

Abstract
Activin A receptor type I or activin receptor-like kinase 2 (ACVRI/ALK2) belongs to type I TGF-β family and plays an important role in bone development. Activating mutations of ALK2 containing the R206 to H mutation, are present in 95% in the rare autosomal genetic disease fibrodysplasiaossificansprogressiva (FOP), which leads to the development of ectopic bone formation in muscle. The effect of AMP-activated protein kinase (AMPK) activation on ALK2R206H-mediated signaling in fibroblast obtained from a FOP patient was assessed in the present study. The activity of the mutated ALK2 was suppressed by pharmacological AMPK activators such as metformin and aspirin, while their actions were blocked by the dominant negative mutant of AMPK and mimicked by the constitutively active mutant of AMPK. Furthermore, activation of AMPK upregulated Smad6 and Smurf1 and thereby enhanced their interactions, resulting in its proteosome-dependent degradation of ALK2. In contrast, knockdown of Smad6 or Smurf1 prevented metformin-induced reduction of ALK2. To evaluate the biological relevance of AMPK action on ALK2 activity, we induced FOP fibroblasts within iPS cells and found that their osteogenic differentiation in vitro was inhibited by metformin. Our studies provide novel insight into potential approaches to treatment of FOP, since several AMPK activators (e.g. metformin, berberine, and aspirin, etc.) are already in clinical use for the treatment of diabetes and metabolic syndromes.
Keywords: Fibrodysplasiaossificansprogressiva, activin A receptor type I/activin receptor like kinase 2, AMP-activated protein kinase, osteogenic differentiation, iPS
Introduction
Bone morphogenetic proteins (BMPs) belong to the TGF-β superfamily. There are approximately 20 different BMPs found in mammals that are grouped into four subfamilies based on their sequence similarity and functions[1]. Seven type I receptors have been identified termed activin receptor-like kinase 1 to 7 (Alk1 to Alk7), among which, BMPs preferentially bind to the Alk1 to 3 and 6, while other members of TGF-β superfamily such as β1, 2, 3 and activins bind to the Alk5 and Alk4[2]. Four type I (Alk1, BMPR-IA/Alk3, BMPR-IB/Alk6, and Alk2) and three type II BMP receptors (BMPRII, activin type II receptor-ACVR-II or Act-II, and ACVR-IIB or Act-IIB) are able to bind to BMPs. Once BMPs bind to the complex of type I and type II receptors, the type II receptor phosphorylates and activates the type I receptor. The phosphorylated receptors in turn activate Smad-dependent and independent signaling cascades that lead to concerted regulation of the osteogenic program. The inhibitory Smad6 blocks signaling pathways through multiple mechanisms including: (1) competitive binding to the type I receptor or Smad1/5/8 to disrupt their interaction; (2) sequestration of Smad1/5/8 to Smad 4; (3) recruitment of Smurf1, a U3 ubiquitin ligase that would target prosomal degradation of either the receptor I, Smad1/5, or Runx2 [3–5].
Fibrodysplasiaossificansprogressiva (FOP) is a devastating genetic disorder that causes progressive heterotopic ossification (HO) that develops through a process of endochondral bone formation, occurring within muscle, tendon, and ligamentous tissues throughout the body, which results over time in the immobilization of the patient[6, 7]. FOP is attributed to autosomal mutations in activin A receptor type I/activin receptor-like kinase 2 (ACVRI/ALK2), a BMP type I receptor [8]. Mutation rate is very rare, one out of two million people. Most of patients carry a heterozygous germ-line mutation in the ACVRI gene with an arginine to histidine substitution at amino acid 206 (R206H) located in the glycine-serine-rich domain of ALK2. Studies have shown that this mutation causes constitutive activation of the receptor. Intriguingly, knock-in studies of activating mutants of ALK2 or transgenic over expression of BMP4 in mice can recapitulate phenotypes of FOP at varying degrees [9–12].
AMPK is an intracellular fuel sensing enzyme consisting of three subunits (α, β, γ). The enzyme is activated under circumstances by which a cell senses energy crisis such as hypoxia and glucose restriction, while it is suppressed in nutrient surplus. When energy is short, AMP or the ratio of AMP to ATP is increased, leading to AMP binding to the γ regulatory subunit of AMPK, enabling phosphorylation of T172 in the activation loop of the α catalytic subunit by upstream kinases including LKB1 and preventing dephosphorylation of this site, causing full activation of the enzyme [13]. The activation of AMPK inhibits glucose production in the liver, stimulates glucose uptake in the muscle and adipose and reduces lipogenesis [14]. Thus, the net effect of AMPK activation is to alleviate hyperglycemia, hyperlipidemia and hyperinsulinemia. As such, AMPK has emerged as a therapeutic target for type 2 diabetes and metabolic syndromes [14]. Indeed, pharmacological activators of AMPK such as metformin are first line anti-diabetic drug. Several other drugs that are used in human, such as berberine, resveratrol, and salicylate [14] have similar actions on AMPK. In addition, AMPK can be activated by many other activators including hormones, cytokines and especially, 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR), the first canonical activator that is a cell permeable agent and is phosphorylated by a nucleoside kinase at the plasma membrane and converted to ZMP, an analog of AMP[15].
