

Abstract
For much of our history, the most basic information about the microbial world has evaded characterization. Next-generation sequencing has led to a rapid increase in understanding of the structure and function of host-associated microbial communities in diverse diseases ranging from obesity to autism. Through experimental systems such as gnotobiotic mice only colonized with known microbes, a causal relationship between microbial communities and disease phenotypes has been supported. Now microbiome research must move beyond correlations and general demonstration of causality to develop mechanistic understandings of microbial influence, including through their metabolic activities. Similar to the microbiome field, advances in technologies for cataloguing small molecules have broadened our understanding of the metabolites that populate our bodies. Integration of microbial and metabolomics data paired with experimental validation has promise for identifying microbial influence on host physiology through production, modification or degradation of bioactive metabolites. Realization of microbial metabolic activities that affect health is hampered by gaps in our understanding of 1) biological properties of microbes and metabolites, 2) which microbial enzymes/pathways produce which metabolites and 3) the effects of metabolites on hosts. Capitalizing upon known mechanistic relationships and filling gaps in our understanding has the potential to enable translational microbiome research across disease contexts.
The Microbiome
An important concept for understanding the state and challenges of microbiome research was articulated by Donald Rumsfeld in 2002, when he said, “There are known knowns; there are things we know we know. We also know there are known unknowns; that is to say we know there are some things we do not know. But there are also unknown unknowns – the ones we don’t know we don’t know…. it is the latter category that tend to be the difficult ones1.”
The modern microbiome field began with a great discovery of “unknown unknowns”. Starting in the mid-1980s when researchers began using targeted sequencing of ribosomal RNA (rRNA) genes to survey microbial populations2, studies began to reveal deep branches of the tree of life previously unknown either because the organisms were invisible to the naked eye or uncultivable in the laboratory3. Considering how many “unknown unknowns” there were about the microbes that live in, on and around us just 30 years ago, the progress made in the field of microbiome research is remarkable.
Several key aspects of microbiome research have contributed to this rapid progress. First, the early adoption of the highly conserved small subunit rRNA gene as a phylogenetic marker allowed for relating all cellular life on earth onto one phylogenetic tree3,4. Examining novel environmental 16S rRNA gene sequences in an evolutionary context allowed for some properties of uncultivated organisms to be predicted. Even when such functional predictions were not possible (e.g. because the 16S rRNA gene sequence was extremely different from any previously characterized organism), simply moving our understanding of microbial biology from one of “unknown unknowns” to “known unknowns,” allowed for the identification of key gaps in understanding. For instance, the 16S rRNA sequences from an uncultivated group of bacteria in the α-proteobacterial lineage called SAR11 was found in nearly every pelagic marine bacterioplankton community, accounting for 26% of all rRNA genes in sea water. Targeted efforts led to the isolation and biological characterization of SAR11 isolates5. Using other –omics techniques coupled with experimental validation, the diverse and complex functions of the SAR116 lineage in the world’s oceans continue to be elucidated7.
A second factor that has contributed to rapid progress in microbiome research is next-generation sequencing. These technologies were used for 16S rRNA targeted characterization of microbial communities through use of the 454 sequencing platform in 20086,8,9. Starting in 2011, the 454 platform was largely replaced with the more high-throughput Illumina platforms10. Coupled with development of bioinformatics pipelines for 16S rRNA targeted microbiome analysis such as QIIME11 and of taxonomic databases for linking 16S rRNA gene sequences to named organisms/lineages (e.g. Silva12 and greengenes13), deep sequencing of microbial populations in many different environments, including many human body sites in disease contexts, has occurred. This has led to a plethora of papers describing associations of human microbial communities with diseases ranging from obesity14,15, depression16, inflammatory bowel disease (IBD)17,18 and heart disease19–21 to autism22–24.
Next generation sequencing has also enabled the sequencing of shotgun metagenomes and transcriptomes. Shotgun metagenomes survey the “functional potential” of a microbial community by mapping the genes present. The metatranscriptome adds an extra layer of information of the genes actively expressed. Effective functional analysis of microbial communities using shotgun metagenomic and metatranscriptomic data is highly dependent on our underlying knowledge of how gene sequences relate to enzymatic or other functions, which is summarized in databases such as KEGG25 and MetaCyc26. However, as for 16S rRNA databases where many known sequences represent organisms of unknown characteristics, the genes detected in shotgun metagenomes and metatranscriptomes contain many genes of unknown function. As an example, only a median of 18.9% percent of the genes present in bacterial genomes can be mapped to a KEGG Orthologue (KO) of known function based on homology. In some cases, only general function is known for a given gene. For instance, Bacteroides thetaiotaomicron has 280 genes predicted to be glycoside hydrolases in the Carbohydrate Active EnZymes database (CAZY)20 because they have sequence domains related to families of structurally-related catalytic and carbohydrate-binding modules, but only 44 of them are in KEGG.
As described for the SAR11 cluster and 16S rRNA, identifying “known genes of unknown function” that may be of particular relevance due to their ubiquity or a strong correlation with disease, can focus targeted efforts to elucidate their functional roles. In one study, a family of nonribosomal peptide synthetase gene clusters in gut bacteria was targeted for functional analysis because they were present in 88% of the National Institutes of Health Human Microbiome Project stool samples and transcribed under conditions of host colonization25. By expressing a subset of these clusters in Escherichia coli or Bacillus subtilis, it was determined that their gene products lead to the production of peptide aldehydes with protease inhibitory activity that selectively targets a subset of cathepsins in human cell proteomes27. The ubiquity of these enzymes indicates that they have the potential to be an important mediator of gut microbiome/host interactions.
While there have been many advances in microbiome sequencing workflows, the key challenge for the microbiome field, and essential for translational research, is to move beyond associations to an understanding of mechanism. One challenge for generating testable hypotheses regarding mechanistic links between microbial communities and disease is a lack of understanding of microbial function in disease causality. A potential way to address this gap is through the application of metabolomics with microbiome analysis.
The Metabolome
An important tool for understanding microbial community function is metabolomics. Metabolomics is a growing field that comprises the measurement of hundreds to thousands of small molecules in a system28. It encompasses metabolite profiling, lipidomics, and metabolic flux measurements28,29 and can also include the targeted analyses of specific small molecules, including sex hormones, lipid mediators30, and amino acids31.
While genomics, transcriptomics, and metadata provide valuable information regarding phenotypes and molecular mechanisms, metabolomics provides a real-time view of dynamic changes as they occur through the quantitation of small molecules. Small molecules play several roles in the cell including membrane structure (lipids), signaling (lipid mediators, neurotransmitters), and building blocks (amino acids). They are also involved in many processes affected by exogenous influences; these include response to oxidative stress (redox potential), inflammatory response, and energy metabolism, to name a few. Overall metabolomics can be thought of as a functional measurement of dozens to hundreds of molecules. This functional knowledge is essential for understanding the links between microbial communities and disease.
Metabolomics can be conducted using either mass spectrometry-based or nuclear magnetic resonance (NMR)-based platforms. NMR offers reproducible, quantitative measurement of approximately 125 small molecules, including carbohydrates, nucleic acids, and fatty acids32,33. NMR is highly robust, can be performed in vivo, for instance to obtain biochemical information about the tissues of the human body in a non-invasive way (without the need for a biopsy), and is non-destructive. However, due to the relatively lower sensitivity of NMR, mass spectrometry-based platforms have become increasingly adopted by clinical and research laboratories wishing to conduct metabolomics.
One advantage of metabolomics is that this technical platform is agnostic to the nature of the sample type or treatment group; hence, it is a valuable tool for both hypothesis generation (untargeted approaches) and testing (targeted approaches). However, numerous challenges exist34 including multiple compound name assignments to a single mass, lack of database search scoring algorithms, limited coverage of important pathways using a single technology, uncharacterized metabolic and degradation products, and lack of standardized nomenclature and analytical methods that compare datasets. High throughput samples and analysis as well as data interpretation pose additional challenges; traditionally, the interpretation step has been challenging for researchers when only long lists of potential biomarkers or compound masses (MS) are provided as the final results. Despite the availability of sophisticated pathway analysis programs35,36, a thorough interpretation of the data has often been beyond the scope of many metabolomics studies; this is especially emphasized in the context of microbiome/host studies, where the origin of metabolites is unclear.
