

ABSTRACT
West Nile virus (WNV) is a neurotropic flavivirus that can cause significant neurological disease. Mouse models of WNV infection demonstrate that a proinflammatory environment is induced within the central nervous system (CNS) after WNV infection, leading to entry of activated peripheral immune cells. We utilized ex vivo spinal cord slice cultures (SCSC) to demonstrate that anti-inflammatory mechanisms may also play a role in WNV-induced pathology and/or recovery. Microglia are a type of macrophage that function as resident CNS immune cells. Similar to mouse models, infection of SCSC with WNV induces the upregulation of proinflammatory genes and proteins that are associated with microglial activation, including the microglial activation marker Iba1 and CC motif chemokines CCL2, CCL3, and CCL5. This suggests that microglia assume a proinflammatory phenotype in response to WNV infection similar to the proinflammatory (M1) activation that can be displayed by other macrophages. We now show that the WNV-induced expression of these and other proinflammatory genes was significantly decreased in the presence of minocycline, which has antineuroinflammatory properties, including the ability to inhibit proinflammatory microglial responses. Minocycline also caused a significant increase in the expression of anti-inflammatory genes associated with alternative anti-inflammatory (M2) macrophage activation, including interleukin 4 (IL-4), IL-13, and FIZZ1. Minocycline-dependent alterations to M1/M2 gene expression were associated with a significant increase in survival of neurons, microglia, and astrocytes in WNV-infected slices and markedly decreased levels of inducible nitric oxide synthase (iNOS). These results demonstrate that an anti-inflammatory environment induced by minocycline reduces viral cytotoxicity during WNV infection in ex vivo CNS tissue.
IMPORTANCE West Nile virus (WNV) causes substantial morbidity and mortality, with no specific therapeutic treatments available. Antiviral inflammatory responses are a crucial component of WNV pathology, and understanding how they are regulated is important for tailoring effective treatments. Proinflammatory responses during WNV infection have been extensively studied, but anti-inflammatory responses (and their potential protective and reparative capabilities) following WNV infection have not been investigated. Minocycline induced the expression of genes associated with the anti-inflammatory (M2) activation of CNS macrophages (microglia) in WNV-infected SCSC while inhibiting the expression of genes associated with proinflammatory (M1) macrophage activation and was protective for multiple CNS cell types, indicating its potential use as a therapeutic reagent. This ex vivo culture system can uniquely address the ability of CNS parenchymal cells (neurons, astrocytes, and microglia) to respond to minocycline and to modulate the inflammatory environment and cytotoxicity in response to WNV infection without peripheral immune cell involvement.
KEYWORDS: West Nile virus, inflammation, microglia, minocycline
INTRODUCTION
Since the introduction of West Nile virus (WNV) to North America in 1999, WNV has become the most common cause of epidemic encephalitis in the United States and is responsible for significant neurological morbidity and mortality (1–3). Mouse models of WNV infection have highlighted the importance of peripheral immune cells in the clearance of WNV from the central nervous system (CNS). Inhibiting peripheral immune cell invasion or function has thus been shown to greatly increase mortality due to increased CNS viral burden (4–6) and/or improper recruitment and activation of antiviral immune cells (7–9).
While the importance of these robust antiviral proinflammatory responses in WNV clearance from the CNS is well known, questions remain as to the extent to which these responses can cause damage to neuronal populations that are normally protected from cytotoxic inflammatory cells by the blood-brain barrier (BBB). Increasing evidence suggests that resolving proinflammatory responses in the CNS increases the extent of CNS tissue injury (10–12). Anti-inflammatory (M2) factors are associated with resolution of proinflammatory (M1) activation and cytotoxic functions with subsequent roles for neuroprotection and trophic support (13–16). However, the ability of M2 factors to modulate the pathology of WNV infections is poorly understood. In mouse models of WNV infection, little is known about how the balance of pro- and anti-inflammatory responses influences neuronal survival and to what extent CNS cells themselves regulate the inflammatory environment.
By utilizing an ex vivo slice culture model system of CNS tissue, our laboratory has replicated critical hallmarks of WNV infection in vivo, including high levels of neuronal infection and death, robust microglial activation and astrogliosis, and significant expression of relevant antiviral M1 cytokines/chemokines (17, 18). This system is ideal for investigating the intrinsic responses of CNS parenchymal cells (including neurons, microglia, and astrocytes) to WNV infection and their ability to engage inflammatory signaling pathways in the absence of potentially confounding contributions from peripheral immune cells or mediators. Although neurons, microglia, and astrocytes all respond to WNV infection, microglia are considered the primary CNS immune cells and likely play the most significant role in modulating inflammatory conditions within the CNS, at least before the entry of peripheral immune cells.
Considering the therapeutic potential of altering inflammatory states in the WNV-infected CNS, it is important to understand the abilities of CNS parenchymal cells in mediating M1 and/or M2 signaling events (particularly for microglia as the resident macrophages). In this report, we treated WNV-infected spinal cord slice cultures (SCSC) with minocycline, a well-characterized anti-inflammatory drug (15, 19, 20) with known inhibitory effects on proinflammatory (M1) microglial activation (21–26). We previously showed in this slice culture system that minocycline could modestly reduce WNV growth (<1 log10 unit) while reducing the expression of some M1 genes (18); here, we further characterized the effect of minocycline on a wider range of inflammatory genes and proteins, as well as investigating its effect on cell death and tissue health during WNV infection.
Our results indicate that minocycline treatment resulted in significant reductions in the RNA and protein expression of genes associated with proinflammatory M1 microglial activation following WNV infection of SCSC and concurrent increases in the expression of genes associated with anti-inflammatory M2 microglial activation. These minocycline-dependent effects coincided with significant reductions in microglial activation, observed via changes in cellular morphology and the expression of ionized calcium binding adaptor molecule 1 (Iba1), a classical marker for M1 microglial activation. In conjunction with these changes in gene expression, WNV-induced cell death was significantly reduced across neuron and glial cell populations, with a notable inhibition of inducible nitric oxide synthase (iNOS) expression.
RESULTS
WNV-induced upregulation of Iba1 gene expression and M1 CC motif chemokine gene and protein expression in SCSC is inhibited by minocycline.
