

SUMMARY
Micro-albuminuria and glomerular hyperfiltration are primary indicators of renal dysfunctions in Sickle Cell Disease (SCD), with more severe manifestations previously associated with variants in APOL1 and HMOX1 among African Americans. We have investigated 413 SCD patients from Cameroon. Anthropometric variables, haematological indices, crude albuminuria, albumin-to-creatinine ratio (ACR) and estimated glomerular filtration rate (eGFR) were measured. Patients were genotyped for 3.7 kB alpha-globin gene (HBA1/HBA2) deletion, and for variants in APOL1 (G1/G2; rs60910145, rs73885319, rs71785313) and HMOX1 (rs3074372, rs743811). The median age was 15 years; the majority presented with micro-albuminuria (60.9%; n = 248), and approximately half with glomerular hyperfiltration (49.5%; n = 200). Age, male sex, haemoglobin level, leucocyte count, mean corpuscular volume, blood pressure, body mass index and creatinine levels significantly affected albuminuria and/or eGFR. Co-inheritance of alpha-thalassaemia was protective against macro-albuminuria (p = 0.03). APOL1 G1/G2 risk variants were significantly associated with the ACR (p = 0.01) and borderline with eGFR (p = 0.07). HMOX1 - rs743811 was borderline associated with micro-albuminuria (p = 0.07) and macro-albuminuria (p = 0.06). The results revealed a high proportion of micro-albuminuria and glomerular hyperfiltration among Cameroonian SCD patients, and support the possible use of targeted genetic biomarkers for risks assessment.
Keywords: Sickle Cell Disease, Albuminuria, Glomerular filtration rate, APOL1, HMOX1
INTRODUCTION
Sickle Cell Disease (SCD) is a monogenic haemoglobinopathy caused by mutation in the β-globin subunit of adult haemoglobin (HbA). An estimated 300 000 SCD-affected births occurs annually, with three quarters of these recorded in sub-Saharan Africa (Piel et al, 2013). SCD patients are susceptible to vaso-occlusion, with the disease characterized by recurrent episodes of ischaemia-reperfusion injury and haemolytic anaemia, cumulatively contributing to progressive end organ damage (Bartolucci & Galacteros, 2012; Rees et al, 2010). The kidney is a particularly susceptible organ, with sickle cell nephropathy (SCN) occurring in 5–18% of patients, conferring increased risk of mortality (Platt et al, 1994; McClellan et al, 2012). Glomerular enlargement is probably the earliest renal abnormality in SCD (Ware et al, 2010), with paediatric patients displaying increased glomerular filtration rate (GFR) and proteinuria (Wigfall et al, 2000; Ware et al, 2010). Micro-albuminuria is the most sensitive early clinical marker of kidney dysfunction (Guasch et al, 1997, 1996), preceding the development of end stage kidney disease (ESKD) in paediatric and adult patients (Becton et al, 2010; Guasch et al, 1999, 2006).
The co-inheritance of genetic factors, specifically alpha-thalassaemia and specific variants in the fetal haemoglobin (HbF) promoting loci, have been proven to delay the clinical progression of kidney disease in SCD patients (Steinberg et al, 2003; Saraf et al, 2017). The co-inheritance of the 3.7kB alpha-thalassaemia deletion and SCD is more specifically associated with a lower prevalence of macro-albuminuria (Guasch et al, 1999; Lamarre et al, 2014; Nebor et al, 2010). In addition, two coding variants of APOL1, G1 (rs73885319, p. Ser358Gly and rs60910145, p.Ile400Met) and G2 (rs71785313, p.Asn404_Tyr405del), have been previously associated with chronic kidney disease (CKD) and focal segmental glomerulosclerosis (FSGS) in African Americans (Genovese et al, 2010; Kopp et al, 2011; Tzur et al, 2010). APOL1 encodes the trypanolytic factor responsible for lysis of Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense, the pathogens that cause human African trypanosomiasis. Moreover, HMOX1 forms the rate-limiting step in the catabolism of free haem (Tenhunen et al, 1968), with two promoter polymorphisms, a GT-dinucleotide repeat (rs3074372) and a single nucleotide polymorphism (SNP) (rs743811), associated with CKD (Bean et al, 2012; Saraf et al, 2015). Genetic variations in both APOL1 and HMOX1 have been associated with SCD nephropathy among adult African Americans (Ashley-Koch et al, 2011; Saraf et al, 2015; Schaefer et al, 2016). The study of associations between these genomic variants and the development of micro-albuminuria has not yet been reported in SCD populations in Africa.
Cameroon is a sub-Saharan African Country that has a population of approximately 20 million, with a SCD carrier frequency range of 8 to 34% (Weatherall and Clegg, 2001). The Cameroon national control programme for SCD has remained a policy document without implementation; there is not yet provision of universal newborn screening, and the median age of SCD diagnosis is 3.3 years, with no development of specialized centres for lifelong medical treatment resulting in very few patients being exposed to hydroxycarbamide (also termed hydroxyurea) treatment (Wonkam et al, 2014a). Moreover, there is no universal medical insurance coverage in Cameroon, and care of SCD patients is therefore dependent on financial support and care giving by family members. However, poverty in Cameroon affects more than 50% of the rural population and up to 30% of the urban population (World Bank, 2010), which in turn means that the financial burden of the necessary medical care for SCD often cannot be met (Wonkam et al, 2014a) and patients frequently suffer exceptionally severe SCD sequelae, such as stroke (Njamnshi et al, 2006) and neurocognitive dysfunctions (Ruffieux et al, 2013).
In this study, we aim to investigate the prevalence of micro-albuminuria and glomerular hyperfiltration in a Cameroonian SCD cohort, and to determine the clinical, biological and genetic predictors of albuminuria and estimated glomerular filtration rate (eGFR).
METHODS
Ethical approval
The study was approved by the University of Cape Town, Faculty of Health Sciences Human Research Ethics Committee (HREC REF: 661/2015), Cape Town, South Africa and the National Ethics Committee Ministry of Public Health, Yaoundé, Republic of Cameroon (No. 033/CNE/DNM/07). All patients older than 18 years signed consent forms, while informed consent was given by the parents or guardians for participants younger than 18 years of age, in accordance with the declaration of Helsinki.
Study Participants
A total of 413 individuals living with sickle anaemia (HbSS) from Cameroon were prospectively investigated. Patients were recruited at the Yaoundé Central Hospital and Laquintinie Hospital in Douala, Cameroon between January 2010 and December 2011. Routine blood counts of patients and haemoglobin (Hb) electrophoresis were conducted on arrival at the hospital, initially using the alkali denaturation test (ADT) in 55.5% of the cohort, and subsequently high performance liquid chromatography (HPLC), when it became available (Wonkam et al, 2014a). Socio-demographic (age, gender), anthropometric (height, weight, body mass index [BMI]), systolic and diastolic blood pressures, clinical variables and haematological indices were collected for these patients.