Metabolomics is still limited by many “known unknowns”. Due to their chemical diversity, it is impossible to quantitate every species or even class of small molecule using a profiling approach. Therefore, targeted small molecule analysis (based on robust mass spectrometry assays) are used to quantitatively pinpoint the roles of individual molecules. For example, inflammatory responses can be elucidated by measuring individual lipid mediators that are part of the arachidonic acid pathway30. Examples of targeted small molecule analyses are most often found in newborn screening, where small molecules are used to determine the presence of many metabolic and genetic disorders34. While a precise and accurate means of quantitating small molecules, targeted assays require knowing the molecule of interest and are often limited in the numbers of molecules that can be quantitated.
Additionally, many metabolites cannot be mapped to current pathways. Even knowing the chemical class or species of these molecules may not be enough to deduce their function. Overall, the challenges in both microbiome and metabolome data interpretation highlights the need for efforts to further develop our knowledge so that the results of multiple ‘omics studies can be used to understand mechanisms and phenotypes in human health and disease.
Microbiome Effects on the Host Metabolome
Metabolomics has been applied to a variety of human sample types where bacterial metabolites may be found. These include sites where the bacteria reside and are active including nasal, feces, and skin37,38, as well as peripheral sites without resident bacterial communities such as blood39–41, urine40,41 and liver42. The latter are of interest because these metabolites have translocated through host barriers (e.g. skin or mucosal sites) and have potential for more systemic health effects. Metabolomics analyses of microbiome-influenced samples often have a primary aim to measure well-characterized metabolites such as short chain fatty acids (SCFAs), siderophores, and biotransformation products from host metabolites and xenobiotics28,43–46. Untargeted analyses aimed at understanding microbiome effects on the metabolome of host-derived samples are complicated because the compounds detected represent a complex milieu of environmental compounds as well as those produced by microbes and/or the host.
Determining the origins of metabolites that cause or influence disease is important for developing targeted therapies. Microbes have highly enhanced metabolic capabilities compared to humans; the gut microbiome encodes an estimated ~ 10 million genes47 compared to an estimated 19,000 in humans48. Microbiomes have the genetic capacity to produce a wide variety of structurally diverse specialized metabolites including natural products, small molecule virulence factors, and signaling metabolites including quorum sensors and autoinducers37,38,49. Most specialized metabolites, their biosynthetic origins, and their roles in microbiome related disease remain understudied37.
A broad understanding of how the presence of a microbiome effects the host metabolome has come from comparing germ free (GF) mice, (i.e. mice with no associated microbial communities), to mice with colonizing microbes including 1) conventional mouse microbiota39 2) the microbiota of different humans (i.e. humanized mice)40 or 3) specific individual bacterial species39,40. Similarly, other studies have compared the metabolomes of mice treated with antibiotics to non-antibiotic treated controls50,51. These studies have found that microbial colonization/composition has a profound influence on both the presence and relative abundances of many metabolites in various sites including the blood 39,40,42, urine40,42, feces40, liver42, and ileum42. Differences observed in GF and/or antibiotic treated mice in the various studies include changes in amino acid metabolism pathways (e.g. a loss of transformation of tryptophan into tryptamine and indole derivatives39–41,51), polysaccharide metabolism (e.g. increases in non-digestible plant-polysaccharides in feces40), decreases in SCFAs, increases in primary bile acids and decreases in secondary bile acids51 and elevation of creatine and creatinine40,52,53. Microbiota colonization also results in differences in many small compounds of unknown identity/function40.
Another way to estimate which metabolites in host samples originate from host versus microbial metabolism is to use metabolic networks described in databases such as KEGG25. These networks capture the relationship between metabolites, the enzymes that produce them, and the organisms (both host and microbial) that contain genes encoding those enzymes (Figure 1A). These networks can, in principle, provide a framework for relating the genes/enzymatic reactions present in the colonizing bacteria (genome/shotgun metagenome), the expression of those genes (meta-transcriptome), and presence of the encoded proteins (meta-proteome), and the metabolites (metabolome) in a sample (Figure 1B). They also provide a framework for investigation of microbiome/host co-metabolism (Figure 1C).
Figure 1.
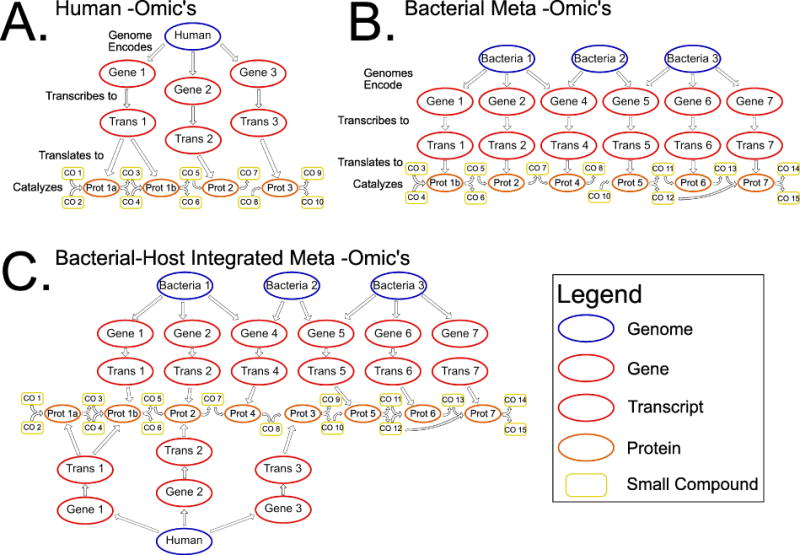
(A) The central dogma of molecular biology is the information flow of genes to transcripts to proteins. In the –omics age, we can observe this information flow through the characterization of genomes, transcriptomes and proteomes. The metabolome, the collection of small compounds present in a biological sample, can be seen as a natural extension of the central dogma. These metabolites can be associated with disease and we can work back from metabolites, to proteins that catalyze the reactions that produce them, to the transcripts that are translated to the genes that encode those transcripts. (B) This model can be extended to include multiple organisms as would be present in a microbial community and the reactions and metabolites that are produced and consumed form connections between the community members. (C) The host can be added back into this network to find host-microbiome interactions.
Linking Microbiome/Metabolome with the Exposome
Another important level of complexity is how the metabolome and microbiome are linked with the exposome (e.g. exogenous factors such pollutants, hygiene products, and the diet). One of the most studied facets of the exposome is diet. Diet composition substantially alters the human microbiome54,55 and diet induced changes can occur as rapidly as within a day54,56. This change is accompanied by alteration in bacterial gene expression54, suggesting that the microbiome is elastic and poised to quickly respond to varied diet exposures.
Studies in both humans57–60 and animal models40 have shown that consumption of different diets is associated with changes in microbially-produced metabolites. For instance, high fiber diets have been associated with the increases in both fecal61,62 and circulating63 levels of SCFAs. Another study of humans on a high protein diets saw an increase in levels of the carcinogenic N-nitroso compounds compared to a maintenance diet, thought to be driven by increased protein fermentation and decreased carbohydrate fermentation by colonic microbiota62. Broader effects of fiber consumption on the fecal and urine metabolome were observed in a study comparing mice on a standard low fat (LF) to a high fat (HF) diet64 which suggests that lack of complex polysaccharides may be a large contributor to the HF-diet associated microbiome/metabolome.
Though a significant facet of the exposome, daily exposures extend beyond diet. A large area of exposure is the skin which is composed of a diverse and rich microbiome, varying by body site65. Spatial mapping of the microbiome and metabolome across 400 sites of the human body revealed associations between microbes and the metabolites present66. Most notably Propionibacterium was associated with sebaceous sites and highly correlated with 491 metabolome features, 73% of which were lipids. Further in vitro analysis revealed that Propionibacterium acnes can oxidize oleic acid, the breakdown products of which were found in sebaceous sites66. These findings suggest that the skin microbiome influences the chemical landscape of the skin metabolome.
The host microbiome is also important in xenobiotic degradation. Gut microbes can metabolize a range of environmental chemicals in ways that can affect their toxicity to the host, including polycyclic aromatic hydrocarbons (PAHs), nitrotoluenes (which are important intermediates in the manufacture of dyes and plastics), and pesticides67. PAH degrading microbes have also been observed on human skin68. Additionally, gut microbes have also been associated with breakdown and reactivation of a variety of drugs including the cardiovascular drug, digoxin and the cancer drug, irinotecan69.
