

SUMMARY
Environmental insults are often detected by multiple sensors that activate diverse signaling pathways and transcriptional regulators, leading to a tailored transcriptional output. To understand how a tailored response is coordinated, we examined the inflammatory response elicited in mouse macrophages by ionizing radiation (IR). RNA-sequencing studies revealed that most radiation-induced genes were strongly dependent on only one of a small number of sensors and signaling pathways, notably the DNA damage-induced kinase ATM, which regulated many IR-response genes, including interferon response genes, via an atypical IRF1-dependent, STING-independent mechanism. Moreover, small, defined sets of genes activated by p53 and NRF2 accounted for the selective response to radiation in comparison to a microbial inducer of inflammation. Our findings reveal that genes comprising an environmental response are activated by defined sensing mechanisms with a high degree of selectivity, and they identify distinct components of the radiation response that may be susceptible to therapeutic perturbation.
eTOC paragraph
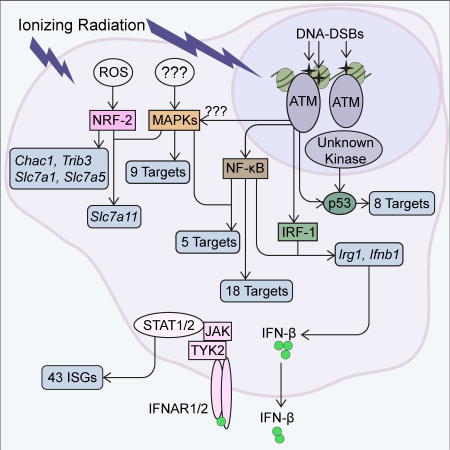
Purbey et al. define the transcriptional response to ionizing radiation in macrophages, revealing a strong dependency on a small number of sensors and signaling pathways, notably the DNA damage-induced kinase ATM, the tumor suppressor p53, and the ROS-induced transcription factor NRF2. Their findings point to selectivity in damage-sensing mechanisms, and identify components of the radiation response as potential therapeutic targets.
INTRODUCTION
Eukaryotes have evolved numerous strategies to respond to a diverse range of environmental insults. The responses require mechanisms to sense the insult and activate signaling pathways, culminating in physiological changes that allow the cell or organism to tolerate and recover from the insult. Many of the physiological changes are coordinated by the induction and repression of gene expression. Although the responses presumably evolved to promote survival and recovery, they can also have negative consequences on human health. One challenge is that many environmental insults are sensed by multiple mechanisms, each of which may regulate genes independently, in synergy with other sensing mechanisms, or in a redundant fashion. Because most studies focus on only one sensing mechanism or signaling pathway, the relationships between the multiple sensing mechanisms activated by an insult have remained poorly understood.
Ionizing irradiation (IR) is among the most ancient environmental insults to which living organisms are exposed. Low dose environmental exposures undoubtedly drove the evolution of radiation response mechanisms. However, the response to higher doses of radiation is now of special interest for the development of strategies to mitigate the negative consequences of radiation therapies and unintended radiation exposures, and to optimize combination cancer therapies in which radiation is combined with other therapeutic approaches, such as checkpoint blockade (Homer et al., 2016; Rosen et al., 2014; Victor et al., 2015). Related goals are to develop improved biomarkers for the accurate detection of radiation exposure, for distinguishing a radiation response from a response to other environmental insults, and for predicting the extent of an exposure (Sullivan et al., 2013).
In mammals, the most severe consequences of IR exposure often result from the death of proliferative cells, including radiosensitive bone marrow stem and progenitor cells (Williams et al., 2010). However, IR exposure can have detrimental effects on a broad range of cell types and organs, through cell death and gene expression changes that promote inflammation and other alterations in cell physiology (Bartoletti-Stella et al., 2013; Rashi-Elkeles et al., 2011; Schaue and McBride, 2010). Macrophages are among the cell types that appear to contribute to pathogenesis following radiation exposure. For example, macrophages and other cells of the innate immune system are thought to modulate Acute Radiation Syndromes (ARS) affecting bone marrow and the gastrointestinal tract, and late fibrosis in lungs and other tissues (Lorimore et al., 2001; Dong et al., 2015; Groves et al., 2015; Heslet et al., 2012). Gene expression changes following IR treatment are also likely to impact the normal functions of macrophages (Williams et al., 2010). Although the exposure of macrophages to IR often has detrimental effects, the macrophage response can also be beneficial. In fact, mice depleted of liver and splenic macrophages exhibit increased susceptibility to IR-induced mortality (Salkowski et al., 1995).