Measurements of renal functions
Routine laboratory tests were performed to measure serum creatinine and urine creatinine concentrations. The level of albumin in the urine was determined using either the Siemens Clinitek Status® test (Siemens Healthcare GmbH, Erlangen, Germany) or the Hemocue Albumin 20® system (Hemocue, Angelholm, Sweden). Urinary albumin quantitation was performed on first morning void samples during a planned hospital visit, when patients were not experiencing SCD vaso-occlusive crisis. The presence of albumin in the urine was defined as normal (<30 mg/l), micro-albuminuria (30–300 mg/l) or macro-albuminuria (>300 mg/l), while the albumin-to-creatinine ratio (ACR), represented as albumin (mg) per mmol creatinine, was defined as normo-albuminuria (<3 mg/mmol), micro-albuminuria (3–30 mg/mmol) or macro-albuminuria (>30 mg/mmol).
The GFR was estimated (eGFR) using three equations: (1) The Modification of Diet in Renal Disease (MDRD) (Levey et al, 1999), (2) Cockcroft-Gault (Cockcroft & Gault, 1976), and (3) the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) (Levey et al, 2009) equation. The proportion of patients with glomerular hyperfiltration (>130 ml/min/1.73m2 for women, and >140 ml/min/1.73m2 for men) (Haymann et al, 2010; Vazquez et al, 2014) was also investigated. The CKD-EPI equation has previously been identified as the most precise equation displaying the least bias in adult SCD patients (Arlet et al, 2012; Asnani et al, 2013), and will therefore be used for illustration in the main text of the present article, but comparative values of factors affecting eGFR using the two other equations are presented as in Table S1.
Molecular methods
Sickle cell anaemia mutation, β-globin gene (HBB) cluster haplotypes, and 3.7 kb α-globin gene (HBA1/HBA2) deletion
DNA was extracted from peripheral blood following the manufacturer’s instructions (Puregene Blood Kit; Qiagen, Hilden, Germany). Molecular analysis to determine the presence of the sickle mutation was carried out on 200 ng DNA by polymerase chain reaction (PCR) to amplify a 770 bp segment of HBB, followed by DdeI restriction analysis of the PCR product (Saiki et al, 1985).
Using published primers and methods, five restriction fragment length polymorphism (RFLP) sites in the HBB cluster were amplified to analyse the XmnI (5′Gγ), HindIII (Gγ), HindIII (Aγ), HincII (3′ψβ) and HinfI (5′β) for the HBB haplotype background (Bitoungui et al, 2015).
Using expand-long template PCR (Roche Diagnostics, Basel, Switzerland), the 3.7 kb HBA1/HBA2 deletion was successfully screened following a previously reported protocol (Rumaney et al, 2014).
Targeted SNPs in APOL1 and HMOX1 genes
SNP genotyping of rs60910145 (APOL1), rs73885319 (APOL1) and rs743811 (HMOX1) was performed using predesigned TaqMan genotyping assays (Applied Biosystems, Foster City, CA, USA). The PCR protocols were performed on the Bio-Rad CFX96 real time PCR system (Bio-Rad Laboratories, Hercules, CA, USA). The rs3074372 (HMOX1) and rs71785313 (APOL1) variants were genotyped using fragment analysis, incorporating fluorescently labelled forward primers. The HMOX1 repeats were classified as short (≤25 repeats) or long (>25 repeats) (Saraf et al, 2015). PCR protocols were performed on the Bio-Rad thermal cycler T100TM, and analysis of the genotype was achieved using the ABI Prism 3130xl Genetic Analyser (Applied Biosystems). Thereafter, Direct Cycle Sequencing using the ABI Prism 3130xl was performed on a subset (10%) of the samples and successfully validated the genotyping results (Fig S1, S2, S3).
Statistical Analysis
Descriptive statistical analysis of the prospectively collected patient data was performed using STATA Software version 14.0.370 for WindowsTM (StataCorp Inc., College Station, TX, USA). Moreover, this software was utilized to investigate associations between clinical variables. A χ2 test with one degree of freedom was used to perform the Hardy-Weinberg equilibrium (HWE). Only the 3.7del HBA1/HBA2 genotypes (p = 0.005) were out of HWE, however this deviation from HWE was expected in view of the strong protective effect of this genetic variant on macro-albuminuria, as displayed in the results. General linear and multinomial regression frameworks, adjusted for age and sex, were performed using the statistical package R (version 3.03, R Foundation for Statistical Computing, Vienna, Austria) to investigate the relationship between genotype results and clinical data. Genotype results were also compared for the various age groups, stages of clinically defined CKD (Levey et al, 2003), and between children/adolescents (<18 years old) and adults (≥18 years old). P values <0.05 were considered statistically significant.
RESULTS
Participants’ descriptions
A total of 413 SCD patients were included. Table I summarizes the participants’ characteristics. There was roughly equal numbers of males and females (210 and 203, respectively), with a median age of 15 years (25th–75th percentiles: 9–23; minimum-maximum: 2 – 58) and a lower proportion of adult patients (40%; n = 162). All participants were homozygous HbSS, and the most prevalent HBB cluster haplotypes were Benin (73.0%, n = 510 chromosomes) and Cameroon (21%, n = 145), with 31.1% (n = 105) and 10.4% (n = 25) of patients having co-inherited a single or double 3.7 kb HBA1/HBA2 deletion, respectively.
Table I.
Description of the studied Cameroonian SCD cohort.
