

Abstract
The design and synthesis of head-to-tail linked artificial macrocycles using the Ugi-reaction has been developed. This synthetic approach of just two steps is unprecedented, short, efficient and works over a wide range of medium (8–11) and macrocyclic (≥12) loop sizes. The substrate scope and functional group tolerance is exceptional. Using this approach, we have synthesized 39 novel macrocycles by two or even one single synthetic operation. The properties of our macrocycles are discussed with respect to their potential to bind to biological targets that are not druggable by conventional, drug-like compounds. As an application of these artificial macrocycles we highlight potent p53-MDM2 antagonism.
Keywords: drug discovery, macrocycles, protein–protein interactions, synthetic methods, Ugi reaction
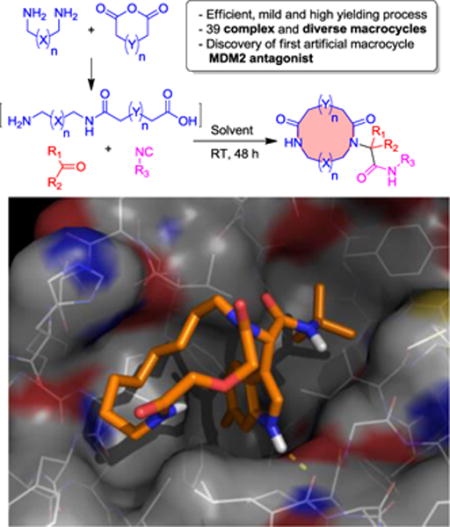
TOC image
Short path, large rings: An unprecedented short and diverse 2-step synthesis is described towards artificial 8–19-membered macrocycles. Artificial macrocycles are useful tools to address difficult biological targets. An example of a designed macrocycle to antagonize the protein–protein interaction p53–MDM2 is given.
Artificial non peptide derived macrocycles are a rather rare and neglected compound class, presumably due to their complex sequential synthesis.[1] Moreover, they have generally not been classified as orally bioavailable and drug-like, until recent advancements in their synthesis and development.[2] However, macrocycles have huge potential in targeting modern postgenomic targets which are difficult to address by small molecules, such as protein–protein interactions (PPI) that are currently a therapeutic domain mostly covered by antibodies.[3–5] As opposed to their natural twins, artificial macrocycles promise to provide better control over synthesizability and their physicochemical properties resulting in drug-like properties. However, very few synthetic methods allow for convergent and fast access to a large macrocyclic chemical space, while not jeopardizing chemical diversity.[6–10]
Owing to increasing interest in macrocycles, many efforts have been made to identify new synthetic methods for their preparation.[11–12] To date, the majority of these methods have focused on sequential multi-step peptide synthesis, followed by stapling via disulfide bridges, cysteine-based cross linkers, RCM, click chemistry, etc.[13] These strategies are not suitable for the elaboration of medium-sized rings or macrocycles with non-natural (non amino acid) side chains and additional ring heteroatoms. Therefore, we introduce here a general synthesis concept for the fast assembly of macrocycles of different size and shape, side-chain and functional-group content. We envisioned a linker moiety using a simple and versatile chemistry. The linker moiety is decorated with orthogonal α,ω-functional groups which can be macrocyclized by another diverse chemistry. Recently, we published an example of this general concept, where we synthesized the linker motive by employing Ugi tetrazole chemistry, followed by a macrocyclization using a second Passerini- or Ugi-multicomponent reaction.[14–15] Here, we wanted to create a manifold of artificial macrocycles by an even shorter sequence involving an initial linear diversification, followed by an exponential diversification step of macrocyclization using Ugi MCR, resulting in an overall 2-step synthesis of complex macrocycles (Figure 1, Scheme 1).
Figure 1.
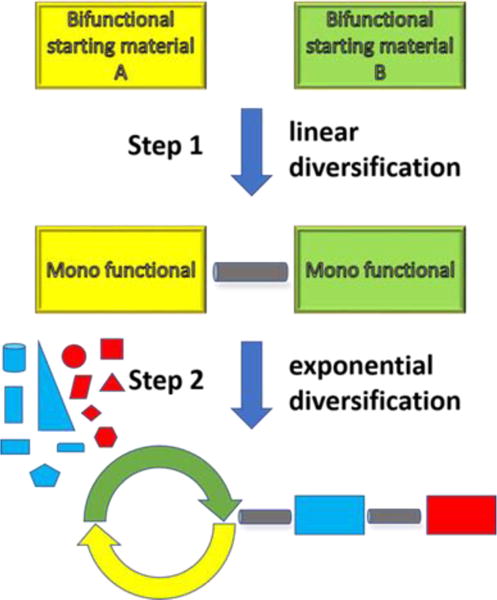
Synthetic concept of rapid generation of macrocycles.
