

Abstract
Severe burn injuries initiate a cascade of downstream events, culminating in multiple organ dysfunction, sepsis, and even death. The elderly are in particular vulnerable to such outcomes, due primarily to a scarcity of knowledge on trauma progression at the biomolecular level in this population. Mitochondria, the cellular powerhouses, have been increasingly scrutinized recently for their contribution to trauma outcomes. We hypothesized that elderly have a worse outcome compared to adult patients due to failed recovery of hepatic mitochondria. Using a murine model of burn injury, Seahorse respirometry and functional proteomic assays, we demonstrate the impact of thermal trauma on hepatic mitochondrial respiration in adult and aged mice. While the mitochondria in adults rebound from the initial insult within 7 days of the injury, the older animals display delayed recovery of mitochondrial bioenergetics accompanied by uncoupling and an oxidative environment. This is associated with a state of increased protein oxidation and nitrosylation, along with increases in circulating mtDNA, a known damage-associated molecular pattern. These findings suggest that hepatic mitochondria fail to normalize after trauma in aged mice and we suggest that this cellular failure is associated with organ damage and subsequently increased morbidity and mortality in elderly burn patients.
Keywords: Trauma, Oxidative phosphorylation, Electron transport chain, Oxidative stress, Elderly
Introduction
Aging is associated with the steady decline of physical and mental faculties, thus rendering the elderly more susceptible to a battery of diseases. Additionally, the risk of accidental injuries from the likes of falls, every day accidents, as well as fires and scald burns increases exponentially [1]. Severe burns, in particular, represent a unique challenge to health care, as the resultant hypermetabolism leads to vast catabolism, sepsis and organ failure [2], [3] and [4]. Although the last decades have brought about great advances in burn care, including meticulous nutritional guidelines and improved wound healing, these disproportionally benefit the young adult population, and survival in the elderly fraction of society significantly lags behind [5]. We and others have recognized the importance of elderly burn care research and development of elderly specific therapies. Approximately 10–15% of patients enrolled in burn units are elderly [3], [4] and [5]. This is even more important in light of a projected elderly population of 1.5 billion by 2050 [1]. It is therefore paramount to take into account the socio-economic impact of burns and severe trauma on the elderly populace.
Aging naturally affects organ structure and function. For instance, it has been shown in numerous animal models that the liver accumulates more products of oxidative damage, such as protein carbonyls, lipid peroxides and 8-oxo-2-deoxyguanosine with age [6] and [7]. The challenge of maintaining organ function after an injury is even more central for survival than previously thought [3]. Not only does trauma affect the liver at the organ level, but also at the cellular and cell organelle levels, with mitochondria particularly prone to disruptions. The detrimental effect trauma has on mitochondrial bioenergetics is multifactorial. Following severe burn injury, increased lipolysis leads to an augmentation in circulating free fatty acids which exert their lipotoxic effects on the electron transport chain [8]. For instance, linoleic acid inhibits mitochondrial complexes I and V, subsequently dropping ATP levels and causing necrosis [9]. Furthermore, increased levels of pro-inflammatory cytokines can possibly induce mitochondrial DNA (mtDNA) damage which correlates with the induction of apoptosis [10]. These inflammatory stimuli provoke the expression of inducible nitric oxide synthase and an increase in the gaseous radical nitric oxide (NO) [11] and [12]. The latter combines via radical-radical coupling with superoxide to produce peroxynitrite, a strong oxidant which further exacerbates mtDNA damage [13]. As complex I, III, IV and V of oxidative phosphorylation are partly encoded by mtDNA, depletion of this biomolecule adversely affects ATP levels and the resultant leak of electrons leads to a surge in nitro-oxidative stress [14]. This is particularly problematic in the elderly population, as an age-associated decline in complex I, complex IV and Mn-superoxide dismutase as well as the number of functional mitochondria in the liver could further exacerbate the systemic dysfunction occurring post-trauma and delay recovery [15] and [16]. For instance, traumatic brain injury in older animals causes increased mitochondrial dysfunction versus their younger counterparts with augmented levels of oxidative stress markers [17].
The liver is the central metabolic hub which governs the fate of intermediary metabolites such as glucose, fatty acids and amino acids. In addition, this organ acts as gatekeeper, limiting the access of noxious compounds to general blood circulation [18]. Indeed, the orchestration of these processes demands intense energy requirements and as such, the presence of functional, coupled mitochondria is imperative. The evidence for the liver necessitating aerobic energy production is stark. This organ generates ATP at a rate of approximately 30 mM/min, which is 3–6 times higher than the levels seen in human skeletal muscle and the visual cortex [19]. When hepatocytes, which make up approximately 70–85% of the liver’s mass, are depleted of mitochondrial DNA, the level of ATP production in the liver drops 10-fold, further emphasizing the importance of oxidative phosphorylation [20]. Hepatic mitochondrial function and the lack thereof have been associated with such pathologies as cirrhosis, cancer and type 2 diabetes mellitus [18]. Aging adversely impacts the liver’s ability to withstand trauma as lower concentrations of cytoprotective heat shock protein 70 renders it vulnerable to hypoxic and ischemic stress [21] and [22]. Furthermore, alterations in the expression of heme oxygenase-1 in the elderly may decrease their stress tolerance [23].
Here, using a murine model of burn injury, we demonstrate how thermal trauma induces hepatic mitochondrial hypometabolism at 24h post-burn in both adults and aged mice. This is associated with changes in enzymes charged with remediating oxidative stress. Within 7 days of injury, mitochondrial bioenergetics in adult mice are restored and higher than their unburned counterparts, perhaps indicative of a compensatory recovery. Aged mice, however, display an inability of mitochondria to fully recover from the initial trauma, with an uncoupled electron transport chain that accompanies an increase in oxidized and nitrosylated proteins and faulty systems to defend against reactive oxygen species (ROS). We postulate that this phenomenon may underlie their increased susceptibility to death from severe burns.
