

Abstract
For long time, the role of LDL and inflammation in the pathogenesis of atherosclerosis have been studied independently from each other and only more recently a common platform has been suggested. Accumulation of excess cholesterol due to the presence of increased circulating LDL promotes endothelium dysfunction and activation, which is associated with increased production of pro‐inflammatory cytokines, overexpression of adhesion molecules, chemokines and C‐reactive protein (CRP), increased generation of reactive oxygen species and reduction of nitric oxide levels and bioavailability. All these processes favour the progressive infiltration of inflammatory cells within the arterial wall where cholesterol accumulates, both extracellularly and intracellularly, and promotes vascular inflammation. According to this, lipid‐lowering therapies should improve inflammation and, indeed, statins decrease circulating inflammatory markers such as CRP and improve endothelial function and plaque burden. Pleiotropic activities have been proposed to explain this effect. However, mendelian randomization studies ruled out a direct role for CRP on coronary artery disease and studies with other lipid lowering drugs, such as ezetimibe showed that the beneficial effect of LDL‐cholesterol‐lowering therapies on systemic inflammatory status, as monitored by changes in CRP plasma levels, could be achieved, independently of the mechanism of action, only in patients presenting with baseline inflamed conditions. These observations strengthen the direct link between cholesterol and inflammation and indicate that decreasing LDL levels is one of the key goals for improving cardiovascular outcome.
Linked Articles
This article is part of a themed section on Targeting Inflammation to Reduce Cardiovascular Disease Risk. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.22/issuetoc and http://onlinelibrary.wiley.com/doi/10.1111/bcp.v82.4/issuetoc
Abbreviations
- CAD
coronary artery disease
- CETP
cholesteryl ester transfer protein
- CHD
coronary heart disease
- CRP
C‐reactive protein
- CV
cardiovascular
- FMD
flow‐mediated dilatation
- HMG‐CoA
3‐hydroxy‐3‐methylglutaryl‐CoA
- hs‐CRP
high‐sensitivity CRP
- IVUS
intravascular ultrasound
- LDL‐C
LDL‐cholesterol
- LDLR
LDL receptor
- Lp(a)
lipoprotein(a)
- NLRP3
nucleotide‐binding domain, leucine‐rich‐containing family, pyrin domain‐containing‐3
- OxLDL
oxidized LDL
- PAV
percent atheroma volume
- PCSK9
proprotein convertase subtilisin/kexin type 9
- SNP
single nucleotide polymorphism
- TG
triglycerides
- VLDL
very low density lipoprotein
Introduction
Inflammation and hypercholesterolemia are linked in a vicious cycle in which the excess of cholesterol that accumulates in the arterial wall induces an inflammatory response that, in turn, accelerates cholesterol deposition and amplifies inflammation.
The role of LDL and inflammation in the pathogenesis of atherosclerosis have been, for a long time, studied independently from each other and only more recently a common platform has been suggested. The role of cholesterol (the ‘cholesterol era’) and that of cholesterol‐carrying LDL (the ‘LDL era’) in the formation of atherosclerotic plaques has been extensively evaluated starting from the beginning of the 20th century and was validated by the discovery of the LDL receptor (Linton et al., 2000; Goldstein and Brown, 2015). In parallel, the concept of a role for inflammation as a key player in atherosclerosis development gained attention, as a result of studies showing that atherosclerotic plaques are characterized by the accumulation of inflammatory cells. This accumulation, by producing pro‐inflammatory cytokines, further promotes the entry of monocytes into the arterial wall, thus further propagating the inflammatory reaction (Ross, 1999; Libby, 2002; Libby, 2012).
The aim of this brief review is to describe the evidence linking cholesterol and LDL to inflammation and discuss the data from clinical trials with lipid‐lowering drugs suggesting that the beneficial impact on inflammation is proportional to the reduction of levels of LDL‐cholesterol (LDL‐C).
Cholesterol promotes inflammation
Accumulation of excess cholesterol within the arteries promotes endothelium dysfunction and activation, which results in increased production of pro‐inflammatory cytokines and reactive oxygen species, overexpression of adhesion molecules, chemokines and reduction of nitric oxide levels and bioavailability (van Diepen et al., 2013; Gimbrone and Garcia‐Cardena, 2016) (Figure 1). These processes contribute to the recruitment and infiltration of monocytes, which differentiate into macrophages and, following the uptake of modified‐LDL via scavenger receptors, become foam cells (Ross, 1999; van Diepen et al., 2013; Sorci‐Thomas and Thomas, 2016) (Figure 1). More recently, cholesterol has been directly linked to inflammation via the activation of the NLRP3 inflammasome (Grebe and Latz, 2013), which might favour the instauration and amplification of a local and systemic immuno‐inflammatory response (Tabas and Bornfeldt, 2016), characterized by the production of several pro‐inflammatory cytokines; among them, C‐reactive protein (CRP), IL‐6 and IL‐1 are well‐established markers of inflammation and their possible causal role in atherosclerosis has been widely investigated (Ridker, 2016). In addition, among genetic traits associated with LDL‐C levels, single nucleotide polymorphisms (SNPs) in the CELSR2/PSRC1/SORT1 locus and in the APOE/APOC1/TOMM40 locus have been associated also with inflammatory‐related phenotypes (Kocarnik et al., 2014), which further supports the strong relationship between cholesterol and inflammation.
Figure 1.
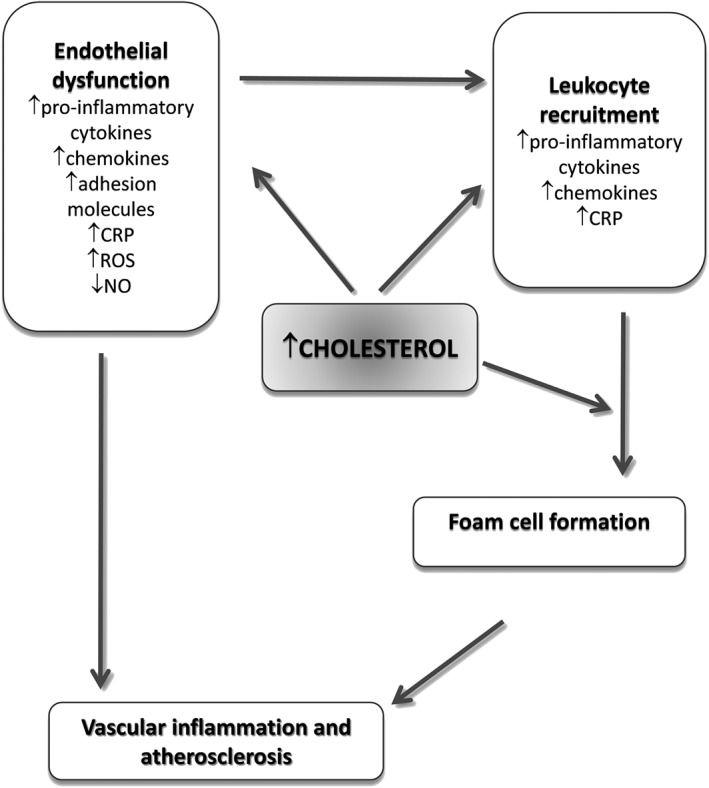
Link between cholesterol and inflammation within the arterial wall.
Cholesterol and CRP
CRP is a plasma protein belonging to the superfamily of pentraxins, proteins involved in acute phase responses. It is synthesized by the liver in response to IL‐6 and is a well‐established marker of inflammation (Pepys and Hirschfield, 2003) whose levels significantly increase in response to inflammatory stimuli (Gabay and Kushner, 1999). A large meta‐analysis from 54 long‐term prospective studies reported a continuous associations of plasma CRP levels with coronary heart disease (CHD) risk (Kaptoge et al., 2010), further supporting the concept that CRP could be a powerful risk biomarker for first and recurrent cardiovascular (CV) events (Koenig, 2013).
CRP is, however, synthesized in cells other than hepatocytes, including cells within the atherosclerotic plaques, as suggested by the co‐localization of CRP and oxidized LDL (OxLDL) and macrophages in atherectomy specimens from patients with stable or unstable angina and acute myocardial infarction (Meuwissen et al., 2006). Also endothelial cells exposed in vitro to modified LDL or to pro‐inflammatory cytokines produce CRP (Venugopal et al., 2005; Chu et al., 2013). The role of the CRP produced by the atherosclerotic plaque, however, is unknown, and it is unclear whether it contributes to CRP plasma levels in CVD patients. CRP has been proposed to play a causal role in atherosclerosis (Zhang et al., 1999; Pasceri et al., 2000; Cirillo et al., 2005). Data from clinical trials indicating that the greatest reduction in CV events in statin‐treated patients is observed in those achieving both LDL‐C and high‐sensitivity CRP (hs‐CRP) reduction (Joshi and Jacobson, 2010) support this hypothesis. However, several other studies, including Mendelian randomization studies, have downplayed the causal role of CRP in CHD (Zacho et al., 2008; Elliott et al., 2009; Wensley et al., 2011; Lane et al., 2014; Noveck et al., 2014) and the field is still open to discussion.