Scheme 1.
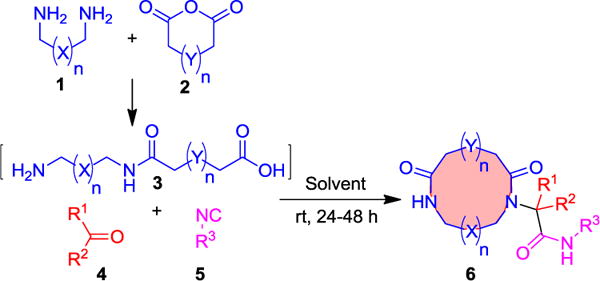
Direct 2-step macrocyclization.
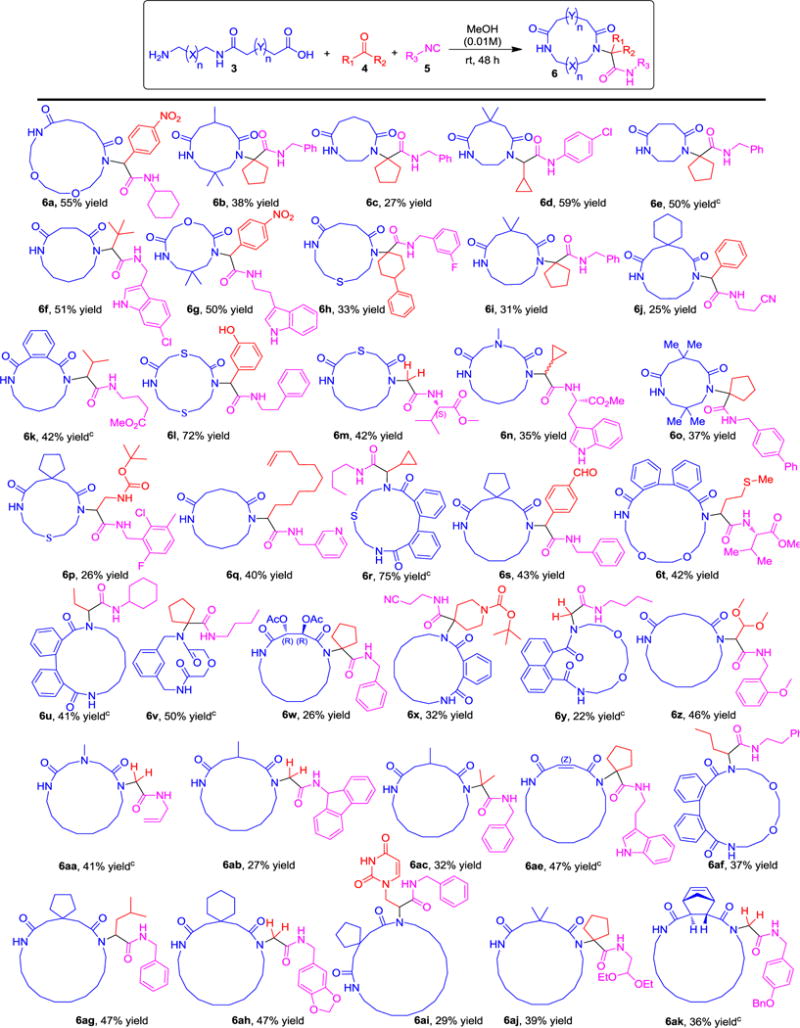
A core feature of our strategy was the use of simple, predictable reactions between diamines, cyclic anhydrides, aldehydes and isocyanides, all of which are starting materials of wide availability. The first step involved the ring opening of cyclic carboxylic acid anhydrides with symmetrical diamines to yield α,ω-amino carboxylic acids. For unambiguous reactivity, we first reacted the mono-Boc protected diamines to yield α,ω-N-Boc amino carboxylic acids, however we soon abandoned this strategy. In order to introduce more flexibility, avoid the use of the halogenated solvent CHCl3 and reduce the number of steps we envisioned performing the direct ring opening reaction of cyclic anhydrides with unprotected alkyl diamines. With this we have synthesized a 36-membered library of terminal amino acids of different chain lengths through the ring opening reaction from commercially available alkyl diamines and cyclic anhydrides in good to excellent yields (see SI_Table 1). Next, we optimized the conditions for the Ugi-macroringclosure of macrocycles (SI_Table 2). The optimized conditions were 1.0 equiv. of 3, 1.0 equiv. of aldehyde/ketone 4, 1.0 equiv. of isocyanide 5 in 0.01M solution of MeOH at room temperature for 24 to 48 h. With this optimized condition in hand, we studied the scope of macrocyclization. We have synthesized a small library of 39, 8-to 19-membered medium to macrocycles in 22 to 75% yields (Table 1, Scheme 2 and 6ad). Different lengths of terminal amino acids including simple methylene chains, heteroatoms, small rings and peripheral groups furnished the macrocycles in good yields. Heteroatoms in a ring, e.g. N-methylated macrocycles are interesting as they can increase hydrophilicity. Phenyl, potentially atropisomeric biphenyl and naphthalene can be conveniently included as ring fragments. Unprotected orthogonally functionalized oxo components such as 3-hydroxybenzaldehyde, terephthalaldehyde and 2-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetaldehyde were tolerated under the reaction conditions and furnished the target macrocycles (6l, 6s and 6ai) in 72%, 43% and 29% yields, respectively. These functional groups can be potentially further elaborated into different products. Similarly, to aldehydes, ketones also worked well and could be incorporated as side chains of different macrocycles in good to excellent yields. The universality of this macrocyclization reaction was further supported by using various functionalized isocyanides derived from aliphatic, aromatic, benzylic, amino acids and heterocycles. Fascinatingly, all these isocyanides reacted very smoothly and resulted in a variety of macrocycles in good yields. The ability of the designed macrocyclization to address complexity was assessed by using tylosin as an oxo component. We have synthesized a tylosin based macrocycle 6al in good yield (53%) with 8:1 ratio of diastereomers that were separated by chromatography. This example nicely underscores the mildness of the procedure being compatible with free hydroxyl groups, α,β-unsaturated ketones, esters, and acetales. Another complex macrocycle is 6am which can be accessed in just two steps, based on a glycosyl isocyanide.[16] Chirality control was introduced in macrocycle 6w, derived from tartaric acid. Our examples indicate that more elaborated substitution patterns and chiral centers can also be employed and lead to complex macrocycles, which are valuable and otherwise difficult to prepare, including spiro cycles, phenyl, biphenyls, naphthalene, pyridine, tylosin and glycosylated macrocycle.