Material and Methods
Mouse model
Male C57BL/6 mice (Jackson Laboratory) were housed at ambient temperature and cared for in accordance with the Guide for the Care and Use of Laboratory Animals. All procedures performed in this study were approved by the Sunnybrook Research Institute Animal Care Committee. Sham mice underwent identical experimental procedures, with the exception of the burn injury. Adult mice were 8 weeks of age, while the aged mice were selected for this study at 50 weeks. Mice were anesthetized by inhalation of 3–5% isoflurane and given a 0.1 mg/kg intraperitoneal (i.p.) injection of buprenorphine. Approximately 40% of the dorsum and ventral regions were shaved with electrical clippers and ~ 1cc of lactated ringers (LR) solution was injected under the skin along the spinal column as well as i.p. Full thickness third degree burns covering 30 % of the total body surface area (TBSA) were achieved by immersing the back of the mice at 98°C for 10 s and the ventral region for 2 s to avoid organ damage. The ventral region was included to help reach a 30% TBSA burn without damaging limbs, the tail or head of the animals. Burned mice were subsequently housed individually in sterile cages and fed ad libitum until sacrifice. Select mice (n = 3 for sham; n = 5 for burn groups) were euthanized at 1 and 7 days post-thermal injury. Livers were harvested for further analysis and blood collected following cardiac puncture.
Mitochondrial isolation and respirometry
Freshly-excised livers were minced in 10 volumes of mitochondrial isolation buffer (MHSE + BSA; 210 mM mannitol, 70 mM sucrose, 5mM HEPES, 1 mM EGTA, 0.5% (w/v) fatty acid-free BSA, pH 7.2). The tissue was then homogenized using a Teflon glass homogenizer with 6 strokes and filtered through three layers of cheesecloth. Mitochondria were isolated via differential centrifugation. Briefly, the homogenate was centrifuged at 600 g for 10 min and the supernatant decanted into a new tube. This fraction was centrifuged at 9000 g for 10 min to afford a mitochondrial pellet. The resultant supernatant which contains soluble proteins was subjected to a BCA protein assay (Thermo Scientific) and set aside for other assays. A clear lipid layer surrounding the mitochondria was carefully aspirated and the pellet re-suspended in 200 uL of MHSE + BSA. BCA assays were performed to gauge protein concentrations. Mitochondrial bioenergetics were assessed using a Seahorse XF96 analyzer (Agilent Technologies). Mitochondrial respiration in a coupled state (10 μg/well) was measured in mitochondrial assay solution (MAS; 220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA and 0.2% (w/v) fatty acid-free BSA, pH 7.2 at 37°C) containing succinate as a substrate (10 mM) and rotenone (2 μM). State 3 respiration (phosphorylating respiration) was triggered via the injection of a cocktail containing 4 mM ADP along with 10 mM pyruvate, 2.5 mM glutamate and 2.5 mM malate. State 4o respiration was assessed by the addition of 2.5 μg/mL oligomycin, while maximal uncoupler-stimulated respiration was observed following the injection of 4 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP). Antimycin A (4 μM), a Complex III inhibitor, was added at the end of the experiment to inhibit mitochondrial respiration, as described [24]. Liver mitochondria from 3 sham mice and 5 burned mice were included in each Seahorse analysis, and every sample analyzed in triplicate. Seahorse XF Stress Test Report Generators were used to analyze coupling percentages. The Seahorse XF Wave software was used to group the respiration data from separate mice into a single representative curve.
Mitochondrial ROS production
Liver mitochondria isolated as described above were tested for ROS emission via the fluorogenic dye 2,7-dichlorodihydrofluorescein-diacetate (DCFDA). Freshly isolated mitochondria were diluted to 0.5 mg/ml in reaction buffer (20 mM Tris base, 125 mM sucrose, 2 mM MgCl2, 4 mM KH2PO4, 1 mM EDTA) in a 96 well plate. Twenty μM DCFDA was added and mitochondria were warmed to 37°C for 5 min, at which point baseline ROS measurements were recorded (λex = 495 nm; λem = 529 nm). Mitochondria were subsequently treated with 5 mM pyruvate and 3 mM malate to drive respiration and DCFDA measurements were taken at 5 min and 15 min (Synergy H4 Hybrid Reader; BioTek). Data were normalized to background fluorescence. All mitochondrial experiments were performed within 4 h of the isolation.