Cholesterol and inflammatory cytokines
Cytokines play a key role in inflammatory diseases and a link with hypercholesterolemia and atherosclerosis has emerged mainly for the IL‐6, IL‐1 and TNFα pathways.
IL‐6 is a pro‐inflammatory cytokine produced by several cell types in response to infections or other conditions, playing a critical role in the pathogenesis of rheumatoid arthritis (RA) (Liu et al., 2015) but also involved in atherogenesis (Schuett et al., 2009). In endothelial cells, modified LDL up‐regulate IL‐6 (Lubrano et al., 2015), which in turn induces the expression of macrophage scavenger receptors involved in the uptake of modified LDL, thus promoting the formation of foam cells (Schuett et al., 2009) and establishing an inflammatory cycle in the plaque. In humans, IL‐6 levels predict future CV risk and correlate with endothelial dysfunction and carotid intima‐media thickness (Ridker, 2016). In addition, carriers of the Asp358Ala SNP in the IL‐6 receptor gene (IL6R) have increased serum levels of IL‐6R (probably due to an increased shedding of the receptor) and a paradoxical increase of IL‐6, but reduced levels of the downstream mediators CRP and fibrinogen. This is suggestive of an attenuation of the IL‐6/IL‐6R axis signalling in carriers of this variant; indeed this IL6R SNP was associated with a decreased CHD risk (Sarwar et al., 2012; Swerdlow et al., 2012). However, data from clinical trials in patients with RA treated with therapies targeting the IL‐6/IL‐6R axis challenged this hypothesis. Indeed the treatment of RA patients with tocilizumab, a monoclonal antibody that blocks both membrane‐bound and circulating IL‐6R, increased LDL‐C levels, an effect observed also with tofacitinib, a JAK inhibitor that blocks intracellular signalling of several cytokines (including IL‐6) (Souto et al., 2015). In vitro tocilizumab reduced the levels of hepatic LDL receptor (LDLR) (Strang et al., 2013), which may result in impaired LDL catabolism and explain the increased LDL‐C plasma levels observed in RA patients treated with this biological agent (Kawashiri et al., 2011; Strang et al., 2013; McInnes et al., 2015). These observations further stress the interplay between cholesterol metabolism and inflammatory signals.
In addition, IL‐1 plays a relevant role in vascular inflammation and atherogenesis (Qamar and Rader, 2012). IL‐1 is able to induce its own production but also up‐regulates the expression of downstream mediators of inflammation such as IL‐6 and CRP (Signorelli et al., 2014). Cholesterol crystals contribute to the activation of IL‐1 dependent pathway by activating the NLRP3 inflammasome, which favours the cleavage and secretion of IL‐1 (Duewell et al., 2010; Rajamaki et al., 2010). Several studies in humans confirmed the involvement of IL‐1 in the development of atherosclerosis (Galea et al., 1996; Fearon and Fearon, 2008; Olofsson et al., 2009), and patients with IL‐1 polymorphisms resulting in higher levels of pro‐inflammatory cytokines were at increased risk for the presence of coronary artery disease (CAD) and CV events (Tsimikas et al., 2014). The central role of IL‐1 in inflammation has been confirmed by studies with agents targeting IL‐1 activity, such as anakinra (an IL‐1 receptor antagonist), which decreased CRP production in acute coronary syndrome patients (Abbate et al., 2010; Abbate et al., 2013; Morton et al., 2015), but failed to reduce the risk of recurrent ischaemic events, whereas it may prevent new‐onset heart failure (Abbate et al., 2015). Canakinumab, a human monoclonal antibody that neutralizes IL‐1ß, reduces CRP and IL‐6 in patients with T2DM and established CVD without affecting plasma cholesterol levels (Choudhury et al., 2016).
TNFα is a pro‐inflammatory cytokine, which contributes to the development of atherosclerosis by inducing endothelial dysfunction and initiating the inflammatory cascade inside the arterial wall (Ross, 1999). Although its increase during acute inflammation is protective, its persistence at high levels during chronic inflammation may results in alterations of both lipid and glucose metabolism with detrimental consequences (Popa et al., 2007). In fact, TNFα may interfere with cholesterol metabolism, by decreasing the secretion of apolipoproteins and reducing cholesterol catabolism and excretion, which results in decreased LDL‐C concentrations (Popa et al., 2007). In addition, TNFα alters the quality of lipoproteins by favouring the generation of pro‐atherogenic small dense LDL and OxLDL due to changes in sphingolipid content (Popa et al., 2007). TNFα also reduces HDL‐C levels and alters HDL composition (Popa et al., 2007). Some SNPs in the TNFα gene are associated with changes in LDL‐C levels; the C‐857T SNP on the TNFα promoter region was associated with higher LDL‐C levels (3.14 mmol·L−1 in the T carriers [TT/CT genotypes] and 2.89 mmol·L−1 in the non‐T carriers [CC genotype], P < 0.05) and increased frequency of carotid plaque in patients with type 2 diabetes mellitus (DM) (87% in the T carriers vs. 63% in the non‐T carriers, P = 0.0358) (Yamashina et al., 2007; Takahashi et al., 2010). Interestingly, when analysed according to statin treatment, LDL‐C levels were higher in the T carriers compared with the C carriers only in statin‐treated subjects, but not in statin‐untreated, and the reduction of LDL‐C levels achieved with statins was lower in the T carriers than in the C carriers (27.6% vs. 36.4%, P = 0.031) (Takahashi et al., 2010). Similarly, among asthmatic patients, the frequency of the promoter region −308G/A polymorphism was higher in subjects having metabolic syndrome and was associated with higher TNFα levels and higher LDL‐C levels in GA/AA genotypes than in GG genotype (3.13 vs. 2.55 mmol·L−1, P = 0.029) (Yang et al., 2015). Despite these observations, the anti‐TNFα therapies seem to be neutral on lipid profile, as reported by several meta‐analyses which could not find major significant changes for LDL‐C or apolipoprotein B following therapy with TNFα antagonists (van Sijl et al., 2011; Daien et al., 2012; Di Minno et al., 2014).
These observations support the concept that hypercholesterolaemia promotes systemic and vascular inflammation through the induction of several mediators. Some of them could contribute to the amplification of the inflammatory response, others, such as CRP, mark the ongoing inflammatory response, while a direct effect of inflammatory cytokines on plasma cholesterol levels is debatable.
Inflammation alters lipid metabolism
Chronic inflammatory diseases (such as RA or systemic lupus erythematosus) are associated with increased CV risk (Haque et al., 2008) and patients present with quantitatively and qualitatively altered lipid and lipoproteins profile that include a reduction of total cholesterol, HDL‐C and apolipoprotein A‐I, and increased levels of small dense LDL, lipoprotein(a) [Lp(a)] and triglycerides (TG) (de Carvalho et al., 2008; Amezaga Urruela and Suarez‐Almazor, 2012; Ammirati et al., 2014; Montecucco et al., 2015). A more severe disease state is associated with more pronounced alterations in lipids and lipoproteins profile.
Inflammatory mediators such as IL‐6, IL‐1ß and TNFα may alter lipid metabolism (Khovidhunkit et al., 2004), by increasing very low density lipoprotein (VLDL) production and secretion by the liver, paralleled by a decreased clearance of TG‐rich lipoproteins, with the net effect of increasing serum TG levels (Khovidhunkit et al., 2004). As a consequence, the activity of the cholesteryl ester transfer protein (CETP), in the attempt to transfer TG from VLDL/LDL to LDL/HDL, increases; these TG‐enriched particles become the substrate of the hepatic lipase and lipoprotein lipase with the generation of small dense LDL and HDL as final products. Small dense LDL enter the arterial intima more easily (Diffenderfer and Schaefer, 2014), and are more prone to oxidation while small dense HDL possess a limited antioxidant and anti‐inflammatory activity (Welty, 2013). In addition, the presence of IL‐6 responsive elements present in the promoter of apo(a) gene contribute to the increased Lp(a) levels observed during inflammation. (Wade et al., 1993).
In addition, LDL levels and composition change during inflammation. On one hand, LDL‐C levels decrease as a consequence of increased LDLR expression, which however fosters the intracellular accumulation of cholesterol (Ruan et al., 2006; Ye et al., 2009) and might induce inflammasome activation. On the other hand, circulating LDL have an increased susceptibility to oxidation (Frostegard et al., 2005; Garcia‐Gomez et al., 2014), which explain the increased plasma levels of OxLDL in patients with chronic inflammatory disease (Ahmad et al., 2014; Nowak et al., 2016). OxLDL are more atherogenic, can amplify the inflammatory response but can also favour the accumulation of cholesterol in lysosomes, which results in increased cellular toxicity by favouring lysosome disruption, because of the presence of cholesterol crystals (Roma et al., 1992). Similarly, apart from a decrease in serum HDL, also HDL particle functions are altered during inflammation, resulting in the deterioration of most steps of the reverse cholesterol transport process and a reduced ability of ‘inflamed’ HDL to protect LDL from oxidation (Namiri‐Kalantari et al., 2015).
Altogether, these observations indicate a convincing link between inflammation and lipids in the process of atherosclerosis. Is there clinical evidence that inflammation can be modified by lipid‐lowering therapies?