Table 1.
|
Unless otherwise noted, the reaction was conducted with 1.0 mmol of amino acid, 1.0 mmol of aldehyde/ketone and 1.0 mmol of isocyanide in MeOH (0.01 M) for 24–48 h at rt.
Yield refers to the column-purified products.
Reaction performed in a cascade one-pot manner (ring opening followed by macrocyclization, see SI procedure C).
Scheme 2.
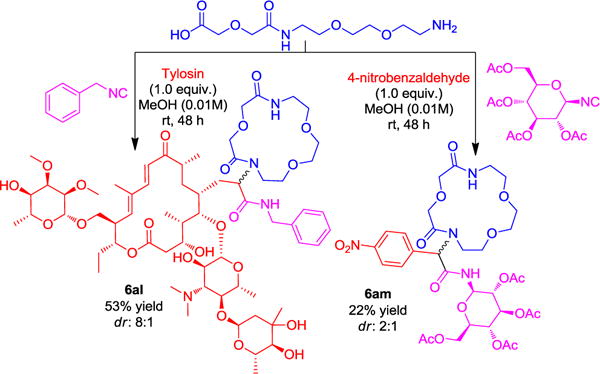
Synthesis of Tylosin and sugar-based macrocycles (6al and 6am).
In some cases, we faced problems in isolating the α,ω-amino acids due to their high polarity (SI_Table 1 red examples). In these cases, we removed the THF solvent of the anhydride ring opening reaction and reacted the crude α,ω-amino acids with the oxo and isocyanide components in the Ugi reaction. Surprisingly, the one pot reaction also produced the desired products (6e, 6k, 6r, 6u, 6v, 6y, 6aa, 6ae and 6ak) in good to moderate yields (Table 1). For a number of the macrocycles we were able to grow crystals and solve the 3D structures. Interestingly in several of the structures the exocyclic amide group is bent back over the macrocycle to form an intramolecular hydrogen bonding with the macrocycles amide group (SI_Figure 2). It has been shown that similar exocyclic amide group arrangements provide structural rigidity within cyclic peptidomimetics and promote the creation of a stabilizing intramolecular hydrogen bonding network.[17] This exocyclic control element can also contribute to increased membrane permeability by allowing the macrocycles to switch between membrane and aqueous solution optimized conformations reminiscent of a chameleonic behavior.[18] Thus, intramolecular hydrogen bonds can contribute to the flexibility of the macrocycles to allows them to adapt to their environment, thereby combining aqueous and lipo solubility, cell permeability, and efficient target binding.[19]
Whitty and coworkers recently analyzed multiple X-ray structures of natural product macrocycles in their protein receptors and proposed specific guidelines for the design of synthetic macrocycle libraries with structural and physicochemical features likely to favor strong binding to protein targets as well as good bioavailability.[20] Thus, we compare the calculated values based on the 38 herein described artificial macrocycles with Whitty’s dataset (SI_Table 3). Macrocyclic heavy atoms (HA) were categorized as (i) ‘ring atoms’, (ii) ‘peripheral atoms’, small groups such as methyl, carbonyl, hydroxyl and halogens that consist of a single HA appending the ring, and (iii) ‘substituent atoms’, comprising larger structures connected to the ring. Structural diversity of all three regions is important for binding to the receptor. All the regions of our artificial macrocycles are within or close to the range of Whitty’s design rules. The balance of polar to non-polar atoms that is important to ensure adequate PSA, is critical for good aqueous solubility. cLogP, MW and other parameters also compare favorably with the dataset of Whitty (SI_Table 4). In summary, our artificial macrocycles fall well into Whitty’s’ design criteria for macrocyclic libraries in most properties.