Microglial M1 activation is commonly assessed by the upregulation of Iba1 (18, 27–30) and CC motif chemokine ligand proteins, including CCL2, CCL3, CCL5, and CCL7 (31–34). We have previously shown that WNV infection of SCSC results in the upregulation of both Iba1 and CC motif chemokines (18). In addition, these chemokines have important roles in WNV-induced neuroinflammation (7, 35, 36). Using reverse transcription-quantitative PCR (RT-qPCR) analysis, we now show that WNV-induced increases in expression of Iba1 and CC motif chemokines in WNV-infected SCSC are reduced by minocycline (Fig. 1 and 2). As expected, Iba1 expression rose dramatically following WNV infection of SCSC (Fig. 1A). Minocycline treatment of WNV-infected SCSC inhibited this effect. At 3 days postinfection (dpi), 5 dpi, and 7 dpi, Iba1 expression was significantly reduced with minocycline treatment compared to time-matched SCSC treated with vehicle alone. To determine if astrocytes were also activated following WNV infection in SCSC, GFAP (glial fibrillary acidic protein) expression was also evaluated via RT-qPCR; GFAP expression rose in WNV-infected SCSC compared to mock-infected SCSC, confirming the presence of WNV-induced astrocyte activation in the system. However, no significant differences were observed between vehicle- and minocycline-treated SCSC (Fig. 1A). These gene expression analyses indicated that minocycline exclusively affected microglial activation, but not astrocyte activation.
FIG 1.
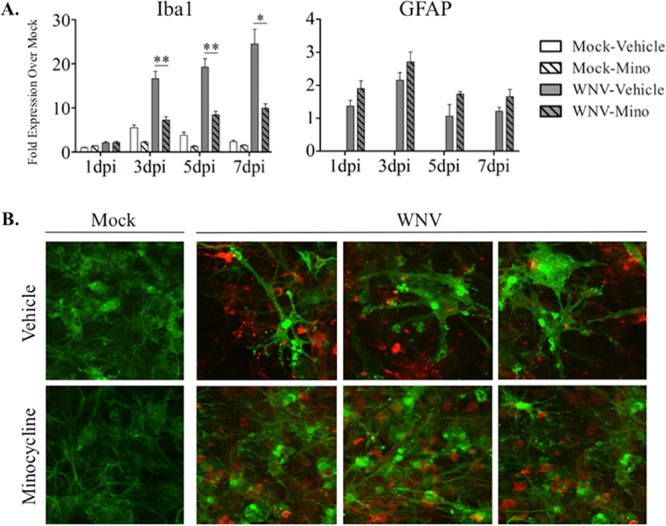
Minocycline treatment inhibits microglial activation during WNV infection of SCSC. (A) RT-qPCR analyses were performed for Iba1 and GFAP expression in SCSC taken at 1 dpi, 3 dpi, 5 dpi, and 7 dpi. Changes in gene expression levels are indicated as fold increase over mock infection, with 1-dpi mock-infected, vehicle-treated SCSC used as the normalized control sample (expression set as 1) and beta-actin used as the normalized control gene. Iba1 gene expression (left) increased in WNV-infected (gray bars) compared to mock-infected (open bars), vehicle-treated SCSC in a time-dependent manner, indicating microglial activation. Minocycline (Mino) treatment of WNV-infected SCSC (hatched gray bars) caused significant decreases in Iba1 expression compared to WNV-infected, vehicle-treated counterparts (gray bars). GFAP gene expression (right) increased in WNV-infected, vehicle treated SCSC (gray bars) compared to mock-infected, vehicle-treated controls (open bars), but not as dramatically nor as high as Iba1. There was no decrease in WNV-induced GFAP expression following minocycline treatment. The asterisks indicate statistically significant differences in Iba1 expression in minocycline-treated versus vehicle-treated, WNV-infected SCSC (*, P < 0.05; **, P < 0.01; unpaired Student t test). The error bars indicate standard errors of the mean. Fifty SCSC (n = 2 mice) were used per experimental condition. (B) Immunohistochemistry of Iba1 (green) and WNV-E (red), imaged with a 60× objective, from mock-infected (top row) and WNV-infected (bottom row) SCSC at 6 dpi. Iba1 expression was increased in WNV-infected SCSC compared to mock-infected samples, but the ameboid and enlarged microglial cells seen in vehicle-treated SCSC were absent from minocycline-treated SCSC.
FIG 2.

Minocycline treatment reduces proinflammatory CC motif chemokine gene and protein expression during WNV infection of SCSC. (A) RT-qPCR analyses were performed for CC motif chemokine gene expression in SCSC taken at 1 dpi, 3 dpi, 5 dpi, and 7 dpi. Each chemokine examined (CCL2, CCL3, CCL5, and CCL7) rose with WNV infection in a time-dependent manner, with significant reductions in expression with minocycline-treated (hatched gray bars) compared to vehicle-treated (gray bars), WNV-infected SCSC. Fifty SCSC (n = 2 mice) were used per experimental condition. (B) ELISA analyses for CCL2, CCL3, CCL5, and CCL7 from SCSC lysates taken at 5 dpi. Each chemokine was elevated in WNV-infected, vehicle-treated SCSC (gray bars) compared to mock-infected controls (open bars), with significant reductions following minocycline treatment (hatched gray bars) for CCL2, CCL3, and CCL5. The asterisks indicate statistically significant differences in minocycline-treated versus vehicle-treated, WNV-infected SCSC (*, P < 0.05; **, P < 0.01; ***, P < 0.001; unpaired Student t test). The error bars indicate standard errors of the mean. One hundred SCSC (n = 4 mice) were used per experimental condition.
We next assessed via immunohistochemistry (IHC) if minocycline had any effect on microglial cell morphology that is stereotypical of microglial activation during WNV infection in SCSC (18) (Fig. 1B). Using antibodies against Iba1 and WNV envelope protein (WNV-E), notable differences in microglial size and ameboid state (indicating proinflammatory chemotaxis) were evident, with microglia in vehicle-treated samples both larger and more ameboid than microglia in minocycline-treated samples. This qualitative assessment suggests that microglial activation states were dramatically altered in minocycline-treated WNV-infected samples.
The expression of CCL2, CCL3, CCL5, and CCL7 was also assessed via RT-qPCR in WNV-infected SCSC with or without minocycline treatment (Fig. 2A). The expression of each chemokine was markedly increased over the 7-day time course in WNV-infected, vehicle-treated SCSC; significant reductions in expression were noted for each chemokine during minocycline treatment at various time points, particularly the later time points (5 dpi and 7 dpi).
To determine if these changes in gene expression reflected changes in protein expression, enzyme-linked immunosorbent assays (ELISA) were performed at 5 dpi for CCL2, CCL3, CCL5, and CCL7. For each chemokine examined, significant increases in protein levels for WNV-infected, vehicle-treated SCSC were observed compared to mock-infected SCSC. With minocycline treatment in WNV-infected SCSC, significant reductions were observed for CCL2, CCL3, and CCL5 (Fig. 2B). These results indicate that at 5 days postinfection, the majority of CC motif chemokines are strongly inhibited in their expression at the RNA and protein levels with minocycline treatment during WNV infection and that these reductions are associated with inhibition of M1 microglial, but not astrocyte, activation.
Minocycline differentially alters inflammatory gene expression during WNV infection of SCSC.