The transcriptional response to IR exposure can be elicited by the sensing of DNA damage, newly generated reactive oxygen species (ROS), or cell death (Candéias and Testard, 2015; Schaue and McBride 2010). In addition, damaged cells have the potential to release damage-associated molecular patterns (DAMPs) that can be sensed by pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) (Candéias and Testard, 2015; Härtlova et al., 2015). At the organism level, IR-induced damage can compromise epithelial barriers, resulting in bacterial translocation and further amplification of inflammatory cascades by pathogen associated molecular patterns (PAMPs) acting through these same PRRs. Genes activated by IR (e.g. Tnf and Il1b) encode proteins that can also contribute to the subsequent response.
The DNA double-strand breaks induced by IR can be sensed by the DNA damage-induced kinases, ATM and DNA-PK (Bensimon et al., 2011). In addition to promoting DNA repair, ATM can activate multiple transcription factors, including NF-κB, p53, and MAP kinase (MAPK)-induced factors (Gannon et al., 2012; Wu et al., 2006; Kool et al., 2003). ROS has been proposed to regulate gene expression by activating ATM, MAPKs, NRF-2, NF-κB, and AP-1 (Kozlov et al., 2016; Ryoo et al., 2016; Gloire et al., 2006). Recently, DNA released into the cytoplasm following IR exposure has been proposed to induce an interferon (IFN) response via activation of the cytosolic DNA sensory pathway that signals through Stimulator of IFN Signaling (STING) (Härtlova et al., 2015).
Although many sensing mechanisms and signaling pathways have the potential to contribute to the IR response, the precise contribution of each mechanism and pathway remains poorly understood. Is the transcriptional output primarily driven by one sensing mechanism, with others playing minimal roles? Do diverse sensing mechanisms act independently to regulate distinct sets of genes, or does the response involve considerable redundancy or synergy? Can the transcriptional output be fully explained by known sensing mechanisms, or do sensing mechanisms, signaling pathways, and transcription factors exist that have not yet been linked to the IR response?
In this study, we explored the above questions through the use of RNA-sequencing (RNA-seq) and chromatin immunoprecipitation-sequencing (ChIP-seq). We used terminally differentiated mouse bone marrow-derived macrophage (BMDMs) to focus on the most fundamental events that occur in the absence of cell-cycle checkpoint effects. By analyzing BMDMs from a variety of mouse strains bearing specific genetic deletions and mutations, we identified dominant regulators of almost all of the 99 most potently induced genes. Remarkably, distinct sensing mechanisms and signaling pathways were found to play major roles in the induction of defined sets of target genes, with little evidence of strong synergy between sensing mechanisms. Of particular note, a careful comparison of the IR response to the response to lipid A, a microbial inducer of inflammation through TLR4, revealed that the selectivity of the IR response can largely be explained by the induction of p53 and the ROS-induced transcription factor, NRF2. Together, the results provide extensive, unbiased insight into the IR response and identify distinct components of the response that may be susceptible to therapeutic perturbation.
RESULTS
Basic Properties of the Transcriptional Cascade Induced by IR
For our analysis of the response to IR, we focused on C57BL/6 BMDMs for three reasons. First, macrophages are thought to contribute to IR-induced pathogenesis. Second, an initial analysis of a non-proliferative cell type allowed us to simplify the study, as a proliferating cell type would reveal many genes that are modulated as an indirect consequence of cell-cycle checkpoints; to fully understand the IR response, it is preferable to first dissect components of the response that are independent of the cell-cycle and to then add additional layers of complexity in the future. Third, the focus on BMDMs facilitated a comparative analysis of the selectivity of inflammatory responses, via a comparison to data previously generated in studies of BMDMs stimulated with Lipid A (Tong et al., 2016).