Identifying Important Microbially-Produced Metabolites for Health and Disease
Studies aimed to explore an association between microbial metabolism and disease states often focus on well-characterized microbial metabolites such as SCFAs70. However, approaches that can identify novel bio-active microbial metabolites as mediators of microbiome-host or microbe-microbe interactions are increasingly being applied71. These include: 1) Bioactivity-guided screening, in which a phenotypic bioassay is performed by testing a collection of microbes, chemical extracts, or cosmid libraries generated from metagenomics DNA against a panel of host pathogens or signaling pathways72,73. 2) Candidate molecule workflows, which mine existing (meta)genomics data to identify Biosynthetic Gene Clusters (BGCs) that may produce metabolites of interest because they are prevalent or correlate with disease49. 3) Integrative analysis of microbiomes and untargeted metabolomes in disease contexts20,50,74–76. All of these approaches can identify molecules with potential roles in mediating host-microbe interactions that can be further validated in in vitro or in vivo systems.
Bioactivity-guided workflows have led to the identification of several metabolites that mediate interactions between microbes and the host or other microbes. For instance, commendamide, which activates the G-protein-coupled receptor G2A/GPR132 (G2A has been implicated in disease models of autoimmunity and atherosclerosis), was discovered initially in a screen of 3,000 megabases of metagenomic DNA for effectors that activate NF-κB, a transcription factor that plays a role in mediating responses to environmental stimuli72. Lugdunin, a novel antibiotic that prohibits colonization by methicillin-resistant Staphylococcus aureus (MRSA), was discovered by screening for the mechanism by which Staphylococcus lugdunensis, a microbe that occupies the same niche as MRSA, could inhibit the growth of MRSA in vitro73. Bioactivity-guided workflows can also include random mutagenesis of the microbe of interest combined with high-throughput phenotypic screening for loss of function, and imaging mass spectrometry of competing microbes in vitro 72,77,78.
Candidate molecule workflows identify BGCs that allow microbes to produce metabolites and capitalize on knowledge of the core enzymatic machinery responsible for their biosynthesis, which has been well-characterized for many classes of specialized metabolites49. This knowledge has been used to develop and implement bioinformatics platforms, such as anti-SMASH and related platforms, to detect widely distributed and closely related gene cluster families, identify potential metabolite producers, and guide purification workflows79–84. Candidate molecule methods have also led to the identification, structural elucidation and/or synthesis of metabolites that mediate interactions between microbes and the host and/or other microbes. These include colibactin, a secondary bacterial metabolite with genotoxic properties85, lactocillin, a potent thiopeptide antibiotic made by a prominent member of the vaginal microbiota 27,49, (dihydro)pyrazinones, which are prevalent in the human gut and that release a peptide aldehyde that inhibits cathepsins in human cell proteomes21, and humimycins, antibiotics with activity against MRSA86. Candidate molecule workflows can also be combined with tandem mass spectrometry (MS/MS) data to link MS/MS spectra of unknown metabolites to their biosynthetic origins, simplifying compound isolation and characterization87,88.
Integrative analysis of microbiomes and untargeted metabolomes in disease contexts have also allowed for the identification of microbially produced compounds found to mediate disease upon further functional characterization. For example, one study used a maternal immune activation (MIA) murine model that developed symptoms commonly associated with Autism Spectrum Disorders (ASD). These MIA mice also had altered gut microbiome and serum metabolome profiles compared to control mice. Oral treatment of the MIA mice with the human commensal Bacteroides fragilis ameliorated ASD-like behaviors. The metabolite 4-ethylphenylsulfate (4EPS) was focused on for functional study because its levels were increased in the MIA mice and restored to control levels by B. fragilis treatment. Interestingly, systemic administration of naïve wild-type mice with 4EPS potassium salt was sufficient to induce anxiety-like behavior similar to that observed in MIA offspring74.
In another study that characterized the fecal microbiome and serum metabolome of non-diabetic Danish individuals, serum levels of branch-chain amino acids (BCAAs) were higher in insulin-resistant individuals and also correlated with fecal microbiomes with higher biosynthetic potential for BCAAs conferred by the species Prevotella copri and Bacteroides vulgatus. These predictions, made based on correlative analyses and functional predictions, were further validated by showing that P. copri could induce insulin resistance while increasing circulating BCAA levels when introduced into High Fat Diet (HFD)-fed mice89.
In some cases, associations between microbially produced metabolites and disease have also been linked with the exposome, including diet. One study showed that a dietary fiber associated reduction in allergic lung inflammation was driven by the SCFA propionate, which exerted its health effects through G protein–coupled receptor 41 mediated signaling in bone marrow72. Other work demonstrated that risk of cardiovascular disease and atherosclerosis was linked with trimethylamine-N-oxide (TMAO), which is produced by host enzymes from trimethylamine (TMA). TMA is a metabolite of dietary phosphatidylcholine and L-carnitine that can be produced by intestinal bacteria but not host enzymes and whose production is associated with particular human gut microbiome compositions20,76.
Metabolites from non-targeted metabolomics can be prioritized for functional exploration using various analytic techniques. As mentioned above, correlative analyses link the presence of a specific metabolite to genotypic and/or phenotypic data and may suggest that a metabolite is produced or induced by the presence of a specific microbe90–94. Other methods include Self Organizing Map generation, which applies multivariate statistics to differentiate samples by identifying the largest changes in metabolite abundance between case and control95–97, and dereplication, or the identification of known molecules. Dereplication by molecular networking uses MS/MS data to visualize the chemical space of samples to pinpoint an in vitro producer of a metabolite produced in vivo 66,98–100. Molecular networking measures relatedness between MS/MS spectra as a proxy for molecular similarity. MS/MS metabolomics data derived from in vitro cultivation of members of the microbiota can be used to seed molecular networks in order to identify either known compounds produced by those members of the community or close analogs99–101
Functional Validation
Once associations between microbiomes and metabolomes in disease have been identified, the roles of specific metabolites or metabolomics patterns must be validated. If a compound is characterized and commercially available, validation is most straightforward. For uncharacterized compounds, molecular biology workflows can be used; for instance, metagenomics-derived BGCs can be expressed in heterologous hosts as was recently used to characterize the (dihydro)pyrazinones from human gut microbiota27. Additionally, if the microbe that produces the metabolite can be grown in pure culture and genetically manipulated, regulators of BGCs can be removed, replaced, or enhanced to increase in vitro metabolite production to levels necessary for isolation, as was recently demonstrated during the characterization of lugdunin73,102.
Despite recent success characterizing a handful of individual metabolites, many specialized metabolites of interest are understudied, in part due to limitations of cultivation. To circumvent these limitations, conventional approaches for microbiota growth have been modified to include environment specific media103–106, optimization of growth conditions based upon (meta)genomic information106, inclusion of helper organisms107,108 and higher throughput cultivation strategies105,109–112. Efforts are also underway to cultivate both native and designer microbiome communities using in vitro and in vivo model systems104 and new model systems continue to be developed113,114.
Animal models are widely used to explore the functional attributes of microbes and their metabolites, including both conventionally raised and GF models of zebrafish, mice, rats, and pigs113. While GF models have been a powerful tool for experimental validation of microbial contributions to disease, use of these models has sometimes been criticized as these animals have alterations in essential physiological processes113,115. Treatment of conventionally raised animals with broad-spectrum antibiotics cocktails followed by repopulation with chosen microbes is a commonly used alterative48,50,116,117 and may circumvent some issues with GF animal model systems, but have the limitation that antibiotics never completely remove indigenous microbes and also may have other effects on the host. Successful use of animal models to functionally validate the effects of microbially-produced metabolites in disease were described above for studies of atherosclerosis20,76,118, ASDs74, and diabetes62. Another elegant example of using molecular biology in conjunction with animal models to identify key metabolites is the elucidation of the role of microcins produced by probiotic E. coli strains in modulating Enterobacteriaceae in the inflamed gut119.
While animal models are most often chosen to explore functional activity of microbes and their metabolites, in vitro systems can provide an intriguing alternative113 due to their higher throughput, flexibility, scaling, and ability to simplify the system for high-resolution molecular analyses. These in vitro models include independent cultivation of the microbiota and interfacing the members of the microbiota with host cell lines using Transwell systems or, eventually, organ-on-a-chip microfluidic devices113. While a number of groups are attempting to systematically construct artificial communities for robust evaluation of microbiome modulation by both organisms and metabolites, such as probiotics and pharmaceuticals respectively, development of environment-specific media has led to some success recapitulating in vivo microbiome communities in vitro for similar purposes103–105,110,112,120. For example, stable oral microbial biofilm communities representing 60–80% of the taxonomic diversity of the original sample has been accomplished121, while fecal communities were recapitulated at 65–95%122. Miniaturized bioreactors and chemostats may provide opportunities to maintain continuous culture of communities removing concerns about sample limitation from patient donors, particularly of microbiome niches that are not as easily accessible as stool113,123. To incorporate host cells, co-culture systems have been developed and models for the context of gut microbiome investigation have been recently reviewed124. These models include co-culture in tubes (HoxBan system), Transwell systems, and microfluidic devices. While microfluidic systems, such as the HuMiX model, are more compatible with multi-omics analyses, it suffers from poor chemical compatibility, challenges with parallelization, and lack of widespread availability113,114,124.