Previous studies have suggested a role of AMPK in osteogenesis. On one hand, several reports have indicated that AMPK promotes osteogenic differentiation in contrast to its negative effect on adipogenesis[16–22]. On the other hand, there are opposite findings; for instances, one study reported that AMPK activity is reduced during differentiation of osteoblasts and forced activation of AMPK suppressed the differentiation [23]. A most recent study reinforces the negative role of AMPK in osteoblast differentiation[24]. This elegant study has shown that the increase of glucose uptake is prerequisite for the differentiation. This is in part through suppression of AMPK, as the latter induces proteasome-dependent degradation of Runx2 through phosphorylation and activation of Smurf1. Additionally, a few studies reported that a negative effect of AMPK on differentiation of osteoclasts[25–27].
We have recently found that AMPK inhibits TGF-β signaling and EMT in cancer cells [28]. This led us to hypothesis that AMPK would inhibit BMP signaling. Thus, we selected fibroblast cells carrying the ALK2 R206H mutation obtained from a FOP patient and treated the cells with different AMPK activators to assess the effect of AMPK on skeletogenic stem cell differentiation.
Material and Methods
Reagents
Metformin, phenformin, aspirin, 5-amino-4-imidazole carboxamide riboside (AICAR) MG132 and Alizarin red S, and monoclonal antibody against flag epitope were purchased from Sigma-Aldrich (St. Louis, MO). A769962 was from LC Laboratories (Woburn, MA). Human recombinant BMP6 was purchased from BioLegend (San Diego, CA). Customized oligonucleotides, siRNAs, Lipofectamine 2000 and StemPro® Osteogenesis Differentiation Kit were from Life Technologies (Grand Island, NE). Antibodies against phospho-smad1/5 (Ser463/465), total smad1, phospho-AMPKα (T172) and total AMPKα were from Cell Signaling Technology, Inc. (Danvers, MA). Monoclonal antibodies against LKB1 and β-actin and Alkaline Phosphates Detection Kit were from EMD Millipore Corporation (Billerica, MA). Antibodies against ALK2, Smad6 and Smurf1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Luciferase kit was from Promega Life Sciences (Madison, WI).
Cell culture
Human dermal fibroblast cells (GM00513) were obtained from Coriell Institute (Camden, NJ) and maintained in Eagle’s alpha minimal essential medium (αMEM) supplemented with 15% fetal bovine serum (FBS) and penicillin/streptomycin. HEK293T cells and mouse embryonic fibroblast (MEF) cells were cultured in DMEM supplemented with 10% FBS and antibiotics. All cells were cultured in 5%CO2 at 37°C.
siRNA Transfection
siRNAs for Smad6 (AM16708, 4390771) and Smurf1 (4390824) and scrambled siRNA (AM4611) were purchased from Life Technologies. siRNAs were transfected into the FOP fibroblast cells using Lipofectamine 2000 according to the manufacturer’s instructions.
Firefly luciferase complementation assay
Complementation plasmids were constructed according to So et al.[29] In brief, luciferase was divided to aminoterminal (NLuc, aa1-475) and carboxyterminal (CLuc, 265–550) portions, and each of ALK2, Smad6, Smurf1, or Smad1 was fused to NLuc (HindIII and BamHI cloning sites) or CLuc (HindIII and BamHI or KpnI sites). cDNAs for NLuc and CLuc were already in pCDNA3.1. Primers were designed and cDNAs for inserts of interest amplified and cloned into the vectors. The plasmids were then transfected into HEK293T cells by the calcium phosphate precipitation method. Luciferase complementation was assayed on different plasmid pairs to select the optimal pairs to be tested. Luciferase activity was assayed according to protocol provided by manufacture.
Generation of iPSCs derived from human dermal fibroblast cells (HDF)
FOP fibroblast cells (GM00513) at low passages were used for iPS cell reprogramming[30]. One microgram each of three episome plasmids, pCXLE-hOCT3/4-shp53 (expressing OCT3/4 and p53 shRNA), pCXLE-hSK (expressing SOX2 and KLF4), and pCXLE-hUL (expressing L-MYC and LIN28), were electroporated into the cells (1× 106) with Nucleofector reagent (Lonza) in 100 μl according to the manufacturer’s instructions. The cells were trypsinized 6 days after transfection and seeded into 6-well plate coated with growth factor-reduced Matrigel (150–300 μg/ml, 30 min coating) (BD Biosciences). Next day, the culture medium was replaced with standard human iPS medium containing FGF(10ng/ml). The colonies started to appear in about 3–4 weeks after plating, which were picked up based on the morphology and used for further amplification and evaluation.
All pluripotent cell clones were maintained in mTeSR1 medium (StemCell Technologies, Vancouver, Canada) on growth factor-reduced Matrigel-coated plates. The ROCK inhibitor Y-27632 (StemCell Technologies, Vancouver, Canada) dissolved in DMSO was added to mTeSR1 at passage and removed on the following day.
Alkaline phosphatase (ALP) activity
The ALP activity was determined using an Alkaline Phosphatase Detection Kit (EMD Millipore, SCR004) following the manufacturer’s instruction. In brief, iPS cells were fixed in 4% paraformaldehyde for 1–2 minutes and washed with PBS. ALP staining solution (Naphthol/Fast Red Violet) was added to cover cells and incubated in dark at room temperature for 15 minutes. The staining solution was removed and washed with distilled water. The cells were examined under a light microscope for ALP activity and images were acquired.
Osteogenic differentiation
Control iPS cells (ALK2 WT) and FOP iPS cells (ALK2 R206H) were cultured in StemPro® Osteogenesis Differentiation medium for different days and the medium replenished every 3 days. The cells were stained in Alizarin red S solution and microscopic photo digital images were taken at 40× magnification. Alizarin deposit was extracted in solution containing 0.5N HCL and 5% SDS, and quantified at OD405 absorbance.