| Variable | Median (25th–75th percentiles) or % | min-max | Observations (n) | |
|---|---|---|---|---|
| Age (years) | 15 (9–23) | 2–58 | 413 | |
| Haematological | Hb (g/l) | 75 (67–84) | 35 - 13.9 | 407 |
| MCV (fl) | 85 (78–92) | 60–117 | 413 | |
| MCHC (g/l) | 335 (316–355) | 215–543 | 413 | |
| WBC (109/l) | 12.7 (10.2–16.2) | 4–49.8 | 413 | |
| Lymphocyte count (109/l) | 5 (3.8 – 6.7) | 1.4 – 22.1 | 406 | |
| Monocyte count (109/l) | 1.4 (0.9–2.1) | 0.1–8.2 | 404 | |
| Platelet count (109/l) | 370 (284–466) | 29 – 1078 | 402 | |
| HbA2 (%) | 3.6 (2.85 – 4.1) | 0–18.2 | 408 | |
| HbF (%) | 8.05 (2.15–13.35) | 0–37.4 | 408 | |
| Anthropometric | BMI (kg/m2) | 17.361 (14.863–20.505) | 10.61–31.029 | 413 |
| Systolic blood pressure | 107 (99–114) | 77–156 | 413 | |
| Diastolic blood pressure | 57 (53–63) | 37–93 | 413 | |
| Clinical Events | VOC crises (n/year) | 2 (1–4) | 0–80 | 412 |
| Consultation (n/year) | 3.1 ± 3.8 | 0–40 | 413 | |
| Hospitalization (n/year) | 1 (0–2) | 0–30 | 405 | |
| Overt stroke (%) | 2.4 | 10/413a | ||
| α Genotypes (%) | αα/αα | 58.6 | 198/338a | |
| αα/α3.7 | 31.1 | 105/338a | ||
| α3.7/α3.7 | 10.3 | 35/338a | ||
| β Haplotypes (%) | Benin/Benin | 50.5 | 195/352a | |
| Benin/Cameroon | 26.1 | 92/352a | ||
| Benin/Atypical | 7.1 | 25/352a | ||
| Cameroon/Cameroon | 6.5 | 23/352a | ||
| Renal functions | Crude albuminuria (mg/l) | 41 (23–83) | 3–1180 | 407 |
| eGFR (CKD-EPI) (ml/min/1.73m2) | 135.1 (112–154.4) | 50.8–250.8 | 404 | |
| Serum creatinine (μmol/l) | 7 (5–8.5) | 2–13.8 | 404 | |
BMI: body mass index; Hb: haemoglobin; MCHC: mean corpuscular haemoglobin concentration; MCV: mean corpuscular volume; RBC: red blood cell count; SCD: sickle cell disease; VOC: vaso-occlusive crises; WBC: white blood cell count. Benin/Benin; Benin/Cameroon; Benin/Atypical; Cameroon/Cameroon.
Number of individuals, not alleles.
Clinical factors affecting renal functions
The general description of renal functions is summarized in Table I, and Table II summarizes the factors affecting crude albuminuria and eGFR respectively, in Cameroonian SCD patients.
Table II.
Factors affecting eGFR and albuminuria levels in Cameroonian SCD patients
| Variables | Correlation coefficients (r) | p-value |
|---|---|---|
| Factors affecting eGFR | ||
| Age (years) | −0.550 | <0.0001 |
| Male Sex | risk | <0.0001 |
| SBP (mm Hg) | −0.367 | <0.0001 |
| DBP (mm Hg) | −0.296 | <0.0001 |
| Haemoglobin (g/l) | −0.142 | 0.0046 |
| BMI (kg/m2) | −0.529 | <0.0001 |
| Serum Creatinine (μmol/l) | −0.901 | <0.0001 |
| Mean arterial pressure (mm Hg) | −0.337 | <0.0001 |
| Factors affecting albuminuria levels | ||
| Age (years) | 0.124 | 0.0041 |
| Haemoglobin (g/l) | −0.091 | 0.0289 |
| BMI (kg/m2) | 0.098 | 0.0489 |
| Leucocyte count (109/l) | 0.116 | 0.0202 |
| Mean arterial pressure (mm Hg) | 0.118 | 0.0175 |
| MCV (fl) | 0.167 | 0.008 |
BMI: body mass index; DBP: diastolic blood pressure; eGFR: estimated glomerular filtration rate; MCV: mean corpuscular volume; SBP: systolic blood pressure; SCD: sickle cell disease.
Glomerular filtration estimates
A total of 404 HbSS individuals had glomerular filtration estimates available. The cohort had a median serum creatinine value of 7 μmol/l (5 – 8.5) and eGFR of 135.1 ml/min/1.73m2 (112–154.4) (Table I). The CKD-EPI equation indicated that 49.5% (n = 200) of SCD patients suffered from glomerular hyperfiltration. The eGFR decreased with age (Fig 1; Fig S3A), as well as the proportion of hyperfiltrating patients per age group (Fig S3B). Moreover, eGFR was significantly increased in children/adolescents compared to adult SCD patients (p<0.0001) (Fig S3C; Table S2). The eGFR was significantly increased in male patients (p<0.0001); and was significantly negatively associated with log transformed systolic and diastolic blood pressure (p<0.0001) (Fig 1). Table S1 displays similar association results with all the equations used to estimate eGFR (MDRD, CKD-EPI and Cockcroft Gault). Isolated hyperfiltration was present in 34.2% (n = 67) of patients, while 61.7% (n = 121) and 4.1% (n = 8) had hyperfiltration in association with micro- and macro-albuminuria, respectively. The eGFR was not associated with crude albuminuria (p = 0.87), but with log(ACR) values (r = 0.022: p = 0.005) (Fig S4).
Figure 1. Clinical Factors affecting eGFR values in SCD: Age, gender, BMI and blood pressure.
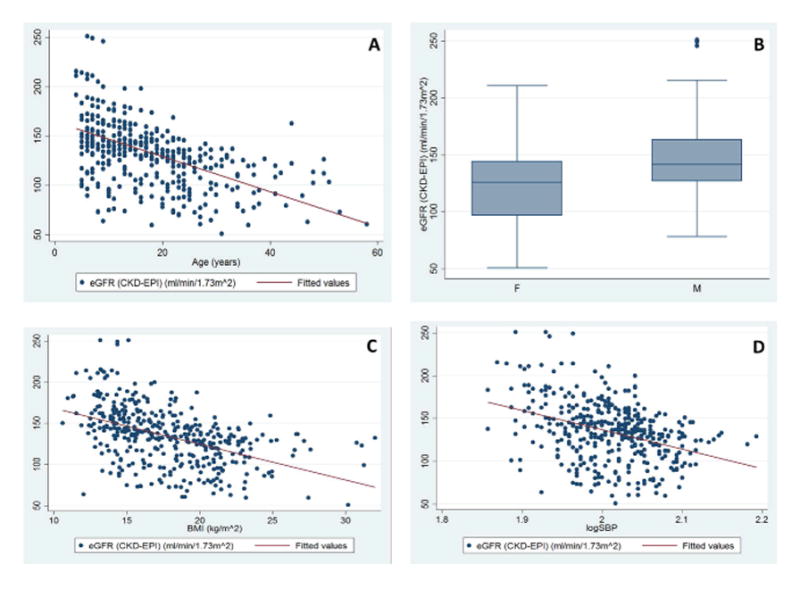
A. Scatter plots illustrating the relationship between eGFR values and age in the SCD patient cohort, using CKD-EPI (r = −0.550, p < 0.0001. Similar results were found for Cockcroft-Gault (r = −0.168, p < 0.0001) and Modification of Diet in Renal Disease (MDRD; r = −0.523, p < 0.0001) equations. The red line indicates a line of best fit, fitted to the data.
B. Box and whisker plots showing the association of eGFR values with gender. Box and whisker plots illustrating the distribution of eGFR values conditioned on gender. The eGFR values were calculated using CKD-EPI. Significant results are indicated using * (p < 0.05); similar significant results were obtained using MDRD equations and Cockcroft-Gault equations. The horizontal lines that constitute the ‘box’ correspond to the lower quartile, median and upper quartile parameters. The length of the ‘whiskers’ that extend from the box in the upwards and downwards direction represent a value of 1.5 times the interquartile range. Values that lie outside this are considered outliers, or extreme values.