Effects of statins on vascular inflammation
As LDL‐C levels are directly correlated with systemic inflammation, which is a key element in the pathogenesis of atherosclerosis (Ross, 1999; Viola and Soehnlein, 2015) targeting lipoprotein metabolism should represent a therapeutic option to reduce the burden of inflammation and of CHD. Indeed, it is well known that statins [3‐hydroxy‐3‐methylglutaryl‐coenzyme A (HMG‐CoA) reductase inhibitors] decrease levels of LDL‐C and reduce CAD. Statins inhibit the biosynthesis of cellular cholesterol in the liver, thus resulting in an increased expression of the LDLR in the hepatocytes, which in turn favours the increased catabolism of LDL from the circulation. By reducing LDL‐C levels, statins decrease the number of LDL particles that can infiltrate the vessel wall and thus limit atherosclerosis progression. Beyond this mechanism, several experimental studies have shown that statins exert additional effects, which, at the molecular level, relate to their ability to influence protein prenylation (Jasinska et al., 2007), thus in turn affecting different intracellular signalling pathways independently of their lipid‐lowering property. These additional functions are grouped under the umbrella of ‘pleiotropic effects of statins’, which have been extensively reviewed elsewhere (Bellosta et al., 2000; Mihos et al., 2014; Satoh et al., 2015). Among them, statins improve endothelial function and reduce platelet aggregation, increase the number and activity of endothelial progenitor cells, inhibit migration and proliferation of smooth muscle cells, stabilize coronary plaques and promote atheroma regression (Bellosta et al., 2000; Mihos et al., 2014; Satoh et al., 2015). Are the beneficial effects of statins on inflammation a consequence of their pleiotropic effects or the consequence of LDL cholesterol lowering?
A key effect of statins is the ability to decrease the levels of inflammatory markers including CRP (Table 1). In the primary prevention setting, lovastatin (20 to 40 mg) (AFCAPS/TexCAPS study) (Ridker et al., 2001) showed to reduce CRP levels by approximately 15%, after 1 year of treatment. The MIRACL and the REVERSAL studies showed a dose‐dependent effect of statins on CRP reduction, with the more aggressive therapy (atorvastatin) to be more effective than the standard therapy (pravastatin) (Kinlay et al., 2003; Nissen et al., 2004).
Table 1.
Effect of statins on CRP levels in clinical trials
| (Ref.) | Drug | Time of intervention | Median baseline CRP level (mg·L−1) | CRP % change (P value) |
|---|---|---|---|---|
| (Ridker et al., 1999) | Pravastatin | 5 years | 2.3 | −17.4 (P = 0.004) |
| (Ridker et al., 2001) | Lovastatin | 1 year | 1.6 | −14.8 (P < 0.001) |
| (Albert et al., 2001) | Pravastatin | 24 weeks | 2.0 | −14.2 (P < 0.001) |
| (Jialal et al., 2001) | Pravastatin | 6 weeks | 2.6 | −20.3 (P < 0.025) |
| Simvastatin | −22.8 (P < 0.025) | |||
| Atorvastatin | −28.3 (P < 0.025) | |||
| (Kinlay et al., 2003) | Atorvastatin | 16 weeks | 11.5 | −34 (P < 0.0001) |
| (Nissen et al., 2004) | Pravastatin | 18 months | 3.0 | −5.2 |
| Atorvastatin | −36.4 (P < 0.001 vs. pravastatin) | |||
| (Ridker et al., 2005) | Atorvastatin | 24 months | 12.2 | −89.3 (P < 0.001) |
| Pravastatin | 11.9 | −82.4 (P < 0.001) | ||
| (Morrow et al., 2006) | Simvastatin | 4 months | 2.4 | −29.2 (P < 0.0001) |
| (Ridker et al., 2008) | Rosuvastatin | 48 months | 4.2 | −57.1 (P < 0.001) |
| (Emberson et al., 2011) | Simvastatin | 5.0 years | 3.07 | −27 (P < 0.0001) |
| (Sever et al., 2013) | Atorvastatin | 6 months | 2.4 w/o events 3.0 w/ events | −25.8 (P < 0.02) |
| (Soedamah‐Muthu et al., 2015) | Atorvastatin | 1 year | 1.3 | −9.8 |
Also in a post hoc analysis of a study with pravastatin in secondary prevention (CARE study), this statin (5 years of treatment) reduced CRP by approximately 17% (Ridker et al., 1999) (Table 1). In both the A to Z trial and the PROVE IT‐TIMI 22 study, the best outcomes were observed in patients who reached both an LDL‐C less than 70 mg·dL−1 and an hs‐CRP less than 2.0 mg·L−1, with even greater benefit in those individuals in which the hs‐CRP was less than 1.0 mg·L−1 (Ridker et al., 2005; Morrow et al., 2006). This observation supported the concept of a ‘dual target therapy’, in which patients benefit from both LDL‐C and hs‐CRP lowering. These observations paved the road for the most important trial which to date examined the effect of statins on hs‐CRP and the resulting clinical outcomes, which is the JUPITER trial. This study investigated approximately 17 800 patients with median LDL‐C of 108 mg·dL−1 but elevated hs‐CRP (>2.0 mg·L−1) (Ridker et al., 2008) which were treated with rosuvastatin (20 mg) or placebo. Major CV events such as stroke, nonfatal myocardial infarction, revascularization, unstable angina or death from CV causes were reduced by 44% (P < 0.00001) in the treatment arm as was the case for both LDL‐C and CRP (Ridker et al., 2008). A major finding of the study was the observation that the greatest reduction in CV events was in the treatment group that achieved both LDL‐C less than 70 mg·dL−1 and hs‐CRP less than 2 mg·L−1 (65% reduction), compared with only a 33% risk reduction in patients that achieved one or neither target (P < 0.0001) (Ridker et al., 2009).
However, other clinical trials (ASCOT and CARDS) (Table 1) have reported different findings. Indeed the lowest risk for CV events was observed in statin‐treated subjects who achieved LDL‐C level below the median independent of on‐treatment CRP levels, suggesting that on‐therapy LDL‐C levels are the major determinant of the beneficial effects of statins (Sever et al., 2013; Soedamah‐Muthu et al., 2015).
It is important to note that baseline CRP levels in the CARDS trial were much lower compared with the JUPITER trial (1.4 vs. 4.3 mg·L−1). Similarly, in the ASCOT trial the median level of CRP in patients without history of CV events (~90% of the studied population) was 2.4 mg·L−1 (Ridker et al., 2008; Sever et al., 2013; Soedamah‐Muthu et al., 2015). In addition, the different characteristics of the subjects included in these studies may have influenced the results. In fact, the JUPITER study included apparently healthy subjects with baseline LDL‐C levels <130 mg·dL−1 (but CRP ≥2 mg·L−1) (Ridker et al., 2008), while CARDS enrolled type 2 diabetes patients (Soedamah‐Muthu et al., 2015) and ASCOT hypertensive patients with other ≥3 other CV risk factors (Sever et al., 2013).
Furthermore, a meta‐analysis, which included 23 studies with statins, reported that 89 ‐ 98% of the CRP reduction was directly related to the degree of LDL‐C reduction obtained (Kinlay, 2007). Thus, although individual statin studies have shown a poor relationship between CRP reduction and LDL‐C reduction, when data are analysed as aggregate, a strong correlation can be observed. The possible relationship between LDL‐C and CRP reductions in different clinical settings might be under‐estimated, as many authors reported a lack of correlation for individual data. Although this may be explained by large measurement error and large intra‐individual variations in CRP levels, we would like to offer a different interpretation that relies on individual variability to the cholesterol and lipid burden‐mediated inflammatory response in tissues that makes each of us unique as responder. This interpretation will also explain why CRP is a very good predictor of event, independent of LDL; because it captures the information derived from individual variability.
Indeed statin therapy was associated also with the reduction of IL‐6, TNF‐α and cell adhesion molecule levels in patients with CV risk factors (van de Ree et al., 2003; Ascer et al., 2004). In the MIRACL study, the reduction of IL‐6 achieved with atorvastatin therapy was associated with a relative reduction of the risk of stroke after an acute coronary syndrome (Kinlay et al., 2008).
Apart from the beneficial effects on inflammatory markers, statins also improve endothelial function in patients at high CV risk or with CAD (Fichtlscherer et al., 2006). Several meta‐analyses have evaluated the benefit of statins on endothelial function in different cohorts. The data indicate that statin therapy is associated with significant improvement in both peripheral and coronary endothelial function (Reriani et al., 2011). Of note, controversies exist among trials reporting the effects of statins on endothelial dysfunction in patients with diabetes mellitus and while the overall finding was that statins improve endothelial function also in diabetics, patients with a more compromised endothelial function are less likely to benefit from statin treatment (Zhang et al., 2012). A key gap in these meta‐analyses is that it is not known whether the benefit of statins on endothelial function is proportional to the reduction of LDL‐C levels.
