

ABSTRACT
NK cells are innate lymphocytes that participate in many immune processes encompassing cancer, bacterial and fungal infection, autoimmunity, and even pregnancy and that specialize in antiviral defense. NK cells express inhibitory and activating receptors and kill their targets when activating signals overpower inhibitory signals. The NK cell inhibitory receptors include a uniquely diverse array of proteins named killer cell immunoglobulin-like receptors (KIRs), the CD94 family, and the leukocyte immunoglobulin-like receptor (LIR) family. The NK cell inhibitory receptors recognize mostly major histocompatibility complex (MHC) class I (MHC-I) proteins. Zika virus has recently emerged as a major threat due to its association with birth defects and its pandemic potential. How Zika virus interacts with the immune system, and especially with NK cells, is unclear. Here we show that Zika virus infection is barely sensed by NK cells, since little or no increase in the expression of activating NK cell ligands was observed following Zika infection. In contrast, we demonstrate that Zika virus infection leads to the upregulation of MHC class I proteins and consequently to the inhibition of NK cell killing. Mechanistically, we show that MHC class I proteins are upregulated via the RIGI-IRF3 pathway and that this upregulation is mediated via beta interferon (IFN-β). Potentially, countering MHC class I upregulation during Zika virus infection could be used as a prophylactic treatment against Zika virus.
IMPORTANCE NK cells are innate lymphocytes that recognize and eliminate various pathogens and are known mostly for their role in controlling viral infections. NK cells express inhibitory and activating receptors, and they kill or spare their targets based on the integration of inhibitory and activating signals. Zika virus has recently emerged as a major threat to humans due to its pandemic potential and its association with birth defects. The role of NK cells in Zika virus infection is largely unknown. Here we demonstrate that Zika virus infection is almost undetected by NK cells, as evidenced by the fact that the expression of activating ligands for NK cells is not induced following Zika infection. We identified a mechanism whereby Zika virus sensing via the RIGI-IRF3 pathway resulted in IFN-β-mediated upregulation of MHC-I molecules and inhibition of NK cell activity. Countering MHC class I upregulation and boosting NK cell activity may be employed as prophylactic measures to combat Zika virus infection.
KEYWORDS: Zika virus, MHC class I, NK cells, NK escape mechanisms
INTRODUCTION
NK cells are lymphocytes belonging to the innate immune system. NK cells participate in many immunological activities, from killing cancerous cells (1–7) and fighting bacteria (8–10) and fungi (11) to playing roles in allergy (12) and graft-versus-host disease (13). NK cells also play roles in autoimmunity (14–18) and pregnancy (19). NK cells are known mostly for their role in controlling viral infections (20, 21) and have been shown to be involved in cytomegalovirus (CMV) (22–24), influenza virus (6, 25–28), Newcastle disease virus (29), vaccinia virus (30), and human metapneumovirus (HMPV) infections (31, 32). NK cells express a large repertoire of inhibitory receptors, which mostly recognize major histocompatibility complex class I (MHC-I) molecules (missing self hypothesis [33]). These include the killer cell immunoglobulin-like receptor (KIR) family, the CD94 family, and the leukocyte immunoglobulin-like receptor (LIR) family (34). NK cells also express inhibitory receptors that do not interact with MHC class I molecules, such as CEACAM1, which recognizes CEACAM1 in a homophilic interaction (35); CD300a, which recognizes phosphatidylserine and phosphatidylethanolamine (36, 37); and TIGIT, which recognizes PVR and several other nectin ligands (38–40).
To execute killing, NK cells use activating receptors, such as NKG2D and the natural cytotoxicity receptors (NCRs) NKp30, NKp44 and NKp46. The activating receptors recognize ligands that are induced by stress or infection, such as MICA, MICB, and ULBP1 to -6, for NKG2D (41, 42), and the hemagglutinins of various viruses for the NCRs (20). Viruses have evolved many immune evasion mechanisms to escape NK cell detection (42).
Zika virus, whose name derives from the Zika forest in Uganda, belongs to the Flaviviridae family, which also includes West Nile virus, dengue virus, yellow fever virus, and Japanese encephalitis virus (43, 44). Zika virus is found in arthropods and is transmitted primarily by the bite of the Aedes mosquito (44). Zika virus is a single-stranded, positive-sense RNA virus, encoding three structural and seven nonstructural proteins (44). Since its first discovery in the 1950s in Africa, this virus had received little attention. Recently, however, Zika virus has emerged as a global concern due to its pandemic potential and its impact on human health. Specifically, since its spread into Brazil in 2015, Zika virus has spread rapidly through vast areas of the Americas (45, 46).
In healthy individuals, Zika virus infection is mostly asymptomatic or causes a self-limited disease, rarely leading to Guillain-Barré syndrome (46). However, congenital Zika virus infection, resulting from transplacental viral transmission, has been associated with microcephaly and an expanding range of neurological abnormalities and birth defects (43, 47, 48).
How Zika virus is sensed by NK cells, and whether it evades NK cell detection, are currently unanswered questions.
RESULTS
Zika virus infection upregulates MHC class I molecules through beta interferon (IFN-β) and inhibits NK cell activity.
It is practically unknown whether and how NK cells recognize cells infected with Zika virus. To test this, we infected ARPE-19 retinal epithelium cells (ARPE cells) with Zika virus. We verified infection by measuring viral RNA accumulation in infected cells (Fig. 1A). Then we stained the infected and mock-infected ARPE cells with a panel of antibodies directed against several NK cell ligands. These include AICL, a ligand for NKp80; B7H6, a ligand for NKp30; CD48, a ligand for 2B4; CEACAM1, which interacts with itself; and MHC class I chain-related proteins A and B (MICA and -B) and UL16 binding proteins (ULBPs), ligands for NKG2D (Fig. 1B). Because the identities of the full spectrum of cellular ligands of the NCRs NKp44 and NKp46 are unknown, we used fusion proteins to detect their expression. Little or no change in the expression of the ligands tested was observed (Fig. 1B). In contrast, an increase in MHC class I expression was observed on the mRNA level (Fig. 1C) and on the cell surface, starting from day 4 postinfection (Fig. 1D).
FIG 1.