C. Scatter plots illustrating the relationship between eGFR values and BMI
Calculated using the CKD-EPI (r = 0.529, p < 0.0001) equation. Similar results were found using MDRD (r = −0.520, p < 0.0001) and Cockcroft-Gault (r = −0.164, p = 0.018) equations. The BMI (kg/m2) variable is displayed on the x-axis, with the eGFR values on the y-axis. The red line indicates a line of best fit, fitted to the data.
D. Scatter plots illustrating the relationship between eGFR values and logSBP calculated using the CKD-EPI (r = −0.367, p < 0.0001) equations. Similar results were found with MDRD (r = − 0.407, p < 0.0001) and Cockcroft-Gault (p = 0.018) equations. The log(SBP) variable is displayed on the x-axis, with the eGFR values on the y-axis. The red line indicates a line of best fit, fitted to the data. Similar significant relationship between eGFR values and log(diastolic blood pressure) was also found, calculated using CKD-EPI (r = −0.296, p < 0.0001), the MDRD (r = −0.300, p < 0.0001) and Cockcroft-Gault (r = −0.164, p = 0.001) equations.
BMI: body mass index; CKD-EPI: Chronic Kidney Disease Epidemiology Collaboration equation; eGFR: estimated glomerular filtration rate; F: female; M: male; SBP: systolic blood pressure; SCD: sickle cell disease.
Albuminuria
The prevalence of micro- and macro-albuminuria was 60.9% (n = 248) and 2.5% (n = 10), respectively. The youngest patient with micro-albuminuria was four years old. Fig S3C displays the median albuminuria values per age group, indicating a general positive trend between albuminuria and increasing age. Crude albuminuria displayed a near significant association with reduced haemoglobin (r = −0.09, p = 0.07), while increased BMI (r = 0.1, p = 0.04), leucocyte count (r = 0.12, p = 0.02), mean arterial pressure (r = 0.12, p = 0.01) and mean corpuscular volume (MCV; r = 0.17, p = 0.008) values were positive predictors of increasing albuminuria (Table II; Fig 2). There was a significant association between albuminuria and increasing age (r = 0.14, p = 0.004) (Fig 2; Fig S3D), and borderline association with decreasing haemoglobin (r = −0.09, p = 0.06) (Fig 2). There was no significant association between patient sex and albuminuria (p=0.18), nor between the median values for children/adolescents and adults (Table S2).
Figure 2. Clinical Factors associated with Albuminuria in SCD: Age, and haematological indices.

Scatter plot illustrating the positive relationship between age (y-axes) and log crude albuminuria, (r = 0.142, p = 0.004) (A), log Leucocyte count (r = 0.116, p = 0.0202) (B); MCV (r = 0.167, p = 0.008) (C) and a nearly significant negative nature of the relationship with Hb level (r = −0.091, p = 0.069) (D). The red lines indicate a line of best fit, fitted to the data.
Hb: haemoglobin; MCV: mean corpuscular volume.
3.7kB HBA1/HBA2 deletion, APOL1 and HMOX1 and renal function
The 3.7kB HBA1/HBA2 deletion was protective against prevalent macro-albuminuria (p = 0.03). APOL1 G1/G2 showed a statistically significant association with ACR (p=0.018), and was borderline with macro-albuminuria (p = 0.08), and eGFR (p = 0.07) values, however not with crude albuminuria (Table III; Fig 3). No significant association was observed between the HMOX1 dinucleotide promoter polymorphism and indicators of renal dysfunction, while the HMOX1 SNP (rs743811) tended to a significant association with crude albuminuria and macro-albuminuria (p = 0.08 and p = 0.06, respectively) in a recessive model, but not with log (ACR) values (Table III). No significant results were observed for the association of genetic variants and clinically defined stages of CKD (Table S3). The frequency of the respective genotypes was compared between the various age groups, and no significant differences were found (Table S4).
Table III.
Targeted genetic factors affecting eGFR and albuminuria levels in Cameroonian SCD patients (Recessive Model)
| Polymorphism | Crude Albuminuria | Macro-albuminuria | Log (ACR) | eGFR (CKD-EPI) |
|---|---|---|---|---|
| APOL1 G1/G2 | 0.923 | 0.085 | 0.018 | 0.072 |
| HMOX1 rs743811 | 0.073 | 0.062 | 0.845 | 0.116 |
| 3.7del HBA1/HBA2 genotypes | 0.137 | 0.034# | 0.809 | 0.231 |
P-values obtained from association analysis using the statistical package ‘R’. Statistically significant results (p<0.05), or results approaching significance (p≅0.05) are highlighted in bold, all in recessive model.
In co-dominant model, the association was stronger, p=0.009.
ACR: albumin/creatinine ratio; CKD-EPI: Chronic Kidney Disease Epidemiology Collaboration equation; eGFR: estimated glomerular filtration rate; SCD: sickle cell disease.
Figure 3. Box and whisker plots showing the association of APOL1 G1/G2 with crude albuminuria, ACR, and eGFR values in the SCD patient cohort.

Box and whisker plots illustrating the distribution of (Panel A) crude albuminuria, (Panel B) ACR and (Panel C) eGFR (calculated using the CKD-EPI equation) values conditioned on the APOL1 G1/G2 polymorphism, based on the presence of zero (n = 333), one (n = 40) or two (n = 5) minor alleles. The horizontal lines that constitute the ‘box’ correspond to the lower quartile, median and upper quartile parameters. The length of the ‘whiskers’ that extend from the box in the upwards and downwards direction represent a distance to the maximum and minimum values, respectively. Significant results are indicated using * (p < 0.05).
ACR: albumin/creatinine ratio; CKD-EPI: Chronic Kidney Disease Epidemiology Collaboration equation; eGFR: estimated glomerular filtration rate; SCD: sickle cell disease.
DISCUSSION
This study is, to our knowledge, the first to investigate targeted genomic variants and their association with clinical renal phenotypes in a sub-Saharan African cohort. The result indicates that the majority of SCD patients in this sample of Cameroonians presented with micro-albuminuria, as well as a high prevalence of glomerular hyperfiltration. Micro-albuminuria is considered the primary marker of early renal dysfunction (Ataga et al, 2014; Gosmanova et al, 2014), conferring increased risk of CKD and ESKD development and, ultimately, premature mortality, indicating the need for routine early detection (Asnani & Reid, 2015a, 2015b). The high prevalence of micro- and macro-albuminuria in our SCD cohort differs with previous occurrence rates of 40% and 19% in adult Americans living with SCD (Haymann et al, 2010), 25.9% and 16.5% in Jamaican SCD patients (Asnani et al, 2011), 28.2% among SCD children in Uganda (Mawanda et al, 2011), and 50% combined prevalence rate reported in Nigerian patients (Bolarinwa et al, 2012). These results could reflect a more severe phenotype in this group of Cameroonian patients living with SCD, as illustrated by a relatively low median Hb level of 74 g/l (Table I). Decreased haemoglobin is a significant predictor of increased albuminuria (Alvarez et al, 2006; Mawanda et al, 2011; McBurney et al, 2002). The relative severity and high proportion of micro-albuminuria in this group could be also attributed to the hospital-based recruitment style, as equally high values were observed in a population of hospital-based SCD patients in Saudi-Arabia (Abo-Zenah et al, 2009).