Aside from affecting endothelial function, statins were also demonstrated to favour atherosclerosis regression, by reducing the percent atheroma volume (PAV), as determined by intravascular ultrasound (IVUS). This was clearly shown in the SATURN and ASTEROID trials (Nicholls et al., 2011), where average on‐treatment LDL‐C levels below 70 mg·dL−1 for 24 months were associated with a significant PAV reduction. A pre‐specified post hoc analysis later indicated that CRP levels, but not LDL‐C levels, were associated with coronary atheroma regression and CV events. However, the absolute change in CRP was not prognostic of major CV events (Puri et al., 2013).
All these observations clearly point to a beneficial effect of statins in inflammation, which is reflected in a decrease of circulating inflammatory markers such as CRP and improvement of endothelial function and plaque burden.
Effects of LDL‐C‐lowering therapies other than statins on vascular inflammation and outcomes
While there is no doubt that statins improve systemic and vascular inflammation, the rationale for using a statin to decrease CRP and therefore to target a causal factor in vascular inflammation associated with atherosclerosis should be carefully considered. Indeed, several analyses have shown that CRP does not play a causal role in the pathogenesis of atherosclerosis (Zacho et al., 2008; Elliott et al., 2009; Wensley et al., 2011) and the acute manifestations of CAD. On the basis of these results, it is likely that the effect of statins on CRP could mirror the beneficial effect of this class of drugs on other atherogenic players, rather than a direct anti‐inflammatory/vasculoprotective effect mediated through CRP reduction. This leaves the room open for considering that statins might exert their anti‐inflammatory activities through the ability of reducing the pro‐inflammatory/pleiotropic effects of cholesterol‐rich lipoproteins. If this is the case, then pharmacological approaches aimed at reducing LDL‐C via mechanisms that are independent of HMG‐CoA reductase inhibition should demonstrate a benefit in terms of CRP reduction and vascular inflammation, which should be proportional to the reduction of LDL‐C achieved. This is the case for some but not all lipid lowering agents.
Ezetimibe, by inhibiting the cholesterol transport protein NPC1‐like 1, reduces the intestinal absorption of cholesterol and is utilized in clinical practice as monotherapy or as an adjunct to statins (Sudhop et al., 2009). This drug has been extensively studied for the ability not only to decrease plasma LDL‐C levels but also to decrease CRP and improve CV outcome. Two‐pooled analyses of randomized, placebo‐controlled trials of ezetimibe in hypercholesterolemic patients have demonstrated that patients treated with placebo had a 5% increase in CRP levels while those treated with ezetimibe had a 1% decrease, with a between‐treatment difference of 6%, that however was not statistically significant (Pearson et al., 2009). LDL‐C decreased significantly with ezetimibe (−18%) compared with placebo (+0.5%) (Pearson et al., 2009). When ezetimibe was added to a statin, CRP was significantly reduced compared with statin monotherapy (−12% vs. −1%), as it was LDL‐C (−27% vs. −3%) (Pearson et al., 2009). When data available from the different clinical trials with ezetimibe are investigated and the two arms (statins only or statins + ezetimibe) are compared, the reduction in CRP levels is proportional to the reduction observed in LDL‐C levels (Figure 2). The correlation between the changes of the two parameters is similar in the statins only and in the statin + ezetimibe group (Figure 2).
Figure 2.
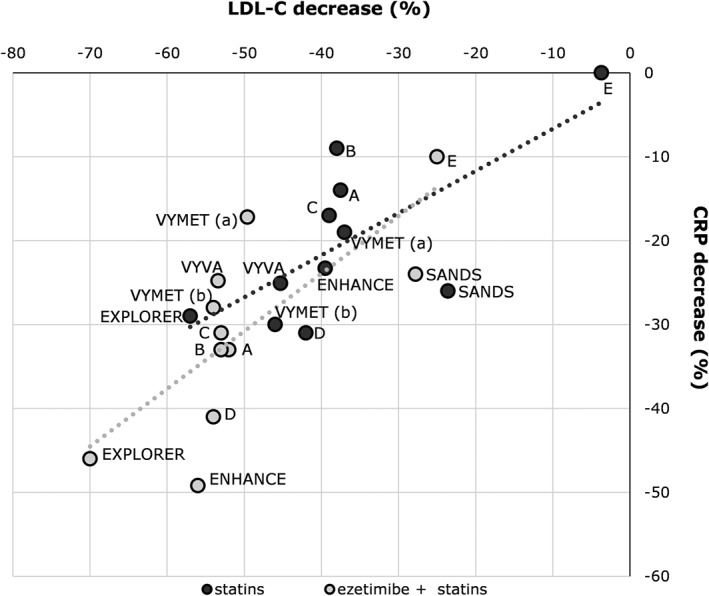
Association between reduction in LDL‐C levels and reduction in CRP levels among clinical trials comparing statin monotherapy with ezetimibe/statin combination. The following studies were included in the figure: VYVA: Ballantyne, Am Heart J, 2005; VYMET: Robinson, Am J Cardiol, 2009; EXPLORER: Ballantyne, Am J Cardiol, 2007; ENHANCE: Kastelein, NEJM, 2008; SANDS: Fleg, J Am Coll Cardiol, 2008; A: Sager, Atherosclerosis, 2005; B: Goldberg, Mayo Clin Proc, 2004; C: Bays, Clin Ther, 2004; D: Ballantyne, Circulation, 2003; E: Gagne, Am J Cardiol, 2002.
Studies with other lipid lowering drugs, including those inhibiting the synthesis of apolipoprotein B containing lipoproteins, such as lomitapide or mipomersen (Norata et al., 2013), as well as those increasing LDL‐C metabolism, such as inhibitors of the proprotein convertase subtilisin/kexin type 9 (PCSK9) (Norata et al., 2014; Norata et al., 2016) or CETP inhibitors have not investigated in detail the effects on CRP reduction and the few available data do not show a robust correlation between the magnitude of LDL‐C reduction and that of CRP (Flaim et al., 2014; Stroes et al., 2014; Cannon et al., 2015; Hovingh et al., 2015; Raal et al., 2015a, b; Nicholls et al., 2016; Sahebkar et al., 2016). While it could appear surprising that PCSK9 inhibitors, which decrease LDL‐C levels up to 60% (Robinson et al., 2015; Sabatine et al., 2015), do not affect CRP levels, also in the placebo arm (statins and/or ezetimibe treated patients) of almost all clinical trials, no significant changes in CRP plasma levels were observed (Figure 3). These findings could be explained by noting that, in clinical trials with anti‐PCSK9 therapies, the median baseline levels of CRP were below 2 mg·L−1 (also in those trials who tested statin‐intolerant patients), thus excluding patients with systemic inflammation from these studies and perhaps limiting the possibility to appreciate a beneficial effect of lipid‐lowering drugs in spite of LDL‐C reduction. Indeed when clinical trials with CRP levels above 2 mg·L−1 are considered, CRP is reduced by lipid‐lowering therapies, independently of the mechanism of action (Figure 3). Furthermore, even in the SATURN study, rosuvastatin, in spite of a LDL‐C reduction of −43.5%, decreased CRP levels only in those patients with CRP baseline level of 2.3 mg·L−1 (0.8 mg·L−1 at follow‐up), while no effect or rather a small increase was reported in the group with CRP baseline level of 1.1 mg·L−1 (1.6 mg·L−1 at follow‐up) (Puri et al., 2013). This further suggests that the beneficial effect of LDL‐C‐lowering therapies on systemic inflammatory status, as monitored by changes in CRP plasma levels, is evident only in patients presenting with increased inflammatory conditions. Could these findings be transferred also to markers of vascular dysfunction and inflammation?
Figure 3.
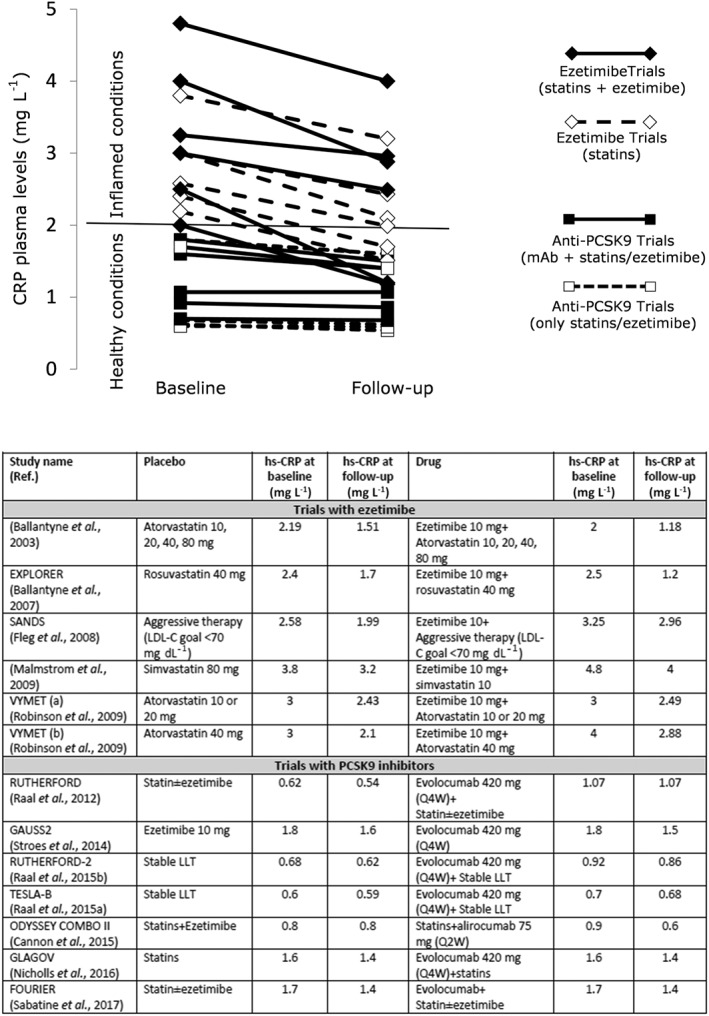
Effects of lipid‐lowering therapies on plasma CRP levels reduction according to CRP baseline levels.