Western blot analysis
Cell extracts were prepared in lysis buffer (25 mM Tris–HCl, pH 7.8, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4 and 25 mM β-glycerol-phosphate, 1 mM DTT, 1% NP-40 and protease inhibitors). The cell debris was removed by centrifugation at 14,000 × g at 4 °C for 15 min and protein concentration measured using a Bio-Rad Protein Assay kit. Protein samples (20 μg) were subjected to SDS-PAGE and electrophoretically transferred to PVDF membranes (EMD Millipore). The membranes were sequentially blotted with the first and second antibodies, and developed by the enhanced chemiluminescence (ECL) method.
Adenovirus Preparation
Adenoviruses encoding the constitutively active mutant, dominant negative mutant of human AMPKα1 subunit, and GFP were prepared as described previously [28].
Statistical analysis
Significance of differences among groups was determined by two tailed student t test. P<0.05 was set for significance.
Results
Inhibition of ALK2 by AMPK
To assess if AMPK plays a role in regulation of BMP-signaling, we obtained fibroblast cells (GM00513) from a FOP patient. Sequencing analysis confirmed heterozygous mutation of ALK2 (R206H). We then treated the cells with different activators of AMPK (metformin, phenformin, A769962, AICAR) at concentrations optimal for activation of AMPK (Figure 1). The data showed that Smad1/5 phosphorylation was inversely related to activation of AMPK (Figure 1A). If the cells were treated with these activators prior to BMP6 incubation, the induction of Smad1/5 phosphorylation was markedly diminished (Figure 1B). Next, we focused on metformin, as it has been used in clinical treatment of type 2 diabetes and metabolic syndromes and informative outcomes might be insightful for expanding its use in the treatment of FOP. Thus, we examined the dose and time effects of metformin and found that it inhibited Smad1/5 phosphorylation in the FOP fibroblast cells in both dose- and time-dependent fashion, in parallel to activation of AMPK (Figure 1C, 1D). Similar but more robust effects were observed with AICAR (Supplementary Figure 1)
Figure 1. Inhibition of ALK2 signaling in FOP fibroblast cells by AMPK activators.
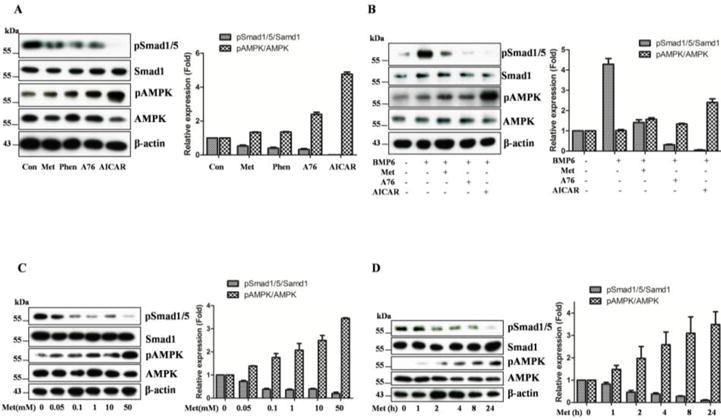
A. FOP fibroblast cells were treated with metformin (Met, 10mM), phenformin (Phen, 1mM), A769962 (A76, 10μM), AICAR (1mM) for 8 hours, or left without treatment (Con). B. The fibroblasts were treated with metformin, A769962, or AICAR for 24 hours, followed by BMP6 (50ng/ml) for 30 min. C. The cells were treated with different doses of metformin. D. The cells were treated with metformin (10mM) for different periods of time. Equal amounts of cell extracts (20μg) were blotted with antibodies as indicated. Representative blots were presented and graphs represent scan densitometric ratio of bands from three independent blots (average ratio±SD).
To ascertain if the effect of metformin is mediated by AMPK, we performed the following assays: first, we assessed the effect of metformin on the response of mouse embryonic fibroblast cells (MEF) to BMP6 (Figure 2A); second, we infected adenovirus expressing a constitutively active AMPK mutant (Figure 2B) or dominant negative AMPK mutant (Figure 2C). The results showed that metformin diminished BMP6-induced phosphorylation of Smad1/5 in MEF cells, which was abolished by ablation of LKB1 or AMPK catalytic subunits (Figure 2A). When FOP fibroblasts were infected by constitutively active AMPK α1 subunit, our data revealed that Smad1/5 phosphorylation was progressively inhibited by increasing volumes of adenovirus, concurrently with decreases in abundance of ALK2, whereas no such effects were observed with adenovirus encoding GFP (Figure 2B). We then infected the dominant negative mutant of AMPKα1 and treated the FOP fibroblasts with metformin overnight, followed with BMP6 for 30 min. As shown in Figure 2C, the inhibitory effect of metformin observed with GFP adenovirus was diminished by the dominant negative mutant of AMPK. Hence, the results from Figures 1 and 2 indicate that AMPK inhibits Smad1/5 phosphorylation induced by mutated ALK2 or BMP6.
Figure 2. AMPK mediates the inhibitory effect of metformin on ALK2 signaling.