Towards Microbiota-targeted Therapeutics
Microbiome/metabolome targeted therapies include both those that inhibit detrimental microbes/metabolites and those that promote beneficial microbes/metabolites (Figure 2). The microbiome/metabolome can also potentially inform personalized medicine, if the treatment of an individual is tailored based on the composition or metabolic capabilities of their microbiome. The complicated and diverse roles in disease of the microbiome are only beginning to be elucidated; thus, most promising research directions have yet to reach their full potential in the clinic.
Figure 2.
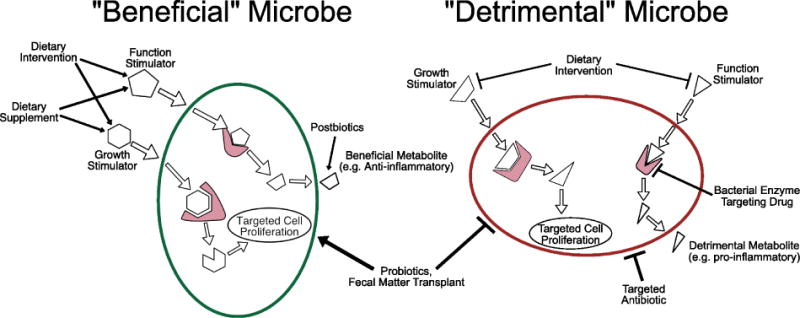
Microbiome-targeted therapies can target detrimental microbes and/or their metabolites (red) or promote beneficial microbes and the production of their metabolites (green). In principle, the presence of beneficial microbial metabolites in the host can be promoted by adding the metabolite itself (dietary intervention or supplementation), the microbe that produces the metabolite (probiotics), the substrate the microbe uses to produce the metabolite (prebiotics) or the beneficial metabolite itself (postbiotics). The presence of detrimental microbial metabolites can be reduced by targeting the microbe that produces it directly (antibiotics) or through competitive exclusion (probiotics/fecal microbiota transplant), by inhibiting the enzymes (shown in pink) that produce the metabolite, or by removing the substrates for that metabolite from the diet.
Targeting detrimental bacteria and metabolites
Although antibiotic treatment can be valuable for discouraging growth of bacteria that produce detrimental metabolites, broad effects on other (beneficial) microbes and rapid acquisition of antibiotic-resistance limits their utility. Thus, another promising approach is competitive exclusion by other “probiotic” bacteria. Perhaps the best evidence that this approach can be successful clinically is the 92% success rate of fecal microbiota transplantation (FMT) for the treatment of recurrent Clostridium difficile infection125. However, understanding metabolic mechanisms of inter-species interaction can allow for better “precision microbiome reconstitution”126 for treatment of C. difficile and potentially of diseases where FMT has had more variable success such as IBD127,128. In the case of C. difficile, protection in mice has been conferred by introducing specific bacteria (e.g. Clostridium scindens) that can produce key metabolites (secondary bile acids) that affect C. difficile sporulation and growth and thus infection126; however, efficacy and safety trials in human populations still need to be conducted.
Another mechanism for preventing detrimental microbial metabolic activity is to inhibit the metabolic pathway rather than the bacteria that contain it. This can be accomplished by limiting inputs to the pathway (e.g. by altering dietary intake) or by inhibiting bacterial enzymatic activity (Figure 2). One promising example is the treatment of cardiovascular disease by inhibiting microbial production of TMA using a structural analog of choline that can inhibit microbial TMA lyases118. In another example, the amino acid arginine can inhibit enzymatic conversion of the cardiac drug digoxin into an inactive form by the gut microbe Egerthella lenta, indicating that co-administering this drug with arginine may prevent its deactivation by microbial enzymes21. However, both of these enzyme-targeted treatment strategies still require validation in clinical cohorts.
Promoting beneficial bacteria and metabolites
Therapies that promote the prevalence/activity of beneficial microbes and their metabolites also have promise for clinical applications that has likely yet to be fully realized. Current probiotic treatments use a small number of intestinal microbes (e.g. various Lactobacillus or Bifidobacteria species, the yeast Saccharomyces boulardii, or E. Nissle) that have already been approved for use in humans and that only represent a small component of bacteria that can colonize the human gut. In some cases, specific probiotics have been shown to affect both microbiome and metabolome composition while promoting health; for instance, a trial in patients with liver cirrhosis found supplementation with Lactobacillus GG improved outcomes (decreased endotoxemia and TNF-α levels) with concomitant changes in microbiome composition and metabolite/microbiome correlations pertaining to amino acid, vitamin and secondary BA metabolism129. As the literature on these probiotics is vast, consensus opinion regarding their utility in the clinic are valuable; the Triennial Yale/Harvard Workshop on Probiotic Recommendations130 gives A–C evidence ratings for the use of specific probiotics in necrotizing enterocolitis, childhood diarrhea, IBD, irritable bowel syndrome, C. difficile diarrhea and liver disease.
There are several bacteria that have shown positive effects in mouse models that may represent a “next generation” of probiotics once safety and efficacy studies in humans can be conducted. For instance Bacteroides uniformis and Akkermansia muciniphila both reduce weight gain and metabolic disease pathology in HFD-fed mice131,132; Bacteroides fragilis, Bacteroides cellulosilyticus, and cocktails of T regulatory cell inducing, spore-forming Clostridia have all been shown to protect against intestinal inflammation in mouse models133–135. The mechanism by which probiotics may confer a health benefit are often not completely understood and include other functions besides the production of small molecules, such as the production of immune-modulatory capsular components133.
Probiotic applications can be challenged by colonization resistance; This challenge can potentially be circumvented by promoting growth and metabolic activity of beneficial bacteria using prebiotics or synbiotics. Prebiotics are broadly defined as any compound that affects the composition or function of the microbiome to exert a beneficial effect upon the host following bacterial metabolism136. Prebiotics are typically fibers and starches extracted from plants or post-fermentation processes with SCFAs being one desirable metabolic product of their digestion 137–142. Synbiotics are formulations that contain both probiotics and prebiotics together. Understanding the metabolic environment necessary for the successful growth of microbes can lead to the intelligent design of prebiotics and synbiotics. For instance, galactooligosaccharide mixtures defined in laboratory culture experiments to have particularly strong prebiotic properties for Bifidobacteria, effectively increased the proportion of Bifidobacteria in the gut in a dose dependent manner when given to healthy human volunteers138. Use of genomic and metabolic data to design prebiotic strategies that target specific microbes and metabolites will become more powerful as our knowledge bases of microbial enzymes and metabolites expand.
Beneficial microbial metabolites can also be directly introduced to the host to circumvent the need for microbial production from dietary substrates, a process referred to as “microbiome-based metabolite treatment” or “postbiotics”143(Figure 2). For instance, the anti-inflammatory microbial fermentation product, butyrate, has been added directly to the colonic environment via enema in efforts to ameliorate inflammation in patients with ulcerative colitis 144,145, although only minor improvements were observed146. The exploration of “postbiotic” therapies are in their infancy and potentially challenged by unintended effects of the compound on gut microbiome composition and activity, and difficulty in determining and delivering physiologically important levels of the metabolite to its active site in the body143.
Personalized medicine
Finally, microbiome/metabolome information has the potential to inform personalized medicine. In one elegant example, gut microbiota composition was included in machine learning-algorithms that also considered blood parameters, dietary habits, anthropometrics, and physical activity, to predict personalized glycemic responses to meals. This model was used to perform personalized dietary intervention to lessen elevated postprandial blood glucose and its metabolic consequences147.
Another promising direction of microbiome/metabolome informed personalized medicine is to consider the role of the microbiome on drug metabolism/pharmacokinetics for proper drug dosing. For instance, evaluation of the presence of digoxin-deactivating strains of the gut bacterium E. lenta has the potential to inform proper administration of this cardiac drug148. Another study suggested that urinary levels of the microbial product p-cresol sulfate (as determined by (1)H NMR spectroscopy) could indicate the metabolic fate of acetaminophen149.
