich plant antimicrobial peptide family.
(G) The Lh VLP hemolymph juvenile hormone binding protein (JHBP)-like (pfam06585 domain, E = 6.10 e−17) protein is aligned with four similar sequences (BLASTp nr, 24 – 30% identity; 1 e−11 <= E <= 6 e−03) from a wheat stem sawfly, the Panamanian leafcutter ant, the diamondback moth, and Drosophila melanogaster. No high identity homologs were found, but moderately similar homologs were found encoded by wasps, ants, and arthropods, in general. Homologs in Drosophila spp. were found, but are not functionally well characterized. The regions of similarities are within 80 to 90% of the length of the VLP protein sequence, but largely only in comparisons to putative homologs from other hymenoptera.
Data S2: Lh VLP GTPase alignments (Related to Figs 2, 3A, & 4A – C)
This document provides alignments of select small and large VLP GTPases (including SmGTPases01/LgGTPases01) with putative homologs/paralogs from (A) Lh; (B) prokaryotic and eukaryotic species; (C) Ganaspis sp.1 (G1); and (D, E) Leptopilina clavipes (Lc). The far N-and C-termini show the most variation among the sequences and were trimmed. The residue range displayed is indicated after the species of origin. The coloring scheme is according to physiochemical properties. ORF = open reading frame.
(A) Eight Lh VLP GTPases are presented here. Additional putative GTPase family members exist, but are not presented here. The first three sequences of ~ 300 residues, are SmGTPases, while the following five sequences of ~ 500 residues are LgGTPases. Among the eight family members shown here, the percent similarities within the N-terminal regions range from ~ 25 to 60% and average pairwise similarity over their full lengths is 45%. Only seven insertion/deletion sites exist in this 222 amino acid alignment and large blocks of identity (> 80% of the alignment length) are present. Highly similar secretion signal peptides are predicted at the N-termini of all GTPases (not shown).
(B) The five most-similar ORFs found in the NCBI TSA transcriptome for G1 (GAIW00000000.1) are shown aligned with Lh VLP Sm/LgGTPase01s. Like the Lh VLP GTPases, several putative G1 GTPases have predicted secretion signal peptides (not shown). Potential G1 homologs demonstrate >= 31% and 35% identity (E <= 3e−03 and 4e−59) to Lh VLP SmGTPase01 and LgGTPase01, respectively (given 75% alignment coverage).
(C) The five most-similar ORFs found in the NCBI TSA transcriptome for Lc (GAXY00000000.2) are shown aligned with Lh VLP Sm/LgGTPase01s. Similar to Lh and G1 GTPases, multiple Lc GTPases have predicted signal sequence peptides. The Lc transcripts identified show similarity in the N-terminal G domain, but are generally much shorter than the Lh VLP GTPases. Lh VLP SmGTPase01 shows <= 34% identity to Lc transcripts (>= 75% alignment criterion, E >= 2e−34) and LgGTPase01 shows <= 44% identity (E >= 3e−99), respectively. Lc also parasitizes Drosophila spp., but its comparative virulence and phylogenetic distance are poorly characterized in comparison to Lh and Lb.
(D) The most similar Lh and Lc sequences have been extracted from (C) and are shown here.