Characterization of NK ligand expression in ARPE cells following Zika virus infection. (A) Levels of Zika virus mRNA, determined by quantitative RT-PCR and normalized to β-actin levels, in mock-infected and virus-infected ARPE cells. The experiment was repeated three times. A one-tailed, heteroscedastic Student t test was used to assess significance. Values are shown as means ± standard errors of the means; *, P < 0.05. (B) FACS staining of mock-infected and Zika virus-infected ARPE cells with antibodies and fusion proteins against various NK cell ligands, as indicated. Shaded histograms represent the background control; black histograms, specific staining of mock-infected ARPE cells; red histograms, specific staining of Zika virus-infected ARPE cells. Median fluorescence intensities are given at the top right of each panel and are color-coded. The histograms combine data from three independent experiments. (C) mRNA levels of MHC class I transcripts in mock-infected and Zika virus-infected ARPE cells, quantified by qRT-PCR. The experiment was repeated three times. A one-tailed, heteroscedastic Student t test was used to assess significance. (D and E) FACS staining of mock-infected and Zika virus-infected ARPE cells with MHC class I MAb W6/32 (D) or with fusion proteins (E). Histograms and median fluorescence intensities are color-coded as explained for panel B. The histograms combine data from three independent experiments. DPI, days postinfection.
To test whether the increase in MHC class I expression observed following Zika virus infection results in increased binding of NK cell inhibitory receptors, we generated fusion proteins composed of the extracellular parts of various class I binding inhibitory receptors fused to human IgG1.
As can be seen in Fig. 1E, the Zika virus-induced upregulation of MHC class I expression resulted in increased binding of KIR2DL2 Ig, which binds HLA-C proteins carrying asparagine at position 80 (49), and LILRB1 Ig, which interacts with HLA-A, -B, and -C proteins (50).
To gain insight into the mechanism by which Zika virus causes the upregulation of MHC class I proteins, we first used quantitative reverse transcription-PCR (qRT-PCR) to assess type I interferons in infected cells. Significant induction of IFN-β expression, but not of IFN-α expression, was observed following infection (Fig. 2A). To test whether IFN-β is involved in the upregulation of MHC class I proteins, we used blocking antibodies against IFN-β. Such blocking significantly reduced the Zika virus-mediated upregulation of MHC class I proteins (detected by W6/32), KIR2DL2 Ig, and LILRB1 Ig, almost to the same level as that in mock-infected cells (Fig. 2B).
FIG 2.

MHC class I upregulation upon Zika virus infection is mediated by IFN-β. (A) mRNA levels of different interferons in mock-infected and Zika virus-infected ARPE cells, quantified by qRT-PCR and normalized to β-actin mRNA levels. The experiment was repeated three times. Analysis of variance was used to identify significant group differences, followed by Fisher protected least-significant-difference comparisons. Values are shown as means ± standard errors of the means. NS, not significant; *, P < 0.05. (B) FACS staining of Zika virus-infected ARPE cells with an MHC class I MAb (W6/32) and fusion proteins, with or without blocking antibodies. Median fluorescence intensities are given at the top right of each panel and are color-coded. The histograms represent two independent experiments. (C) Primary IL-2-activated NK cells were incubated with the indicated targets at a 1:1 ratio. CD107a degranulation (as a percentage of the value for the no-target baseline) was assessed. The experiment was conducted three independent times and included NK cells obtained from five independent donors. (D) Primary IL-2-activated NK cells were incubated with the indicated targets at a 1:1 ratio, with or without blocking antibodies against IFN-αβ receptors that were included during the infection period. CD107a degranulation (as a percentage of the value for the no-target baseline) was assessed. The experiment was conducted twice and included five independent NK cell donors. For panels C and D, significant differences were determined as described for panel A.
Next, we wanted to test whether the IFN-β-mediated increase in MHC class I expression leads to inhibition of NK cell activity. To this end, we used primary interleukin 2 (IL-2)-activated NK cells, propagated from several independent donors, and observed significantly lower levels of CD107a degranulation following incubation with Zika virus-infected ARPE cells (Fig. 2C). This effect was abolished when blocking antibodies against IFN-β were used during Zika virus infection (Fig. 2D).
Zika virus-mediated upregulation of MHC class I molecules is not restricted to a particular cell line.
To test whether similar findings are observed in additional cells, we infected A549 lung carcinoma cells with Zika virus and verified the infection by determining viral RNA accumulation in infected cells (Fig. 3A). As with ARPE cells, Zika virus infection of A549 cells had little or no effect on the expression of any of the NK ligands tested (Fig. 3B), except for MHC class I molecules and the inhibitory protein CEACAM1 (Fig. 3B); the latter was not expressed on ARPE cells (Fig. 1). Accordingly, the activation of NK cells, as measured by CD107a degranulation, was significantly inhibited by incubation with Zika virus-infected A549 cells (Fig. 3C).
FIG 3.

Zika virus-mediated upregulation of MHC class I molecules is recapitulated in the A549 cell model and in primary human foreskin fibroblasts (HFF). (A) Zika virus mRNA levels in mock-infected and Zika virus-infected A549 cells, quantified by qRT-PCR and normalized to β-actin mRNA levels. The experiment was repeated three times. A one-tailed, heteroscedastic Student t test was used to assess significance. Values are shown as means ± standard errors of the means; *, P < 0.05. (B) FACS staining of mock-infected and Zika virus-infected A549 cells for various NK cell ligands, as indicated. Gray histograms represent the background control; black and red histograms, specific staining of mock-infected and Zika virus-infected A549 cells, respectively. For ligands for which significant changes were observed following infection, median fluorescence intensities are indicated at the top (color-coded). The histograms combine data from three independent experiments. (C) Primary IL-2-activated NK cells were incubated with the indicated targets at a 1:1 ratio. CD107a degranulation (as a percentage of the value for the no-target baseline) was assessed. The experiment was conducted twice and included four independent NK cell donors. Analysis of variance was used to identify significant group differences, followed by Fisher protected least-significant-difference comparisons. (D) Levels of Zika virus mRNA, determined by quantitative RT-PCR and normalized to β-actin levels, in mock-infected and Zika virus-infected HFF. A one-tailed, heteroscedastic Student t test was used to assess significance. (E) FACS staining of mock-infected and Zika virus-infected HFF. Black and red histograms represent specific staining of mock-infected and Zika virus-infected HFF, respectively. Median fluorescence intensities are indicated at the upper right and are color-coded. (F) IFN-β mRNA levels, quantified by qRT-PCR and normalized to β-actin mRNA levels, in mock-infected and Zika virus-infected HFF.