Electrophoresis and in-gel activity assays
Blue native (BN) polyacrylamide gel electropohoresis (PAGE) was performed as described [25]. Briefly, membrane and cytosolic fractions were isolated and prepared in a non-denaturing buffer (50 mM Bis-Tris, 500 mM ε-aminocaproic acid, pH 7.0, 4°C) at a concentration of 4 μg/μL. Ten percent n-dodecyl β-D-maltoside was added to mitochondrial fractions to solubilize membrane bound proteins. A 2–10% or 4–16% gradient gel was prepared [Bio-Rad MiniProteanTM 2 system] using 1 mm spacers to ensure optimal protein separation. Sixty micrograms of protein was loaded into each well and electrophoresed under native conditions in anode buffer (50 mM Bis-Tris, pH 7 at 4°C) at 80 V to ensure proper stacking, then at 300 V for proper migration through the gel, ensuring the current does not surpass 25 mA. The blue cathode buffer (50 mM Tricine, 15 min Bis-Tris, 0.02% (w/v) Coomassie G-250, pH 7 at 4°C) was utilized to help visualize the running front and was exchanged to a colourless cathode buffer (50 mM Tricine, 15 mM Bis-Tris, pH 7 at 4°C) when the running front was halfway through the gel. Upon completion, the gel was equilibrated in reaction buffer (25 mM Tris-HCl, 5 mM MgCl2, pH 7.4) for 15–30 min. Complex I was detected by the addition of 5 mM KCN, 1 mM NADH and 0.4 mg/mL iodonitrotetrazolium chloride (INT). Analysis of complex II was achieved with a reaction mixture containing 20 mM succinate, 0.2 mM phenazine methosulfate and 25 mg nitrotetrazolium blue in 10 mL of 5 mM Tris/HCl, pH 7.4. Complex III activity was assessed with 5 mg diaminobenzidine (DAB) in 10 mL of 50 mM sodium phosphate, pH 7.2. Complex IV was assayed by the addition of 10 mg/mL of DAB, 10 mg/mL cytochrome C, and 562.5 mg/mL of sucrose. A reaction mixture containing 35 mM Tris, 270 mM glycine, 14 mM MgCl2, 0.2% Pb(NO3)2 and 8 mM ATP permitted the visualization of Complex V activity [26].
For in-gel glutathione peroxidase (GPx) activity, gels were washed 3 times for 10 min in ddH2O containing 1 mM GSH. Afterwards, they were incubated in a 0.008% cumene hydroperoxide solution for 10 min. Following two 5 min ddH2O washes, GPx enzyme activity was elucidated by mixing in 1% ferric chloride and 1% potassium ferricyanide solutions on the gel. The reaction mixture for catalase was similar, but GSH was omitted and 2% ferric chloride and potassium ferricyanide solutions were used, as described [27]. Reactions were stopped using a destaining solution (40% methanol, 10% glacial acetic acid) once the activity bands had reached the desired intensity. Acetic acid was omitted for the complex V reaction, as the acid would dissolve the lead phosphate precipitate. Reactions performed without the addition of a substrate or cofactor in the reaction mixture ensured specificity. Densitometry was performed using Image J for Windows.
Oxidized protein assays
Total protein carbonyl content was assessed by first mixing 1 mg of soluble protein with 0.2% (w/v) 2,4-dinitrophenylhydrazine (DNPH). After 60 mins of incubation at room temperature, 200 μL of 50 % (w/v) TCA was added to each sample. The resulting precipitate was removed by centrifugation at 18,000 g for 10 min. The pellet was washed 3 times with 10 % (w/v) TCA followed by centrifugation at 18,000 g for 10 min. The protein pellet was then washed thrice with a solution of ethylacetate:ethanol (1:1). The pellet was dissolved in 600 μL of 6 M guanidine and the absorbance was measured at 370 nm (ε = 21.5 M−1 cm−1).
Immunoblot experiments
To characterize the expression of mitochondria-localized superoxide dismutase (MnSOD) and nitrosylated tyrosine (NO-Tyr) residues, Western blots were performed. Following SDS-PAGE of liver lysate (30 ug) from sham and burned mice, the proteins were transferred using a Trans-Blot Turbo Transfer System (Bio-Rad). Upon completion, the nonspecific binding sites on the membrane were blocked using 5% (w/v) non-fat skim milk for 1 h. The membrane was then washed twice in TTBS (12.1 g Tris, 40 g NaCl, 2% Tween 20; pH 7.6 at 4°C) for 5 min. Blots were incubated overnight with the primary antibody (1:1000 α/β- tubulin; 1:5000 GAPDH; 1:1000 MnSOD; 1:1000 NO-Tyr). The membrane was washed twice in TTBS for 5 min, followed by incubation with secondary antibody, which consisted of horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies (1:3000) for 1 h. The membrane was washed in TTBS twice for a period of 5 min and detection of the desired protein was achieved with a BioRad ChemiDoc Imaging System. All antibodies were obtained from Abcam. SuperSignal West Pico Chemiluminescent Substrate was a product of Thermo Scientific Inc. Densitometry was performed using Image J for Windows.
Quantification of plasma mtDNA
Fresh blood was collected in EDTA tubes and centrifuged for 20 min at 3000 rpm. Mitochondrial DNA was quantified with real-time qPCR using a StepONE Plus PCR System (Applied Biosystems, California, USA) and Biorad SsoAdvanced Universal SYBR® Green Supermix (Bio-Rad, California, USA) according to manufacturer’s directions. Primers were selected that amplify the NADH dehydrogenase subunit 1 motif on the mitochondrial genome. Primers used were as follows; NAD1 (F′: CCC ATT CGC GTT ATT CTT, R′; AAG TTG ATC GTA ACG GAA GC). Optimization was performed to determine a 1:20 dilution ratio of plasma in nuclease free sterile water and 2 ul of starting material was used per reaction. Relative ND1 copy number was calculated based on the threshold cycle. All samples were run in duplicate, simultaneously with negative controls. For liver, 50–100 mg of tissue was homogenized in 1 mL of TRIzol® reagent and we then proceeded to phase separation as per manufacturer’s directions. The upper aqueous phase (containing RNA) was separated from the lower red phenol phase and interphase (containing DNA). Five hundred μL of back enzyme extraction buffer (BEB; 4 M guanidine thiocyanate, 50 mM sodium citrate, 1 M tris free base) was added to the interphase and lower red phenol phase. Samples were mixed well by inverting several times and then allowed to sit at room temp for 10 min. Following this step, samples were centrifuged at 3000 g for 30 min at 4°C and the resulting upper phase was transferred to fresh tubes. DNA was precipitated by adding 400 uL of isporopanol and mixed by inverting several times. Samples were allowed to sit at room temp for 5 min and then centrifuged at 12,000 g for 5 min. Supernatant was discarded and the resulting pellet was washed with 75% ethanol and this step was repeated 3 times. The pellet was allowed to dry for 5–10 min and re-suspended in ~50 ul of sterile ddH2O. Spectrophotometry was performed to assess DNA quantity and samples were then diluted to a concentration of 2 ng/ul. Relative levels of mtDNA were assessed via qPCR (as above) using approximately 20 ng of template with the aforementioned NAD1 primers.