The prevalence of glomerular hyperfiltration was comparable to previous studies of patients in French and West Indian cohorts (Arlet et al, 2012; Haymann et al, 2010), and Congolese HbSS children also displayed a similar prevalence of hyperfiltration (Aloni et al, 2014, 2017). Among Jamaicans living with SCD, up to half of the hyper-filtrating individuals might also have micro-albuminuria (Asnani & Reid, 2015b, 2015a), further supporting the fact that the state of hyperfiltration is an early pathology in those likely to progress to CKD. Vazquez et al (2014) have similarly described increasing rates of development of microalbuminuria with increasing levels of hyperfiltration.
The present study confirms important clinical predictors of renal dysfunctions. Specifically, our findings replicate the positive association of crude albuminuria with increasing age, reduced haemoglobin and increased BMI (Guasch et al, 2006; Saraf et al, 2014; Eke et al, 2012; Ranque et al, 2014), while high leucocyte count, high mean arterial pressure and higher MCV values were identified as positive correlates of increasing albuminuria in the present Cameroonian SCD cohort (Table II). We did not observe a significant association between crude albuminuria and rising blood pressure, as reported previously in Jamaica (Asnani et al, 2016; Aygun et al, 2011), or between crude albuminuria and hyperfiltration, as observed previously among a French population with African ancestry and African Americans (Arlet et al, 2012; Thompson et al, 2007). However, these other studies investigated adult patients, while in the present study there is a majority of children and adolescent individuals, and consequently a lower proportion of macro-albuminuria that may explain these differences. Nevertheless, in a subset of the present cohort, we observed a positive relationship between eGFR values and log(ACR) (fig S4B). The surprising positive association of albuminuria and GFR was also recently shown in a prospective longitudinal study (Asnani et al, 2016). The inconsistent relationship between albuminuria and glomerular filtration remains controversial and deserves further investigation (Mawanda et al, 2011; Barros et al, 2006; Arlet et al, 2012; Thompson et al, 2007), using prospective studies to unravel the role of albuminuria as a predictor of GFR and the progression of sickle glomerulopathy and worsening CKD in SCD. As found in the present study, elevated haemoglobin, systolic and diastolic blood pressure, BMI, serum creatinine and mean arterial pressure were found to be significant predictors for reduced eGFR (Asnani et al, 2016; Becker et al, 2014).
The 3.7kB HBA1/HBA2 deletion was co-inherited in 41.4% (n = 141) of patients, significantly higher than in the previously reported non-SCD individuals (Wonkam et al, 2014b). As replicated in the present study, and associated with lower MCV, co-inheritance of SCD and α-thalassaemia had protective effects against macro-albuminuria development (Day et al, 2012; Lamarre et al, 2014; Nebor et al, 2010). Co-inheritance of SCD and α-thalassaemia was associated with reduced clinical severity and, possibly, increased survival rates in a group of Cameroonian patients (Rumaney et al, 2014; Wonkam et al, 2014b). The lack of association between the targeted genes investigated with clinically defined stages of CKD, as well as the lack of differential frequencies of targeted genetic variants studied among various age groups, child/adolescent and adult patients, are probably due to the small number of homozygote individuals for the minor alleles in the respective sub-groups. These associations require further elucidation through the investigation of a larger cohort of adult patients.
Previous studies indicated an association of homozygous or compound heterozygous APOL1 G1/G2 variants with kidney disease in SCD patients among adult African American (Ashley-Koch et al, 2011; Saraf et al, 2015), possibly through increased risk of haemoglobinuria (Saraf et al, 2015). In addition, associations of HMOX1 with ESKD, possibly through reduced protection of the kidney from haemoglobin-mediated toxicity, was also observed (Saraf et al, 2015). Indeed, intravascular haemolysis is a potential cause of oxidative injury and endothelial damage in SCD (Gladwin et al, 2012; Tracz et al, 2007; Nath & Katusic, 2012). Exposing human proximal tubular cells to increasing cell-free haemoglobin led to increasing concentrations of supernatant kidney injury molecule, reduced viability and induction of HMOX1 (Saraf et al, 2015). HMOX1 rs743811 was associated with chronic kidney disease stage and longer HMOX1 GT-tandem repeats (>25) were associated with lower eGFR in African Americans (Saraf et al, 2015). In the present study, the dinucleotide promoter polymorphism showed no significant association with crude albuminuria, macro-albuminuria, increased ACR levels or eGFR for all equations, which may be due to a lack of statistical power due to the young age of our cohort, with relatively low proportion of adults. Moreover, in African American children with SCD, shorter HMOX1 GT repeats were associated with lower rates of hospitalization for acute chest syndrome, consistent with the shorter GT-tandem repeat length polymorphism having a protective effect in SCD (Bean et al, 2012), and deserves further investigation in Africa.
The significant association of variants at APOL1 and near-significant association of HMOX1 rs743811 with crude albuminuria or eGFR, both early indicators of renal dysfunction, are novel findings requiring further investigation in more adult Cameroonian SCD patients. In total, the present study indicates that the 3.7kB HBA1/HBA2 deletion, HMOX1 and APOL1 G1/G2 polymorphisms, in addition to other variants, such as HbF-promoting loci (Saraf et al, 2017), and variants that still need validations (Schaefer et al, 2016), could lead to early identification of high-risk SCD patients, and contribute to a better screening strategies, leading to targeted preventive and therapeutic interventions.
Possible limitations of the present study are the cross-sectional nature and the hospital based recruitment style that could have selected the sickest SCD patients. Moreover, owing to the young age of the cohort, another possible limitation is the use of the CKD-EPI, Cockcroft-Gault and MDRD equations to estimate the GFR, where the Schwartz formula could have been applied. This formula is postulated to be most accurate in patients younger than 19 years (Schwartz et al, 2009), however new reports indicate the original formula overestimates eGFR values, with the new adjusted equation including variables not measured for this cohort (cystatin C and blood urea nitrogen), and not yet validated in an SCD cohort (Selistre et al, 2016); while the CKD-EPI equation has been reported as the most appropriate for SCD populations (Arlet et al, 2012; Asnani et al, 2013). The young age of the cohort is probably indicative of the severity and related low life expectancy of SCD patients in Cameroon, and the high prevalence of renal dysfunctions could be partly attributed to the associated poor health care system and the lack of a comprehensive programme for SCD, that is also accompanied with SCD being a great burden for Cameroonian families and patients (Wonkam et al, 2014a, c). Future studies will need to investigate a prospective large cohort of adult SCD patients, in steady state.