To test whether similar findings are observed in primary cells, we infected primary human foreskin fibroblasts (HFF) with Zika virus. We verified the infection (Fig. 3D) and observed upregulation of MHC-I molecules (Fig. 3E) and IFN-β levels (Fig. 3F) in the infected cells.
Characterization of interactions between Zika virus-infected cells and NK cells.
Next, we wanted to study the kinetics of clearance of Zika virus-infected targets by NK cells and the NK cell response in terms of cytokine secretion. To this end, we infected ARPE and A549 cells with Zika virus and incubated NK cells with the mock-infected and Zika virus-infected cells at a ratio of 1:1. Figure 4A shows the ratios of NK cells to ARPE cells at different time points. While mock-infected ARPE cells, which express several NK cell activating ligands (Fig. 1 and 3), were killed efficiently by NK cells (the ratio of NK cells to target cells was significantly increased [Fig. 4A, black lines]), Zika virus-infected cells were unaffected over time, indicating that they were protected from NK cell killing (Fig. 4A, red lines). In parallel, cytokine secretion by the NK cells was assessed at each time point. We focused on IFN-γ and tumor necrosis factor alpha (TNF-α), since these are the main NK cell cytokines. TNF-α secretion did not exceed baseline levels, irrespective of Zika virus infection (data not shown). Surprisingly we observed a strong, time-dependent secretion of IFN-γ from NK cells incubated with Zika virus-infected cells, as early as 5 h following coincubation (Fig. 4B). Very low levels of IFN-γ were secreted by NK cells that were incubated with mock-infected cells, probably because the mock-infected cells had already been eliminated by the NK cells after 5 h of incubation (Fig. 4A). Therefore, the NK cells cannot continuously secrete cytokines. Similar results, both in direct killing (Fig. 4C) and in cytokine secretion (Fig. 4D), were observed when mock-infected and Zika virus-infected A549 cells were used.
FIG 4.
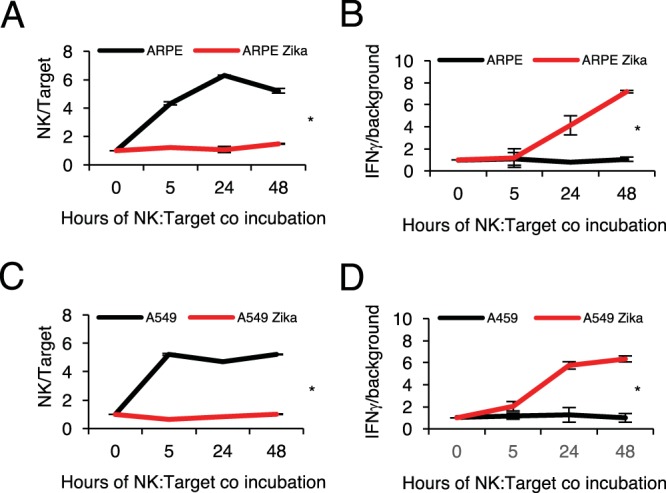
Characterization of interactions of Zika virus-infected cells with NK cells. (A and C) NK cells were incubated with target cells at a ratio of 1:1. Target cell and NK cell numbers were assessed by FACS at different time points (as indicated), and the NK/target ratio is presented. (B and D) IFN-γ secretion was assessed under the same conditions as those described for panels A and C using standard ELISA. A repeated-measures Student t test was used to assess significance. Results for one NK line representative of the three used are shown. Values are means ± standard errors of the means; *, P < 0.05.
MHC class I upregulation in Zika virus-infected cells is mediated via the RIGI-IRF3 pathway.
We have established previously that the IFI16 sensor, which senses DNA, and the RIGI sensor, which senses RNA, are involved in CEACAM1 upregulation following HCMV, HMPV, and influenza virus infection (32, 51). Because we observed CEACAM1 upregulation in A549 cells, and because Zika virus is a single-stranded, positive-sense RNA virus, we suspected that Zika virus is sensed by the RIGI-IRF3 pathway and that this leads to IFN-β induction and MHC class I upregulation. To test this, we used short hairpin RNA (shRNA) to knock down (KD) RIGI and IRF3. Scrambled shRNA (Scramble) was used as a control. KD evaluation using qRT-PCR was performed before (data not shown) and after infection and was found comparable for infected and uninfected cells (Fig. 5A and B show IRF3 and RIGI expression levels in IRF3 KD and RIGI KD infected cells, respectively, compared to those in infected cells with scrambled shRNA). We next infected the Scramble, RIGI KD, and IRF3 KD A549 cells with Zika virus and verified the infection in all cells, using qRT-PCR (Fig. 5C [please note that the values for uninfected cells are zero]). We observed that the infection levels of all cells were similar. As described above, little or no change in NK cell ligand expression was observed in the various A549 lines upon infection with Zika virus (Fig. 5D). In contrast, the induction of CEACAM1 and the upregulation of MHC class I molecules that are observed following Zika virus infection were abrogated following RIGI KD and IRF3 KD (Fig. 6A and B).
FIG 5.

Expression of NK cell activating ligands following KD of IRF3 and RIGI. (A and B) mRNA levels, quantified by qRT-PCR, of IRF3 (A) and RIGI (B) in A549 cells transduced with the indicated shRNA or a scrambled sequence. A one-tailed, heteroscedastic Student t test was used to assess significance. (C) mRNA levels of Zika virus transcripts, quantified by qRT-PCR and normalized to β-actin mRNA levels, in mock-infected or Zika virus-infected IRF3 KD and RIGI KD A549 cells. Analysis of variance was used to identify significant group differences, followed by Fisher protected least-significant-difference comparisons. For panels A to C, the experiment was repeated three times. Values are shown as means ± standard errors of the means; *, P < 0.05. (D) FACS staining of mock-infected and Zika virus-infected Scramble, IFR3 KD, and RIGI KD A549 cells with various NK cell ligands, as indicated. Gray histograms represent background control staining; black and red histograms, specific staining of mock-infected and Zika virus-infected A549 cells, respectively. The histograms combine data from three independent experiments.