We initially performed RNA-seq with mRNA from mouse C57BL/6 BMDMs collected 0, 0.5, 1, 2, 6, and 24 hrs after 6 Gray (Gy) of irradiation. An analysis of three biological replicates revealed that, on average, 9,201 (31.8%) of the 29,082 annotated Refseq genes (prior to removal of duplicate isoforms) reached an average transcript level of at least 3 RPKM (Reads Per Kilobase per Million mapped reads) in at least one time point. We chose this relatively high 3 RPKM threshold to increase the accuracy of the fold-induction calculations described below.
Of the 9,201 expressed genes, 387 (4.2%) were induced by at least 2-fold (p<0.01, FDR<0.05) (Figure 1A). Importantly, however, 70.8% of these genes were induced by an average of less than 4-fold (Figure 1B); only 99 genes, all of which are protein-coding genes, were induced more than 4-fold (Figure 1B,C). These genes span a broad range of basal and induced transcript levels (Figure 1D). We focused on these 99 potently induced genes to increase the accuracy of the analysis. In particular, when performing loss-of-function experiments, it is difficult to accurately quantify the extent to which induction of a weakly induced gene is impacted by the absence of a regulatory factor.
Figure 1. Basic Properties of the Transcriptional Cascade Induced by IR.

(A) A maximum average fold induction profile is shown for Refseq genes in either untreated or irradiated (6Gy) samples over a 24-hr time period. 6-Gy induced 9201 unique Refseq genes. The dashed gray line represents the 2-fold mark. The results represent average values from three independent experiments with the full radiation-induced time courses. See also Figure S1.
(B) A maximum average fold induction profile is shown for genes induced >2-fold (n=387) with p<0.01 and FDR<0.05. Approximately 75% of these genes were induced <4-fold.
(C) A maximum average fold induction profile is shown for the 99 genes induced >4-fold.
(D) Basal (bottom) and peak (top) RPKM values are shown for the 99 genes induced >4-fold.
(E) Kinetic profiles are shown for the 99 genes induced by 6-Gy ionizing radiation over a 24-hr time period. The genes are separated into ERG and LRG classes.
A dose-response analysis revealed that the number of induced genes was much higher with 6 Gy IR than 2 Gy, whereas the number was only moderately increased with 20 Gy (Figure S1A,B). Hierarchical clustering revealed that the activation kinetics was similar in the 3 biological replicates of the 6 Gy experiments (Figure S1C).
The 99 genes that were strongly induced with 6 Gy IR were first grouped according to the time point at which their expression peaked (Figure 1E; see Figure S2 for gene names). Genes whose mRNAs peaked at 0.5, 1 or 2 hrs were classified as Early Response Genes (ERGs), whereas most genes whose mRNAs peaked at the 24-hr time point were classified as Late Response Genes (LRGs). Six genes that exhibited peak mRNA levels at 24 hrs were included in the ERG class because they were induced by at least 3-fold by 2 hrs. Notably, no genes exhibited peak mRNA levels at the 6-hr time point.
IR Induces All Genes Independently of the Myd88 and Trif Pathways
IR can lead to the release of DAMPs, which can promote a transcriptional response through TLRs (Candéias and Testard, 2015). To evaluate the extent to which TLR sensors contribute to the transcriptional response in our experiments, we analyzed irradiated BMDMs from Myd88−/− Trif−/− mice. In these cells, the radiation response remained intact, with mRNA levels reduced greater than 50% in comparison to WT cells for only 1 gene (data not shown). Thus, although TLR pathways may contribute to the response in other physiological settings, they do not appear to be dominant regulators of the BMDM response to IR.
Most LRGs Correspond to Interferon-Stimulated Genes (ISGs)
A promoter motif analysis revealed a strong enrichment of Interferon-Sensitive Response Elements (ISREs) and Interferon Regulatory Factor (IRF) binding motifs in the promoters of LRGs (Figure S3A), raising the possibility that the LRG cluster is dominated by Type 1 IFN-induced genes. Consistent with this possibility and with previous reports (Mboko et al., 2012; Hartlova et al., 2015), the Ifnb1 gene, encoding IFN-β1, was strongly induced by IR (Figure S3B), although its transcript level did not reach the 3 RPKM threshold needed for inclusion in the set of 99 genes monitored in this study.