Minocycline is known to broadly inhibit proinflammatory activity while promoting anti-inflammatory activity (15, 19, 37, 38). We used RT-qPCR analysis to evaluate the expression of a series of canonical pro- and anti-inflammatory genes during a 7-day time course following WNV infection of SCSC (Fig. 3). Via literature review, we determined a panel of genes to assess pro- and anti-inflammatory activation (13–16, 39–47). As examples of proinflammatory genes with relevance to WNV infections, we investigated the expression of CXC motif chemokine 10 (CXCL10), interferon alpha (IFN-α), interleukin 1-beta (IL-1β), IL-6, interferon regulatory factor 1 (IRF1), and tumor necrosis factor alpha (TNF-α) (Fig. 3A), and for anti-inflammatory genes, we investigated the expression found in inflammatory zone 1 (FIZZ1), IL-4, IL-7, IL-10, and IL-13 and triggering receptor expressed on myeloid cells 2 (TREM2) (Fig. 3B). In order to better understand the overall impact of minocycline treatment on the balance between pro- and anti-inflammatory gene expression, we examined the percent change in individual gene expression with minocycline treatment compared to the control for all tested genes in each separate category (Fig. 4). This comparison makes it clear that minocycline broadly and significantly reduced proinflammatory gene expression and led to an enhancement in the expression of anti-inflammatory genes.
FIG 3.

Minocycline treatment broadly modulates proinflammatory and anti-inflammatory gene expression during WNV infection of SCSC. RT-qPCR analyses were performed for proinflammatory (A) and anti-inflammatory (B) gene expression in SCSC taken at 1 dpi, 3 dpi, 5 dpi, and 7 dpi. (A) Expression of proinflammatory genes rose with WNV infection in a time-dependent manner, with varying levels of minocycline-dependent decreases (or no effect) in expression. (B) Expression of anti-inflammatory genes rose with WNV infection in a time-dependent manner, with broad minocycline-dependent increases in expression at different time points. The asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01; unpaired Student t test). The error bars indicate standard errors of the mean. Fifty SCSC (n = 2 mice) were used per experimental condition.
FIG 4.
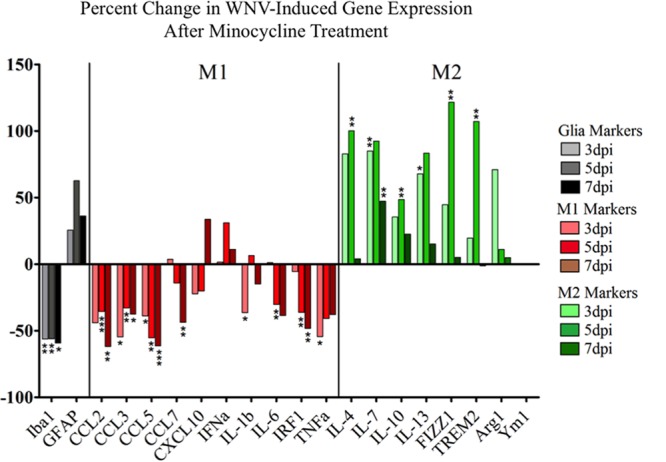
Minocycline treatment reduces proinflammatory (M1) gene expression and increases anti-inflammatory (M2) gene expression during WNV infection in SCSC. RT-qPCR analysis data were graphed to show the percent change in expression with minocycline treatment at each time point for each gene. When categorized as M1 or M2, a clear pattern was observed with minocycline treatment, with large-scale reductions in M1 gene expression (red bars) and increases in M2 gene expression (green bars). Iba1 and GFAP expression are also indicated (black bars) to show minocycline effects on glial activation. The asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001; unpaired Student t test).
Minocycline is neuroprotective during WNV infection in SCSC.
We have previously shown that at 6 dpi, WNV infection causes substantial neuronal death in SCSC; over 90% of cells stained with the neuronal marker microtubule-associated protein 2 (Map2+ cells) had WNV-E cytoplasmic staining, which is indicative of widespread WNV neuronal infection (18). To assess if minocycline treatment is neuroprotective during WNV infection, we stained cells with WNV-E and Map2 at 6 dpi in SCSC. WNV infection caused a dramatic loss of Map2+ cells in WNV-infected compared to vehicle-treated SCSC (Fig. 5A and B). Minocycline treatment reduced, but did not fully prevent, this neuronal loss (Fig. 5B). A similar significant increase in neuronal survival after minocycline treatment was also seen with another neuronal marker, NeuN, at the same time point (data not shown). These results indicate that minocycline is neuroprotective during WNV SCSC infection.
FIG 5.

Minocycline treatment is protective for neurons, microglia, and astrocytes during WNV infection in SCSC. (A) Immunohistochemistry of Map2 (green) and WNV-E (red) from mock-infected (top row) and WNV-infected (bottom row) SCSC at 6 dpi. Mock-infected SCSC showed dispersed Map2 expression throughout tissue, while WNV-infected SCSC had greatly reduced Map2 expression and cell bodies. Minocycline-treated SCSC with WNV infection had noticeably more Map2+ cells than their vehicle-treated counterparts. (B) Quantification of Map2+ cell bodies imaged with a 10× objective. Across multiple FOV, there was a large loss of Map2+ cells in WNV-infected SCSC compared to mock-infected SCSC but a statistical difference between minocycline- and vehicle-treated SCSC with WNV infection. Mock infected, n = 8 SCSC; WNV infected, n = 12 SCSC. (C) Immunohistochemistry of Iba1 (green) and WNV-E (red) from mock-infected (top row) and WNV-infected (bottom row) SCSC at 6 dpi. WNV infection causes substantial microglial loss in vehicle-treated SCSC, with less microglial loss observed with minocycline treatment. (D) Quantification of Iba1+ cell bodies imaged with a 10× objective. Large loss of Iba1+ cell bodies was observed for WNV-infected SCSC, but cell loss was more prominent for vehicle-treated SCSC than for minocycline-treated SCSC. For each condition, n = 12 SCSC. (E) Immunohistochemistry of GFAP (green) and WNV-E (red) from mock-infected (top row) and WNV-infected (bottom row) SCSC at 6 dpi. WNV infection caused significant astrogliosis (increased GFAP expression and thickened processes) compared to mock-infected SCSC. Increased astrocyte death was apparent in vehicle-treated WNV-infected SCSC (small GFAP+ particles) but not as widespread in minocycline-treated, WNV-infected SCSC. (F) Quantification of the percentage of each FOV in which astrocytes were normal, astrogliotic, or dead in images taken with a 20× objective. Dead astrocytes were observed in WNV-infected SCSC but to a greater extent in vehicle-treated versus minocycline-treated SCSC. Both WNV-infected SCSC had similar levels of astrogliosis. For each condition, n = 8 SCSC. The asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01; unpaired Student t test). The error bars indicate standard errors of the mean.
Minocycline protects glial cells during WNV infection in SCSC.