FIG 6.

Zika virus sensing is mediated by IRF3 and RIGI. (A and B) FACS staining of mock-infected and Zika virus-infected Scramble, IFR3 KD, and RIGI KD A549 cells with CEACAM1 (A) or MHC class I molecules (B). The black Scramble histograms in the IRF3 KD and RIGI KD panels are identical and are shown twice for the sake of clarity. Median fluorescence intensities are given at the top right and are color-coded. The histograms combine data from three independent experiments. (C to E) mRNA levels, quantified by qRT-PCR, of CEACAM1 (C), MHC class I (D), and IFN-β (E) in IFR3 KD and RIGI KD A549 cells, compared to levels in A549 cells transduced with a scrambled sequence (C and D) or normalized to β-actin mRNA levels (E). Each experiment was repeated three times. (F) Primary IL-2-activated NK cells were incubated with the indicated targets at a 1:1 ratio. CD107a degranulation (as a percentage of the baseline value) was assessed. The experiment was conducted twice and included four independent NK donors. For panels C to F, analysis of variance was used to identify significant group differences, followed by Fisher protected least-significant-difference comparisons. Values are shown as means ± standard errors of the means; *, P < 0.05.
In agreement with these results, the mRNA level of CEACAM1 (which was increased following Zika virus infection), remained unchanged in the RIGI KD and IRF3 KD A549 cells (Fig. 6C). The increase in MHC class I expression that was observed following Zika virus infection was smaller when RIGI and IRF3 were knocked down (Fig. 6D). Furthermore, the level of IFN-β mRNA induction upon infection was decreased following RIGI and IRF3 KD (Fig. 6E). Finally, the inhibition of CD107a degranulation that was observed when primary IL-2-activated NK cells were incubated with Zika virus-infected Scramble A549 cells was not observed when NK cells were incubated with Zika virus-infected IRF3 KD or RIGI KD A549 cells (Fig. 6F).
DISCUSSION
Flaviviruses were long ago shown to upregulate MHC class I molecules during infection, in part by inducing IFN-β expression in mice (52) and humans (53), and it was further demonstrated that this process is driven by NF-κB (54). Elevation of MHC class I expression resulted in enhanced T cell lysis (55) and inhibition of NK cell killing (56). Here we show that similar mechanisms are utilized by Zika virus.
In view of the recent emergence and rapid spread of the Zika virus epidemic and the severe consequences of congenital Zika virus infection, it is crucial to learn how Zika virus interacts with the innate and adaptive arms of the immune system. Indeed, several works have already investigated the recognition of Zika virus by T cells (57, 58), and others have shown that Zika virus infection can induce type I IFNs and downstream interferon-stimulated genes (ISGs). Zika virus infection of human skin fibroblasts induced the transcription of Toll-like receptor 3 (TLR3), RIGI, and MDA5, as well as other ISGs, such as OAS2, ISG15, and MX1 (59). Zika virus infection was also shown to induce a type I interferon response in human placental macrophages and placental tissues (48, 60). Moreover, recent studies have demonstrated the importance of the interferon response in modulating Zika virus infection and disease outcome (61–64).
In response to these mechanisms, Zika virus inhibits RIGI and type I IFN signaling in dendritic cells (DCs) by antagonizing type I IFN-mediated phosphorylation of STAT1 and STAT2 (65). Another study showed that Zika virus infection impairs the induction of type I IFNs and that the viral NS5 protein mediates the degradation of STATs in the proteasome (66). Zika virus nonstructural proteins NS1, NS4B, and NS2B3 have been found to inhibit the induction of IFN downstream ISGs through several pathways, including inhibition of IFN-β and JAK STAT signaling (67).
In this study, we observed that Zika virus infection is sensed by RIGI and IRF3, which lead to type I IFN secretion, and that this results in MHC class I and CEACAM1 upregulation. Blocking IFN-β significantly reduced MHC-I upregulation. Thus, despite the mechanisms developed by the virus to avoid sensing, it is still sensed by the host to a certain extent. We propose that Zika virus does not completely shut down sensing but rather exploits the interferon response, because it is beneficial for the virus to upregulate MHC class I proteins and to inhibit NK cell activity (it is still possible that other pathways of upregulation may exist, as has been shown previously for West Nile virus [68]). This idea is further supported by the observation of increased IFN-γ secretion by NK cells following their interaction with Zika virus-infected cells. IFN-γ secretion by NK cells in the presence of Zika virus-infected cells probably contributes to the ability of target cells to upregulate MHC class I molecules and escape direct killing by NK cells.
The fact that MHC class I molecules were significantly upregulated indicates that the virus escape mechanisms are directed mainly against NK cells, with little regard for avoiding T cell detection. We think that the virus did not evolve mechanisms to inhibit T cell activity because of its rapid replication kinetics. T cells are simply not fast enough to react. NK cells, on the other hand, respond within hours, and therefore, the virus evolved mechanisms to inhibit their activity.
Thus, developing a vaccine against the virus that can activate T cells is desirable, since MHC class I molecules are upregulated. Indeed, breakthroughs have been achieved in the pursuit of a vaccine against Zika virus (69, 70). However, we propose that for Zika virus treatment, once patients are infected, a therapeutic approach directed to boosting NK cell activity should be employed.
MATERIALS AND METHODS
Cells and Zika virus infection.
Vero cells, ARPE-19 retinal epithelium cells, and A549 lung carcinoma cells were obtained from the American Type Culture Collection. Primary human foreskin fibroblasts (HFF) were provided by A. Lifshitz, Hadassah Medical Center Clinical Virology Laboratory. Vero cells were used for Zika virus propagation. Cells were infected with Zika virus strain PRVABC59, a low-passage-number, sequence-verified strain isolated from an infected patient in Puerto Rico in December 2015 (generously provided by S. Lanciotti, U.S. Centers for Disease Control and Prevention), at a multiplicity of infection (MOI) of 0.01. At 2 days postinfection (dpi), supernatants were collected, centrifuged to remove cellular debris, and frozen at 80°C. Virus titers were determined by a standard plaque assay.