To test the role of IFN signaling, WT cells and cells lacking the Type 1 IFN receptor, IFNAR, were collected 0, 0.5, 1, 2, 6, and 24 hrs after IR, followed by RNA-seq analysis. Although none of the ERGs exhibited IFNAR dependence, 43 of the 48 LRGs exhibited substantially reduced maximum transcript levels (<30% of WT) in the irradiated Ifnar−/− cells (Figure 2A, column 7; Figure 2B). The basal transcript level for many of these genes was also reduced in the Ifnar−/− cells (Figure 2A, column 8), consistent with previous evidence that a low level of tonic IFN signaling occurs in cultured cells (Gough et al., 2012). These results demonstrate that the late response of BMDMs to IR is dominated by a Type 1 IFN response.
Figure 2. Dependence of Radiation-Induced Genes on IFNAR and ATM.

(A) Columns 1–6 show relative percent expression of each gene (maximum expression in WT cells=100%) in WT BMDMs. ERGs are arranged according to degree of dependence on ATM, and LRGs are arranged according to dependence on IFNAR. Column 7 shows peak expression in irradiated Ifnar−/− cells in comparison to WT cells. Column 8 shows basal expression in Ifnar−/− cells in comparison to WT. Columns 9–14 show the kinetic profiles of genes in Atm−/− cells (percent expression relative to WT). Column 15 shows peak expression in Atm−/− cells in comparison to WT. Column 16 shows basal expression in Atm−/− cells in comparison to WT. All values represent averages from 2 independent RNA-seq experiments performed with different BMDM preparations; each experiment with mutant BMDMs was performed with a parallel and full time-course with WT BMDMs. See also Figures S2 and S3.
(B) A dot plot displays the dependence of each ERG and LRG on IFNAR.
(C) A dot plot displays the dependence of ERGs and of both IFNAR-dependent (IFNAR-D) and IFNAR-independent (IFNAR-I) LRGs on ATM.
(D) The graphs show that, after taking into account the impact of ATM on basal expression of each LRG, virtually all LRGs exhibit strong dependence of ATM for their activation by IR. Genes exhibiting greatly elevated basal expression in Atm−/− cells in comparison to WT cells (>3-fold) are shown in the left graph. Genes for which ATM plays only a minimal role in basal expression are shown in the right graph. Blue circles represent maximum fold induction in WT cells. Red circles and black crosses represent basal and maximum fold-induction values, respectively, for each gene relative to the basal WT expression level.
ATM Mediates Induction of Many ERGs and LRGs
The strong enrichment of NF-κB motifs in the promoters of ERGs (Figure S3A) could not be explained by signaling through the MyD88 and TRIF pathways. However, NF-κB is also activated by ATM, which helps sense DNA damage (Shiloh and Ziv, 2013; Lee et al., 1998). To evaluate the extent to which ATM contributes to the IR response, WT and Atm−/− macrophages were collected 0, 0.5, 1, 2, 6, and 24 hrs after irradiation, followed by RNA-seq analysis. The results revealed that 45% and 30% of the ERGs and LRGs, respectively, exhibited strong ATM dependence (<30% of WT expression in Atm−/− cells) (Figure 2A, columns 9–15; Figure 2C; Figure S2). Consistent with the ATM-dependence of the IFNAR-dependent LRGs, Ifnb1 mRNA also exhibited strong ATM-dependence (Figure S3C).
One challenge in interpreting the Atm−/− data is that many genes, especially LRGs, exhibited significantly increased basal transcript levels in unstimulated Atm−/− cells (Figure 2A, columns 9 and 16). The high basal levels were usually maintained in the irradiated cells but without further induction (Figure 2A, columns 9–14). These results are consistent with previous evidence that ATM plays an important role in minimizing basal activation of an IFN response by the cytosolic DNA sensory pathway that relies on STING (Hartlova et al., 2015). Consistent with predictions, Ifnb1 mRNA was slightly increased in the Atm−/− cells prior to irradiation (Figure S3C); however, basal Ifnb1 transcript levels were generally very low and their measurement was less reliable than the measurement of mRNAs for IFN target genes. To summarize, ATM was critical for the induction of many radiation-induced genes, but its absence also led to the aberrant induction of the IFN gene program both before and after irradiation.