To further understand the effect of minocycline on WNV-induced cytotoxicity, we assessed the health of microglia and astrocytes during WNV infection in SCSC. To determine the extent of microglial loss during WNV in SCSC, we examined the expression of Iba1 via IHC (Fig. 5C and D). In WNV-infected SCSC, Iba1+ cells were markedly decreased compared to mock-infected controls. However, in minocycline-treated SCSC, WNV infection did not reduce microglial cell populations as dramatically as in vehicle-treated SCSC (Fig. 5D), indicating that minocycline treatment protected microglia from WNV-induced cell death.
To determine how WNV infection induced astrogliosis and astrocyte death, we stained for GFAP in WNV-infected SCSC via IHC (Fig. 5E and F). In WNV-infected SCSC, astrocytes are highly reactive to WNV with notable astrogliosis (a process that involves increased GFAP expression and thickened cellular processes) compared to mock-infected SCSC. Astrocyte death was also apparent in WNV-infected SCSC, as depicted by small GFAP+ particles. Minocycline treatment significantly enhanced astrocyte survival but did not alter WNV-induced astrogliosis (Fig. 5F).
Minocycline reduces iNOS expression during WNV infection in SCSC.
Minocycline has been extensively shown to reduce the expression of cytotoxic factors and to promote neuronal survival across disease models (15, 19, 20, 25, 26). To determine if minocycline treatment could reduce the expression of cytotoxic factors during WNV infection of CNS tissue, we observed the expression of iNOS via IHC in SCSC infected with WNV at 6 dpi. iNOS is a classic cytotoxic factor produced by microglia (48–50) that is expressed during WNV infections (51), and its expression can be inhibited by minocycline (52, 53).
In 6-dpi WNV-infected SCSC treated with vehicle alone, iNOS expression was markedly increased compared to mock-infected SCSC (Fig. 6). In contrast, minocycline-treated SCSC infected with WNV did not show increased levels of iNOS expression.
FIG 6.
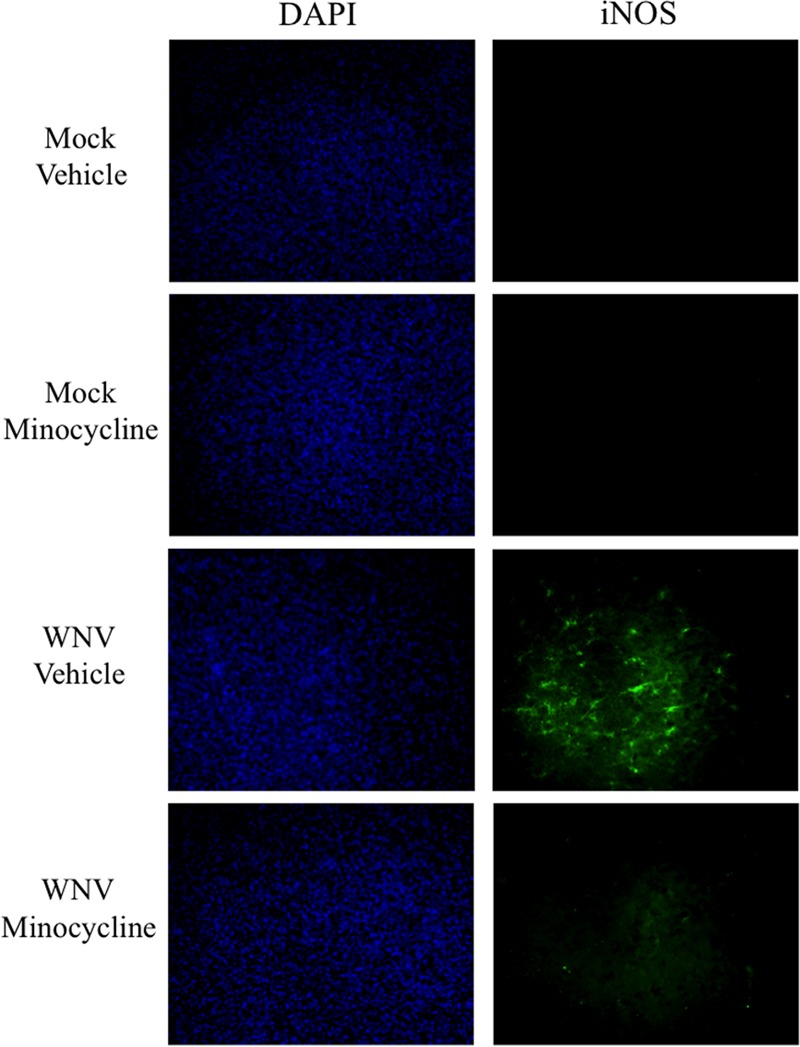
Minocycline treatment reduces iNOS expression during WNV infection in SCSC. (Right column) Immunohistochemistry of iNOS (green) from mock-infected and WNV-infected SCSC at 6 dpi. Clusters of iNOS+ cells were delineated in vehicle-treated SCSC with WNV infection, whereas no similar cells were seen in minocycline-treated SCSC with WNV infection or any mock-infected SCSC. (Left column) DAPI (4′,6-diamidino-2-phenylindole) imaging showing equivalent numbers of cells for each field of vision.
DISCUSSION
Our findings provide further evidence to a growing body of work that shows that, under certain conditions, skewing the balance of inflammatory mediators toward an anti-inflammatory versus a proinflammatory environment can have beneficial effects for cell survival during cytotoxic events in the CNS. For WNV infections, having the ability to efficiently induce M2 signals to repair neurons and return homeostatic conditions after viral clearance via M1 neuroinflammation would likely be important for improving recovery and avoiding complications from chronic inflammation. Across disease models, there is continuing debate over whether distinct M1 and M2 phenotypes exist in vivo (54–57), but the fact that microglia can produce both pro- and anti-inflammatory mediators is widely accepted (13, 14, 16, 58). The ability of minocycline to specifically inhibit M1 microglial activation while also appearing to promote M2 signaling and cell survival during WNV infection, as described in this report, is consistent with other studies that have shown protective effects with minocycline administration for other neuroinvasive viral infections, including Japanese encephalitis virus (59–61), Venezuelan equine encephalitis virus (62), and experimental reovirus (63) infections. Whether minocycline would have therapeutic potential in treatment of human WNV infections or mouse models remains unclear; delayed administration of minocycline could conceivably allow early antiviral immune responses to occur but prevent their later deleterious effects.
There is prior evidence that anti-inflammatory responses can confer beneficial effects in relation to WNV infections. Elevated levels of IL-4 are found in human patient populations that have better long-term recovery compared to patients with worse overall outcomes; this coincides with reduced proinflammatory gene expression in their peripheral blood mononuclear cells (PBMC) (64). Additionally, in mouse models of WNV infection, combinatorial treatment with antiviral medication and the histone deacetylase inhibitor vorinostat (which can have anti-inflammatory properties) led to improved survival, and the therapeutic effects of vorinostat on WNV infection were dependent on the timing of drug administration during WNV infection (65).