ARPE-19 and A549 cells and HFF were infected at an MOI of 1 and were harvested upon the appearance of a viral cytopathic effect, at 4, 3, and 6 dpi, respectively.
Antibodies and fusion proteins.
Fusion proteins were generated in HEK 293T cells, as described previously (4). The staining by all fusion proteins was visualized using a secondary allophycocyanin (APC)-conjugated goat anti-human antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). Secondary anti rabbit or anti-mouse antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) were used to visualize staining with the antibodies used in this study.
CD107a degranulation, killing, and cytokine secretion assays.
Primary IL-2-activated NK cells were coincubated with the target cells at a ratio of 1:1 at 37°C. For CD107a degranulation, the assay was conducted in the presence of an APC-conjugated CD107a antibody and a CD56 antibody (both from Biotest, Israel) for 2 h. CD107a levels in NK cells were determined by flow cytometry. To determine killing and proliferation over time, targets were irradiated (6,000 rads) prior to coincubation with the NK cells. To determine NK cell and target cell numbers, fluorescence-activated cell sorter (FACS) staining was used. Cells (NK and target) were distinguished based on forward- and side-scatter profiles over the time points indicated in the figures, and cell numbers were assessed in a 30-s reading time frame. Cytokines were measured using a standard enzyme-linked immunosorbent assay (ELISA) (Biolegend, Petach Tikvah, Israel).
RNA purification and quantification.
Infected and mock-infected cells were washed, harvested in lysis buffer, and stored at −80°C until assayed. DNA-free RNA was extracted (NucleoSpin RNA isolation kit; Macherey-Nagel) and subjected to reverse transcription (GoScript; ABI), followed by qRT-PCR (on a 7900HT system; Applied Biosystems). Zika virus oligonucleotide sequences were as follows: forward, 5′-CCACTAACGTTCTTTTGCAGACAT-3′; reverse, 5′-CCGCTGCCCAACACAAG-3′; probe, 5′-6-carboxyfluorescein (6-FAM)/AG CCT ACC T/ZEN/T GAC AAG CAG TCA GAC ACT CA (48)A/3′-Iowa Black fluorescence quencher (IABkFQ). β-Actin oligonucleotide sequences were as follows: forward, 5′-CCTGGCACCCAGCACAAT-3′; reverse, 5′-GCCGATCCACACGGAGTACT-3′; probe, 5′-6-FAM/AT CAA GAT C/ZEN/A TTG CTC CTC CTG AGC GC/3′-IABkFQ. Interferon (IFN) oligonucleotide sequences were as follows: for IFN-α, 5′-GGCTGTGAGGAAATACTTCCAAAGAA-3′ (forward) and 5′-GATCTCATGATTTCTGCTCTGACAAC-3′ (reverse); for IFN-β, 5′-TGGGAGGCTTGAATACTGCCTCAA-3′ (forward) and 5′-TGCGGCGTCCTCCTTCTGGA-3′ (reverse).
For CEACAM1 and MHC-I qRT PCR, mRNA was isolated using an R1055 Quick-RNA MiniPrep kit (Eisenberg Bros., Israel) and was reverse transcribed to cDNA using Moloney murine leukemia virus (M-MLV) reverse transcriptase (catalog no. 28025-013; Invitrogen, Thermo Fisher Scientific, MA). qRT-PCR was performed with Platinum SYBR green qPCR SuperMix-UDG w/ROX (Thermo Fisher Scientific) in triplicate, normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and ACTB. Primer sequences may be found in reference 51.
ACKNOWLEDGMENTS
This work was supported by the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013) (ERC grant agreement 320473-BacNK). Further support came from the Israel Science Foundation, the GIF, the ICRF professorship grant, the Helmholtz Foundation, and the Rosetrees Trust (all to O.M.). O.M. is a Crown Professor of Molecular Immunology. The work was also supported by grants from the Israel Science Foundation and the European Union Seventh Framework Programme (562 FP7/2012–2016) (grant agreement 316655) to D.G.W.
REFERENCES
- 1.Elboim M, Gazit R, Gur C, Ghadially H, Betser-Cohen G, Mandelboim O. 2010. Tumor immunoediting by NKp46. J Immunol 184:5637–5644. doi: 10.4049/jimmunol.0901644. [DOI] [PubMed] [Google Scholar]
- 2.Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, Enk J, Mandelboim O. 2012. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol 188:2509–2515. doi: 10.4049/jimmunol.1102461. [DOI] [PubMed] [Google Scholar]
- 3.Glasner A, Isaacson B, Mandelboim O. 2017. Expression and function of NKp46 W32R: the human homologous protein of mouse NKp46 W32R (Noe). Sci Rep 7:40944. doi: 10.1038/srep40944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glasner A, Roth Z, Varvak A, Miletic A, Isaacson B, Bar-On Y, Jonjic S, Khalaila I, Mandelboim O. 2015. Identification of putative novel O-glycosylations in the NK killer receptor Ncr1 essential for its activity. Cell Discov 1:15036. doi: 10.1038/celldisc.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glasner A, Simic H, Miklic K, Roth Z, Berhani O, Khalaila I, Jonjic S, Mandelboim O. 2015. Expression, function, and molecular properties of the killer receptor Ncr1-Noe. J Immunol 195:3959–3969. doi: 10.4049/jimmunol.1501234. [DOI] [PubMed] [Google Scholar]
- 6.Glasner A, Zurunic A, Meningher T, Lenac Rovis T, Tsukerman P, Bar-On Y, Yamin R, Meyers AF, Mandeboim M, Jonjic S, Mandelboim O. 2012. Elucidating the mechanisms of influenza virus recognition by Ncr1. PLoS One 7:e36837. doi: 10.1371/journal.pone.0036837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halfteck GG, Elboim M, Gur C, Achdout H, Ghadially H, Mandelboim O. 2009. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J Immunol 182:2221–2230. doi: 10.4049/jimmunol.0801878. [DOI] [PubMed] [Google Scholar]