Statistical analysis
Data were expressed as mean and standard error of the mean (SEM). Statistical correlations of the data were checked for significance using the Student t test or one-way ANOVA followed by Tukey post-hoc (p ≤ 0.05). Three sham mice and five burned mice were included in each analysis.
Results
Mitochondrial function at 24 h post-burn
Burns, similar to other traumas, are initially characterized by an ‘ebb’ phase, where metabolism and resting energy expenditure (REE) are diminished [28]. Therefore, we observed the impact of severe thermal trauma (30% TBSA) on mitochondrial respiration at an acute time point (1 day post-burn). We initially observed that, while 100% of the adult mice receiving a 30% TBSA burn survived until the 7 day time point, the survival rate for the aged mice was 90%, with the deaths occurring within the 24h time point (Fig. 1A). At 24 h post-burn, adult mice displayed a significantly lower oxygen consumption rate (OCR) (Fig. 1B; Suppl. Fig. 1A–C), a finding that was ascertained via in-gel activity assays for each individual complex of the electron transport chain (ETC; Fig. 1C). While the initial decrease of mitochondrial activity in the aged mice was not as severe as their adult counterparts, there is a clear effect on respiration rates (Fig. 1D; Suppl. Fig. 1A–C), with significant decreases in basal as well as FCCP-stimulated maximal respiration and lowered activity of each complex, with the exception of III, which was not significantly affected (Fig. 1E). Despite the lowered activity of the respiratory complexes, there were surprisingly no significant differences in either the aged and adult cohorts in the production of ROS by isolated mitochondria, as measured fluorescently by the oxidation of DCFDA (Fig. 2A, adult; 2B, aged). We next sought to analyze the activity of mitochondria-localized glutathione peroxidase (mGPx; Fig. 2C, adult; 2D, aged) and the expression of MnSOD (Fig. 2E, adult; 2F, aged) to determine if these were contributing to the control of ROS production. While in the adult mitochondria, these enzymes appear to be diminished, coinciding with the decreased activity of mitochondrial complexes, no significant differences were noted in the aged mice, likely due to a less drastic drop in mitochondrial respiration (Fig. 1C, D). Catalase activity was decreased in both the adult and aged burn groups (Fig. 3A, adult; 3B, aged) but no changes were evident in the activity of cytoplasmic glutathione peroxidase (cGPx) (Fig. 3C, adult; 3D, aged).
Figure 1.

An analysis of mitochondrial dynamics in adult and aged mice at 24 h post-burn. (A) Respiration profiles of adult sham (■) and burn (30% TBSA; ■) hepatic mitochondria from a Seahorse XF96 analyzer. (B) Representative images from in-gel enzyme activity assays for complexes I–V of the ETC in adult mice. (C) Respiration profiles of aged sham (■) and burn (■) hepatic mitochondria from a Seahorse XF96 analyzer (D) Representative images from in-gel enzyme activity assays and densitometric measurements of complexes I–V of the ETC in aged mice. (E) Survival curve of adult and aged mice following a 30% TBSA burn. n=3 and SEM for sham mice; n=5 and SEM for burnt mice. *p ≤ 0.05; **p≤ 0.01.
Figure 2.

ROS production and oxidative defense in adult and aged mice at 24 h post-burn. DCFDA oxidation of adult (A) and aged (B) hepatic mitochondria at basal levels (DCFDA; no substrate), 5 min with substrate (5 mM pyruvate, 3 mM malate) and 15 min with substrate. Densitometric measurements and representative images of mitochondrial glutathione peroxidase (mGPx) from adult (C) and aged (D) mice. Representative Western blots and densitometry for MnSOD from adult (E) and aged (F) mice. n=3 and SEM for sham mice; n=5 and SEM for burnt mice. *p ≤ 0.05 (□, Sham; ■, Burn).
Figure 3.

Activity of cytoplasmic enzymes mediating defense against ROS in adult and aged mice at 24 h post-burn. Representative in-gel activity assays and densitometric measurements for catalase from adult (A) and aged (B) mice post-burn. Densitometric measurements and representative activity gel images for cytoplasmic glutathione peroxidase (cGPx) from adult (C) and aged (D) mice post-burn. n=3 and SEM for sham mice; n=5 and SEM for burnt mice. *p ≤ 0.05 (□, Sham; ■, Burn).
Adult mitochondrial function at 7 days post-burn
At one week, the respiration rate of hepatic mitochondria was increased over the sham mice, perhaps indicative that these mice have entered the hypermetabolic or ‘flow’ phase of metabolic progression post-trauma (Fig. 4A; Suppl. Fig. 1D–F) [29]. The coupling percentage, a measure of how much of the ETC is dedicated to the synthesis of ATP, is unchanged despite this observed increase in the OCR (Fig. 4B). Isolated mitochondria were assessed for ROS formation using DCFDA, and no significant differences were observed over a 15 min time course with malate and pyruvate as substrates (Fig. 4C). In-gel activity assays were performed to confirm the results obtained by respirometry and indeed, each individual complex was more active as ascertained by densitometric measurements (Fig. 4D). Although respiratory parameters appear to be increased, the concentration and expression of protein carbonyl and NO–Tyr residues, respectively, remain unchanged from the sham mice (Fig. 5A, B). As there are no variances in the production of ROS or the coupling percentage between adult sham and burned mice, this is to be expected. Furthermore, there is a marked increase in the activity and expression of mGPx (Fig. 5C) and MnSOD (Fig. 5D), respectively, coinciding with the increased mitochondrial respiration at day 7 post-burn. Changes in mitochondrial respiration were not accompanied by variations in the activity of the cytoplasmic enzymes cGPx or catalase in these mice (Fig. 5E, F).