Despite the complexity of the Atm−/− data, a quantitative analysis of each LRG revealed that ATM was consistently critical for the radiation-induced component of the transcript level for each gene (Figure 2D). For this analysis, the LRGs were first separated into two groups: those with a greatly elevated basal transcript level in Atm−/− cells (left), and those with a relatively normal basal transcript level (right). Then, the basal and radiation-induced transcript levels for each gene in Atm−/− cells were compared to the radiation-induced transcript level in WT cells. The results revealed that ATM was important for the radiation-dependent component of the induction of each LRG, after taking into consideration the impact of ATM deficiency on basal transcript levels.
These results support previous evidence that DNA damage is a critical sensor for IR, and that ATM plays a dominant role in the inflammatory response induced by IR in BMDMs. Importantly, several ERGs exhibited little or no ATM dependence, and a subset of the LRGs exhibited only partial ATM dependence, demonstrating the involvement of other radiation sensors.
STING and DNA-PKcs Contribute to Basal Expression of IFNAR-dependent LRGs, but not to IR-Induced Transcription
The finding that ATM plays a critical role in the IFN response is interesting to consider in relation to previous findings suggesting that double-strand breaks induced by IR activate an IFN response via the STING pathway (Hartlova et al., 2015). At face value, these results suggest that ATM and STING may function together to promote an IFN response. However, one limitation of the prior study is that it lacked a time course to distinguish the impact of STING on basal expression from its impact on radiation-induced transcription.
To examine the role of STING in the radiation response in greater depth, BMDMs from WT and StingGt/Gt mice (which cannot signal through STING) were collected 0, 0.5, 1, 2, 6, and 24 hr after irradiation, followed by RNA-seq analysis. The results revealed strongly reduced basal transcript levels for most of the IFNAR-dependent, ATM-dependent LRGs before irradiation (0-hr time point) and between 0.5 and 2 hrs post-irradiation (Figure 3A, columns 1–4, 11–14, 18). Since these time points precede the induction of LRGs, the results support prior evidence that tonic activation of a Type I IFN response is due to STING activation by cytosolic nucleic acids (Hartlova et al., 2015). Surprisingly, however, the STING deficiency did not reduce radiation-induced transcript levels at the 6- and 24-hr time points (Figure 3A, columns 5, 6, 15–17; Figure 3B). This effect on basal but not induced transcript levels in the StingGt/Gt cells was highlighted in the gene expression kinetics for three representative IFNAR-dependent LRGs (Figure 3D). Thus, despite its important role in regulating the tonic IFN response, STING does not appear to contribute to radiation-induced transcription.
Figure 3. Roles of STING and DNA-PKcs in Basal and Radiation-Induced Gene Expression.

(A) Similar to Figure 2A, columns 1–10 display the expression kinetics of each of the 99 induced genes, as well as the impact of ATM and IFNAR deficiency on their basal and maximum expression. Columns 11–16 display expression kinetics in StingGt/Gt cells (percent expression relative to WT). Column 17 and 18 display peak and basal expression (low basal highlighted) in StingGt/Gt cells (percent relative to WT). Column 19 and 20 display peak and basal expression in Scid macrophages. All values represent averages from 2 independent RNA-seq experiments performed with different BMDM preparations; each experiment with mutant BMDMs was performed with a parallel and full time-course with WT BMDMs.
(B) A dot plot displays the dependence of each ERG, IFNAR-D and IFNAR-I LRG on STING.
(C) A dot plot displays the dependence of each ERG, IFNAR-D and IFNAR-I LRG on DNA-PKcs.
(D) Gene expression kinetics are shown for selected IFNAR-dependent genes (Irf7, Ccl5, Ifi44) in WT and StingGt/Gt cells. The maximum RPKM in WT cells equals 100%.
(E) A dot plot displays the basal expression level of IFNAR-D LRGs in Atm−/−, Ifnar−/−, StingGt/Gt, and Scid cells (% relative to WT basal expression level).
(F) The graph shows a gene-by-gene analysis of the basal expression levels of IFNAR-D LRGs in Atm−/−, Ifnar−/−, StingGt/Gt, and Scid cells (percent relative to WT basal expression level). The results show the proportional effects of IFNAR, STING, and SCID deficiency, and the inversely proportional effect of ATM deficiency. See also Figure S4.
We also examined BMDMs from Scid mice, which contain a point mutation in the gene encoding the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), which has been reported to contribute to STING-dependent transcription (Ferguson et al., 2012; Dempsey and Bowie, 2015). Consistent with the published reports and with our findings, reduced basal transcript levels were observed for most IFNAR-dependent genes (Figure 3A, column 20). However, the Scid mutation had little effect on the radiation-induced transcript levels for these genes (Figure 3A, column 19; Figure 3C).