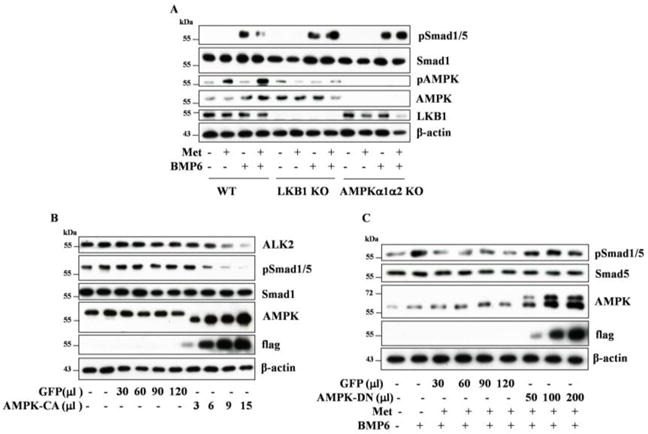
A. MEF cells, wildtype, LKB1 KO, or AMPKα1 & α2 KO, were treated with metformin (10mM) for 24 hours, followed by BMP6 (25ng/ml) for 30 min. B–C. FOP fibroblasts were infected with adenovirus in varying volumes for 48 hours. The virus expresses an active mutant of AMPKα1 (AMPK-CA) (B) or a dominant negative mutant of AMPKα1 (AMPK-DN) (C), both tagged with the flat epitope, or GFP as a control. The cells were treated with or without metformin (10mM) for 24 hours and followed with BMP6 (50ng/ml) for 30 min. Equal amounts of cell extracts (20μg) were blotted with antibodies, as indicated.
AMPK upregulates Smad6 and Smurf1
Figure 2B demonstrates that ALK2 abundance is decreased by the active mutant of AMPK. This was not due to inhibition of RNA transcription as we found no differences in qPCR analysis (data not shown). Thus, we tested if AMPK regulates Smad6 and/or Smurf1 abundance as this complex binds to ALK2 and targets it for proteasome degradation. Toward this end, the FOP fibroblast cells were treated with metformin in different doses for 24 hours or different periods of time at 10mM, and alternatively with aspirin which is used as a second AMPK activatorat different doses for 24 hours. Our data revealed that reduction of ALK2 by treatment with metformin or aspirin was associated with increases in Smad6 and Smurf1 (Figure 3).
Figure 3. AMPK upregulates Smad6 and Smurf1.
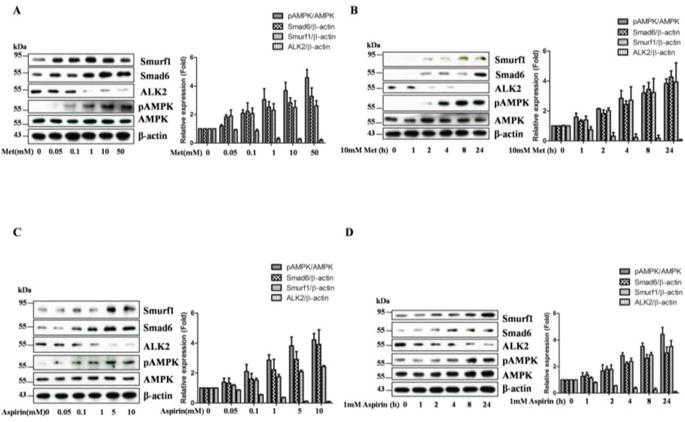
FOP fibroblast cells were treated with metformin (A, B) or aspirin (C) at varying concentrations for 24 hours (A, C) or different times (B, D). Equal cell extracts (20μg) were resolved by Western blot with antibodies, as indicated. Representative blots were presented and graphs represent scan densitometric ratio of bands from three independent blots (average ratio±SD).
To assess if increased abundance of Smad6 and Smurf1 leads to changes in their interaction, we engineered two constructs; the first one fusing Smad6 to the aminoterminal portion of firefly luciferase (Smad6-NLuc) and Smurf1 to the carboxy terminal portion of the luciferase (Smurf1-CLuc) (Supplementary Figure 2). The two constructs were cotransfected into HEK293T cells and then treated with or without phenformin, a potent AMPK activator with similar mechanism to metformin (but the latter failed to activate AMPK in HEK293T cells), for 8 hours. The cell extracts were then prepared for the luciferase activity assay. As shown in Figure 4A, the luciferase activity reconstitution assays revealed that the interaction between Smad6 and Smurf1 was enhanced significantly by phenformin (p<0.05). Similar assay was conducted with ALK2 and Smad1. Figure 4B shows that the interaction between ALK2 and Smad1 was markedly diminished by phenformin (p<0.01).
Figure 4. AMPK regulates the interaction of key components of the ALK signaling pathway.
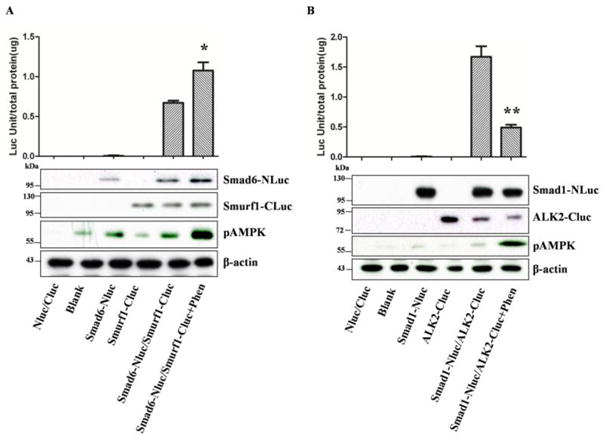
Transfection of plasmid pairs as indicated was performed with plasmids into HEK293T cells and 36 hours later, the cells were treated with or without phenformin (1mM) for 8 hours. Luciferase assay was conducted and normalized with total protein (units/μg). A. phenformin enhances Smad6 and Smurf1 interaction. B. phenformmin inhibits ALK2 and Smad1 interaction. The results represent averages of a triplicate experiment (means±SD). *P=0.0482<0.05, **p=0.0007<0.01. Representative blots show expression of recombinant proteins.