Figure 4.

An analysis of mitochondrial dynamics in adult mice at 7 days post-burn. (A) Respiration profiles of adult sham (■) and burn (■) hepatic mitochondria from a Seahorse XF96 analyzer. (B) Coupling percentage of sham and burn mitochondria as determined by the Seahorse XF Stress Test Report Generator. (C) DCFDA oxidation of adult hepatic mitochondria at basal levels (DCFDA; no substrate), 5 min with substrate (5 mM pyruvate, 3 mM malate) and 15 min with substrate. (D) Representative images from in-gel activity assays and densitometric measurements for complexes I–V of the ETC in adult mice. n=3 and SEM for sham mice; n=5 and SEM for burnt mice. *p ≤ 0.05; **p≤ 0.01.
Figure 5.

ROS markers and enzymes mediating defense against ROS in adult mice at 7 days post-burn. (A) Protein carbonyl concentrations in hepatic mitochondria. (B) Representative Western blot image and densitometric measurements of nitro-tyrosine levels normalized to GAPDH. (C) Representative image and densitometric measurements of mGPx following in-gel enzyme activity assays. (D) Densitometric measurements and representative Western blot image of MnSOD (E) Densitometric measurements and representative image of catalase following in-gel enzyme activity assays. (F) Representative image from in-gel enzyme activity assays for cGPx with associated densitometric measurements. n=3 and SEM for sham mice; n=5 and SEM for burnt mice. *p ≤ 0.05 (□, Sham; ■, Burn).
Mitochondrial function of aged mice at 7 days post-burn
While the adult mice appear to exhibit a compensatory recovery of mitochondrial bioenergetics at 7 days post-burn, aged mice possess severely dysfunctional hepatic mitochondria, as evidenced by the lowered OCR of these isolated organelles in the burned group (Fig. 6A; Suppl. Fig. 1D–F). Indeed, it seems as though these mitochondria fail to respond to ADP, malate, pyruvate and glutamate, a cocktail designed to stimulate respiration, and there is a significant drop in the coupling percentage (Fig. 6B), indicative of lowered ATP production compared to the sham group. In line with the diminished coupling of the ETC, these mitochondria produce significantly more ROS, as demonstrated via incubation with substrate and DCFDA (Fig. 6C). To ascertain the respirometry results, individual complexes were probed following BN-PAGE and the densitometric measurements are presented here, showing a significant decrease in the activities of complex II, IV and V. While no changes were observed in complex I, there is an inexplicable increase in the activity of complex III in the aged burnt mice (Fig. 6D). This is an interesting finding, as the latter two complexes are the major sites of ROS production within the mitochondria, thus explaining the observed increase in DCFDA oxidation [30].
Figure 6.
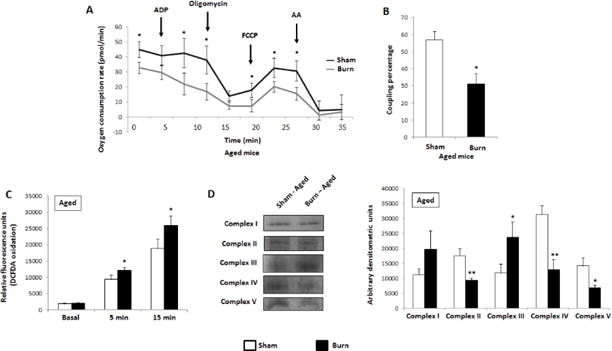
An analysis of mitochondrial dynamics in aged mice at 7 days post-burn. (A) Respiration profiles of aged sham (■) and burn (■) hepatic mitochondria from a Seahorse XF96 analyzer. (B) Coupling percentage of sham and burn mitochondria as determined by the Seahorse XF Stress Test Report Generator. (C) DCFDA oxidation of aged hepatic mitochondria at basal levels (DCFDA; no substrate), 5 min with substrate (5 mM pyruvate, 3 mM malate) and 15 min with substrate. (D) Representative images and densitometric measurements from in-gel enzyme activity assays for complexes I–V of the ETC in aged mice. n=3 and SEM for sham mice; n=5 and SEM for burnt mice. *p ≤ 0.05; **p≤ 0.01
Nitro-oxidative stress and mitochondrial damage in the aged animals
Given the defects in these mitochondria, we expected to find an increase in damage markers in the liver of aged burnt mice. Indeed, there is an increase in protein carbonyl (Fig. 7A) and NO-Tyr residues (Fig. 7B), markers of ROS formation and nitro-oxidative stress. To assess systemic mitochondrial dysfunction, the levels of circulating mitochondrial DNA (mtDNA), a known damage-associated molecular pattern (DAMP), were measured in plasma [31]. While mtDNA was barely detectable in the adult mice, there was a large increase in this biomolecule in both aged sham and burn mice (Fig. 7C). In addition, mtDNA from liver mitochondria was analyzed and there was a significant decrease in the aged burn group versus the uninjured sham mice (Fig. 7D). Furthermore, the total activity of GPx in the burn cohort was significantly lowered compared to the sham, indicating a decreased ability of these mice to protect against oxidative damage (Fig. 7E). No significant variations were detected in the activity and expression of catalase and MnSOD, respectively (Fig. 7F, G).