Macrocycles possess a larger surface area than small molecules and have been proposed to be a suitable molecular class to antagonize protein-protein interactions (PPIs). Specifically, loops and their binding partners represent new and promising PPIs for the development of macrocyclic and constrained peptide inhibitors.[21] Based on our previous knowledge of p53-MDM2 antagonists and as an application of our macrocycles we have designed 15-membered molecule 6ad which should mimic the hot-spot triad (F19, W23 and L26) of p53 interacting with the negative regulator protein MDM2 (see Figure 2).[22] We tested the binding of 6ad towards MDM2 by 2D HSQC-NMR and fluorescence polarization and found a binding constant Ki of 2.25 μM (see SI_Table 5). In accordance with the peak shifts of MDM2 and the modelling we speculate that the macrocycle binds into the L26 pocket, the 6-chloro indole moiety into the W23 and the tert-butyl group into the F19 pocket of the triad binding site. To the best of our knowledge this is the first artificial macrocycle shown to bind to the MDM2 receptor. Stapled peptides bind also to MDM2 but their staple is oriented to a binding site adjacent to the hot-spot.[23] Future reporting will be done on detailed SAR of our artificial macrocycles binding to MDM2 and other protein protein interactions.
Figure 2.
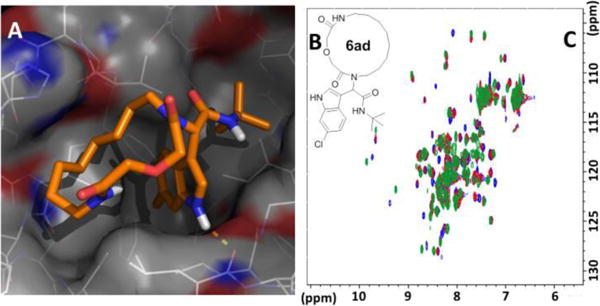
Macrocycle 6ad binding to MDM2 receptor (details SI). A: Modelling of 6ad into MDM2 receptor (PDB ID 1YCR); B: 2D structure of 6ad; C: Superimposed 1H-15N HSQC NMR spectra 15N-labelled MDM2 titrated with 6ad: blue - reference MDM2 spectrum, red – 4:1 (MDM2:6ad) titration step, green – 1:5 (MDM2:6ad; overtitration).
In summary, we have developed a general method that can quickly and accurately convert simple, broadly available, small molecule building blocks into macrocycles via a one-pot Ugi-reaction. This robust and operationally simple 2-step, air- and moisture-tolerant procedure is a valuable addition to MCR chemistry and expands its unique scaffold diversity by giving access to 8–19 membered rings. Recent research suggests that passive cell permeability in cyclic peptides and non peptidic macrocycles sharply decreases at a MW of approximately 1000 Da.[24] However, the macrocyclic space of compounds less than 1000 Da remains largely unexplored and represents an interesting opportunity to chart new territories of drug discovery. The herein described unprecedented short 2-step synthesis of artificial macrocycles with 4 points of diversity covers this interesting chemical space and provides a fast entry to test binding hypothesis towards difficult intracellular targets.