The PANACEA study showed that, in obese patients with metabolic syndrome, a low‐dose statin/ezetimibe combination (10/10 mg) not only resulted in a similar of LDL‐C reduction compared to high‐dose statin monotherapy (simvastatin 80 mg) but also in a comparable endothelial function as determined by both flow‐mediated dilatation (FMD) and peripheral arterial tonometry (EndoPAT) (Westerink et al., 2013). Similar observations were previously reported also in other studies. Stable CAD patients with dysglycemia that commonly are dyslipidemic but also present endothelial dysfunction and vascular inflammation, were treated with simvastatin 80 mg or ezetimibe/simvastatin 10/10 mg for 6 weeks. Similar LDL‐C reductions from the baseline were obtained as well as similar improvement of FMD (Settergren et al., 2008). Similarly, obese women with LDL‐C ≥100 mg·dL−1 treated with simvastatin 80 mg or ezetimibe/simvastatin 10/10 mg for 8 weeks showed similar reductions of LDL‐C levels (−27% and −30%, respectively) and similar increases of FMD (+39% and +41%, respectively) (Garcia et al., 2016). These observations, suggesting that lipid‐lowering per se rather than pleiotropic properties of statins plays a key role in the improvement of endothelial function, were further supported by the results of a meta‐analysis including six trials with 213 participants which compared high‐dose statin versus low‐dose statin combined with ezetimibe (Ye et al., 2012). The two lipid‐lowering regimens induced similar reductions of LDL‐C (P = 0.12 between treatments) and CRP levels (P = 0.89) and similar increments of FMD (P = 0.68), suggesting similar beneficial effects. In line with these findings, we have reported that pravastatin and ezetimibe reduced LDL‐C levels similarly and increased FMD at a comparable extent in subjects with moderate hypercholesterolemia (Grigore et al., 2013).
Not all studies are in agreement with these findings. Treatment of dyslipidemic subjects without signs of CAD with simvastatin 40 mg or ezetimibe/simvastatin 10/10 mg for 4 weeks resulted in the comparable decreases in LDL‐C levels (−38.5% vs. −34.8%), while FMD was improved in subjects treated with simvastatin 40 mg but not in those who received the combination therapy (Liu et al., 2009). Patients with CAD treated with atorvastatin 40 mg (either de novo therapy or dose escalation from chronic 10 to 40 mg) had improvement in their endothelial function, whereas those treated with ezetimibe 10 mg alone or added to chronic simvastatin 10 mg did not show changes, despite LDL‐C levels decreased similarly with all considered therapeutic regimens (Fichtlscherer et al., 2006). Similar results were observed in patients with chronic heart failure, in which simvastatin 10 mg or ezetimibe 10 mg reduced LDL‐C levels to a similar extent but only simvastatin improved endothelial function (Landmesser et al., 2005).
Of note, the results from the GLAGOV trial, which was aimed at investigating the effects of PCSK9 inhibition in patients with angiographic coronary disease, showed a greater decrease in PAV after 76 weeks of treatment with evolocumab compared with statins alone (Nicholls et al., 2016). Baseline CRP levels were 1.6 mg·L−1 and were unaffected by the treatment, further supporting a beneficial effect of LDL‐C lowering on atherosclerosis, which was independent of systemic inflammatory status (Nicholls et al., 2016).
In summary, most studies indicate that lipid‐lowering per se may play a key role in reducing vascular inflammation, although the well‐established pleiotropic effects of statins still leave the question open.
Conclusion
Historically, atherosclerosis has been seen as the consequence of impaired lipid metabolism, which promotes endothelial dysfunction and cholesterol deposition into the plaque, thus resulting in the recruitment of inflammatory cells. This response to injury becomes uncontrolled and generates the inflammatory response. On this basis, therapeutic approaches aimed at controlling either the excess of lipoproteins or the dampening the inflammatory response were tested. While most of the studies with anti‐inflammatory agents did not show a relevant benefit in terms of CV risk reduction, statins, by promoting LDL‐R expression and LDL‐C reduction, demonstrated a significant and robust benefit in terms of CV risk reduction. Later this benefit was, at least in part, ascribed to the ability of statins to reduce CRP. However, Mendelian randomization studies have clearly demonstrated that CRP is not a causal factor for atherosclerosis but rather a marker of systemic inflammation. The question of why statins by decreasing CRP levels, independently of LDL‐C reduction, improved CV risk has been involving the scientific community for the last 15 years. Lipid lowering treatments such as ezetimibe or anti‐PCSK9 monoclonal antibodies, which reduce LDL‐C through mechanisms independent of the inhibition of HMG‐CoA reductase, result in CRP reduction only in those patients with baseline levels above 2 mg·L−1, which is the threshold indicating the presence of an inflammatory condition. On the contrary, in patients with CRP below 2 mg·L−1, any type of LDL‐C‐lowering treatment, from statins to ezetimibe to anti‐PCSK9 antibodies, do not change CRP levels in spite of a substantial LDL‐C reduction, probably because these patients do not present a relevant systemic inflammation. Of note, even in patients where CRP is below 2 mg·L−1 and not altered by the therapy, such as in the GLAGOV study, LDL‐C reduction with anti‐PCSK9 antibodies results in atherosclerotic plaque regression as determined by IVUS (Nicholls et al., 2016). As a further proof of concept directly linking LDL‐C with vascular impairment, data demonstrating that LDL‐C reduction via different approaches will translate into a decrease of CV events also when baseline CRP levels are below 2 mg·L−1 (thus not expected to be modulated by the therapy) have been recently published (Sabatine et al., 2017). The FOURIER trial showed that, in more than 27 500 patients with atherosclerotic disease, the addition of evolocumab to an optimized regimen of lipid‐lowering therapy significantly reduced the risk of CV events as a consequence of LDL‐C reduction (from 92 to 30 mg·dL−1), compared to that of patients maintaining the optimized regimen of lipid‐lowering therapy (Sabatine et al., 2017). Of note, the level of CRP was 1.7 mg·L−1 (IQR 0.9–3.6) at baseline and 1.4 mg·L−1 (IQR 0.7–3.1) after 48 weeks of treatment in both arms (Sabatine et al., 2017).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Conflict of interest
The authors has received research funding, and/or honoraria for advisory boards, consultancy or speaker bureau from Aegerion (ALC, GDN), Amgen (ALC, GDN), AstraZeneca (ALC), Eli Lilly (ALC), Genzyme (ALC), Mediolanum (ALC), Merck or MSD (ALC), Pfizer (ALC, GDN), Recordati (ALC, GDN), Rottapharm (ALC), Sanofi‐Regeneron (ALC, GDN) and Sigma‐Tau (ALC). A.P. reports no disclosures.
Acknowledgements
The work of the authors is supported by Fondazione Cariplo 2015‐0524 and 2015‐0564 (ALC), and 2016‐0852 (GDN); H2020 REPROGRAM PHC‐03‐2015/667837‐2 (ALC); Telethon Foundation (GGP13002) (GDN), Ministero della Salute GR‐2011‐02346974 (GDN); Aspire Cardiovascular grant 2016‐WI218287 (GDN).
Catapano, A. L. , Pirillo, A. , and Norata, G. D. (2017) Vascular inflammation and low‐density lipoproteins: is cholesterol the link? A lesson from the clinical trials. British Journal of Pharmacology, 174: 3973–3985. doi: 10.1111/bph.13805.