Figure 7.

ROS markers, mitochondrial damage and enzymes mediating defense against ROS in aged mice at 7 days post-burn. (A) Protein carbonyl concentrations in hepatic mitochondria. (B) Representative Western blot image and densitometric measurements of nitro-tyrosine levels normalized to GAPDH. (C) A comparison of plasma mtDNA levels in sham and burned animals from adult and aged cohorts. (D) A comparison of liver mtDNA levels in sham and burned animals from adult and aged cohorts. (E) Densitometric measurements of GPx with a representative image following in-gel enzyme activity assays. (F) Densitometric measurements of catalase activity with a representative image following in-gel enzyme activity assays. (F) Representative Western blot of MnSOD with associated densitometry. n=3 and SEM for sham mice; n=5 and SEM for burnt mice. *p ≤ 0.05 (□, Sham; ■, Burn).
Discussion
This study describes functional alterations in hepatic mitochondria following severe-burn injury in both adult and aged mice over time. The early impact of trauma on oxidative phosphorylation induces a decrease in energy production and respiration while stifling the activities of enzymes designed to control nitro-oxidative stress. Although the hepatic mitochondrial profile of adult mice exhibits a compensatory recovery, this biomolecular event is severely delayed in an aged murine model of burn injury. The importance of functional hepatic mitochondria is multifactorial. Although no significant increase in apoptosis was noted at the time points reported here, prolonged mitochondrial abnormalities may be associated with increased nitro-oxidative stress and sensitize hepatocytes to apoptosis/necrosis as well as the subsequent recruitment of infiltrating immune cells and consequently, inflammation [32]. Furthermore, faulty mitochondrial mechanisms pertaining to the mobilization and beta-oxidation of fatty acids precede the development of fatty liver disorders [33]. Insulin resistance, a key outcome of severe burn trauma, may be preceded by faulty mitochondrial bioenergetics [34]. Prolonged defects in mitochondrial ATP production and mtDNA mutations may lead to the development of hepatocellular carcinoma [35] and [36]. The findings described here are consistent with other conditions, such as sepsis, where it has been demonstrated that mitochondrial function in aged animals and elderly humans is significantly worse than their younger counterparts [37] and [38].
While the initial insult following severe burns and trauma is localized, the cascade of events which initiates downstream complications begins immediately. For instance, the coupling of ETC activity to ATP production diminishes in skeletal muscle by 3 h post-burn in a 30% TBSA mouse model [39]. In humans, circulating free fatty acids in elderly burn patients are significantly higher than what is seen in adult burns [3]. Taken together, the evidence points to an exaggerated inflammatory response and increased lipolysis in the elderly being primary factors for hepatic dysfunction following severe burns. These phenomena likely contribute to the increased mortality seen in burn patients. Indeed, a burn injury covering 20% TBSA leads to a 95% survival rate in adult patients, a figure which drops to approximately 25% in the elderly populace [5].
Here we show that, during the acute phase of burn injury, hepatic mitochondrial function is diminished in both adult and aged mice. The drop in mitochondrial respiratory activity coincides with the reported decrease in oxygen consumption during the ‘ebb’ phase of trauma [40]. Furthermore, mitochondrial anti-oxidative stress enzymes are also decreased, particularly in the adult cohort. At day 7 post-burn, liver mitochondria from the injured adult mice have fully recovered from the initial insult, and are respiring more than the non-burnt cohort. This compensatory recovery is supported by both the Seahorse data and in-gel enzyme activity assays and may help generate sufficient ATP to remediate the noxious effects of the burn injury [41]. While this increase in oxidative phosphorylation may contribute to the hypermetabolic phenotype post-burn, we postulate that the lack of significant differences with regards to ROS production and downstream effects of nitro-oxidative stress may be indicative of healthy liver energetics.
In the aged mice, however, liver mitochondria remain severely uncoupled and faulty even at a week post-injury. This is reflected in the increased levels of both oxidized and nitrosylated proteins. It’s of interest to note that, even in sham mice, the coupling percentage and response to substrate are both attenuated compared to adult sham mice, likely a result of the natural aging process which is known to exert its effects on mitochondrial bioenergetics. For instance, Short et al. have demonstrated that the capacity of skeletal muscle mitochondria to produce ATP declines by approximately 5% per decade [42]. While no changes are observed in the activity of catalase and MnSOD, there is a decline in GPx functionality in the burnt cohort, perhaps indicative of the exhaustion of this tripeptide in these mice. One particularly interesting finding from this research is the increase in activity of complex III at day 7 in aged mice with severe thermal injury. As in-gel enzyme activity assays are semi-quantitative, it cannot be ruled out that this finding is an artefact of this technique, yet the unique role of this protein makes that unlikely. As opposed to complexes I and II which produce superoxide in the mitochondrial matrix, complex III can release this moiety to the intermembrane space where it infiltrates the cytosol. Considerable evidence has implicated complex III-derived ROS in the stabilization of hypoxia-inducible factor 1-alpha (HIF-1α). Given the role of HIF-1α in regulating glucose uptake and metabolism, the increased activity of this complex in the burned mice and its plausible signalling role merit further investigation [43]. An important limitation of the work presented here is the age of the mice (50 weeks), which is not geriatric, but was chosen as a model for the biomolecular processes which underlie aging without the extreme mortality that would result from inducing burns in older mice. Indeed, others have shown that in mice around 50 weeks of age, mitochondrial processes begin to decline, and we hypothesize that the failed recovery of these organelles would be more pronounced in geriatric animals [44] and [45].