Future Directions
It’s been almost nine years since we were all invited to the marriage of metagenomics and metabolomics150 and progress in integrating these data types and elucidating the function of key small-molecules that are produced by the microbiome is being made. Understanding the metabolic capabilities of the microbes that inhabit all areas of the human body will be key to determining their effects on health and disease. To advance this knowledge, more work remains to be done. While 16S rRNA can be used to conveniently survey the composition of microbial communities, its ability to predict metabolic effects is limited both by the incomplete knowledge present in bacterial genomic databases and strain level variation. Metagenomic sequencing gives more knowledge of the genes present, but the majority of these genes still have unknown function. KEGG is still the most used database for orthologous gene group assignment and is the most comprehensive database for linking orthologous gene groups to reactions and annotated compounds25. KEGG has moved to a subscription service model since 2011 and as such cannot be integrated into new open source tools. MetaCyc26 is free to use and redistribute but is missing features from KEGG such as a universal gene orthology. As the gaps in these databases are filled through genome sequencing and both large-scale and targeted biochemistry and/or functional studies, our power to understand the functional potential of host associated microbial communities will increase.
Metabolomics has perhaps even greater challenges than the evaluation of microbial community structure and function using DNA and RNA sequencing. Extraction techniques are not standardized and subject to technical variability. Peak extraction and annotation of mass spectroscopy data is challenged by multiple metabolites matching to the same mass34. Current best practices do not give confident results without further experimental validation and quantification which is not currently possible on the scale of detected metabolites in untargeted experiments. As the technology for untargeted mass spec metabolomics becomes more mature, these issues will hopefully become less prominent allowing understanding of metabolomes across studies. Paired with an increased biochemical understanding of the gene products encoded by microbiomes, the ability to develop a mechanistic understanding of how microbial communities influence the host via their metabolic activities will be enhanced. To rapidly advance microbiome and metabolome integration to understand roles for microbial metabolism in diverse disease contexts, advanced yet “user friendly” methods for multi-omic integration need to be developed.
Even with these formidable challenges, many studies have identified key microbes and their enzymes and metabolites that can be targeted therapeutically in the context of varied diseases. A key challenge in the field is to move many of these novel discoveries from the bench to the bedside. By understanding the metabolic interplay between microbiome and host, novel prebiotics and probiotics can be explored, as well as personalized treatment of disease that capitalize on knowledge of microbiomes and their interactions with the host.
Acknowledgments
All authors have no conflicts of interest related to the content of this paper and have read the journal’s authorship statement.
Abbreviations
- SCFA
Short chain fatty acids
- BGC
Biosynthetic Gene Cluster
- MS/MS
Tandem mass spectrometry
- rRNA
Ribosomal RNA
- GF
Germ Free
- KO
KEGG Orthologue
- LC/MS
liquid chromatography/mass spectroscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rumsfeld D, Myers R. DoD News Briefing. 2002 http://archive.defense.gov/Transcripts/Transcript.aspx?TranscriptID=2636.
- 2.Stahl DA, Lane DJ, Olsen GJ, Pace NR. Characterization of a Yellowstone hot spring microbial community by 5S rRNA sequences. Appl Environ Microbiol. 1985;49(6):1379–1384. doi: 10.1128/aem.49.6.1379-1384.1985. http://www.ncbi.nlm.nih.gov/pubmed/2409920. Accessed April 6, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pace NR. A Molecular View of Microbial Diversity and the Biosphere. Science (80-) 1997;276(5313) doi: 10.1126/science.276.5313.734. http://science.sciencemag.org/content/276/5313/734.full. Accessed April 6, 2017. [DOI] [PubMed] [Google Scholar]
- 4.Pace NR. Mapping the tree of life: progress and prospects. Microbiol Mol Biol Rev. 2009;73(4):565–576. doi: 10.1128/MMBR.00033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rappé MS, Connon SA, Vergin KL, Giovannoni SJ. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature. 2002;418(6898):630–633. doi: 10.1038/nature00917. [DOI] [PubMed] [Google Scholar]
- 6.Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. Exploring Microbial Diversity and Taxonomy Using SSU rRNA Hypervariable Tag Sequencing. In: Eisen JA, editor. PLoS Genet. 11. Vol. 4. 2008. p. e1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsementzi D, Wu J, Deutsch S, et al. SAR11 bacteria linked to ocean anoxia and nitrogen loss. Nature. 2016;536(7615):179–183. doi: 10.1038/nature19068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dethlefsen L, Huse S, Sogin ML, Relman DA, Weightman A. The Pervasive Effects of an Antibiotic on the Human Gut Microbiota, as Revealed by Deep 16S rRNA Sequencing. In: Eisen JA, editor. PLoS Biol. 11. Vol. 6. 2008. p. e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenna P, Hoffmann C, Minkah N, et al. The Macaque Gut Microbiome in Health, Lentiviral Infection, and Chronic Enterocolitis. PLoS Pathog. 2008;4(2):e20. doi: 10.1371/journal.ppat.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;(Supplement 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raman M, Ahmed I, Gillevet PM, et al. Fecal Microbiome and Volatile Organic Compound Metabolome in Obese Humans With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol. 2013;11(7):868–875.e3. doi: 10.1016/j.cgh.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 15.Martinez KB, Leone V, Chang EB. Western diets, gut dysbiosis, and metabolic diseases: Are they linked? Gut Microbes. 2017:1–13. doi: 10.1080/19490976.2016.1270811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evrensel A, Ceylan ME. The Gut-Brain Axis: The Missing Link in Depression. Clin Psychopharmacol Neurosci. 2015;13(3):239–244. doi: 10.9758/cpn.2015.13.3.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halfvarson J, Brislawn CJ, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2:17004. doi: 10.1038/nmicrobiol.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickson I. Gut microbiota: Diagnosing IBD with the gut microbiome. Nat Rev Gastroenterol Hepatol. 2017;14(4):195–195. doi: 10.1038/nrgastro.2017.25. [DOI] [PubMed] [Google Scholar]
- 19.Kitai T, Kirsop J, Tang WHW. Exploring the Microbiome in Heart Failure. Curr Heart Fail Rep. 2016;13(2):103–109. doi: 10.1007/s11897-016-0285-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta + doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clayton TA. Metabolic differences underlying two distinct rat urinary phenotypes, a suggested role for gut microbial metabolism of phenylalanine and a possible connection to autism. FEBS Lett. 2012;586(7):956–961. doi: 10.1016/j.febslet.2012.01.049. [DOI] [PubMed] [Google Scholar]
- 23.Buie T. Potential Etiologic Factors of Microbiome Disruption in Autism. Clin Ther. 2015;37(5):976–983. doi: 10.1016/j.clinthera.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Rosenfeld CS. Microbiome Disturbances and Autism Spectrum Disorders. Drug Metab Dispos. 2015;43(10) doi: 10.1124/dmd.115.063826. http://dmd.aspetjournals.org/content/43/10/1557.long. Accessed April 7, 2017. [DOI] [PubMed] [Google Scholar]
- 25.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caspi R, Altman T, Billington R, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42(D1):D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo C-J, Chang F-Y, Wyche TP, et al. Discovery of Reactive Microbiota-Derived Metabolites that Inhibit Host Proteases. Cell. 2017;168(3):517–526.e18. doi: 10.1016/j.cell.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wishart DS. Emerging applications of metabolomics in drug discovery and precision medicine. Nat Rev Drug Discov. 2016;15(7):473–484. doi: 10.1038/nrd.2016.32. http://dx.doi.org/10.1038/nrd.2016.32. [DOI] [PubMed] [Google Scholar]