Conclusion
The present study has revealed a high proportion of patients with micro-albuminuria (60.9%; n = 248) and glomerular hyperfiltration (49.5%; n = 200) among this group of patients living with SCD in Cameroon. Age, male sex, haemoglobin level, leucocyte count, MCV, mean blood pressure, systolic and diastolic blood pressure, BMI and creatinine levels significantly affected micro-albuminuria and/or eGFR. The study has confirmed that co-inheritance of alpha-thalassaemia is protective against macro-albuminuria, while APOL1 G1/G2 risk variants are significantly associated with the ACR. This indicates a possible role of both APOL1 G1/G2 and, possibly, HMOX1-rs743811 as useful biomarkers for risk assessment of renal dysfunctions in SCD.
Supplementary Material
Acknowledgments
FUNDING
The molecular experiments of the study were funded by the National Health Laboratory Services (NHLS), South Africa; and the National Institutes of Health, USA, grant number 1U01HG007459-01. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conceived and designed the experiments: AG, GP, AW. Performed the experiments: AG, VJN. Patient recruitment, sample and clinical data collection and processing: VJNB, BCC. Analysed the data: AG, APK, EC, AW. Contributed reagents/materials/analysis tools: VJNB, BCC, APK, AW. Wrote the paper: AG, AW. Revised and approved the manuscript: AG, VJNB, BCC, GP, APK, EC, AW.
Footnotes
DISCLOSURE STATEMENT
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Abo-Zenah H, Moharram M, El Nahas AM. Cardiorenal risk prevalence in sickle cell hemoglobinopathy. Nephron Clinical Practice. 2009;112:c98–c106. doi: 10.1159/000213897. [DOI] [PubMed] [Google Scholar]
- Aloni MN, Ngiyulu RM, Gini-Ehungu JL, Nsibu CN, Ekila MB, Lepira FB, Nseka NM. Renal function in children suffering from sickle cell disease: Challenge of early detection in highly resource-scarce settings. PLOS ONE. 2014;9:1–5. doi: 10.1371/journal.pone.0096561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloni MN, Ngiyulu RM, Nsibu CN, Ekulu PM, Makulo JR, Gini-Ehungu J-L, Nseka NM, Lepira FB. Congolese children with sickle cell trait may exhibit glomerular hyperfiltration: A case control study. Journal of Clinical Laboratory Analysis. 2017:e22143. doi: 10.1002/jcla.22143. (electronic publication ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez O, Montane B, Lopez G, Wilkinson J, Miller T. Early blood transfusions protect against microalbuminuria in children with sickle cell disease. Pediatric Blood & Cancer. 2006;47:71–76. doi: 10.1002/pbc.20645. [DOI] [PubMed] [Google Scholar]
- Arlet JB, Ribeil JA, Chatellier G, Eladari D, De Seigneux S, Souberbielle JC, Friedlander G, de Montalembert M, Pouchot J, Prie D, Courbebaisse M. Determination of the best method to estimate glomerular filtration rate from serum creatinine in adult patients with sickle cell disease: a prospective observational cohort study. BMC Nephrology. 2012;13:83. doi: 10.1186/1471-2369-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley-Koch AE, Okocha EC, Garrett ME, Soldano K, De Castro LM, Jonassaint JC, Orringer EP, Eckman JR, Telen MJ. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. British Journal of Haematology. 2011;155:386–394. doi: 10.1111/j.1365-2141.2011.08832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnani M, Reid M. Cystatin C: A useful marker of glomerulopathy in sickle cell disease? Blood Cells, Molecules, and Diseases. 2015a;54:65–70. doi: 10.1016/j.bcmd.2014.07.018. [DOI] [PubMed] [Google Scholar]
- Asnani M, Serjeant G, Royal-Thomas T, Reid M. Predictors of renal function progression in adults with homozygous sickle cell disease. British Journal of Haematology. 2016;173:461–468. doi: 10.1111/bjh.13967. [DOI] [PubMed] [Google Scholar]
- Asnani MR, Reid ME. Renal function in adult Jamaicans with homozygous sickle cell disease. Hematology. 2015b;20:422–428. doi: 10.1179/1607845414Y.0000000213. [DOI] [PubMed] [Google Scholar]
- Asnani MR, Fraser RA, Reid ME. Higher rates of hemolysis are not associated with albuminuria in Jamaicans with sickle cell disease. PLOS ONE. 2011;6:e18863. doi: 10.1371/journal.pone.0018863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnani MR, Lynch O, Reid ME. Determining Glomerular Filtration Rate in Homozygous Sickle Cell Disease: Utility of Serum Creatinine Based Estimating Equations. PLOS ONE. 2013;8:e69922. doi: 10.1371/journal.pone.0069922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataga KI, Derebail VK, Archer DR. The glomerulopathy of sickle cell disease. American Journal of Hematology. 2014;89:907–914. doi: 10.1002/ajh.23762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygun B, Mortier NA, Smeltzer MP, Hankins JS, Ware RE. Glomerular hyperfiltration and albuminuria in children with sickle cell anemia. Pediatric Nephrology. 2011;26:1285–1290. doi: 10.1007/s00467-011-1857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros FB, Lima CSP, Santos AO, Mazo-Ruiz MFC, Lima MCL, Etchebehere ECSC, Costa FF, Saad STO, Camargo EE, Ramos CD. 51Cr-EDTA measurements of the glomerular filtration rate in patients with sickle cell anaemia and minor renal damage. Nuclear Medicine Communications. 2006;27:959–62. doi: 10.1097/01.mnm.0000243373.03636.6e. [DOI] [PubMed] [Google Scholar]
- Bartolucci P, Galacteros F. Clinical management of adult sickle-cell disease. Current Opinion in Hematology. 2012;19:149–155. doi: 10.1097/MOH.0b013e328351c35f. [DOI] [PubMed] [Google Scholar]
- Bean CJ, Boulet SL, Ellingsen D, Pyle ME, Barron-Casella E, Casella JF, Payne B, Driggers J, Trau H, Yang G, Jones K, Ofori-Acquah SF, Hooper WC, DeBaun MR. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood. 2012;120:3822–3828. doi: 10.1182/blood-2011-06-361642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AM, Goldberg JH, Henson M, Ahn C, Tong L, Baum M, Buchanan GR. Blood pressure abnormalities in children with sickle cell anemia. Pediatric Blood & Cancer. 2014;61:518–522. doi: 10.1002/pbc.24843. [DOI] [PubMed] [Google Scholar]
- Becton LJ, Kalpatthi RV, Rackoff E, Disco D, Orak JK, Jackson SM, Shatat IF. Prevalence and clinical correlates of microalbuminuria in children with sickle cell disease. Pediatric Nephrology. 2010;25:1505–1511. doi: 10.1007/s00467-010-1536-8. [DOI] [PubMed] [Google Scholar]