We transfected Smad6 siRNA into the FOP fibroblast cells in order to examine if knockdown of Smad6 prevented ALK2 degradation triggered by AMPK. The results showed that ALK2 was upregulated and appeared to be resistant to metformin treatment after Smad6 knockdown (Figure 5A). A similar result was obtained with Smurf1 siRNA. To assess if the effect of AMPK is associated with proteosomal degradation of ALK2, we incubated the FOP fibroblast cells with MG132, a proteosome inhibitor, along with metformin for 24 hours. We observed that MG132 increased abundance of ALK2 and abrogated the effect of metformin (Figure 5C). Similarly, MG132 also blunted the effect of the active mutant of AMPKα1 (Figure 5D).
Figure 5. AMPK promotes proteosomal degradation of ALK2.
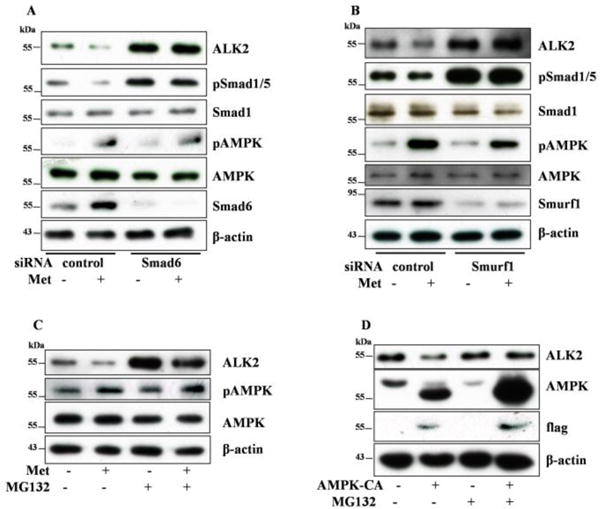
A–B. Scrambled siRNA (control) and siRNAs for Smad6 (A) or Smurf1 (B) were transfected into FOP fibroblast cells and then treated with or without metformin (10mM) for 24 hours. C–D. FOP fibroblast cells were treated with or without MG132 and/or metformin for 24 hours (C), or after infection with adenovirus expressing the active mutant of AMPK (AMPK-CA), the cells were incubated with MG132 for 24 hours (D). Cell extracts were then blotted with antibodies, as indicated.
AMPK inhibits osteogenic differentiation of iPS cells derived from FOP fibroblast cells
Weinduced the FOP fibroblast cells to iPS cells by electroporation of episomal plasmids carrying OCT3/4 (plus p53 shRNA), SOX2, KLF4, C-MYC, and LIN28. The iPS clones were isolated, amplified and characterized by Western blot, clonal morphology, alkaline phosphatase staining (Supplementary Figure 3). After confirmation of successful induction and isolation of the iPS clones, they were induced to differentiate into osteogenic cells in the differentiation medium for 21 days. The cells were collected and assayed for mineralization using alizarin red S and expression of osteoblastic markers during the course of induction (Supplementary Figure 3). The results showed that the cells with ALK2 R206H exhibited slightly better differentiation than that with wild type ALK2, as reflected by mineralization assay and Western blot analysis of differentiation markers. To test the effect of AMPK, metformin was added during induction of differentiation. As shown in Figure 6, the data revealed that metformin greatly diminished mineralization. They also showed that metformin significantly inhibited osteogenic differentiation of iPS cells bearing wild type ALK2, which was probably due to the inhibition of BMP signaling, as previous studies reported that BMP2 was induced during osteogenic differentiation [2, 31].
Figure 6. Metformin inhibits osteogenic differentiation of iPS cells.
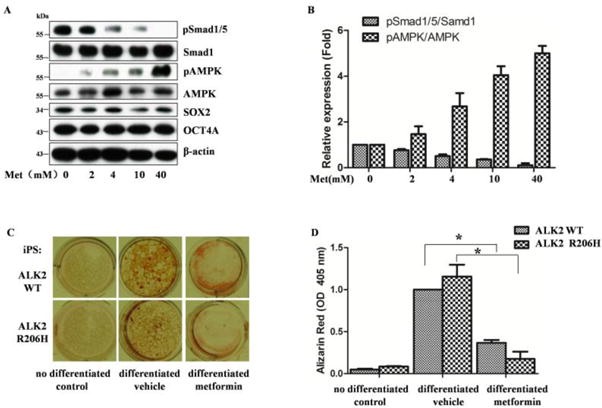
A. metformin inhibits Smad1/5 phosphorylation of iPS cells. The cells were treated with metformin at different doses for 24 hours and blotted with antibodies as indicated. B. Graphs represent scan densitometric unit ratio of related bands in A from three independent experiments (average ratio±SD).C. osteogenic differentiation was induced in the presence or absence of metformin (10 mM). Mineralization is detected as red staining with Alizarin Red S. D. Graph represents alizarin deposit quantitation from three independent experiments using spectrophotometer. ALK2WT (metformin vs vehicle,*p=0.0009), ALK2R206H (metformin vs vehicle, *p=0.0012).