To what extent the timing and efficacy of M1 versus M2 responses make a functional difference in various disease models remains to be fully elucidated. Nonetheless, understanding how proinflammatory effects are regulated and subsequently resolved in the CNS is of vital importance in pinpointing how best to intervene against neurological diseases that remain difficult to treat. In particular, at least for the WNV slice culture model, the evidence presented here indicates a potential role for minocycline-dependent protection for neurons and glial cells with a concurrent balance shift in gene expression favoring M2 over M1. While the mechanism by which minocycline confers anti-inflammatory effects and inhibits microglial activation is unknown, this system provides an effective platform to further investigate how these events occur.
Our results demonstrate that minocycline treatment causes specific decreases in proinflammatory gene expression and concurrent increases in anti-inflammatory gene expression, giving credence to the theory of different (M1 versus M2) microglial phenotypes. The effect of minocycline on the expression of the CC motif chemokines (in particular CCL2, CCL3, and CCL5) during WNV infection was notable. These chemokines have important roles in WNV-induced neuroinflammation (7, 35, 36) and are prominently produced by activated microglia (31–34); the genes were significantly downregulated after minocycline administration during WNV infection. Some proinflammatory genes do not change significantly with minocycline treatment (CXCL10 and IFN-α), which is likely due to nonmicroglial production; notably, WNV-infected neurons are able to produce CXCL10 (66), and neurons and astrocytes have the ability to produce significant amounts of proinflammatory cytokines/chemokines during viral infections (67–70).
The relevance of elevated M2 gene expression during WNV infection in SCSC is unclear; with minocycline administration, the expression of some M2 genes rose further, but whether this was the primary catalyst for the modest increase in cell survival versus the decrease in M1 gene expression (or a combination of the two) remains to be determined. Combined with the modest reduction in WNV growth with minocycline treatment reported previously in this system (18) and the strong inhibition of iNOS expression, several possible mechanisms might be involved in the reduced tissue injury observed in this study.
Attempts to detect M2 proteins at 5 dpi were unsuccessful, but the observed changes in gene expression should not detract from their potential relevance, given that most M1 proteins (besides the CC motif chemokines) were also difficult to detect with ELISA methods. One of the advantages of the SCSC model is the ability to garner significant amounts of data from small samples, so future pursuits concerning protein expression may need to come from brain slice cultures (BSC) of WNV infection.
A highlight of this study is the dichotomy seen in M1 and M2 gene expression following minocycline treatment during WNV infection (Fig. 3); while much controversy surrounds the concept of M1 versus M2 categorization, minocycline clearly influenced the expression of these inflammatory genes in a stereotypical manner that would be expected given its status as a well-characterized anti-inflammatory reagent. The fact that these alterations were observed in a WNV disease model that had not previously been part of the wider M1/M2 debate adds to the discussion as to what the role of balancing M1 and M2 effects is in the course of neuropathology and how microglia influence neuroinflammation more broadly.
Many questions remain as to how microglia and astrocytes interact with each other to influence neuronal survival during cytotoxic events (71–74). While our evidence here suggests minocycline inhibited microglial activation (but not astrocyte activation) and reduced cell death, the specific contributions of individual cell types to pro- and anti-inflammatory gene expression are not completely clear. Further studies using the ex vivo model system could prove useful in determining specific contributions made by the support cells of the CNS during WNV infection and in testing pharmacologic reagents that can alter their activity.
MATERIALS AND METHODS
SCSC.
SCSC were prepared from neonatal (5- to 6-day-old) NIH Swiss Webster mice in compliance with IACUC protocols and institutional guidelines at the University of Colorado, Denver (U. C. Denver). The protocol has been previously described in detail (18). Briefly, spinal cords were removed and embedded in 2% agarose and mounted onto a Vibratome cutting instrument (VT1000S; Leica). Transverse sections (400 μm) of thoracic and lumbar regions of the spinal cord were sliced and collected in medium ((Dulbecco's modified Eagle's medium [DMEM]) (10 mM Tris, 28 mM d-glucose, pH 7.2, 95% O2-5% CO2). Spinal cord sections were removed from the agarose and plated onto 30-mm 0.4-μm-pore-size cell culture membrane inserts (Millipore). The inserts were placed into 35-mm well plates with 1.1 ml culture medium (neurobasal A: 10 mM HEPES, 400 μM l-glutamine, 600 μM GlutaMAX, 1× B27 (Invitrogen), 60 μg/ml streptomycin, 60 U/ml penicillin, 6 U/ml nystatin) supplemented with 10% fetal bovine serum (FBS) and maintained at 5% CO2 in a cell culture incubation chamber. The next day, new culture medium was added with 5% FBS, and 2 days later, the medium was changed without FBS supplementation. Medium changes were performed every 2 days.
WNV infection.
NY99 strain (clone-derived strain 382-99) stocks were procured as previously described (75). Three days after preparing SCSC (when the medium was changed without FBS supplement), SCSC were inoculated directly with 1 × 104 PFU/slice in a volume of 20 μl culture medium. SCSC were washed 12 h later to remove excess medium and virus.
Minocycline administration.
Minocycline HCl (Sigma) was prepared at a stock concentration of 1 mM in slice culture medium and then diluted to 20 μM in final slice culture medium to be applied to SCSC during medium changes. Minocycline treatment began at the time of WNV infection; the WNV inoculum was directly applied to SCSC, whereas minocycline was not directly applied to SCSC and had access via medium only.
IHC.
SCSC were washed in PBS and fixed in 10% neutral buffered formalin for at least 1 h. The fixed SCSC were rewashed in PBS and immersed in block solution (PBS, 4% normal goat serum, 2% bovine serum albumin, 0.3% Triton X) for 1 h. The SCSC were then incubated overnight at room temperature with primary antibodies diluted in block solution. The next day, the SCSC were washed 3 times with wash solution (PBS, 0.3% Triton X) and then incubated for 2 h at room temperature with secondary antibodies in block solution. After being washed 3 times with wash solution, the SCSC were briefly rinsed with distilled water (dH2O) before being mounted onto microscopy slides with ProLong Gold antifade reagent (Molecular Probes). For cell assessments and quantification experiments, the slides were imaged using a Nikon PCM-2000 laser scanning confocal microscope, with image procurement conducted with SimplePCI software (v4.6; Compix). For iNOS detection, the slides were imaged using a Zeiss Axiocam on a Nikon Eclipse E800 epifluorescence microscope, with image procurement conducted with Axiovision software (v4.8; Zeiss). The primary antibodies used were mouse anti-WNV envelope protein (1:200; ATCC), rabbit anti-Map2 (1:100; Millipore), rabbit anti-Iba1 (1:500; Wako), rabbit anti-GFAP (1:900; Abcam), and rabbit anti-iNOS (1:300; Abcam). The secondary antibodies used were goat anti-mouse Alexa Fluor 568 and goat anti-rabbit Alexa Fluor 488 (1:1,000; Invitrogen).