- 8.Chaushu S, Wilensky A, Gur C, Shapira L, Elboim M, Halftek G, Polak D, Achdout H, Bachrach G, Mandelboim O. 2012. Direct recognition of Fusobacterium nucleatum by the NK cell natural cytotoxicity receptor NKp46 aggravates periodontal disease. PLoS Pathog 8:e1002601. doi: 10.1371/journal.ppat.1002601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gur C, Coppenhagen-Glazer S, Rosenberg S, Yamin R, Enk J, Glasner A, Bar-On Y, Fleissig O, Naor R, Abed J, Mevorach D, Granot Z, Bachrach G, Mandelboim O. 2013. Natural killer cell-mediated host defense against uropathogenic E. coli is counteracted by bacterial hemolysin A-dependent killing of NK cells. Cell Host Microbe 14:664–674. doi: 10.1016/j.chom.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, Shussman N, Almogy G, Cuapio A, Hofer E, Mevorach D, Tabib A, Ortenberg R, Markel G, Miklic K, Jonjic S, Brennan CA, Garrett WS, Bachrach G, Mandelboim O. 2015. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42:344–355. doi: 10.1016/j.immuni.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vitenshtein A, Charpak-Amikam Y, Yamin R, Bauman Y, Isaacson B, Stein N, Berhani O, Dassa L, Gamliel M, Gur C, Glasner A, Gomez C, Ben-Ami R, Osherov N, Cormack BP, Mandelboim O. 2016. NK cell recognition of Candida glabrata through binding of NKp46 and NCR1 to fungal ligands Epa1, Epa6, and Epa7. Cell Host Microbe 20:527–534. doi: 10.1016/j.chom.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghadially H, Horani A, Glasner A, Elboim M, Gazit R, Shoseyov D, Mandelboim O. 2013. NKp46 regulates allergic responses. Eur J Immunol 43:3006–3016. doi: 10.1002/eji.201343388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghadially H, Ohana M, Elboim M, Gazit R, Gur C, Nagler A, Mandelboim O. 2014. NK cell receptor NKp46 regulates graft-versus-host disease. Cell Rep 7:1809–1814. doi: 10.1016/j.celrep.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gur C, Doron S, Kfir-Erenfeld S, Horwitz E, Abu-Tair L, Safadi R, Mandelboim O. 2012. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut 61:885–893. doi: 10.1136/gutjnl-2011-301400. [DOI] [PubMed] [Google Scholar]
- 15.Gur C, Enk J, Kassem SA, Suissa Y, Magenheim J, Stolovich-Rain M, Nir T, Achdout H, Glaser B, Shapiro J, Naparstek Y, Porgador A, Dor Y, Mandelboim O. 2011. Recognition and killing of human and murine pancreatic β cells by the NK receptor NKp46. J Immunol 187:3096–3103. doi: 10.4049/jimmunol.1101269. [DOI] [PubMed] [Google Scholar]
- 16.Gur C, Enk J, Weitman E, Bachar E, Suissa Y, Cohen G, Schyr RB, Sabanay H, Horwitz E, Glaser B, Dor Y, Pribluda A, Hanna JH, Leibowitz G, Mandelboim O. 2013. The expression of the beta cell-derived autoimmune ligand for the killer receptor NKp46 is attenuated in type 2 diabetes. PLoS One 8:e74033. doi: 10.1371/journal.pone.0074033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gur C, Porgador A, Elboim M, Gazit R, Mizrahi S, Stern-Ginossar N, Achdout H, Ghadially H, Dor Y, Nir T, Doviner V, Hershkovitz O, Mendelson M, Naparstek Y, Mandelboim O. 2010. The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol 11:121–128. doi: 10.1038/ni.1834. [DOI] [PubMed] [Google Scholar]
- 18.Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Stimac D, Wunderlich FT, Bruning JC, Mandelboim O, Polic B. 2015. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol 16:376–385. doi: 10.1038/ni.3120. [DOI] [PubMed] [Google Scholar]
- 19.Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, Gazit R, Yutkin V, Benharroch D, Porgador A, Keshet E, Yagel S, Mandelboim O. 2006. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med 12:1065–1074. doi: 10.1038/nm1452. [DOI] [PubMed] [Google Scholar]
- 20.Koch J, Steinle A, Watzl C, Mandelboim O. 2013. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol 34:182–191. doi: 10.1016/j.it.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Lam VC, Lanier LL. 2017. NK cells in host responses to viral infections. Curr Opin Immunol 44:43–51. doi: 10.1016/j.coi.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babic M, Pyzik M, Zafirova B, Mitrovic M, Butorac V, Lanier LL, Krmpotic A, Vidal SM, Jonjic S. 2010. Cytomegalovirus immunoevasin reveals the physiological role of “missing self” recognition in natural killer cell dependent virus control in vivo. J Exp Med 207:2663–2673. doi: 10.1084/jem.20100921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nachmani D, Zimmermann A, Oiknine Djian E, Weisblum Y, Livneh Y, Khanh Le VT, Galun E, Horejsi V, Isakov O, Shomron N, Wolf DG, Hengel H, Mandelboim O. 2014. MicroRNA editing facilitates immune elimination of HCMV infected cells. PLoS Pathog 10:e1003963. doi: 10.1371/journal.ppat.1003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seidel E, Le VT, Bar-On Y, Tsukerman P, Enk J, Yamin R, Stein N, Schmiedel D, Oiknine Djian E, Weisblum Y, Tirosh B, Stastny P, Wolf DG, Hengel H, Mandelboim O. 2015. Dynamic co-evolution of host and pathogen: HCMV downregulates the prevalent allele MICA *008 to escape elimination by NK cells. Cell Rep doi: 10.1016/j.celrep.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnon TI, Achdout H, Lieberman N, Gazit R, Gonen-Gross T, Katz G, Bar-Ilan A, Bloushtain N, Lev M, Joseph A, Kedar E, Porgador A, Mandelboim O. 2004. The mechanisms controlling the recognition of tumor- and virus-infected cells by NKp46. Blood 103:664–672. doi: 10.1182/blood-2003-05-1716. [DOI] [PubMed] [Google Scholar]