Discussion
FOP is a debilitating disease with the development of extensive systemic HO. Although the incidence is very low, it is incurable and lethal. Current clinical management relies mostly on anti-inflammatory drugs that are at best only partially effective. An ideal treatment strategy would be to target components of the BMP signaling pathway inasmuch as a critical pathogenic factor is the activating mutations of the ACVRI allele. However, inhibitors targeting activated ALK2 and downstream components are still in scarcity and at the experimental stage. Therefore, an innovative approach would be to identify clinically available drugs having well defined safety profiles that are used in treating other diseases and re-tasking them to fulfill this clinical need. Toward this end, the present study is designed to assess the impact of some of the clinically used AMPK activators that have proven to be safe, such as metformin and aspirin, on the ALK2-mediated signaling events. We showed that AMPK activated by these agents or genetic manipulations inhibited Smad1/5 phosphorylation in FOP fibroblast cells carrying the ALK2 R206H mutation. Furthermore, we found that the inhibitory effect of AMPK was mediated by upregulation of Smurf1 and Smad6, which leads to increased interaction of these two molecules and subsequent degradation of ALK2. Finally, our data revealed AMPK activation also suppressed BMP-signaling in iPS cells derived from the FOP fibroblasts and iPS osteogenic differentiation in vitro. These data therefore are the first to show that AMPK suppresses BMP signaling, which might have an important implication in the treatment of FOP patients.
Since heterozygous mutation of the ACVR1 gene was identified to be responsible for FOP in 2006, several attempts have been made to target this signaling pathway in treating this disease using animal models or cell based assays. Thus, two small molecular compounds, dorsomorphin and LDN-193189, have been shown to bind and inhibit ALK2 and reduce the incidence of HO in transgenic mice carrying ALK2 mutation[32]. Secondly, siRNA specifically targeting the mutated site of ALK2 R206H has been used to suppress osteoblast differentiation [33, 34]. Thirdly, all-trans retinoic acid (RA) and PARγ agonists have also been reported to inhibit HO in transgenic mice [35]. These agents reduce the level of Smads and BMP-induced expression of Id1 [35] All these agents are currently at the experimental stage, and their toxicity, efficacy and pharmacodynamics have not as yet been fully tested. Hence, an alternative approach to the development of totally novel inhibitors to this pathway, would be to identify compounds that are already in use clinically and have proven safety profiles. A recent study of Cappato et al [36] using high throughput screen identified dipyridamole, which was shown to inhibit the expression of ALK2, inhibit osteoblast differentiation in vitro, and block heterotopic bone formation in vivo. This study could be immediately translated into clinical application as dipyridamole is in clinical use as a platelet anti-aggregate.
It has been known that AMPK can be activated by a variety of pharmacological agents, such as metformin, aspirin, berberine, and resveratrol that are used in treatment of type 2 diabetes and metabolic syndromes. These agents are inexpensive and have been proven to be safe. Hence, our finding that AMPK suppresses BMP signaling would lead to opening of a new avenue for the potential treatment of FOP and other types of heterotopic ossifications. The next step will be to extend our study to animal models. In fulfilling this task, there are several available conditional knock-in or transgenic mouse models in which constitutively active ALK2 is expressed [9, 11]. In addition, induction of iPS cells derived from FOP to osteoblast differentiation and transplantation of the differentiated cells into nude mice has emerged as another type of model that is useful in studying the mechanism of HO, and for testing efficacy of drugs in the treatment of FOP [37, 38]. As such, it will be intriguing to apply clinically available AMPK activators to these FOP models. A positive outcome would prompt fast route of clinical trial.
We have shown that increased Smad1/5 phosphorylation in FOP fibroblasts bearing ALK2 R206H while activated AMPK suppresses ALK2. However, we have not tested if other ALKs are regulated by AMPK in the fibroblast cells used in the present study. It is not surprising if other BMP-regulated ALKs are also downregulated in fibroblast cells, as they are regulated by Smad6/Smurf1. It has been known that different ALKs execute different functions[1]. For example, ALK1 plays an important role in angiogenesis and vascularization, while ALK2 mediates the effect of BMPs on osteogenesis. In fact, our recent publication has shown that AMPK activation inhibits ALK1-mediated angiogenesis via upregulation of Smad6 and Smurf1[39]. Our present study adds new information on AMPK regulation of osteogenic differentiation through induction of ALK2 degradation.
Conclusion
Our present study has shown that metformin and other AMPK activators can inhibit ALK2-mediated signaling and associated osteogenic differentiation of iPS cells derived from FOP fibroblast cells. The inhibition was ascribed to upregulation of Smurf1 and Smad1 and increased interaction of these two molecules, leading to proteosomal degradation of ALK2. Our finding is of translational potential inasmuch as several pharmacological AMPK activators have been used in clinical treatment of other diseases such as diabetes, obesity and cancer.
Supplementary Material
Highlights.