The findings discussed herein demonstrate how the recovery of hepatic mitochondrial bioenergetics in an aged murine model of burn injury is delayed versus their younger counterparts. While it is known that dysfunctional mitochondria are a staple of grave injuries, this is the first comparison of these organelles and their timed reaction to severe trauma between age groups. The persistence of faulty mitochondrial dynamics in the elderly population may contribute to the development of liver disorders and adversely affect the ability of this organ to detoxify noxious compounds. Furthermore, the increased circulation of mtDNA serves as a DAMP which can regulate the innate immune response via such paths as the activation of the NLRP3 inflammasome or TLR9 pathway, thus exacerbating the inflammatory response to trauma [46]. Pharmaceutical stratagems to negate mitochondrial damage, such as the antioxidants Coenzyme Q10 and metalloporphyrins, or bolster its function via the biogenesis of this organelle, may be of particular benefit to the elderly populace for whom trauma outcomes are significantly worse than adults.
Supplementary Material
Highlights.
Fluctuations in mitochondrial bioenergetics underlie metabolic progression post-burn.
Both adult and elderly mice have lower hepatic bioenergetics 24 h after thermal trauma.
Adult mice bioenergetics go through a compensatory recovery by 7 days following injury.
Elderly mice exhibit a delay in their recovery of mitochondrial dynamics at this time point.
Elderly mice mitochondria produce more reactive oxygen species and the products thereof.
Acknowledgments
Abdikarim Abdullahi is a recipient of the Vanier Canada Graduate Scholarship from the Canadian Institutes of Health Research.
Funding: This work was supported by grants from the Canadian Institutes of Health Research (#123336), the Canada Foundation for Innovation Leader’s Opportunity Fund (#25407) and National Institutes of Health (2R01GM087285-05A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors have no conflict of interest to declare.
Author’s contributions: CA performed experiments and wrote portions of the manuscript; TS, AA and AP performed experiments; MJ guided the experiments and wrote portions of the manuscript.
References
- 1.Poulose N, Raju R. Aging and injury: alterations in cellular energetics and organ function. Aging Dis. 2014;5:101–108. doi: 10.14336/AD.2014.0500101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jeschke MG, Gauglitz GG, Kulp GA, Finnerty CC, Williams FN, Kraft R, Suman OE, et al. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS One. 2011;6:e21245. doi: 10.1371/journal.pone.0021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeschke MG, Patsouris D, Stanojcic M, Abdullahi A, Rehou S, Pinto R, Chen P, et al. Pathophysiologic Response to Burns in the Elderly. EBioMedicine. 2015;2:1536–1548. doi: 10.1016/j.ebiom.2015.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeschke MG, Pinto R, Costford SR, Amini-Nik S. Threshold age burn size associated with poor outcomes in the elderly after burn injury. Burns. 2016;42:276–281. doi: 10.1016/j.burns.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pham TN, Kramer CB, Wang J, Rivara FP, Heimbach DM, Gibran NS, Klein MB. Epidemiology outcomes of older adults with burn injury: an analysis of the National Burn Repository. J Burn Care Res. 2014;30:30–36. doi: 10.1097/BCR.0b013e3181921efc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age. Proc Natl Aca Sci USA. 2001;98:10469–10474. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salmon AB. Oxidative stress in the etiology of age-associated decline in glucose metabolism. Longev Healthspan. 2012;1:1–7. doi: 10.1186/2046-2395-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeschke MG, Abdullahi A, Burnett M, Rehou S, Stanojcic M. Glucose Control in Severely Burned Patients Using Metformin: An Interim Safety and Efficacy Analysis of a Phase II Randomized Controlled Trial. Ann Surg. 2016;264:518–527. doi: 10.1097/SLA.0000000000001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran JH, Mon T, Hendrickson TL, Mitchell LA, Grant DF. Defining mechanisms of toxicity for linoleic acid monoepoxides and diols in Sf-21 cells. Chem Res Toxicol. 2001;14:431–437. doi: 10.1021/tx000200o. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Xu M, Xo R, Mates A, Wilson GL, Pearsall AW, Grishko V. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthritis Cartilage. 2010;18:424–432. doi: 10.1016/j.joca.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Bhat NR, Zhang P, Bhat AN. Cytokine induction of inducible nitric oxide synthase in an oligodendrocyte cell line: role of p38 mitogen-activated protein kinase activation. J Neurochem. 1999;72:472–478. doi: 10.1046/j.1471-4159.1999.0720472.x. [DOI] [PubMed] [Google Scholar]
- 12.Lange M, Hamahata A, Enkhbaatar P, Cox RA, Nakano Y, Westphal M, Traber LD, et al. Beneficial effects of concomitant neuronal and inducible nitric oxide synthase inhibition in ovine burn and inhalation injury. Shock. 2011;35:626–631. doi: 10.1097/SHK.0b013e31820fe671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calcerrada P, Peluffo G, Radi R. Nitric oxide-derived oxidants with a focus on peroxynitrite: molecular targets, cellular responses and therapeutic implications. Curr Pharm Des. 2011;17:3905–3932. doi: 10.2174/138161211798357719. [DOI] [PubMed] [Google Scholar]
- 14.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmuker DL. Age-related changes in liver structure and function: Implications for disease? Exp Gerontol. 2005;40:650–659. doi: 10.1016/j.exger.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 16.Navarro A, Gomez C, López-Cepero JM, Boveris A. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physiol Regul Integr Comp Physiol. 2004;286:505–511. doi: 10.1152/ajpregu.00208.2003. [DOI] [PubMed] [Google Scholar]