- Bitoungui VJN, Pule GD, Hanchard N, Ngogang J, Wonkam A. Beta-globin gene haplotypes among cameroonians and review of the global distribution: is there a case for a single sickle mutation origin in Africa? Omics: a Journal of Integrative Biology. 2015;19:171–179. doi: 10.1089/omi.2014.0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolarinwa RA, Akinlade KS, Kuti MaO, Olawale OO, Akinola NO. Renal disease in adult Nigerians with sickle cell anemia: a report of prevalence, clinical features and risk factors. Saudi Journal of Kidney Diseases and Transplantation. 2012;23:171–175. [PubMed] [Google Scholar]
- Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- Day TG, Drasar ER, Fulford T, Sharpe CC, Thein SL. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica. 2012;97:201–205. doi: 10.3324/haematol.2011.050336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eke CB, Okafor HU, Ibe BC. Prevalence and correlates of microalbuminuria in children with sickle cell anaemia: Experience in a tertiary health facility in Enugu, Nigeria. International Journal of Nephrology. 2012 doi: 10.1155/2012/240173. Article ID 240173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of Trypanolytic ApoL1 Variants with Kidney Disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladwin MT, Kanias T, Kim-Shapiro DB. Hemolysis and cell-free hemoglobin drive an intrinsic mechanism for human disease. The Journal of Clinical Investigation. 2012;122:1205–8. doi: 10.1172/JCI62972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosmanova EO, Zaidi S, Wan JY, Adams-Graves PE. Prevalence and progression of chronic kidney disease in adult patients with sickle cell disease. Journal of Investigative Medicine. 2014;62:804–7. doi: 10.1097/01.JIM.0000446836.75352.72. [DOI] [PubMed] [Google Scholar]
- Guasch A, Cua M, Mitch WE. Early detection and the course of glomerular injury in patients with sickle cell anemia. Kidney International. 1996;49:786–791. doi: 10.1038/ki.1996.109. [DOI] [PubMed] [Google Scholar]
- Guasch A, Cua M, You W, Mitch WE. Sickle cell anemia causes a distinct pattern of glomerular dysfunction. Kidney International. 1997;51:826–833. doi: 10.1038/ki.1997.116. [DOI] [PubMed] [Google Scholar]
- Guasch A, Zayas CF, Eckman JR, Muralidharan K, Zhang W, Elsas LJ. Evidence that microdeletions in the alpha globin gene protect against the development of sickle cell glomerulopathy in humans. Journal of the American Society of Nephrology: JASN. 1999;10:1014–1019. doi: 10.1681/ASN.V1051014. [DOI] [PubMed] [Google Scholar]
- Guasch A, Navarrete J, Nass K, Zayas CF. Glomerular involvement in adults with sickle cell hemoglobinopathies: Prevalence and clinical correlates of progressive renal failure. Journal of the American Society of Nephrology: JASN. 2006;17:2228–2235. doi: 10.1681/ASN.2002010084. [DOI] [PubMed] [Google Scholar]
- Haymann JP, Stankovic K, Levy P, Avellino V, Tharaux PL, Letavernier E, Grateau G, Baud L, Girot R, Lionnet F. Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clinical Journal of the American Society of Nephrology: CJASN. 2010;5:756–761. doi: 10.2215/CJN.08511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. Journal of the American Society of Nephrology: JASN. 2011;22:2129–37. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarre Y, Romana M, Lemonne N, Hardy-Dessources MD, Tarer V, Mougenel D, Waltz X, Tressi??res B, Lalanne-Mistrih ML, Etienne-Julan M, Connes P. Alpha thalassemia protects sickle cell anemia patients from macro-albuminuria through its effects on red blood cell rheological properties. Clinical Hemorheology and Microcirculation. 2014;57:63–72. doi: 10.3233/CH-131772. [DOI] [PubMed] [Google Scholar]
- Levey AS, Bosch J, Lewis J, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Annals of Internal Medicine. 1999;130:461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- Levey AS, Coresh J, Balk E, Kausz AT, Levin A, Steffes MW, Hogg RJ, Perrone RD, Lau J, Eknoyan G. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Annals of Internal Medicine. 2003;139:137–147. doi: 10.7326/0003-4819-139-2-200307150-00013. [DOI] [PubMed] [Google Scholar]
- Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) A new equation to estimate glomerular filtration rate. Annals of Internal Medicine. 2009;150:604–12. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawanda M, Ssenkusu JM, Odiit A, Kiguli S, Muyingo A, Ndugwa C. Micro-albuminuria in Ugandan children with sickle cell anaemia: a cross-sectional study. Annals of Tropical Paediatrics. 2011;31:115–121. doi: 10.1179/1465328111Y.0000000013. [DOI] [PubMed] [Google Scholar]
- McBurney PG, Hanevold CD, Hernandez CM, Waller JL, McKie KM. Risk factors for microalbuminuria in children with sickle cell anemia. J Pediatr Hematol Oncol. 2002;24:473–477. doi: 10.1097/00043426-200208000-00013. [DOI] [PubMed] [Google Scholar]
- McClellan AC, Luthi JC, Lynch JR, Soucie JM, Kulkarni R, Guasch A, Huff ED, Gilbertson D, McClellan WM, DeBaun MR. High one year mortality in adults with sickle cell disease and end-stage renal disease. British Journal of Haematology. 2012;159:360–367. doi: 10.1111/bjh.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath Ka, Katusic ZS. Vasculature and Kidney Complications in Sickle Cell Disease. Journal of the American Society of Nephrology. 2012;23:781–784. doi: 10.1681/ASN.2011101019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebor D, Broquere C, Brudey K, Mougenel D, Tarer V, Connes P, Elion J, Romana M. Alpha-thalassemia is associated with a decreased occurrence and a delayed age-at-onset of albuminuria in sickle cell anemia patients. Blood Cells, Molecules, and Diseases. 2010;45:154–158. doi: 10.1016/j.bcmd.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Njamnshi AK, Mbong EN, Wonkam A, Ongolo-Zogo P, Djientcheu VDP, Sunjoh FL, Wiysonge CS, Sztajzel R, Mbanya D, Blackett KN, Dongmo L, Muna WFT. The epidemiology of stroke in sickle cell patients in Yaounde, Cameroon. Journal of the Neurological Sciences. 2006;250:79–84. doi: 10.1016/j.jns.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH, Williams TN, Weatherall DJ, Hay SI. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. The Lancet. 2013;381:142–151. doi: 10.1016/S0140-6736(12)61229-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in Sickle Cell Disease. The New England Journal of Medicine. 1994;330:1639–16344. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- Ranque B, Menet A, Diop IB, Thiam MM, Diallo D, Diop S, Diagne I, Sanogo I, Kingue S, Chelo D, Wamba G, Diarra M, Anzouan JB, N’Guetta R, Diakite CO, Traore Y, Legueun G, Deme-Ly I, Belinga S, Boidy K, Kamara I, Tharaux PL, Jouven X. Early renal damage in patients with sickle cell disease in sub-Saharan Africa: A multinational, prospective, cross-sectional study. The Lancet Haematology. 2014;1:e64–e73. doi: 10.1016/S2352-3026(14)00007-6. [DOI] [PubMed] [Google Scholar]