References
- Abbate A, Kontos MC, Abouzaki NA, Melchior RD, Thomas C, Van Tassell BW et al. (2015). Comparative safety of interleukin‐1 blockade with anakinra in patients with ST‐segment elevation acute myocardial infarction (from the VCU‐ART and VCU‐ART2 pilot studies). Am J Cardiol 115: 288–292. [DOI] [PubMed] [Google Scholar]
- Abbate A, Kontos MC, Grizzard JD, Biondi‐Zoccai GG, Van Tassell BW, Robati R et al. (2010). Interleukin‐1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU‐ART] Pilot study). Am J Cardiol 105: 1371–1377 e1371. [DOI] [PubMed] [Google Scholar]
- Abbate A, Van Tassell BW, Biondi‐Zoccai G, Kontos MC, Grizzard JD, Spillman DW et al. (2013). Effects of interleukin‐1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University‐Anakinra Remodeling Trial (2) (VCU‐ART2) pilot study]. Am J Cardiol 111: 1394–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad HM, Sarhan EM, Komber U (2014). Higher circulating levels of OxLDL % of LDL are associated with subclinical atherosclerosis in female patients with systemic lupus erythematosus. Rheumatol Int 34: 617–623. [DOI] [PubMed] [Google Scholar]
- Albert MA, Danielson E, Rifai N, Ridker PM (2001). Effect of statin therapy on C‐reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 286: 64–70. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amezaga Urruela M, Suarez‐Almazor ME (2012). Lipid paradox in rheumatoid arthritis: changes with rheumatoid arthritis therapies. Curr Rheumatol Rep 14: 428–437. [DOI] [PubMed] [Google Scholar]
- Ammirati E, Bozzolo EP, Contri R, Baragetti A, Palini AG, Cianflone D et al. (2014). Cardiometabolic and immune factors associated with increased common carotid artery intima‐media thickness and cardiovascular disease in patients with systemic lupus erythematosus. Nutr Metab Cardiovasc Dis: NMCD 24: 751–759. [DOI] [PubMed] [Google Scholar]
- Ascer E, Bertolami MC, Venturinelli ML, Buccheri V, Souza J, Nicolau JC et al. (2004). Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis 177: 161–166. [DOI] [PubMed] [Google Scholar]
- Bellosta S, Ferri N, Arnaboldi L, Bernini F, Paoletti R, Corsini A (2000). Pleiotropic effects of statins in atherosclerosis and diabetes. Diabetes Care 23 (Suppl 2): B72–B78. [PubMed] [Google Scholar]
- Cannon CP, Cariou B, Blom D, McKenney JM, Lorenzato C, Pordy R et al. (2015). Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J 36: 1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury RP, Birks JS, Mani V, Biasiolli L, Robson MD, L'Allier PL et al. (2016). Arterial effects of canakinumab in patients with atherosclerosis and type 2 diabetes or glucose intolerance. J Am Coll Cardiol 68: 1769–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CS, Wang YC, Lu LS, Walton B, Yilmaz HR, Huang RY et al. (2013). Electronegative low‐density lipoprotein increases C‐reactive protein expression in vascular endothelial cells through the LOX‐1 receptor. PLoS One 8: e70533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo P, Golino P, Calabro P, Cali G, Ragni M, De Rosa S et al. (2005). C‐reactive protein induces tissue factor expression and promotes smooth muscle and endothelial cell proliferation. Cardiovasc Res 68: 47–55. [DOI] [PubMed] [Google Scholar]
- Daien CI, Duny Y, Barnetche T, Daures JP, Combe B, Morel J (2012). Effect of TNF inhibitors on lipid profile in rheumatoid arthritis: a systematic review with meta‐analysis. Ann Rheum Dis 71: 862–868. [DOI] [PubMed] [Google Scholar]
- de Carvalho JF, Bonfa E, Borba EF (2008). Systemic lupus erythematosus and “lupus dyslipoproteinemia”. Autoimmun Rev 7: 246–250. [DOI] [PubMed] [Google Scholar]
- Di Minno MN, Ambrosino P, Peluso R, Di Minno A, Lupoli R, Dentali F (2014). Lipid profile changes in patients with rheumatic diseases receiving a treatment with TNF‐alpha blockers: a meta‐analysis of prospective studies. Ann Med 46: 73–83. [DOI] [PubMed] [Google Scholar]
- Diffenderfer MR, Schaefer EJ (2014). The composition and metabolism of large and small LDL. Curr Opin Lipidol 25: 221–226. [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464: 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF et al. (2009). Genetic Loci associated with C‐reactive protein levels and risk of coronary heart disease. JAMA 302: 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberson J, Bennett D, Link E, Parish S, Danesh J, Armitage J et al. (2011). C‐reactive protein concentration and the vascular benefits of statin therapy: an analysis of 20,536 patients in the Heart Protection Study. Lancet 377: 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon WF, Fearon DT (2008). Inflammation and cardiovascular disease: role of the interleukin‐1 receptor antagonist. Circulation 117: 2577–2579. [DOI] [PubMed] [Google Scholar]
- Fichtlscherer S, Schmidt‐Lucke C, Bojunga S, Rossig L, Heeschen C, Dimmeler S et al. (2006). Differential effects of short‐term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for 'pleiotropic' functions of statin therapy. Eur Heart J 27: 1182–1190. [DOI] [PubMed] [Google Scholar]
- Flaim JD, Grundy JS, Baker BF, McGowan MP, Kastelein JJ (2014). Changes in mipomersen dosing regimen provide similar exposure with improved tolerability in randomized placebo‐controlled study of healthy volunteers. J Am Heart Assoc 3: e000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frostegard J, Svenungsson E, Wu R, Gunnarsson I, Lundberg IE, Klareskog L et al. (2005). Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum 52: 192–200. [DOI] [PubMed] [Google Scholar]
- Gabay C, Kushner I (1999). Acute‐phase proteins and other systemic responses to inflammation. N Engl J Med 340: 448–454. [DOI] [PubMed] [Google Scholar]
- Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM (1996). Interleukin‐1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol 16: 1000–1006. [DOI] [PubMed] [Google Scholar]
- Garcia‐Gomez C, Bianchi M, de la Fuente D, Badimon L, Padro T, Corbella E et al. (2014). Inflammation, lipid metabolism and cardiovascular risk in rheumatoid arthritis: a qualitative relationship? World J Orthop 5: 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MM, Varela CG, Silva PF, Lima PR, Goes PM, Rodrigues MG et al. (2016). Endothelial effect of statin therapy at a high dose versus low dose associated with ezetimibe. Arq Bras Cardiol 106: 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbrone MA Jr, Garcia‐Cardena G (2016). Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 118: 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS (2015). A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebe A, Latz E (2013). Cholesterol crystals and inflammation. Curr Rheumatol Rep 15: 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigore L, Raselli S, Garlaschelli K, Redaelli L, Norata GD, Pirillo A et al. (2013). Effect of treatment with pravastatin or ezetimibe on endothelial function in patients with moderate hypercholesterolemia. Eur J Clin Pharmacol 69: 341–346. [DOI] [PubMed] [Google Scholar]
- Haque S, Mirjafari H, Bruce IN (2008). Atherosclerosis in rheumatoid arthritis and systemic lupus erythematosus. Curr Opin Lipidol 19: 338–343. [DOI] [PubMed] [Google Scholar]
- Hovingh GK, Kastelein JJ, van Deventer SJ, Round P, Ford J, Saleheen D et al. (2015). Cholesterol ester transfer protein inhibition by TA‐8995 in patients with mild dyslipidaemia (TULIP): a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet 386: 452–460. [DOI] [PubMed] [Google Scholar]
- Jasinska M, Owczarek J, Orszulak‐Michalak D (2007). Statins: a new insight into their mechanisms of action and consequent pleiotropic effects. Pharmacol Rep 59: 483–499. [PubMed] [Google Scholar]
- Jialal I, Stein D, Balis D, Grundy SM, Adams‐Huet B, Devaraj S (2001). Effect of hydroxymethyl glutaryl coenzyme a reductase inhibitor therapy on high sensitive C‐reactive protein levels. Circulation 103: 1933–1935. [DOI] [PubMed] [Google Scholar]
- Joshi PH, Jacobson TA (2010). Therapeutic options to further lower C‐reactive protein for patients on statin treatment. Curr Atheroscler Rep 12: 34–42. [DOI] [PubMed] [Google Scholar]
- Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R et al. (2010). C‐reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta‐analysis. Lancet 375: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashiri SY, Kawakami A, Yamasaki S, Imazato T, Iwamoto N, Fujikawa K et al. (2011). Effects of the anti‐interleukin‐6 receptor antibody, tocilizumab, on serum lipid levels in patients with rheumatoid arthritis. Rheumatol Int 31: 451–456. [DOI] [PubMed] [Google Scholar]
- Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR et al. (2004). Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 45: 1169–1196. [DOI] [PubMed] [Google Scholar]
- Kinlay S (2007). Low‐density lipoprotein‐dependent and ‐independent effects of cholesterol‐lowering therapies on C‐reactive protein: a meta‐analysis. J Am Coll Cardiol 49: 2003–2009. [DOI] [PubMed] [Google Scholar]
- Kinlay S, Schwartz GG, Olsson AG, Rifai N, Leslie SJ, Sasiela WJ et al. (2003). High‐dose atorvastatin enhances the decline in inflammatory markers in patients with acute coronary syndromes in the MIRACL study. Circulation 108: 1560–1566. [DOI] [PubMed] [Google Scholar]
- Kinlay S, Schwartz GG, Olsson AG, Rifai N, Szarek M, Waters DD et al. (2008). Inflammation, statin therapy, and risk of stroke after an acute coronary syndrome in the MIRACL study. Arterioscler Thromb Vasc Biol 28: 142–147. [DOI] [PubMed] [Google Scholar]
- Kocarnik JM, Pendergrass SA, Carty CL, Pankow JS, Schumacher FR, Cheng I et al. (2014). Multiancestral analysis of inflammation‐related genetic variants and C‐reactive protein in the population architecture using genomics and epidemiology study. Circ Cardiovasc Genet 7: 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig W (2013). High‐sensitivity C‐reactive protein and atherosclerotic disease: from improved risk prediction to risk‐guided therapy. Int J Cardiol 168: 5126–5134. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S et al. (2005). Simvastatin versus ezetimibe: pleiotropic and lipid‐lowering effects on endothelial function in humans. Circulation 111: 2356–2363. [DOI] [PubMed] [Google Scholar]
- Lane T, Wassef N, Poole S, Mistry Y, Lachmann HJ, Gillmore JD et al. (2014). Infusion of pharmaceutical‐grade natural human C‐reactive protein is not proinflammatory in healthy adult human volunteers. Circ Res 114: 672–676. [DOI] [PubMed] [Google Scholar]
- Libby P (2002). Inflammation in atherosclerosis. Nature 420: 868–874. [DOI] [PubMed] [Google Scholar]
- Libby P (2012). Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 32: 2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton MF, Yancey PG, Davies SS, Jerome WGJ, Linton EF, Vickers KC (2000). The role of lipids and lipoproteins in atherosclerosis [Updated 2015 Dec 24 ]In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000‐. Available from:. https://www.ncbi.nlm.nih.gov/books/NBK343489/.
- Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK (2009). Evidence for statin pleiotropy in humans: differential effects of statins and ezetimibe on rho‐associated coiled‐coil containing protein kinase activity, endothelial function, and inflammation. Circulation 119: 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Teichtahl AJ, Wicks IP (2015). Interleukin‐6 in rheumatoid arthritis – from the laboratory to the bedside. Curr Pharm Des 21: 2187–2197. [DOI] [PubMed] [Google Scholar]
- Lubrano V, Gabriele M, Puntoni MR, Longo V, Pucci L (2015). Relationship among IL‐6, LDL cholesterol and lipid peroxidation. Cell Mol Biol Lett 20: 310–322. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Thompson L, Giles JT, Bathon JM, Salmon JE, Beaulieu AD et al. (2015). Effect of interleukin‐6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo‐controlled study. Ann Rheum Dis 74: 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen M, van der Wal AC, Niessen HW, Koch KT, de Winter RJ, van der Loos CM et al. (2006). Colocalisation of intraplaque C reactive protein, complement, oxidised low density lipoprotein, and macrophages in stable and unstable angina and acute myocardial infarction. J Clin Pathol 59: 196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihos CG, Pineda AM, Santana O (2014). Cardiovascular effects of statins, beyond lipid‐lowering properties. Pharmacol Res 88: 12–19. [DOI] [PubMed] [Google Scholar]
- Montecucco F, Favari E, Norata GD, Ronda N, Nofer JR, Vuilleumier N (2015). Impact of systemic inflammation and autoimmune diseases on apoA‐I and HDL plasma levels and functions. Handb Exp Pharmacol 224: 455–482. [DOI] [PubMed] [Google Scholar]
- Morrow DA, de Lemos JA, Sabatine MS, Wiviott SD, Blazing MA, Shui A et al. (2006). Clinical relevance of C‐reactive protein during follow‐up of patients with acute coronary syndromes in the Aggrastat‐to‐Zocor Trial. Circulation 114: 281–288. [DOI] [PubMed] [Google Scholar]
- Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B et al. (2015). The effect of interleukin‐1 receptor antagonist therapy on markers of inflammation in non‐ST elevation acute coronary syndromes: the MRC‐ILA Heart Study. Eur Heart J 36: 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namiri‐Kalantari R, Gao F, Chattopadhyay A, Wheeler AA, Navab KD, Farias‐Eisner R et al. (2015). The dual nature of HDL: Anti‐Inflammatory and pro‐Inflammatory. Biofactors 41: 153–159. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P et al. (2011). Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med 365: 2078–2087. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ et al. (2016). Effect of evolocumab on progression of coronary disease in statin‐treated patients: the GLAGOV randomized clinical trial. JAMA 316: 2373–2384. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA et al. (2004). Effect of intensive compared with moderate lipid‐lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 291: 1071–1080. [DOI] [PubMed] [Google Scholar]
- Norata GD, Ballantyne CM, Catapano AL (2013). New therapeutic principles in dyslipidaemia: focus on LDL and Lp(a) lowering drugs. Eur Heart J 34: 1783–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norata GD, Tavori H, Pirillo A, Fazio S, Catapano AL (2016). Biology of proprotein convertase subtilisin kexin 9: beyond low‐density lipoprotein cholesterol lowering. Cardiovasc Res 112: 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norata GD, Tibolla G, Catapano AL (2014). Targeting PCSK9 for hypercholesterolemia. Annu Rev Pharmacol Toxicol 54: 273–293. [DOI] [PubMed] [Google Scholar]
- Noveck R, Stroes ES, Flaim JD, Baker BF, Hughes S, Graham MJ et al. (2014). Effects of an antisense oligonucleotide inhibitor of C‐reactive protein synthesis on the endotoxin challenge response in healthy human male volunteers. J Am Heart Assoc. doi: 10.1161/JAHA.114.001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak B, Madej M, Luczak A, Malecki R, Wiland P (2016). Disease activity, oxidized‐LDL fraction and anti‐oxidized LDL antibodies influence cardiovascular risk in rheumatoid arthritis. Adv Clin Exp Med 25: 43–50. [DOI] [PubMed] [Google Scholar]
- Olofsson PS, Sheikine Y, Jatta K, Ghaderi M, Samnegard A, Eriksson P et al. (2009). A functional interleukin‐1 receptor antagonist polymorphism influences atherosclerosis development. The interleukin‐1beta:interleukin‐1 receptor antagonist balance in atherosclerosis. Circ J 73: 1531–1536. [DOI] [PubMed] [Google Scholar]
- Pasceri V, Willerson JT, Yeh ET (2000). Direct proinflammatory effect of C‐reactive protein on human endothelial cells. Circulation 102: 2165–2168. [DOI] [PubMed] [Google Scholar]
- Pearson TA, Ballantyne CM, Veltri E, Shah A, Bird S, Lin J et al. (2009). Pooled analyses of effects on C‐reactive protein and low density lipoprotein cholesterol in placebo‐controlled trials of ezetimibe monotherapy or ezetimibe added to baseline statin therapy. Am J Cardiol 103: 369–374. [DOI] [PubMed] [Google Scholar]
- Pepys MB, Hirschfield GM (2003). C‐reactive protein: a critical update. J Clin Invest 111: 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF (2007). The role of TNF‐alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res 48: 751–762. [DOI] [PubMed] [Google Scholar]
- Puri R, Nissen SE, Libby P, Shao M, Ballantyne CM, Barter PJ et al. (2013). C‐reactive protein, but not low‐density lipoprotein cholesterol levels, associate with coronary atheroma regression and cardiovascular events after maximally intensive statin therapy. Circulation 128: 2395–2403. [DOI] [PubMed] [Google Scholar]
- Qamar A, Rader DJ (2012). Effect of interleukin 1beta inhibition in cardiovascular disease. Curr Opin Lipidol 23: 548–553. [DOI] [PubMed] [Google Scholar]
- Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R et al. (2015a). Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double‐blind, placebo‐controlled trial. Lancet 385: 341–350. [DOI] [PubMed] [Google Scholar]
- Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L et al. (2015b). PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD‐2): a randomised, double‐blind, placebo‐controlled trial. Lancet 385: 331–340. [DOI] [PubMed] [Google Scholar]
- Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT et al. (2010). Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One 5: e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reriani MK, Dunlay SM, Gupta B, West CP, Rihal CS, Lerman LO et al. (2011). Effects of statins on coronary and peripheral endothelial function in humans: a systematic review and meta‐analysis of randomized controlled trials. Eur J Cardiovasc Prev Rehabil 18: 704–716. [DOI] [PubMed] [Google Scholar]
- Ridker PM (2016). From C‐reactive protein to interleukin‐6 to interleukin‐1: moving upstream to identify novel targets for atheroprotection. Circ Res 118: 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH et al. (2005). C‐reactive protein levels and outcomes after statin therapy. N Engl J Med 352: 20–28. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ et al. (2008). Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med 359: 2195–2207. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ et al. (2009). Reduction in C‐reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet 373: 1175–1182. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS et al. (2001). Measurement of C‐reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 344: 1959–1965. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E (1999). Long‐term effects of pravastatin on plasma concentration of C‐reactive protein. Cholesterol Recurrent Events (CARE) Investigators Circ 100: 230–235. [DOI] [PubMed] [Google Scholar]
- Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M et al. (2015). Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 372: 1489–1499. [DOI] [PubMed] [Google Scholar]
- Roma P, Bernini F, Fogliatto R, Bertulli SM, Negri S, Fumagalli R et al. (1992). Defective catabolism of oxidized LDL by J774 murine macrophages. J Lipid Res 33: 819–829. [PubMed] [Google Scholar]
- Ross R (1999). Atherosclerosis – an inflammatory disease. N Engl J Med 340: 115–126. [DOI] [PubMed] [Google Scholar]
- Ruan XZ, Moorhead JF, Tao JL, Ma KL, Wheeler DC, Powis SH et al. (2006). Mechanisms of dysregulation of low‐density lipoprotein receptor expression in vascular smooth muscle cells by inflammatory cytokines. Arterioscler Thromb Vasc Biol 26: 1150–1155. [DOI] [PubMed] [Google Scholar]
- Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA et al. (2017). Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med https://doi.org/10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J et al. (2015). Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 372: 1500–1509. [DOI] [PubMed] [Google Scholar]
- Sahebkar A, Di Giosia P, Stamerra CA, Grassi D, Pedone C, Ferretti G et al. (2016). Effect of monoclonal antibodies to PCSK9 on high‐sensitivity C‐reactive protein levels: a meta‐analysis of 16 randomized controlled treatment arms. Br J Clin Pharmacol 81: 1175–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN et al. (2012). Interleukin‐6 receptor pathways in coronary heart disease: a collaborative meta‐analysis of 82 studies. Lancet 379: 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Takahashi Y, Tabuchi T, Minami Y, Tamada M, Takahashi K et al. (2015). Cellular and molecular mechanisms of statins: an update on pleiotropic effects. Clin Sci (Lond) 129: 93–105. [DOI] [PubMed] [Google Scholar]
- Schuett H, Luchtefeld M, Grothusen C, Grote K, Schieffer B (2009). How much is too much? Interleukin‐6 and its signalling in atherosclerosis. Thromb Haemost 102: 215–222. [DOI] [PubMed] [Google Scholar]
- Settergren M, Bohm F, Ryden L, Pernow J (2008). Cholesterol lowering is more important than pleiotropic effects of statins for endothelial function in patients with dysglycaemia and coronary artery disease. Eur Heart J 29: 1753–1760. [DOI] [PubMed] [Google Scholar]
- Sever PS, Poulter NR, Chang CL, Thom SA, Hughes AD, Welsh P et al. (2013). Evaluation of C‐reactive protein before and on‐treatment as a predictor of benefit of atorvastatin: a cohort analysis from the Anglo‐Scandinavian Cardiac Outcomes Trial lipid‐lowering arm. J Am Coll Cardiol 62: 717–729. [DOI] [PubMed] [Google Scholar]
- Signorelli SS, Fiore V, Malaponte G (2014). Inflammation and peripheral arterial disease: the value of circulating biomarkers (Review). Int J Mol Med 33: 777–783. [DOI] [PubMed] [Google Scholar]
- Soedamah‐Muthu SS, Livingstone SJ, Charlton‐Menys V, Betteridge DJ, Hitman GA, Neil HA et al. (2015). Effect of atorvastatin on C‐reactive protein and benefits for cardiovascular disease in patients with type 2 diabetes: analyses from the collaborative atorvastatin diabetes trial. Diabetologia 58: 1494–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci‐Thomas MG, Thomas MJ (2016). Microdomains, inflammation, and atherosclerosis. Circ Res 118: 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souto A, Salgado E, Maneiro JR, Mera A, Carmona L, Gomez‐Reino JJ (2015). Lipid profile changes in patients with chronic inflammatory arthritis treated with biologic agents and tofacitinib in randomized clinical trials: a systematic review and meta‐analysis. Arthritis Rheumatol 67: 117–127. [DOI] [PubMed] [Google Scholar]
- Strang AC, Bisoendial RJ, Kootte RS, Schulte DM, Dallinga‐Thie GM, Levels JH et al. (2013). Pro‐atherogenic lipid changes and decreased hepatic LDL receptor expression by tocilizumab in rheumatoid arthritis. Atherosclerosis 229: 174–181. [DOI] [PubMed] [Google Scholar]
- Stroes E, Colquhoun D, Sullivan D, Civeira F, Rosenson RS, Watts GF et al. (2014). Anti‐PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS‐2 randomized, placebo‐controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol 63: 2541–2548. [DOI] [PubMed] [Google Scholar]
- Sudhop T, Reber M, Tribble D, Sapre A, Taggart W, Gibbons P et al. (2009). Changes in cholesterol absorption and cholesterol synthesis caused by ezetimibe and/or simvastatin in men. J Lipid Res 50: 2117–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, Sofat R et al. (2012). The interleukin‐6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet 379: 1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Bornfeldt KE (2016). Macrophage phenotype and function in different stages of atherosclerosis. Circ Res 118: 653–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Takahashi K, Yamashina M, Maesawa C, Kajiwara T, Taneichi H et al. (2010). Association of the TNF‐{alpha}‐C‐857T polymorphism with resistance to the cholesterol‐lowering effect of HMG‐CoA reductase inhibitors in type 2 diabetic subjects. Diabetes Care 33: 463–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsimikas S, Duff GW, Berger PB, Rogus J, Huttner K, Clopton P et al. (2014). Pro‐inflammatory interleukin‐1 genotypes potentiate the risk of coronary artery disease and cardiovascular events mediated by oxidized phospholipids and lipoprotein(a). J Am Coll Cardiol 63: 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Ree MA, Huisman MV, Princen HM, Meinders AE, Kluft C (2003). Strong decrease of high sensitivity C‐reactive protein with high‐dose atorvastatin in patients with type 2 diabetes mellitus. Atherosclerosis 166: 129–135. [DOI] [PubMed] [Google Scholar]
- van Diepen JA, Berbee JF, Havekes LM, Rensen PC (2013). Interactions between inflammation and lipid metabolism: relevance for efficacy of anti‐inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis 228: 306–315. [DOI] [PubMed] [Google Scholar]
- van Sijl AM, Peters MJ, Knol DL, de Vet RH, Sattar N, Dijkmans BA et al. (2011). The effect of TNF‐alpha blocking therapy on lipid levels in rheumatoid arthritis: a meta‐analysis. Semin Arthritis Rheum 41: 393–400. [DOI] [PubMed] [Google Scholar]
- Venugopal SK, Devaraj S, Jialal I (2005). Macrophage conditioned medium induces the expression of C‐reactive protein in human aortic endothelial cells: potential for paracrine/autocrine effects. Am J Pathol 166: 1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola J, Soehnlein O (2015). Atherosclerosis – A matter of unresolved inflammation. Semin Immunol 27: 184–193. [DOI] [PubMed] [Google Scholar]
- Wade DP, Clarke JG, Lindahl GE, Liu AC, Zysow BR, Meer K et al. (1993). 5′ control regions of the apolipoprotein(a) gene and members of the related plasminogen gene family. Proc Natl Acad Sci U S A 90: 1369–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welty FK (2013). How do elevated triglycerides and low HDL‐cholesterol affect inflammation and atherothrombosis? Curr Cardiol Rep 15: 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T et al. (2011). Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ 342: d548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerink J, Deanfield JE, Imholz BP, Spiering W, Basart DC, Coll B et al. (2013). High‐dose statin monotherapy versus low‐dose statin/ezetimibe combination on fasting and postprandial lipids and endothelial function in obese patients with the metabolic syndrome: the PANACEA study. Atherosclerosis 227: 118–124. [DOI] [PubMed] [Google Scholar]
- Yamashina M, Kaneko Y, Maesawa C, Kajiwara T, Ishii M, Fujiwara F et al. (2007). Association of TNF‐alpha gene promoter C‐857T polymorphism with higher serum LDL cholesterol levels and carotid plaque formation in Japanese patients with type 2 diabetes. Tohoku J Exp Med 211: 251–258. [DOI] [PubMed] [Google Scholar]
- Yang YH, Liu YQ, Zhang L, Li H, Li XB, Ouyang Q et al. (2015). Genetic polymorphisms of the TNF‐alpha‐308G/A are associated with metabolic syndrome in asthmatic patients from Hebei province, China. Int J Clin Exp Pathol 8: 13739–13746. [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Chen Y, Lei H, Liu Q, Moorhead JF, Varghese Z et al. (2009). Inflammatory stress increases unmodified LDL uptake via LDL receptor: an alternative pathway for macrophage foam‐cell formation. Inflamm Res 58: 809–818. [DOI] [PubMed] [Google Scholar]
- Ye Y, Zhao X, Zhai G, Guo L, Tian Z, Zhang S (2012). Effect of high‐dose statin versus low‐dose statin plus ezetimibe on endothelial function: a meta‐analysis of randomized trials. J Cardiovasc Pharmacol Ther 17: 357–365. [DOI] [PubMed] [Google Scholar]
- Zacho J, Tybjaerg‐Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG (2008). Genetically elevated C‐reactive protein and ischemic vascular disease. N Engl J Med 359: 1897–1908. [DOI] [PubMed] [Google Scholar]
- Zhang L, Gong D, Li S, Zhou X (2012). Meta‐analysis of the effects of statin therapy on endothelial function in patients with diabetes mellitus. Atherosclerosis 223: 78–85. [DOI] [PubMed] [Google Scholar]
- Zhang YX, Cliff WJ, Schoefl GI, Higgins G (1999). Coronary C‐reactive protein distribution: its relation to development of atherosclerosis. Atherosclerosis 145: 375–379. [DOI] [PubMed] [Google Scholar]