A closer examination of basal transcript levels for IFNAR-dependent LRGs in the Atm−/−, Ifnar−/−, StingGt/Gt, and Scid macrophages revealed variable but consistent increases in Atm−/− cells, with variable but consistent decreases in the other three mutants (Figure 3E). Interestingly, ordering the 43 IFNAR-dependent LRGs according to the magnitude of the elevated basal transcript levels in Atm−/− cells revealed that the magnitude of elevation was roughly proportional to the magnitude of suppression in the other three mutants (Figures 3F, S4). Suppression was less pronounced in the Scid cells than in the Ifnar−/− or StingGt/Gt cells.
Together, these results suggest that the tonic IFN response observed in cultured BMDMs requires a pathway that includes STING, DNA-PK, and IFNAR, with ATM keeping this tonic IFN response at a low level. However, STING and DNA-PK do not contribute to the radiation-induced transcription of LRGs. Furthermore, despite ATM’s role in suppressing the tonic IFN response, it is critical for the potent induction of IFNAR-dependent genes by IR.
Differential Roles of IRF3 and IRF1 in Tonic and Radiation-Induced IFN Responses
We next examined BMDMs deficient in the gene encoding the transcription factor IRF3, which is known to be critical for Ifnb1 induction by multiple pathways, including the STING and TRIF pathways (Liu et al., 2015; Tamura et al., 2008). Consistent with the important role for STING in the tonic expression of IFNAR-dependent genes, the basal expression of these genes was reduced in Irf3−/− BMDMs (Figure 4A). In fact, the impact of IRF3 deficiency on the basal transcript level for each LRG correlated with the impact of STING deficiency (Figure 4B; Figure S4). However, IRF3 did not participate in the radiation-induced transcription of the IFNAR-dependent LRGs 24 hrs after irradiation (Figure S2). We also examined BMDMs lacking the gene encoding mitochondrial antiviral-signaling protein (MAVS), which contributes to the IFN response to intracellular RNA (Yoneyama et al., 2015); by qRT-PCR, no effect on either the tonic or radiation-induced transcript levels for representative LRGs was observed in Mavs−/− cells (data not shown).
Figure 4. Basal and Radiation-Induced Expression of IFNAR-Dependent LRGs Depend on IRF3 and IRF1, Respectively.

(A) A dot plot displays basal expression of IFNAR-dependent LRGs relative to WT in Atm−/−, Ifnar−/−, StingGt/Gt and Irf3−/− BMDMs. All values represent averages from 2 independent RNA-seq experiments performed with different BMDM preparations.
(B) The graph shows a gene-by-gene analysis of the basal expression levels of IFNAR-D LRGs in Atm−/−, StingGt/Gt and Irf3−/− BMDMs. The results show the proportional effects of STING and IRF3 deficiency, and the inversely proportional effect of ATM deficiency.
(C) Similar to Figure 2A, columns 1–8 display the expression kinetics of each of the 99 induced genes, as well as the impact of ATM and IFNAR deficiency on their maximum expression. Columns 9–14 display the expression kinetics of each gene in Irf1−/− cells (percent expression relative to WT). Column 15 displays peak expression in Irf1−/− cells (percent relative to WT). All values represent averages from 2 independent RNA-seq experiments performed with different BMDM preparations; each experiment with mutant BMDMs was performed with a parallel and full time-course with WT BMDMs.
(D) The scatter plot shows comparative maximum expression relative to WT of ERGs, IFNAR-D LRGs, and IFNAR-I LRGs in Irf1−/− (y-axis) and Ifnar−/− (x-axis) BMDMs.
(E) UCSC Genome Browser tracks from IRF3 and/or IRF1 ChIP-seq experiments are shown for the Ifnb1 locus in BMDMs treated with 6 Gy IR (top) and Lipid A (bottom). Antibodies used and time points are shown at the left).