- 29.Sethi S, Brietzke E. Recent advances in lipidomics: Analytical and clinical perspectives. Prostaglandins Other Lipid Mediat. 2017;128:8–16. doi: 10.1016/j.prostaglandins.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Armstrong M, Liu AH, Harbeck R, Reisdorph R, Rabinovitch N, Reisdorph N. Leukotriene-E4 in human urine: Comparison of online purification and liquid chromatography–tandem mass spectrometry to affinity purification followed by enzyme immunoassay. J Chromatogr B. 2009;877(27):3169–3174. doi: 10.1016/j.jchromb.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armstrong M, Jonscher K, Reisdorph NA. Analysis of 25 underivatized amino acids in human plasma using ion-pairing reversed-phase liquid chromatography/time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2007;21(16):2717–2726. doi: 10.1002/rcm.3124. [DOI] [PubMed] [Google Scholar]
- 32.Spratlin JL, Serkova NJ, Eckhardt SG. Clinical Applications of Metabolomics in Oncology: A Review. Clin Cancer Res. 2009;15(2) doi: 10.1158/1078-0432.CCR-08-1059. http://clincancerres.aacrjournals.org/content/15/2/431.long. Accessed April 6, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Markley JL, Brüschweiler R, Edison AS, et al. The future of NMR-based metabolomics. Curr Opin Biotechnol. 2017;43:34–40. doi: 10.1016/j.copbio.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohler I, Verhoeven A, Derks RJ, Giera M. Analytical pitfalls and challenges in clinical metabolomics. Bioanalysis. 2016;8(14):1509–1532. doi: 10.4155/bio-2016-0090. [DOI] [PubMed] [Google Scholar]
- 35.Persicke M, Rückert C, Plassmeier J, et al. MSEA: metabolite set enrichment analysis in the MeltDB metabolomics software platform: metabolic profiling of Corynebacterium glutamicum as an example. Metabolomics. 2012;8(2):310–322. doi: 10.1007/s11306-011-0311-6. [DOI] [Google Scholar]
- 36.Xia J, Wishart DS, Xia J, Wishart DS. Current Protocols in Bioinformatics. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2016. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis; pp. 14.10.1–14.10.91. [DOI] [PubMed] [Google Scholar]
- 37.Garg N, Luzzatto-Knaan T, Melnik AV, et al. Natural products as mediators of disease. Nat Prod Rep. 2017;34(2):194–219. doi: 10.1039/C6NP00063K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donia MS, Fischbach MA. Small molecules from the human microbiota. Science (80-) 2015;349(6246) doi: 10.1126/science.1254766. http://science.sciencemag.org/content/349/6246/1254766.full. Accessed April 6, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wikoff WR, Anfora AT, Liu J, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106(10):3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcobal A, Kashyap PC, Nelson TA, et al. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 2013;7(10):1933–1943. doi: 10.1038/ismej.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin F-PJ, Dumas M-E, Wang Y, et al. A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol Syst Biol. 2007;3(1):112. doi: 10.1038/msb4100153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin F-PJ, Wang Y, Sprenger N, et al. Probiotic modulation of symbiotic gut microbial-host metabolic interactions in a humanized microbiome mouse model. Mol Syst Biol. 2008;4:157. doi: 10.1038/msb4100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbert JA, Quinn RA, Debelius J, et al. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature. 2016;535(7610):94–103. doi: 10.1038/nature18850. http://dx.doi.org/10.1038/nature18850. [DOI] [PubMed] [Google Scholar]
- 44.Clarke G, Stilling RM, Kennedy PJ, Stanton C, Cryan JF, Dinan TG. Minireview: Gut Microbiota: The Neglected Endocrine Organ. Mol Endocrinol. 2014;28(8):1221–1238. doi: 10.1210/me.2014-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sridharan GV, Choi K, Klemashevich C, et al. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat Commun. 2014;5:5492. doi: 10.1038/ncomms6492. [DOI] [PubMed] [Google Scholar]
- 46.Ursell LK, Haiser HJ, Van Treuren W, et al. The Intestinal Metabolome: An Intersection Between Microbiota and Host. Gastroenterology. 2014;146(6):1470–1476. doi: 10.1053/j.gastro.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Jia H, Cai X, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotech. 2014;32(8):834–841. doi: 10.1038/nbt.2942. http://dx.doi.org/10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 48.Theriot CM, Koenigsknecht MJ, Carlson PE, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun. 2014;5:781–791. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Donia MS, Cimermancic P, Schulze CJ, et al. A Systematic Analysis of Biosynthetic Gene Clusters in the Human Microbiome Reveals a Common Family of Antibiotics. Cell. 2014;158(6):1402–1414. doi: 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Theriot CM, Bowman AA, Young VB. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. mSphere. 2016;1(1) doi: 10.1128/mSphere.00045-15. http://msphere.asm.org/content/1/1/e00045-15. Accessed April 6, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antunes LCM, Han J, Ferreira RBR, Lolić P, Borchers CH, Finlay BB. Effect of antibiotic treatment on the intestinal metabolome. Antimicrob Agents Chemother. 2011;55(4):1494–1503. doi: 10.1128/AAC.01664-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Swann JR, Tuohy KM, Lindfors P, et al. Variation in Antibiotic-Induced Microbial Recolonization Impacts on the Host Metabolic Phenotypes of Rats. J Proteome Res. 2011;10(8):3590–3603. doi: 10.1021/pr200243t. [DOI] [PubMed] [Google Scholar]
- 53.Romick-Rosendale LE, Goodpaster AM, Hanwright PJ, et al. NMR-based metabonomics analysis of mouse urine and fecal extracts following oral treatment with the broad-spectrum antibiotic enrofloxacin (Baytril) Magn Reson Chem. 2009;47(S1):S36–S46. doi: 10.1002/mrc.2511. [DOI] [PubMed] [Google Scholar]
- 54.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2013;505(7484):559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruengsomwong S, Korenori Y, Sakamoto N, Wannissorn B, Nakayama J, Nitisinprasert S. Senior Thai Fecal Microbiota Comparison Between Vegetarians and Non-Vegetarians Using PCR-DGGE and Real-Time PCR. J Microbiol Biotechnol. 2014;24(248):1026–1033. doi: 10.4014/jmb.1310.10043. [DOI] [PubMed] [Google Scholar]
- 56.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu GD, Compher C, Chen EZ, et al. Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut. 2016;65(1):63–72. doi: 10.1136/gutjnl-2014-308209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scalbert A, Brennan L, Manach C, et al. The food metabolome: a window over dietary exposure. Am J Clin Nutr. 2014;99(6):1286–1308. doi: 10.3945/ajcn.113.076133. [DOI] [PubMed] [Google Scholar]
- 59.Edmands WM, Ferrari P, Rothwell JA, et al. Polyphenol metabolome in human urine and its association with intake of polyphenol-rich foods across European countries. Am J Clin Nutr. 2015;102(4):905–913. doi: 10.3945/ajcn.114.101881. [DOI] [PubMed] [Google Scholar]
- 60.De Angelis M, Montemurno E, Vannini L, et al. Effect of Whole-Grain Barley on the Human Fecal Microbiota and Metabolome. Appl Environ Microbiol. 2015;81(22):7945–7956. doi: 10.1128/AEM.02507-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinritz SN, Weiss E, Eklund M, et al. Intestinal Microbiota and Microbial Metabolites Are Changed in a Pig Model Fed a High-Fat/Low-Fiber or a Low-Fat/High-Fiber Diet. In: Zoetendal EG, editor. PLoS One. 4. Vol. 11. 2016. p. e0154329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Russell WR, Gratz SW, Duncan SH, et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr. 2011;93(5):1062–1072. doi: 10.3945/ajcn.110.002188. [DOI] [PubMed] [Google Scholar]
- 63.Trompette A, Gollwitzer ES, Yadava K, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20(2):159–166. doi: 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- 64.Montoliu I, Cominetti O, Boulangé CL, et al. Modeling Longitudinal Metabonomics and Microbiota Interactions in C57BL/6 Mice Fed a High Fat Diet. Anal Chem. 2016;88(15):7617–7626. doi: 10.1021/acs.analchem.6b01343. [DOI] [PubMed] [Google Scholar]
- 65.Grice EA, Kong HH, Conlan S, et al. Topographical and Temporal Diversity of the Human Skin Microbiome. Science (80-) 2009;324(5931):1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bouslimani A, Porto C, Rath CM, et al. Molecular cartography of the human skin surface in 3D. Proc Natl Acad Sci U S A. 2015;112(17):E2120–9. doi: 10.1073/pnas.1424409112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Claus SP, Guillou H, Ellero-Simatos S, Cerniglia CE, Chen H. The gut microbiota: a major player in the toxicity of environmental pollutants? npj Biofilms Microbiomes. 2016;2:16003. doi: 10.1038/npjbiofilms.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sowada J, Schmalenberger A, Ebner I, Luch A, Tralau T. Degradation of benzo[a]pyrene by bacterial isolates from human skin. FEMS Microbiol Ecol. 2014;88(1):129–139. doi: 10.1111/1574-6941.12276. [DOI] [PubMed] [Google Scholar]