Fibrodysplasiaossificansprogressiva is a devastating genetic disease which is caused by a R206 to H mutation in ALK2
AMPK inhibits either mutant ALK2, or wild ALK activated with BMP6
AMPK causes proteosomal degradation of ALK2 by upregulation of Smurf1 and Smad6
AMPK inhibits osteogenic differentiation of iPS cells derived from FOP fibroblast cells
Metformin may be useful in the treatment of FOP patients
Acknowledgments
This work was supported in part by grants from National Eye Institute (R21EY024388) and National Natural Science Foundation of China (81272926, 81572753, 31660332). HL was supported by graduate scholarship from Nanchang University. YY was supported in part by Jiangxi Province Yuan Hang Scholarship Program. YYW was supported by PhD scholarship from China Scholar Council (201507040075), SSJ was supported by PhD scholarship from China Scholar Council (201506820020). We are thankful to grant support from Jiangxi Province Bureau of Foreign Experts. We are also thankful to Drs. Carmela Abraham and Cidi Chen for the firefly luciferase cloning plasmid.
Abbreviations
- ACVRI/ALK2
activin A receptor type I/activin receptor-like kinase 2
- ALP
alkaline phosphatase
- AMP
adenosine monophosphate
- AMPK
AMP-activated protein kinase
- BMP
bone morphogenetic protein
- AICAR
5-Aminoimidazole-4-carboxamide ribonucleotide
- FGF
fibroblast growth factor
- FOP
Fibrodysplasiaossificansprogressiva
- HDF
human dermal fibroblast cells
- HO
heterotopic ossifications
- iPS
induced pluripotent stem cell
- LKB1
Liver kinase B1
- Smurf1
SMAD Specific E3 Ubiquitin Protein Ligase 1
- TGF-β
Transforming growth factor beta
- Smad6
SMAD Family Member 6
- SOX2
sex determining region Y)-box 2
- KLF4
Kruppel-like factor 4
- C-MYC
a protooncogene, V-Myc Avian Myelocytomatosis Viral Oncogene Homolog
- Lin28
Lin-28 homolog A
- RA
retinoic acid
- ZMP
5- amino-4-imidazolecarboxamide ribotide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
All authors declare no conflict of interest in this work.
References
- 1.Carreira AC, Alves GG, Zambuzzi WF, Sogayar MC, Granjeiro JM. Bone Morphogenetic Proteins: structure, biological function and therapeutic applications. Arch Biochem Biophys. 2014;561:64–73. doi: 10.1016/j.abb.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 2.Oryan A, Alidadi S, Moshiri A, Bigham-Sadegh A. Bone morphogenetic proteins: a powerful osteoinductive compound with non-negligible side effects and limitations. Biofactors. 2014;40:459–481. doi: 10.1002/biof.1177. [DOI] [PubMed] [Google Scholar]
- 3.Li Q. Inhibitory SMADs: potential regulators of ovarian function. Biol Reprod. 2015;92:50. doi: 10.1095/biolreprod.114.125203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen R, Chen M, Wang YJ, Kaneki H, Xing L, O’Keefe RJ, Chen D. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J Biol Chem. 2006;281:3569–3576. doi: 10.1074/jbc.M506761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murakami G, Watabe T, Takaoka K, Miyazono K, Imamura T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol Biol Cell. 2003;14:2809–2817. doi: 10.1091/mbc.E02-07-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaplan FS, Chakkalakal SA, Shore EM. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis Model Mech. 2012;5:756–762. doi: 10.1242/dmm.010280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramirez DM, Ramirez MR, Reginato AM, Medici D. Molecular and cellular mechanisms of heterotopic ossification. Histol Histopathol. 2014;29:1281–1285. doi: 10.14670/HH-29.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 9.Agarwal S, Loder SJ, Brownley C, Eboda O, Peterson JR, Hayano S, Wu B, Zhao B, Kaartinen V, Wong VC, Mishina Y, Levi B. BMP signaling mediated by constitutively active Activin type 1 receptor (ACVR1) results in ectopic bone formation localized to distal extremity joints. Dev Biol. 2015;400:202–209. doi: 10.1016/j.ydbio.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS, Shore EM. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res. 2012;27:1746–1756. doi: 10.1002/jbmr.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, Economides AN. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015;7:303ra137. doi: 10.1126/scitranslmed.aac4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kan L, Hu M, Gomes WA, Kessler JA. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am J Pathol. 2004;165:1107–1115. doi: 10.1016/S0002-9440(10)63372-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2015;33:1–7. doi: 10.1016/j.ceb.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes. 2013;62:2164–2172. doi: 10.2337/db13-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010;6:457–470. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah M, Kola B, Bataveljic A, Arnett TR, Viollet B, Saxon L, Korbonits M, Chenu C. AMP-activated protein kinase (AMPK) activation regulates in vitro bone formation and bone mass. Bone. 2010;47:309–319. doi: 10.1016/j.bone.2010.04.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pantovic A, Krstic A, Janjetovic K, Kocic J, Harhaji-Trajkovic L, Bugarski D, Trajkovic V. Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone. 2013;52:524–531. doi: 10.1016/j.bone.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 18.Jeyabalan J, Shah M, Viollet B, Chenu C. AMP-activated protein kinase pathway and bone metabolism. J Endocrinol. 2012;212:277–290. doi: 10.1530/JOE-11-0306. [DOI] [PubMed] [Google Scholar]