Cell count quantification.
Cells were counted by hand on Microsoft Paint software. Each image constituted a field of vision (FOV) for a specified magnification; FOV were of similar regions for each SCSC (i.e., the anterior horn) to ensure proper comparisons of cell populations.
RT-qPCR.
SCSC were removed from culture membranes and homogenized in RLT buffer (Qiagen) with 1% beta-mercaptoethanol and loaded into RNeasy spin columns (Qiagen). Following the manufacturer's protocols for the RNeasy minikit (Qiagen), purified RNA was collected from the spin columns. The RNA quality and concentration were measured with an Agilent 2100 bioanalyzer, and cDNA was prepared with iScript (Bio-Rad) following the manufacturer's directions. The cDNA was mixed with primers (Table 1) and 2× SYBR green master mix (SABiosciences) to 20-μl volumes in individual wells of a 96-well PCR plate, and PCR amplification was performed with a CFX96 thermocycler (Bio-Rad). Relative gene expression was determined via threshold cycle analysis using Bio-Rad CFX Manager software. Beta-actin expression was the control gene setting, and 1-dpi mock vehicle SCSC was the normalization set point for individual gene analyses.
TABLE 1.
Gene primers used in RT-qPCR analysis

ELISA.
SCSC were collected in lysis buffer (R&D) and homogenized/sonicated to create lysate to screen with a custom Mouse Mix & Match Cytokine ELISArray strip kit (Signosis). Lysate was added to ELISArray plate wells for 2 h of binding incubation, and the plates were washed 3 times and then incubated for 1 h with streptavidin-bound detection antibody. After 3 additional washes, streptavidin-horseradish peroxidase (HRP) solution was incubated for 45 min. Detection solution was then added to each well for 30 min before stop solution was applied, turning the solution yellow, depending on the amount of bound detection antibody. The strength of the yellow color was quantified colorimetrically at 450 nm with an Emax spectrometer (Molecular Devices).
Figures and statistical analysis.
All graphs and statistical analyses were created and assessed with GraphPad Prism software.
ACKNOWLEDGMENTS
The study described in this publication was supported by NIH grants R01 NS076512 (K.L.T.), T32 AI052066 (E.D.Q.), and R21/R33 AI101064 (K.L.T.) and by a VA merit grant (K.L.T.). K.L.T. is also supported by the Reuler-Lewin Family Professorship. P.C. is supported by the Colorado Clinical and Translational Sciences Institute (U.C. Denver). S.S. is a recipient of the RNA Bioscience Initiative Graduate Scholars Award (U.C. Denver).
REFERENCES
- 1.Petersen LR, Brault AC, Nasci RS. 2013. West Nile virus: review of the literature. JAMA 310:308–315. doi: 10.1001/jama.2013.8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Debiasi R, Tyler KL. 2006. West Nile virus meningoencephalitis. Nat Clin Pract Neurol 2:264–275. doi: 10.1038/ncpneuro0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis LE, DeBiasi R, Goade DE, Haaland KY, Harrington JA, Harnar JB, Pergam SA, King MK, DeMasters BK, Tyler KL. 2006. West Nile virus neuroinvasive disease. Ann Neurol 60:286–300. doi: 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- 4.Shrestha B, Samuel MA, Diamond MS. 2006. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol 80:119–121. doi: 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shrestha B, Diamond MS. 2007. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J Virol 81:11749–11757. doi: 10.1128/JVI.01136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shrestha B, Pinto AK, Green S, Bosch I, Diamond MS. 2012. CD8+ T cells use TRAIL to restrict West Nile virus pathogenesis by controlling infection in neurons. J Virol 86:8937–8948. doi: 10.1128/JVI.00673-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bardina SV, Michlmayr D, Hoffman KW, Obara CJ, Sum J, Charo IF, Lu W, Pletnev AG, Lim JK. 2015. Differential roles of chemokines CCL2 and CCL7 in monocytosis and leukocyte migration during West Nile virus infection. J Immunol 195:4306–4318. doi: 10.4049/jimmunol.1500352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrestha B, Wang T, Samuel MA, Whitby K, Craft J, Fikrig E, Diamond MS. 2006. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J Virol 80:5338–5348. doi: 10.1128/JVI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, Gale M Jr. 2012. IL-1b signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog 8:e1003039. doi: 10.1371/journal.ppat.1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minogue AM. 2017. Role of infiltrating monocytes/macrophages in acute and chronic neuroinflammation: effects on cognition, learning, and affective behaviour. Prog Neuropsychopharmacol Biol Psychiatry 79:15–18. doi: 10.1016/j.pnpbp.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. 2014. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 83:1098–1116. doi: 10.1016/j.neuron.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 12.Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, Ji X, Leak RK, Gao Y, Chen J, Hu X. 2016. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke 47:498–504. doi: 10.1161/STROKEAHA.115.012079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biber K, Owens T, Boddeke E. 2014. What is microglia neurotoxicity (not)? Glia 62:841–854. doi: 10.1002/glia.22654. [DOI] [PubMed] [Google Scholar]
- 14.Orihuela R, McPherson CA, Harry GJ. 2016. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 173:649–665. doi: 10.1111/bph.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergold PJ. 2016. Treatment of traumatic brain injury with anti-inflammatory drugs. Exp Neurol 275:367–380. doi: 10.1016/j.expneurol.2015.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jha MK, Lee WH, Suk K. 2016. Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem Pharmacol 103:1–16. doi: 10.1016/j.bcp.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Clarke P, Leser JS, Quick ED, Dionne KR, Beckham JD, Tyler KL. 2014. Death receptor-mediated apoptotic signaling is activated in the brain following infection with West Nile virus in the absence of a peripheral immune response. J Virol 88:1080–1089. doi: 10.1128/JVI.02944-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quick ED, Leser JS, Clarke P, Tyler KL. 2014. Activation of intrinsic immune responses and microglial phagocytosis in an ex vivo spinal cord slice culture model of West Nile virus infection. J Virol 88:13005–13014. doi: 10.1128/JVI.01994-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrido-Mesa N, Zarzuelo A, Galvez J. 2013. Minocycline: far beyond an antibiotic. Br J Pharmacol 169:337–352. doi: 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li C, Yuan K, Schluesener H. 2013. Impact of minocycline on neurodegenerative disease in rodents: a meta-analysis. Rev Neurosci 24:553–562. doi: 10.1515/revneuro-2013-0040. [DOI] [PubMed] [Google Scholar]