- 69.Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Micro. 2016;14(5):273–287. doi: 10.1038/nrmicro.2016.17. http://dx.doi.org/10.1038/nrmicro.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee W-J, Hase K. Gut microbiota-generated metabolites in animal health and disease. Nat Chem Biol. 2014;10(6):416–424. doi: 10.1038/nchembio.1535. http://dx.doi.org/10.1038/nchembio.1535. [DOI] [PubMed] [Google Scholar]
- 71.Wilson MR, Zha L, Balskus EP. Natural Product Discovery from the Human Microbiome. J Biol Chem. 2017 Apr; doi: 10.1074/jbc.R116.762906. jbc.R116.762906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cohen LJ, Kang H-S, Chu J, et al. Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proc Natl Acad Sci U S A. 2015;112(35):E4825–34. doi: 10.1073/pnas.1508737112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zipperer A, Konnerth MC, Laux C, et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature. 2016;535(7613):511–516. doi: 10.1038/nature18634. http://dx.doi.org/10.1038/nature18634. [DOI] [PubMed] [Google Scholar]
- 74.Hsiao EY, McBride SW, Hsien S, et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell. 2013;155(7):1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neis E, Dejong C, Rensen S. The Role of Microbial Amino Acid Metabolism in Host Metabolism. Nutrients. 2015;7(4):2930–2946. doi: 10.3390/nu7042930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14(2):111–129. doi: 10.1038/nrd4510. http://dx.doi.org/10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 78.Moree WJ, Phelan VV, Wu C-H, et al. Interkingdom metabolic transformations captured by microbial imaging mass spectrometry. Proc Natl Acad Sci U S A. 2012;109(34):13811–13816. doi: 10.1073/pnas.1206855109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blin K, Medema MH, Kazempour D, et al. antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41(W1):W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Medema MH, Blin K, Cimermancic P, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39(suppl):W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weber T, Blin K, Duddela S, et al. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43(W1):W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blin K, Wolf T, Chevrette MG, et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017 Apr; doi: 10.1093/nar/gkx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zierep PF, Padilla N, Yonchev DG, Telukunta KK, Klementz D, Günther S. SeMPI: a genome-based secondary metabolite prediction and identification web server. Nucleic Acids Res. 2017 Apr; doi: 10.1093/nar/gkx289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khater S, Gupta M, Agrawal P, et al. SBSPKSv2: structure-based sequence analysis of polyketide synthases and non-ribosomal peptide synthetases. Nucleic Acids Res. 2017 Apr; doi: 10.1093/nar/gkx344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Balskus EP, Medema MH, Claesen J, et al. Colibactin: understanding an elusive gut bacterial genotoxin. Nat Prod Rep. 2015;32(11):1534–1540. doi: 10.1039/C5NP00091B. [DOI] [PubMed] [Google Scholar]
- 86.Chu J, Vila-Farres X, Inoyama D, et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat Chem Biol. 2016;12(12):1004–1006. doi: 10.1038/nchembio.2207. http://dx.doi.org/10.1038/nchembio.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kersten RD, Ziemert N, Gonzalez DJ, et al. Glycogenomics as a mass spectrometry-guided genome-mining method for microbial glycosylated molecules. Proc Natl Acad Sci U S A. 2013;110(47):E4407–16. doi: 10.1073/pnas.1315492110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kersten RD, Yang Y-L, Xu Y, et al. A mass spectrometry–guided genome mining approach for natural product peptidogenomics. Nat Chem Biol. 2011;7(11):794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pedersen HK, Gudmundsdottir V, Nielsen HB, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535(7612):376–381. doi: 10.1038/nature18646. [DOI] [PubMed] [Google Scholar]
- 90.Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10(8):538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 91.Faust K, Sathirapongsasuti JF, Izard J, et al. Microbial Co-occurrence Relationships in the Human Microbiome. In: Ouzounis CA, editor. PLoS Comput Biol. 7. Vol. 8. 2012. p. e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kelder T, Stroeve JHM, Bijlsma S, Radonjic M, Roeselers G. Correlation network analysis reveals relationships between diet-induced changes in human gut microbiota and metabolic health. Nutr Diabetes. 2014;4(6):e122. doi: 10.1038/nutd.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lozupone C, Faust K, Raes J, et al. Identifying genomic and metabolic features that can underlie early successional and opportunistic lifestyles of human gut symbionts. Genome Res. 2012;22(10):1974–1984. doi: 10.1101/gr.138198.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weiss S, Van Treuren W, Lozupone C, et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016;10(7):1669–1681. doi: 10.1038/ismej.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Derewacz DK, Covington BC, McLean JA, Bachmann BO. Mapping Microbial Response Metabolomes for Induced Natural Product Discovery. ACS Chem Biol. 2015;10(9):1998–2006. doi: 10.1021/acschembio.5b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goodwin CR, Covington BC, Derewacz DK, et al. Structuring Microbial Metabolic Responses to Multiplexed Stimuli via Self-Organizing Metabolomics Maps. Chem Biol. 2015;22(5):661–670. doi: 10.1016/j.chembiol.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goodwin CR, Sherrod SD, Marasco CC, et al. Phenotypic Mapping of Metabolic Profiles Using Self-Organizing Maps of High-Dimensional Mass Spectrometry Data. Anal Chem. 2014;86(13):6563–6571. doi: 10.1021/ac5010794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Quinn RA, Phelan VV, Whiteson KL, et al. Microbial, host and xenobiotic diversity in the cystic fibrosis sputum metabolome. ISME J. 2016;10(6):1483–1498. doi: 10.1038/ismej.2015.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang M, Carver JJ, Phelan VV, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotech. 2016;34(8):828–837. doi: 10.1038/nbt.3597. http://dx.doi.org/10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang JY, Sanchez LM, Rath CM, et al. Molecular Networking as a Dereplication Strategy. J Nat Prod. 2013;76(9):1686–1699. doi: 10.1021/np400413s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Watrous J, Roach P, Alexandrov T, et al. Mass spectral molecular networking of living microbial colonies. Proc Natl Acad Sci U S A. 2012;109(26):E1743–52. doi: 10.1073/pnas.1203689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rutledge PJ, Challis GL. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Micro. 2015;13(8):509–523. doi: 10.1038/nrmicro3496. http://dx.doi.org/10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- 103.Browne HP, Forster SC, Anonye BO, et al. Culturing of “unculturable” human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533(7604):543–546. doi: 10.1038/nature17645. http://dx.doi.org/10.1038/nature17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stewart CS, Hold GL, Duncan SH, Flint HJ, Harmsen HJM. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int J Syst Evol Microbiol. 2002;52(6):2141–2146. doi: 10.1099/00207713-52-6-2141. [DOI] [PubMed] [Google Scholar]
- 105.Goodman AL, Kallstrom G, Faith JJ, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A. 2011;108(15):6252–6257. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Oberhardt MA, Zarecki R, Gronow S, et al. Harnessing the landscape of microbial culture media to predict new organism–media pairings. Nat Commun. 2015;6:8493. doi: 10.1038/ncomms9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bertrand S, Bohni N, Schnee S, Schumpp O, Gindro K, Wolfender J-L. Metabolite induction via microorganism co-culture: A potential way to enhance chemical diversity for drug discovery. Biotechnol Adv. 2014;32(6):1180–1204. doi: 10.1016/j.biotechadv.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 108.D’Onofrio A, Crawford JM, Stewart EJ, et al. Siderophores from Neighboring Organisms Promote the Growth of Uncultured Bacteria. Chem Biol. 2010;17(3):254–264. doi: 10.1016/j.chembiol.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barkal LJ, Theberge AB, Guo C-J, et al. Microbial metabolomics in open microscale platforms. Nat Commun. 2016;7:10610. doi: 10.1038/ncomms10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lagier J-C, Armougom F, Million M, et al. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012;18(12):1185–1193. doi: 10.1111/1469-0691.12023. [DOI] [PubMed] [Google Scholar]