- 19.Kim EK, Lim S, Park JM, Seo JK, Kim JH, Kim KT, Ryu SH, Suh PG. Human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by AMP-activated protein kinase. J Cell Physiol. 2012;227:1680–1687. doi: 10.1002/jcp.22892. [DOI] [PubMed] [Google Scholar]
- 20.Chen T, Wu YW, Lu H, Guo Y, Tang ZH. Adiponectin enhances osteogenic differentiation in human adipose-derived stem cells by activating the APPL1-AMPK signaling pathway. Biochem Biophys Res Commun. 2015;461:237–242. doi: 10.1016/j.bbrc.2015.03.168. [DOI] [PubMed] [Google Scholar]
- 21.Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Sugimoto T. Metformin enhances the differentiation and mineralization of osteoblastic MC3T3-E1 cells via AMP kinase activation as well as eNOS and BMP-2 expression. Biochem Biophys Res Commun. 2008;375:414–419. doi: 10.1016/j.bbrc.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 22.Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Sugimoto T. Activation of AMP kinase and inhibition of Rho kinase induce the mineralization of osteoblastic MC3T3-E1 cells through endothelial NOS and BMP-2 expression. Am J Physiol Endocrinol Metab. 2009;296:E139–146. doi: 10.1152/ajpendo.90677.2008. [DOI] [PubMed] [Google Scholar]
- 23.Kasai T, Bandow K, Suzuki H, Chiba N, Kakimoto K, Ohnishi T, Kawamoto S, Nagaoka E, Matsuguchi T. Osteoblast differentiation is functionally associated with decreased AMP kinase activity. J Cell Physiol. 2009;221:740–749. doi: 10.1002/jcp.21917. [DOI] [PubMed] [Google Scholar]
- 24.Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, Takarada T, Lezaki T, Pessin JE, Hinoi E, Karsenty G. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell. 2015;161:1576–1591. doi: 10.1016/j.cell.2015.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeyabalan J, Shah M, Viollet B, Roux JP, Chavassieux P, Korbonits M, Chenu C. Mice lacking AMP-activated protein kinase alpha1 catalytic subunit have increased bone remodelling and modified skeletal responses to hormonal challenges induced by ovariectomy and intermittent PTH treatment. J Endocrinol. 2012;214:349–358. doi: 10.1530/JOE-12-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee YS, Kim YS, Lee SY, Kim GH, Kim BJ, Lee SH, Lee KU, Kim GS, Kim SW, Koh JM. AMP kinase acts as a negative regulator of RANKL in the differentiation of osteoclasts. Bone. 2010;47:926–937. doi: 10.1016/j.bone.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Kang H, Viollet B, Wu D. Genetic deletion of catalytic subunits of AMP-activated protein kinase increases osteoclasts and reduces bone mass in young adult mice. J Biol Chem. 2013;288:12187–12196. doi: 10.1074/jbc.M112.430389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin H, Li N, He H, Ying Y, Sunkara S, Luo L, Lv N, Huang D, Luo Z. AMPK Inhibits the Stimulatory Effects of TGF-beta on Smad2/3 Activity, Cell Migration, and Epithelial-to-Mesenchymal Transition. Mol Pharmacol. 2015;88:1062–1071. doi: 10.1124/mol.115.099549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.So PP, Zeldich E, Seyb KI, Huang MM, Concannon JB, King GD, Chen CD, Cuny GD, Glicksman MA, Abraham CR. Lowering of amyloid beta peptide production with a small molecule inhibitor of amyloid-beta precursor protein dimerization. Am J Neurodegener Dis. 2012;1:75–87. [PMC free article] [PubMed] [Google Scholar]
- 30.Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, Shibata T, Kunisada T, Takahashi M, Takahashi J, Saji H, Yamanaka S. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 31.Edgar CM, Chakravarthy V, Barnes G, Kakar S, Gerstenfeld LC, Einhorn TA. Autogenous regulation of a network of bone morphogenetic proteins (BMPs) mediates the osteogenic differentiation in murine marrow stromal cells. Bone. 2007;40:1389–1398. doi: 10.1016/j.bone.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med. 2008;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowery JW, Rosen V. Allele-specific RNA interference in FOP silencing the FOP gene. Gene Ther. 2012;19:701–702. doi: 10.1038/gt.2011.190. [DOI] [PubMed] [Google Scholar]
- 35.Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, Chandraratna RA, Mishina Y, Enomoto-Iwamoto M, Pacifici M, Iwamoto M. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat Med. 2011;17:454–460. doi: 10.1038/nm.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cappato S, Tonachini L, Giacopelli F, Tirone M, Galietta LJ, Sormani M, Giovenzana A, Spinelli AE, Canciani B, Brunelli S, Ravazzolo R, Bocciardi R. High throughput screening for modulators of ACVR1 transcription potentially applicable to the treatment of Fibrodysplasia Ossificans Progressiva. Dis Model Mech. 2016 doi: 10.1242/dmm.023929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto Y, Ikeya M, Hino K, Horigome K, Fukuta M, Watanabe M, Nagata S, Yamamoto T, Otsuka T, Toguchida J. New Protocol to Optimize iPS Cells for Genome Analysis of Fibrodysplasia Ossificans Progressiva. Stem Cells. 2015;33:1730–1742. doi: 10.1002/stem.1981. [DOI] [PubMed] [Google Scholar]
- 38.Kim BY, Jeong S, Lee SY, Lee SM, Gweon EJ, Ahn H, Kim J, Chung SK. Concurrent progress of reprogramming and gene correction to overcome therapeutic limitation of mutant ALK2-iPSC. Exp Mol Med. 2016;48:e237. doi: 10.1038/emm.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ying Y, Ueta T, Jiang S, Lin H, Wang Y, Vavvas D, Wen R, Chen YG, Luo Z. Metformin inhibits ALK1-mediated angiogenesis via activation of AMPK. Oncotarget. 2017;8:32794–32806. doi: 10.18632/oncotarget.15825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.