- 21.Zhu F, Zheng Y, Liu Y, Zhang X, Zhao J. 2014. Minocycline alleviates behavioral deficits and inhibits microglial activation in the offspring of pregnant mice after administration of polyriboinosinic-polyribocytidilic acid. Psychiatry Res 219:680–686. doi: 10.1016/j.psychres.2014.06.046. [DOI] [PubMed] [Google Scholar]
- 22.Stokes JA, Arbogast TE, Moya EA, Fu Z, Powell FL. 2017. Minocycline blocks glial cell activation and ventilatory acclimatization to hypoxia. J Neurophysiol 117:1625–1635. doi: 10.1152/jn.00525.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, Hirakawa A, Takeuchi H, Suzumura A, Ishiguro N, Kadomatsu K. 2013. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis 4:e525. doi: 10.1038/cddis.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao TV, Forehand CC, Hess DC, Fagan SC. 2013. Minocycline repurposing in critical illness: focus on stroke. Curr Top Med Chem 13:2283–2290. doi: 10.2174/15680266113136660160. [DOI] [PubMed] [Google Scholar]
- 25.Markovic DS, Vinnakota K, van Rooijen N, Kiwit J, Synowitz M, Glass R, Kettenmann H. 2011. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain Behav Immun 25:624–628. doi: 10.1016/j.bbi.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Peng B, Xiao J, Wang K, So KF, Tipoe GL, Lin B. 2014. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J Neurosci 34:8139–8150. doi: 10.1523/JNEUROSCI.5200-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. 2001. Enchanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 32:1208–1215. doi: 10.1161/01.STR.32.5.1208. [DOI] [PubMed] [Google Scholar]
- 28.Gao M, Dong Q, Yao H, Zhang Y, Yang Y, Dang Y, Zhang H, Yang Z, Xu M, Xu R. 2017. Induced neural stem cells modulate microglia activation states via CXCL12/CXCR4 signaling. Brain Behav Immun 59:288–299. doi: 10.1016/j.bbi.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 29.Go M, Kou J, Lim JE, Yang J, Fukuchi KI. 2016. Microglial response to LPS increases in wild-type mice during aging but diminishes in an Alzheimer's mouse model: implication of TLR4 signaling in disease progression. Biochem Biophys Res Commun 479:331–337. doi: 10.1016/j.bbrc.2016.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori I, Imai Y, Kohsaka S, Kimura Y. 2000. Upregulated expression of Iba1 molecules in the central nervous system of mice in response to neurovirulent influenza A virus infection. Microbiol Immunol 44:729–735. doi: 10.1111/j.1348-0421.2000.tb02556.x. [DOI] [PubMed] [Google Scholar]
- 31.Piotrowska A, Kwiatkowski K, Rojewska E, Slusarczyk J, Makuch W, Basta-Kaim A, Przewlocka B, Mika J. 2016. Direct and indirect pharmacological modulation of CCL2/CCR2 pathway results in attenuation of neuropathic pain—in vivo and in vitro evidence. J Neuroimmunol 297:9–19. doi: 10.1016/j.jneuroim.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Hanson C, Cmarik JL, Ruscetti S. 2009. Neurodegeneration induced by PVC-211 murine leukemia virus is associated with increased levels of vascular endothelial growth factor and macrophage inflammatory protein 1a and is inhibited by blocking activation of microglia. J Virol 83:4912–4922. doi: 10.1128/JVI.02343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanisch UK, van Rossum D, Xie Y, Gast K, Misselwitz R, Auriola S, Goldsteins G, Koistinaho J, Kettenmann H, Moller T. 2004. The microglia-activating potential of thrombin: the protease is not involved in the induction of proinflammatory cytokines and chemokines. J Biol Chem 279:51880–51887. doi: 10.1074/jbc.M408318200. [DOI] [PubMed] [Google Scholar]
- 34.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Petersen PK. 2004. Role of microglia in central nervous system infections. Clin Microbiol Rev 17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Getts DR, Terry RL, Getts MT, Muller M, Rana S, Strestha B, Radford J, van Rooijen N, Campbell IL, King NJ. 2008. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med 205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar M, Roe K, Orillo B, Muruve DA, Nerurkar VR, Gale M Jr, Verma S. 2013. Inflammasome adaptor protein apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in West Nile virus encephalitis. J Virol 87:3655–3667. doi: 10.1128/JVI.02667-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng LS, Kaneko N, Sawamoto K. 2015. Minocycline treatment ameliorates interferon-alpha-induced neurogenic defects and depression-like behaviors in mice. Front Cell Neurosci 9:5. doi: 10.3389/fncel.2015.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levkovitch-Verbin H, Waserzoog Y, Vander S, Makarovsky D, Piven I. 2014. Minocycline upregulates pro-survival genes and downregulates pro-apoptotic genes in experimental glaucoma. Graefes Arch Clin Exp Ophthalmol 252:761–772. doi: 10.1007/s00417-014-2588-4. [DOI] [PubMed] [Google Scholar]
- 39.Hofmann U, Knorr S, Vogel B, Weirather J, Frey A, Ertl G, Frantz S. 2014. Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circ Heart Fail 7:822–830. doi: 10.1161/CIRCHEARTFAILURE.113.001020. [DOI] [PubMed] [Google Scholar]
- 40.Zhang B, Gensel JC. 2014. Is neuroinflammation in the injured spinal cord different than in the brain? Examining intrinsic differences between the brain and spinal cord. Exp Neurol 258:112–120. doi: 10.1016/j.expneurol.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 41.Kiyota T, Ingraham KL, Swan RJ, Jacobsen MT, Andrews SJ, Ikezu T. 2012. AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in AAP+PS1 mice. Gene Ther 19:724–733. doi: 10.1038/gt.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klein B, Mrowetz H, Thalhamer J, Scheiblhofer S, Weiss R, Aigner L. 2016. Allergy enhances neurogenesis and modulates microglial activation in the hippocampus. Front Cell Neurosci 10:169. doi: 10.3389/fncel.2016.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Linnartz B, Wang Y, Neumann H. 2010. Microglial immunoreceptor tyrosine-based activation and inhibition motif signaling in neuroinflammation. Int J Alzheimers Dis 2010:587463. doi: 10.4061/2010/587463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hellwig S, Heinrich A, Biber K. 2013. The brain's best friend: microglial neurotoxicity revisited. Front Cell Neurosci 7:71. doi: 10.3389/fncel.2013.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loane DJ, Kumar A. 2016. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol 275:316–327. doi: 10.1016/j.expneurol.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang W, Vodovotz Y, Kusturiss MB, Barclay D, Greenwald K, Boninger ML, Coen PM, Brienza D, Sowa G. 2014. Identification of distinct monocyte phenotypes and correlation with circulating cytokine profile in acute response to spinal cord injury: a pilot study. PM R 6:332–341. doi: 10.1016/j.pmrj.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merchant MS, Bernstein D, Amoako M, Baird K, Fleisher TA, Morre M, Steinberg SM, Sabatino M, Stroncek DF, Venkatasan AM, Wood BJ, Wright M, Zhang H, Mackall CL. 2016. Adjuvant immunotherapy to improve outcome in high-risk pediatric sarcomas. Clin Cancer Res 22:3182–3191. doi: 10.1158/1078-0432.CCR-15-2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawano T, Watanabe F, Ishiguchi M, Doe N, Furuyama T, Inagaki S. 2015. Effect of Sema4D on microglial function in middle cerebral artery occlusion mice. Glia 63:2249–2259. doi: 10.1002/glia.22890. [DOI] [PubMed] [Google Scholar]