- 111.Ling LL, Schneider T, Peoples AJ, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517(7535):455–459. doi: 10.1038/nature14098. http://dx.doi.org/10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zengler K, Toledo G, Rappe M, et al. Cultivating the uncultured. Proc Natl Acad Sci U S A. 2002;99(24):15681–15686. doi: 10.1073/pnas.252630999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fritz JV, Desai MS, Shah P, et al. From meta-omics to causality: experimental models for human microbiome research. Microbiome. 2013;1(1):14. doi: 10.1186/2049-2618-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shah P, Fritz JV, Glaab E, et al. A microfluidics-based in vitro model of the gastrointestinal human–microbe interface. Nat Commun. 2016;7:11535. doi: 10.1038/ncomms11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. 2016;16(6):341–352. doi: 10.1038/nri.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lundberg R, Toft MF, August B, Hansen AK, Hansen CHF. Antibiotic-treated versus germ-free rodents for microbiota transplantation studies. Gut Microbes. 2016;7(1):68–74. doi: 10.1080/19490976.2015.1127463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Winston JA, Theriot CM. Impact of microbial derived secondary bile acids on colonization resistance against Clostridium difficile in the gastrointestinal tract. Anaerobe. 2016;41:44–50. doi: 10.1016/j.anaerobe.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang Z, Roberts AB, Buffa JA, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163(7):1585–1595. doi: 10.1016/j.cell.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sassone-Corsi M, Nuccio S-P, Liu H, et al. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature. 2016;540(7632):280–283. doi: 10.1038/nature20557. http://dx.doi.org/10.1038/nature20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tan J, Zuniga C, Zengler K. Unraveling interactions in microbial communities – from co-cultures to microbiomes. J Microbiol. 2015;53(5):295–305. doi: 10.1007/s12275-015-5060-1. [DOI] [PubMed] [Google Scholar]
- 121.Edlund A, Yang Y, Hall AP, et al. An in vitro biofilm model system maintaining a highly reproducible species and metabolic diversity approaching that of the human oral microbiome. Microbiome. 2013;1(1):25. doi: 10.1186/2049-2618-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Auchtung JM, Robinson CD, Britton RA. Cultivation of stable, reproducible microbial communities from different fecal donors using minibioreactor arrays (MBRAs) Microbiome. 2015;3(1):42. doi: 10.1186/s40168-015-0106-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Robinson CD, Auchtung JM, Collins J, Britton RA. Epidemic Clostridium difficile strains demonstrate increased competitive fitness compared to nonepidemic isolates. Infect Immun. 2014;82(7):2815–2825. doi: 10.1128/IAI.01524-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.von Martels JZH, Sadaghian Sadabad M, Bourgonje AR, et al. The role of gut microbiota in health and disease: In vitro modeling of host-microbe interactions at the aerobe-anaerobe interphase of the human gut. Anaerobe. 2017;44:3–12. doi: 10.1016/j.anaerobe.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 125.Rao K, Young VB. Fecal microbiota transplantation for the management of Clostridium difficile infection. Infect Dis Clin North Am. 2015;29(1):109–122. doi: 10.1016/j.idc.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Buffie CG, Bucci V, Stein RR, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517(7533):205–208. doi: 10.1038/nature13828. http://dx.doi.org/10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389(10075):1218–1228. doi: 10.1016/S0140-6736(17)30182-4. [DOI] [PubMed] [Google Scholar]
- 128.Moayyedi P, Surette MG, Kim PT, et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology. 2015;149(1):102–109.e6. doi: 10.1053/j.gastro.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 129.Bajaj JS, Heuman DM, Hylemon PB, et al. Randomised clinical trial: Lactobacillus GG modulates gut microbiome, metabolome and endotoxemia in patients with cirrhosis. Aliment Pharmacol Ther. 2014;39(10):1113–1125. doi: 10.1111/apt.12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Floch MH, Walker WA, Sanders ME, et al. Recommendations for Probiotic Use–2015 Update: Proceedings and Consensus Opinion. J Clin Gastroenterol. 2015;49(Suppl 1):S69–73. doi: 10.1097/MCG.0000000000000420. [DOI] [PubMed] [Google Scholar]
- 131.Gauffin Cano P, Santacruz A, Moya Angela, Sanz Y. Bacteroides uniformis CECT 7771 ameliorates metabolic and immunological dysfunction in mice with high-fat-diet induced obesity. PLoS One. 2012;7(7) doi: 10.1371/journal.pone.0041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Everard A, Belzer C, Geurts L, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110(22):9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Neff CP, Rhodes ME, Arnolds KL, et al. Diverse Intestinal Bacteria Contain Putative Zwitterionic Capsular Polysaccharides with Anti-inflammatory Properties. Cell Host Microbe. 2016;20(4):535–547. doi: 10.1016/j.chom.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453(7195):620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 135.Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500(7461):232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 136.Bindels LB, Delzenne NM, Cani PD, Walter J. Towards a more comprehensive concept for prebiotics. Nat Rev Gastroenterol Hepatol. 2015;12(5):303–310. doi: 10.1038/nrgastro.2015.47. [DOI] [PubMed] [Google Scholar]
- 137.Tzortzis G, Goulas AK, Gibson GR. Synthesis of prebiotic galactooligosaccharides using whole cells of a novel strain, Bifidobacterium bifidum NCIMB 41171. Appl Microbiol Biotechnol. 2005;68(3):412–416. doi: 10.1007/s00253-005-1919-0. [DOI] [PubMed] [Google Scholar]
- 138.Depeint F, Tzortzis G, Vulevic J, I’anson K, Gibson GR. Prebiotic evaluation of a novel galactooligosaccharide mixture produced by the enzymatic activity of Bifidobacterium bifidum NCIMB 41171, in healthy humans: a randomized, double-blind, crossover, placebo-controlled intervention study. Am J Clin Nutr. 2008;87(3):785–791. doi: 10.1093/ajcn/87.3.785. http://www.ncbi.nlm.nih.gov/pubmed/18326619. Accessed April 7, 2017. [DOI] [PubMed] [Google Scholar]
- 139.O’Mahony L, McCarthy J, Kelly P, et al. Lactobacillus and bifidobacterium in irritable bowel syndrome: Symptom responses and relationship to cytokine profiles. Gastroenterology. 2005;128(3):541–551. doi: 10.1053/j.gastro.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 140.Whorwell PJ, Altringer L, Morel J, et al. Efficacy of an Encapsulated Probiotic Bifidobacterium infantis 35624 in Women with Irritable Bowel Syndrome. Am J Gastroenterol. 2006;101(7):1581–1590. doi: 10.1111/j.1572-0241.2006.00734.x. [DOI] [PubMed] [Google Scholar]
- 141.Guyonnet D, Chassany O, Ducrotte P, et al. Effect of a fermented milk containing Bifidobacterium animalis DN-173 010 on the health-related quality of life and symptoms in irritable bowel syndrome in adults in primary care: A multicentre, randomized, double-blind, controlled trial. Aliment Pharmacol Ther. 2007;26(3):475–486. doi: 10.1111/j.1365-2036.2007.03362.x. [DOI] [PubMed] [Google Scholar]
- 142.Silk DBA, Davis A, Vulevic J, Tzortzis G, Gibson GR. Clinical trial: The effects of a trans-galactooligosaccharide prebiotic on faecal microbiota and symptoms in irritable bowel syndrome. Aliment Pharmacol Ther. 2009;29(5):508–518. doi: 10.1111/j.1365-2036.2008.03911.x. [DOI] [PubMed] [Google Scholar]
- 143.Suez J, Elinav E. The path towards microbiome-based metabolite treatment. Nat Microbiol. 2017;2(6):17075. doi: 10.1038/nmicrobiol.2017.75. [DOI] [PubMed] [Google Scholar]
- 144.Canani RB, Di Costanzo M, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol. 2011;17(12):1519–1528. 1519. doi: 10.3748/wjg.v17.i12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zimmerman MA, Singh N, Martin PM, et al. Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am J Physiol – Gastrointest Liver Physiol. 2012;302(12) doi: 10.1152/ajpgi.00543.2011. http://ajpgi.physiology.org/content/302/12/G1405. Accessed June 19, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hamer HM, Jonkers DMAE, Vanhoutvin SALW, et al. Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clin Nutr. 2010;29(6):738–744. doi: 10.1016/j.clnu.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 147.Zeevi D, Korem T, Zmora N, et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell. 2015;163(5):1079–1094. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 148.Haiser HJ, Seim KL, Balskus EP, Turnbaugh PJ. Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes. 2014;5(2):233–238. doi: 10.4161/gmic.27915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A. 2009;106(34):14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Turnbaugh PJ, Gordon JI, Rusch DB, et al. An Invitation to the Marriage of Metagenomics and Metabolomics. Cell. 2008;134(5):708–713. doi: 10.1016/j.cell.2008.08.025. [DOI] [PubMed] [Google Scholar]