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. The Lancet. 2010;376:2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- Ruffieux N, Njamnshi AK, Wonkam A, Hauert CA, Chanal J, Verdon V, Fonsah JY, Eta SC, Doh RF, Ngamaleu RN, Kengne AM, Fossati C, Sztajzel R. Association between biological markers of sickle cell disease and cognitive functioning amongst Cameroonian children. Child Neuropsychology. 2013;19:143–60. doi: 10.1080/09297049.2011.640932. [DOI] [PubMed] [Google Scholar]
- Rumaney MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Kengne AP, Ngogang J, Wonkam A. The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLOS ONE. 2014;9:1–10. doi: 10.1371/journal.pone.0100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- Saraf SL, Zhang X, Kanias T, Lash JP, Molokie RE, Oza B, Lai C, Rowe JH, Gowhari M, Hassan J, Desimone J, Machado RF, Gladwin MT, Little JA, Gordeuk VR. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anaemia. British Journal of Haematology. 2014;164:729–739. doi: 10.1111/bjh.12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraf SL, Zhang X, Shah B, Kanias T, Gudehithlu KP, Kittles R, Machado RF, Arruda JAL, Gladwin MT, Singh AK, Gordeuk VR. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica. 2015;100:1275–1284. doi: 10.3324/haematol.2015.124875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraf SL, Shah BN, Zhang X, Han J, Tayo BO, Abbasi T, Ostrower A, Guzman E, Molokie RE, Gowhari M, Hassan J, Jain S, Cooper RS, Machado RF, Lash JP, Gordeuk VR. APOL1, α-thalassemia, and BCL11A variants as a genetic risk profile for progression of chronic kidney disease in sickle cell anemia. Haematologica. 2017;102:e1–e6. doi: 10.3324/haematol.2016.154153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer BA, Flanagan JM, Alvarez OA, Nelson SC, Aygun B, Nottage KA, George A, Roberts CW, Piccone CM, Howard TA, Davis BR, Ware RE. Genetic Modifiers of White Blood Cell Count, Albuminuria and Glomerular Filtration Rate in Children with Sickle Cell Anemia. PLOS ONE. 2016;11:e0164364. doi: 10.1371/journal.pone.0164364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GJ, Munoz A, Scheider MF, Mak RH, Kaskel F, Warady BA, Furth SL. New Equations to Estimate GFR in Children with CKD. J Am Soc Nephrol. 2009;20:629–637. doi: 10.1681/ASN.2008030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selistre L, Rabilloud M, Cochat P, de Souza V, Iwaz J, Lemoine S, Beyerle F, Polide-Figueiredo CE, Dubourg L. Comparison of the Schwartz and CKD-EPI Equations for Estimating Glomerular Filtration Rate in Children, Adolescents, and Adults: A Retrospective Cross-Sectional Study. PLOS Medicine. 2016;13:1–18. doi: 10.1371/journal.pmed.1001979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Eckman J, Varma M, Ramirez G, Adler B, Smith W, Carlos T, Ataga K, DeCastro L, Bigelow C, Saunthararajah Y, Telfer M, Vichinsky E, Claster S, Shurin S, Bridges K, Waclawiw M, Bonds D, Terrin M. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–1651. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proceedings of the National Academy of Sciences of the United States of America. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, Reid M, Hambleton I, GRS Albuminuria and renal function in homozygous sickle cell disease: Observations from a cohort study. Archives of Internal Medicine. 2007;167:701–708. doi: 10.1001/archinte.167.7.701. [DOI] [PubMed] [Google Scholar]
- Tracz MJ, Alam J, Nath KA. Physiology and Pathophysiology of Heme: Implications for Kidney Disease. Journal of the American Society of Nephrology. 2007;18:414–420. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Human Genetics. 2010;128:345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez B, Shah B, Zhang X, Lash JP, Gordeuk VR, Saraf SL. Hyperfiltration is associated with the development of microalbuminuria in patients with sickle cell anemia. American Journal of Hematology. 2014;89:1156–1157. doi: 10.1002/ajh.23817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware RE, Rees RC, Sarnaik SA, Iyer RV, Alvarez OA, Casella JF, Shulkin BL, Shalaby-Rana E, Strife CF, Miller JH, Lane PA, Wang WC, Miller ST. Renal Function in Infants with Sickle Cell Anemia: Baseline Data from the BABY HUG Trial. The Journal of Pediatrics. 2010;156:66–70. e1. doi: 10.1016/j.jpeds.2009.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bulletin of the World Health Organization. 2001;79:704–12. [PMC free article] [PubMed] [Google Scholar]
- Wigfall DR, Ware RE, Burchinal MR, Kinney TR, Foreman JW. Prevalence and Clinical Correlates of Glomerulopathy Children With Sickle Cell Disease. The Journal of Pediatrics. 2000;136:749–753. [PubMed] [Google Scholar]
- Wonkam A, Ngo Bitoungui VJ, Vorster Aa, Ramesar R, Cooper RS, Tayo B, Lettre G, Ngogang J. Association of variants at BCL11A and HBS1L-MYB with hemoglobin F and hospitalization rates among sickle cell patients in Cameroon. PLOS ONE. 2014a;9:e92506. doi: 10.1371/journal.pone.0092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonkam A, Rumaney MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Ngogang J. Coinheritance of sickle cell anemia and α-thalassemia delays disease onset and could improve survival in cameroonian’s patients (Sub-Saharan Africa) American Journal of Hematology. 2014b;89:664–665. doi: 10.1002/ajh.23711. [DOI] [PubMed] [Google Scholar]
- Wonkam A, Mba CZ, Mbanya D, Ngogang J, Ramesar R, Angwafo FF., 3rd Psychosocial Burden of Sickle Cell Disease on Parents with an Affected Child in Cameroon. Journal of Genetic Counseling. 2014c;23:192–201. doi: 10.1007/s10897-013-9630-2. [DOI] [PubMed] [Google Scholar]
- World Bank. Education for all-fast track initiative: support to the education sector. The World Bank; Washington, DC, USA: 2010. pp. 1–2. Report No. 48373-CM. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.