- 26.Bar-On Y, Glasner A, Meningher T, Achdout H, Gur C, Lankry D, Vitenshtein A, Meyers AF, Mandelboim M, Mandelboim O. 2013. Neuraminidase-mediated, NKp46-dependent immune-evasion mechanism of influenza viruses. Cell Rep 3:1044–1050. doi: 10.1016/j.celrep.2013.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bar-On Y, Seidel E, Tsukerman P, Mandelboim M, Mandelboim O. 2014. Influenza virus uses its neuraminidase protein to evade the recognition of two activating NK cell receptors. J Infect Dis 210:410–418. doi: 10.1093/infdis/jiu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, Davis DM, Strominger JL, Yewdell JW, Porgador A. 2001. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409:1055–1060. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- 29.Jarahian M, Watzl C, Fournier P, Arnold A, Djandji D, Zahedi S, Cerwenka A, Paschen A, Schirrmacher V, Momburg F. 2009. Activation of natural killer cells by Newcastle disease virus hemagglutinin-neuraminidase. J Virol 83:8108–8121. doi: 10.1128/JVI.00211-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarahian M, Fiedler M, Cohnen A, Djandji D, Hammerling GJ, Gati C, Cerwenka A, Turner PC, Moyer RW, Watzl C, Hengel H, Momburg F. 2011. Modulation of NKp30- and NKp46-mediated natural killer cell responses by poxviral hemagglutinin. PLoS Pathog 7:e1002195. doi: 10.1371/journal.ppat.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diab M, Glasner A, Issacson B, Bar-On Y, Drori Y, Yamin R, Duev-Cohen A, Danziger O, Zamostiano R, Mandelboim M, Jonjic S, Bacharach E, Mandelboim O. 2017. NK-cell receptors NKp46 and NCR1 control human metapneumovirus infection. Eur J Immunol doi: 10.1002/eji.201646756. [DOI] [PubMed] [Google Scholar]
- 32.Diab M, Vitenshtein A, Drori Y, Yamin R, Danziger O, Zamostiano R, Mandelboim M, Bacharach E, Mandelboim O. 2016. Suppression of human metapneumovirus (HMPV) infection by the innate sensing gene CEACAM1. Oncotarget doi: 10.18632/oncotarget.11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kärre K. 2002. NK cells, MHC class I molecules and the missing self. Scand J Immunol 55:221–228. doi: 10.1046/j.1365-3083.2002.01053.x. [DOI] [PubMed] [Google Scholar]
- 34.Vilches C, Parham P. 2002. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol 20:217–251. doi: 10.1146/annurev.immunol.20.092501.134942. [DOI] [PubMed] [Google Scholar]
- 35.Markel G, Lieberman N, Katz G, Arnon TI, Lotem M, Drize O, Blumberg RS, Bar-Haim E, Mader R, Eisenbach L, Mandelboim O. 2002. CD66a interactions between human melanoma and NK cells: a novel class I MHC-independent inhibitory mechanism of cytotoxicity. J Immunol 168:2803–2810. doi: 10.4049/jimmunol.168.6.2803. [DOI] [PubMed] [Google Scholar]
- 36.Cantoni C, Bottino C, Augugliaro R, Morelli L, Marcenaro E, Castriconi R, Vitale M, Pende D, Sivori S, Millo R, Biassoni R, Moretta L, Moretta A. 1999. Molecular and functional characterization of IRp60, a member of the immunoglobulin superfamily that functions as an inhibitory receptor in human NK cells. Eur J Immunol 29:3148–3159. doi:. [DOI] [PubMed] [Google Scholar]
- 37.Lankry D, Simic H, Klieger Y, Levi-Schaffer F, Jonjic S, Mandelboim O. 2010. Expression and function of CD300 in NK cells. J Immunol 185:2877–2886. doi: 10.4049/jimmunol.0903347. [DOI] [PubMed] [Google Scholar]
- 38.Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, Enk J, Jonjic S, Mandelboim O. 2013. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol 43:2138–2150. doi: 10.1002/eji.201243072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, Stern-Ginossar N, Tsukerman P, Jonjic S, Mandelboim O. 2009. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A 106:17858–17863. doi: 10.1073/pnas.0903474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, Eaton D, Grogan JL. 2009. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol 10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 41.Raulet DH. 2003. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 42.Seidel E, Glasner A, Mandelboim O. 2012. Virus-mediated inhibition of natural cytotoxicity receptor recognition. Cell Mol Life Sci 69:3911–3920. doi: 10.1007/s00018-012-1001-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lazear HM, Diamond MS. 2016. Zika virus: new clinical syndromes and its emergence in the Western Hemisphere. J Virol 90:4864–4875. doi: 10.1128/JVI.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Musso D, Gubler DJ. 2016. Zika virus. Clin Microbiol Rev 29:487–524. doi: 10.1128/CMR.00072-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brasil P, Pereira JP Jr, Moreira ME, Ribeiro Nogueira RM, Damasceno L, Wakimoto M, Rabello RS, Valderramos SG, Halai UA, Salles TS, Zin AA, Horovitz D, Daltro P, Boechat M, Raja Gabaglia C, Carvalho de Sequeira P, Pilotto JH, Medialdea-Carrera R, Cotrim da Cunha D, Abreu de Carvalho LM, Pone M, Machado Siqueira A, Calvet GA, Rodrigues Baiao AE, Neves ES, Nassar de Carvalho PR, Hasue RH, Marschik PB, Einspieler C, Janzen C, Cherry JD, Bispo de Filippis AM, Nielsen-Saines K. 2016. Zika virus infection in pregnant women in Rio de Janeiro. N Engl J Med 375:2321–2334. doi: 10.1056/NEJMoa1602412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parra B, Lizarazo J, Jimenez-Arango JA, Zea-Vera AF, Gonzalez-Manrique G, Vargas J, Angarita JA, Zuniga G, Lopez-Gonzalez R, Beltran CL, Rizcala KH, Morales MT, Pacheco O, Ospina ML, Kumar A, Cornblath DR, Munoz LS, Osorio L, Barreras P, Pardo CA. 2016. Guillain-Barré syndrome associated with Zika virus infection in Colombia. N Engl J Med 375:1513–1523. doi: 10.1056/NEJMoa1605564. [DOI] [PubMed] [Google Scholar]
- 47.Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. 2016. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med 374:1981–1987. doi: 10.1056/NEJMsr1604338. [DOI] [PubMed] [Google Scholar]