Supplementary Material
Acknowledgments
The work was financially supported by NIH 2R01GM097082-05 (A.D.), the Innovative Medicines Initiative (grant agreement No. 115489), also European Union’s Seventh Framework Programme (FP7/2007–2013) and EFPIA companies’ in-kind contribution (A.D.) and by the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (Contract No. POIG.02.01.00-12-023/08, to K.K.). Funding came also from the European Union’s Horizon 2020 research and innovation programme under MSC ITN “Accelerated Early stage drug dIScovery” (No 675555, to A.D.), CoFund ALERT (No 665250, to A.D.), UMO-2012/06/A/ST5/00224 and UMO-2014/12/W/NZ1/00457 from the National Science Centre, Poland (to T.A.H.). E.M.M.A. was supported by the Egyptian government.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Yu XF, Sun DQ. Molecules. 2013;18:6230–6268. doi: 10.3390/molecules18066230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doak BC, Zheng J, Dobritzsch D, Kihlberg J. J Med Chem. 2016;59:2312–2327. doi: 10.1021/acs.jmedchem.5b01286. [DOI] [PubMed] [Google Scholar]
- 3.Marsault E, Peterson ML. J Med Chem. 2011;54:1961–2004. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- 4.Heinis C. Nat Chem Biol. 2014;10:696–698. doi: 10.1038/nchembio.1605. [DOI] [PubMed] [Google Scholar]
- 5.Driggers EM, Hale SP, Lee J, Terrett NK. Nat Rev Drug Discov. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 6.Failli A, Immer H, Gotz M. Can J Chem. 1979;57:3257–3261. [Google Scholar]
- 7.Beck B, Larbig G, Mejat B. Org Lett. 2003;5:1047–1050. doi: 10.1021/ol034077e. [DOI] [PubMed] [Google Scholar]
- 8.Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. Science. 2004;305:1601–1605. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jebrail MJ, Ng AHC, Rai V, Hili R, Yudin AK, Wheeler AR. Angew Chem, Int Ed. 2010;49:8625–8629. doi: 10.1002/anie.201001604. [DOI] [PubMed] [Google Scholar]
- 10.Koopmanschap G, Ruijter E, Orru RVA. J Org Chem. 2014;10:544–598. doi: 10.3762/bjoc.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim H, Chang S. Angew Chem, Int Ed. 2017;56:3344–3348. doi: 10.1002/anie.201700113. [DOI] [PubMed] [Google Scholar]
- 12.Vasco AV, Perez CS, Morales FE, Garay HE, Vasilev D, Gavin JA, Wessjohann LA, Rivera DG. J Org Chem. 2015;80:6697–6707. doi: 10.1021/acs.joc.5b00858. [DOI] [PubMed] [Google Scholar]
- 13.White CJ, Yudin AK. Nat Chem. 2011;3:509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- 14.Abdelraheem EMM, Kurpiewska K, Kalinowska-Tluscik J, Dömling A. J Org Chem. 2016;81:8789–8795. doi: 10.1021/acs.joc.6b01430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao GP, et al. Org Lett. 2015;17:4980–4983. doi: 10.1021/acs.orglett.5b02419. [DOI] [PubMed] [Google Scholar]
- 16.Neochoritis CG, Zhang J, Dömling A. Synthesis. 2015;47:2407–2413. [Google Scholar]
- 17.Hickey JL, Zaretsky S, Denis MAS, Chakka SK, Morshed MM, Scully CCG, Roughton AL, Yudin AK. J Med Chem. 2016;59:5368–5376. doi: 10.1021/acs.jmedchem.6b00222. [DOI] [PubMed] [Google Scholar]
- 18.Whitty A, Zhong M, Viarengo L, Beglov D, Hall DR, Vajda S. Drug Discovery Today. 2016;21:712–717. doi: 10.1016/j.drudis.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsson P, Doak BC, Over B, Kihlberg J. Advanced Drug Delivery Reviews. 2016;101:42–61. doi: 10.1016/j.addr.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 20.Villar EA, Beglov D, Chennamadhavuni S, Porco JA, Kozakov D, Vajda S, Whitty A. Nat Chem Biol. 2014;10:723–731. doi: 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gavenonis J, Sheneman BA, Siegert TR, Eshelman MR, Kritzer JA. Nat Chem Biol. 2014;10:716–722. doi: 10.1038/nchembio.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang YJ, et al. ACS Chem Biol. 2014;9:802–811. doi: 10.1021/cb400728e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baek S, Kutchukian PS, Verdine GL, Huber R, Holak TA, Lee KW, Popowicz GM. J Am Chem Soc. 2012;134:103–106. doi: 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]
- 24.Pye CR, Hewitt WM, Schwochert J, Haddad TD, Townsend CE, Etienne L, Lao Y, Limberakis C, Furukawa A, Mathiowetz AM, Price DA, Liras S, Lokey RS. J Med Chem. 2017;60:1665–1672. doi: 10.1021/acs.jmedchem.6b01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.