- 17.Gilmer LK, Ansari MA, Roberts KN, Scheff SW. Age-related mitochondrial changes after traumatic brain injury. J Neurotrauma. 2010;27:939–950. doi: 10.1089/neu.2009.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Auger C, Alhasawi A, Contavadoo M, Appanna VD. Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front Cell Dev Biol. 2015;3:40. doi: 10.3389/fcell.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmid AI, Chmelik M, Szendroedi J, Krssak M, Brehm A, Moser E, Roden M. Quantitative ATP synthesis in human liver measured by localized 31P spectroscopy using the magnetization transfer experiment. NMR Biomed. 2008;21:437–443. doi: 10.1002/nbm.1207. [DOI] [PubMed] [Google Scholar]
- 20.Park KS, Nam KJ, Kim JW, Lee YB, Han CY, Jeong JK, Lee HK, et al. Depletion of mitochondrial DNA alters glucose metabolism in SK-Hep1 cells. Am J Physiol Endocrinol Metab. 2001;280:E1007–1014. doi: 10.1152/ajpendo.2001.280.6.E1007. [DOI] [PubMed] [Google Scholar]
- 21.Okaya T, Blanchard J, Schuster R, Kuboki S, Husted T, Caldwell CC, Zingarelli B, Wong H, Solomkin JS, Lentsch AB. Age-dependent responses to hepatic ischemia/reperfusion injury. Shock. 2005;24:421–427. doi: 10.1097/01.shk.0000181282.14050.11. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Behrns KE, Leeuwenburgh C, Kim J. Critical role of autophagy in ischemia/reperfusion injury to aged livers. Autophagy. 2012;8:140–141. doi: 10.4161/auto.8.1.18391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bloomer SA, Zhang HJ, Brown KE, Kregel KC. Differential regulation of hepatic heme oxygenase-1 protein with aging and heat stress. J Gerontol A Biol Sci Med Sci. 2009;64:419–425. doi: 10.1093/gerona/gln056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auger C, Appanna VD. A novel ATP-generating machinery to counter nitrosative stress is mediated by substrate-level phosphorylation. Biochim Biophys Acta. 2015;1850:43–50. doi: 10.1016/j.bbagen.2014.09.028. [DOI] [PubMed] [Google Scholar]
- 26.Wittig I, Karas M, Schagger H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol Cell Proteomics. 2007;6:1215–1225. doi: 10.1074/mcp.M700076-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc. 2010;5:51–66. doi: 10.1038/nprot.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks NC, Marshall AH, Qa’aty N, Hiyama Y, Boehning D, Jeschke MG. XBP-1s is linked to suppressed gluconeogenesis in the Ebb phase of burn injury. Mol Med. 2013;19:72–78. doi: 10.2119/molmed.2012.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hall KL, Shahrokhi S, Jeschke MG. Enteral nutrition support in burn care: a review of current recommendations as instituted in the Ross Tilley Burn Centre. Nutrients. 2012;4:1554–1565. doi: 10.3390/nu4111554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song BJ, Akbar M, Abdelmegeed MA, Byun K, Lee B, Yoon SK, Hardwick JP. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014;3:109–123. doi: 10.1016/j.redox.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. 2008;14:193–199. doi: 10.3748/wjg.14.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, Mikus CR, Laye MJ, Laughlin MH, Booth FW, Ibdah JA. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52:727–736. doi: 10.1016/j.jhep.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong L, Tan D, Bai R, Yeh K, Chang J. Molecular alterations in mitochondrial DNA of hepatocellular carcinomas: is there a correlation with clinicopathological profile? J Med Genet. 2004;41:e65. doi: 10.1136/jmg.2003.013532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- 37.Lorente L, Martín MM, López-Gallardo E, Blanquer J, Solé-Violán J, Labarta L, Díaz C, Jiménez A, Montoya J, Ruiz-Pesini E. Decrease of oxidative phosphorylation system function in severe septic patients. J Crit Care. 2015;30:935–939. doi: 10.1016/j.jcrc.2015.05.031. [DOI] [PubMed] [Google Scholar]
- 38.Coletta C, Módis K, Oláh G, Brunyánszki A, Herzig DS, Sherwood ER, Ungvári Z, Szabo C. Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: preclinical studies in a murine model of cecal ligation and puncture. Crit Care. 2014;18:511. doi: 10.1186/s13054-014-0511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porter C, Herndon DN, Bhattarai N, Ogunbileje JO, Szczesny B, Szabo C, Toliver-Kinsky T, Sidossis LS. Differential acute and chronic effects of burn trauma on murine skeletal muscle bioenergetics. Burns. 2016;42:112–122. doi: 10.1016/j.burns.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stahel PF, Flierl MA, Moore EE. “Metabolic staging” after major trauma - a guide for clinical decision making? Scand J Trauma Resusc Emerg. 2010;18:34. doi: 10.1186/1757-7241-18-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szczesny B, Brunyanszki A, Ahmad A, Olah G, Porter C, Toliver-Kinsky T, Sidossis L, et al. Time-Dependent and Organ-Specific Changes in Mitochondrial Function, Mitochondrial DNA Integrity, Oxidative Stress and Mononuclear Cell Infiltration in a Mouse Model of Burn Injury. PLoS One. 2015;10:e0143730. doi: 10.1371/journal.pone.0143730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci USA. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–666. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 44.Rogers NH, Landa A, Park S, Smith RG. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell. 2012;11:120–130. doi: 10.1111/acel.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nunes-Souza V, César-Gomes CJ, Da Fonseca LJ, Guedes Gda S, Smaniotto S, Rabelo LA. Aging increases susceptibility to high fat diet-induced metabolic syndrome in C57BL/6 mice: improvement in glycemic and lipid profile after antioxidant therapy. Oxid Med Cell Longev. 2016:1987960. doi: 10.1155/2016/1987960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang C, Wei X, Wei Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell. 2016;7:11–16. doi: 10.1007/s13238-015-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.