- 48.Weisblum Y, Oiknine-Djian E, Vorontsov OM, Haimov-Kochman R, Zakay-Rones Z, Meir K, Shveiky D, Elgavish S, Nevo Y, Roseman M, Bronstein M, Stockheim D, From I, Eisenberg I, Lewkowicz AA, Yagel S, Panet A, Wolf DG. 2017. Zika virus infects early- and midgestation human maternal decidual tissues, inducing distinct innate tissue responses in the maternal-fetal interface. J Virol 91:e01905-16. doi: 10.1128/JVI.01905-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mandelboim O, Reyburn HT, Vales-Gomez M, Pazmany L, Colonna M, Borsellino G, Strominger JL. 1996. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J Exp Med 184:913–922. doi: 10.1084/jem.184.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirayasu K, Arase H. 2015. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. J Hum Genet 60:703–708. doi: 10.1038/jhg.2015.64. [DOI] [PubMed] [Google Scholar]
- 51.Vitenshtein A, Weisblum Y, Hauka S, Halenius A, Oiknine-Djian E, Tsukerman P, Bauman Y, Bar-On Y, Stern-Ginossar N, Enk J, Ortenberg R, Tai J, Markel G, Blumberg RS, Hengel H, Jonjic S, Wolf DG, Adler H, Kammerer R, Mandelboim O. 2016. CEACAM1-mediated inhibition of virus production. Cell Rep 15:2331–2339. doi: 10.1016/j.celrep.2016.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King NJ, Kesson AM. 1988. Interferon-independent increases in class I major histocompatibility complex antigen expression follow flavivirus infection. J Gen Virol 69(Part 10):2535–2543. doi: 10.1099/0022-1317-69-10-2535. [DOI] [PubMed] [Google Scholar]
- 53.Shen J, T-To SS, Schrieber L, King NJ. 1997. Early E-selectin, VCAM-1, ICAM-1, and late major histocompatibility complex antigen induction on human endothelial cells by flavivirus and comodulation of adhesion molecule expression by immune cytokines. J Virol 71:9323–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kesson AM, King NJ. 2001. Transcriptional regulation of major histocompatibility complex class I by flavivirus West Nile is dependent on NF-κB activation. J Infect Dis 184:947–954. doi: 10.1086/323603. [DOI] [PubMed] [Google Scholar]
- 55.Liu Y, King N, Kesson A, Blanden RV, Mullbacher A. 1989. Flavivirus infection up-regulates the expression of class I and class II major histocompatibility antigens on and enhances T cell recognition of astrocytes in vitro. J Neuroimmunol 21:157–168. doi: 10.1016/0165-5728(89)90171-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Momburg F, Mullbacher A, Lobigs M. 2001. Modulation of transporter associated with antigen processing (TAP)-mediated peptide import into the endoplasmic reticulum by flavivirus infection. J Virol 75:5663–5671. doi: 10.1128/JVI.75.12.5663-5671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elong Ngono A, Vizcarra EA, Tang WW, Sheets N, Joo Y, Kim K, Gorman MJ, Diamond MS, Shresta S. 2017. Mapping and role of the CD8+ T cell response during primary Zika virus infection in mice. Cell Host Microbe 21:35–46. doi: 10.1016/j.chom.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pardy RD, Rajah MM, Condotta SA, Taylor NG, Sagan SM, Richer MJ. 2017. Analysis of the T cell response to Zika virus and identification of a novel CD8+ T cell epitope in immunocompetent mice. PLoS Pathog 13:e1006184. doi: 10.1371/journal.ppat.1006184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao-Lormeau VM, Choumet V, Briant L, Despres P, Amara A, Yssel H, Misse D. 2015. Biology of Zika virus infection in human skin cells. J Virol 89:8880–8896. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O'Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, Schinazi RF, Chakraborty R, Suthar MS. 2016. Zika virus infects human placental macrophages. Cell Host Microbe 20:83–90. doi: 10.1016/j.chom.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr, Cherry S, Sadovsky Y, Coyne CB. 2016. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 19:705–712. doi: 10.1016/j.chom.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. 2016. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pierson TC, Graham BS. 2016. Zika virus: immunity and vaccine development. Cell 167:625–631. doi: 10.1016/j.cell.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie X, Shan C, Shi PY. 2016. Restriction of Zika virus by host innate immunity. Cell Host Microbe 19:566–567. doi: 10.1016/j.chom.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 65.Bowen JR, Quicke KM, Maddur MS, O'Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, Suthar MS. 2017. Zika virus antagonizes type I interferon responses during infection of human dendritic cells. PLoS Pathog 13:e1006164. doi: 10.1371/journal.ppat.1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC. 2016. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 17:1766–1775. doi: 10.15252/embr.201642627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, Yang H, Cui J. 2017. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov 3:17006. doi: 10.1038/celldisc.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng Y, King NJ, Kesson AM. 2004. Major histocompatibility complex class I (MHC-I) induction by West Nile virus: involvement of 2 signaling pathways in MHC-I up-regulation. J Infect Dis 189:658–668. doi: 10.1086/381501. [DOI] [PubMed] [Google Scholar]
- 69.Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, Dowd KA, Sutherland LL, Scearce RM, Parks R, Wagner W, Granados A, Greenhouse J, Walker M, Willis E, Yu JS, McGee CE, Sempowski GD, Mui BL, Tam YK, Huang YJ, Vanlandingham D, Holmes VM, Balachandran H, Sahu S, Lifton M, Higgs S, Hensley SE, Madden TD, Hope MJ, Kariko K, Santra S, Graham BS, Lewis MG, Pierson TC, Haynes BF, Weissman D. 2017. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543:248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, Ciaramella G, Diamond MS. 2017. Modified mRNA vaccines protect against Zika virus infection. Cell 169:176. doi: 10.1016/j.cell.2017.03.016. [DOI] [PubMed] [Google Scholar]