- 49.Moro MA, Cardenas A, Hurtado O, Leza JC, Lizasoain I. 2004. Role of nitric oxide after brain ischemia. Cell Calcium 36:265–275. doi: 10.1016/j.ceca.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 50.Zhao YY, Yan DJ, Chen ZW. 2013. Role of AIF-1 in the regulation of inflammatory activation and diverse disease processes. Cell Immunol 284:75–83. doi: 10.1016/j.cellimm.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 51.Uddin MJ, Suen WW, Prow NA, Hall RA, Bielefeldt-Ohmann H. 2015. West Nile virus challenge alters the transcriptional profiles of innate immune genes in rabbit peripheral blood mononuclear cells. Front Vet Sci 2:76. doi: 10.3389/fvets.2015.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scholz R, Sobotka M, Caramoy A, Stempfl T, Moehle C, Langmann T. 2015. Minocycline counter-regulates pro-inflammatory microglia responses in the retina and protects from degeneration. J Neuroinflammation 12:209. doi: 10.1186/s12974-015-0431-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun JS, Yang YJ, Zhang YZ, Huang W, Li ZS, Zhang Y. 2015. Minocycline attenuates pain by inhibiting spinal microglia activation in diabetic rats. Mol Med Rep 12:2677–2682. doi: 10.3892/mmr.2015.3735. [DOI] [PubMed] [Google Scholar]
- 54.Morganti JM, Riparip LK, Rosi S. 2016. Call off the dog(ma): M1/M2 polarization is concurrent following traumatic brain injury. PLoS One 11:e0148001. doi: 10.1371/journal.pone.0148001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moehle MS, West AB. 2015. M1 and M2 immune activation in Parkinson's disease: foe and ally? Neuroscience 302:59–73. doi: 10.1016/j.neuroscience.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malyshev I, Malyshev Y. 2015. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage “switch” phenotype. Biomed Res Int 2015:341308. doi: 10.1155/2015/341308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ransohoff RM. 2016. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19:987–991. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- 58.Herder V, Iskandar CD, Kegler K, Hansmann F, Elmarabet SA, Khan MA, Kalkhul A, Deschl U, Baumgartner W, Ulrich R, Beineke A. 2015. Dynamic changes of microglia/macrophage M1 and M2 polarization in Theiler's murine encephalomyelitis. Brain Pathol 25:712–723. doi: 10.1111/bpa.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Das S, Dutta K, Kumawat KL, Ghoshal A, Adhya D, Basu A. 2011. Abrogated inflammatory response promotes neurogenesis in a murine model of Japanese encephalitis. PLoS One 6:e17225. doi: 10.1371/journal.pone.0017225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dutta K, Kumawat KL, Nazmi A, Mishra MK, Basu A. 2010. Minocycline differentially modulates viral infection and persistence in an experimental model of Japanese encephalitis. J Neuroimmune Pharmacol 5:553–565. doi: 10.1007/s11481-010-9233-8. [DOI] [PubMed] [Google Scholar]
- 61.Mishra MK, Basu A. 2008. Minocycline neuroprotects, reduces microglial activation, inhibits caspase 3 induction, and viral replication following Japanese encephalitis. J Neurochem 105:1582–1595. doi: 10.1111/j.1471-4159.2008.05238.x. [DOI] [PubMed] [Google Scholar]
- 62.Valero N, Mosquera J, Alcocer S, Bonilla A, Salazar J, Alvarez-Mon M. 2015. Melatonin, minocycline, and ascorbic acid reduce oxidative stress and viral titers and increase survival rate in experimental Venezuelan equine encephalitis. Brain Res 1622:368–376. doi: 10.1016/j.brainres.2015.06.034. [DOI] [PubMed] [Google Scholar]
- 63.Richardson-Burns SM, Tyler KL. 2005. Minocycline delays disease onset and mortality in reovirus encephalitis. Exp Neurol 192:331–339. doi: 10.1016/j.expneurol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 64.Qian F, Thakar J, Yuan X, Nolan M, Murray KO, Lee WT, Wong SJ, Meng H, Fikrig E, Kleinstein SH, Montgomery RR. 2014. Immune markers associated with host susceptibility to infection with West Nile virus. Viral Immunol 27:39–47. doi: 10.1089/vim.2013.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nelson J, Roe K, Orillo B, Shi PY, Verma S. 2015. Combined treatment of adenosine nucleoside inhibitor NITD008 and histone deacetylase inhibitor vorinostat represents an immunotherapy strategy to ameliorate West Nile virus infection. Antiviral Res 122:39–45. doi: 10.1016/j.antiviral.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol 79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chakraborty S, Nazmi A, Dutta K, Basu A. 2010. Neurons under viral attack: victims or warriors? Neurochem Int 56:727–735. doi: 10.1016/j.neuint.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramesh G, MacLean AG, Philipp MT. 2013. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm 2013:480739. doi: 10.1155/2013/480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Russo MV, McGavern DB. 2015. Immune surveillance of the CNS following infection and injury. Trends Immunol 36:637–650. doi: 10.1016/j.it.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Asano S, Chantler PD, Barr TL. 2016. Gene expression profiling in stroke: relevance of blood-brain interaction. Curr Opin Pharmacol 26:80–86. doi: 10.1016/j.coph.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jo M, Kim JH, Song GJ, Seo M, Hwang EM, Suk K. 2017. Astrocytic orosomucoid-2 modulates microglia activation and neuroinflammation. J Neurosci 37:2878–2894. doi: 10.1523/JNEUROSCI.2534-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Popiolek-Barczyk K, Mika J. 2016. Targeting the microglial signaling pathways: new insights in the modulation of neuropathic pain. Curr Med Chem 23:2908–2928. doi: 10.2174/0929867323666160607120124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Su Y, Wang Y, Zhou Y, Zhu Z, Zhang Q, Zhang X, Wang W, Gu X, Guo A, Wang Y. 2017. Macrophage migration inhibitory factor activates inflammatory responses of astrocytes through interaction with CD74 receptor. Oncotarget 8:2719–2730. doi: 10.18632/oncotarget.13739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brault AC, Huang CY, Langevin SA, Kinney RM, Bowen RA, Ramey WN, Panella NA, Holmes EC, Powers AM, Miller BR. 2007. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat Genet 39:1162–1166. doi: 10.1038/ng2097. [DOI] [PMC free article] [PubMed] [Google Scholar]