In contrast to the results obtained with Irf3−/− BMDMs, RNA-seq analysis of BMDMs lacking another IRF family member, IRF1, revealed a critical role for this factor in the induction of IFNAR-dependent LRGs by IR (Figure 4C,D). The IRF1 dependence reflected an IRF1 requirement for induction of the Ifnb1 gene itself (Figure S3D). Although IRF3 and IRF7 are often responsible for the induction of Ifnb1 transcription, IRF1 plays a partial role in Ifnb1 induction in other settings (Tamura et al., 2008). To examine whether IRF1 might serve as a direct activator of Ifnb1 transcription in response to IR, ChIP-seq was performed with antibodies directed against IRF1 and IRF3 in BMDMs collected 0, 1, 2, and 6 hrs after irradiation. IRF1 but not IRF3 bound the Ifnb1 promoter in response to IR (Figure 4E, top), whereas IRF3 bound the promoter following LPS stimulation (Figure 4E, bottom). Thus, there appears to be selectivity in terms of the IRF family member that is activated by each insult.
To summarize, these results suggest that IRF1 acts downstream of ATM in response to IR and serves as a direct activator of Ifnb1, thereby promoting a potent Type I IFN response. However, the pathway regulating this IR response activity of ATM is entirely distinct from the pathway regulating ATM’s tonic function in cells that have not been exposed to IR, and in IR-treated cells monitored at early time points (0–2 hrs).
ERK and p38 MAP Kinases Regulate Many ATM-Dependent and -Independent ERGs
The above results provide insight into the roles of ATM, IRF1, IFNAR, IRF3, STING, and DNA-PK in regulating LRGs. Importantly, 24 of the 51 ERGs also exhibited strong ATM-dependence. To better define the requirements for induction of the 51 ERGs, WT macrophages were irradiated in the presence of the ERK inhibitor, PD0325901, and the p38 inhibitor, BIRB0796, either alone or together. RNA was then isolated 0, 0.5, 1, and 2 hrs after irradiation and examined by RNA-seq. The individual inhibitors had relatively modest effects on their own, with far more pronounced effects when the inhibitors were combined (Figure S5A). These findings suggest considerable redundancy between the two MAPKs, similar to our previous observations in Lipid A-treated BMDMs (Tong et al. 2016).
The analysis in Figure 5A (column 2) demonstrates that the radiation-induced expression of 16 of the 51 ERGs was strongly inhibited by the combined MAPK inhibitors. Only 6 of these genes also exhibited strong dependence on ATM. Moreover, 19 of the ATM-dependent genes exhibited little or no dependence on the MAPKs. These results suggest that one or more sensors of IR exposure in addition to ATM must exist that can activate MAPKs and contribute to the transcriptional output. For the genes that exhibited dependence on both ATM and MAPKs, ATM may serve as the critical activator of the MAPKs (Tang et al., 2002), or a different sensor may be responsible for the MAPK-dependence of these genes.
Figure 5. Roles of MAPKs and NRF2 in the Induction of ERGs.

(A) An expression heat map shows peak expression of ERGs in Atm−/− cells, WT cells pretreated with both Erk and p38 inhibitors, Nrf2−/− cells, WT pretreated with NAC, and Irf1−/− cells. The genes were separated into 7 classes on the basis of their dependencies. All values represent averages from 2 independent RNA-seq experiments performed with different BMDM preparations; each experiment with mutant or inhibitor-treated WT BMDMs was performed with a parallel and full time-course with WT BMDMs or with control solvent-treated WT BMDMs, respectively. See also Figure S5.
(B) A scatter plot shows the maximum expression of ERGs, IFNAR-dependent LRGs, and IFNAR-independent LRGs in Nrf2−/− BMDMs (y-axis) and in WT BMDMs pretreated with NAC (x-axis).
(C) The expression kinetics of the 5 genes exhibiting the greatest NRF2-dependence is shown in WT and Nrf2−/− BMDMs and in WT BMDMS treated with NAC.
Interestingly, only one ERG, Irg1, exhibited IRF1 dependence (Figure 5A, column 5). In Lipid A-stimulated macrophages, Ifnb1 and Irg1 were among a small group of 9 strongly induced primary response genes that exhibited strong IRF3 dependence (Tong et al. 2016). In response to IR, both of these genes instead depend on ATM and IRF1 for their induction (although, as mentioned above, Ifnb1 is not among the group of 51 ERGs because it did not reach the 3 RPKM threshold needed for inclusion in this group).
ROS Act through NRF2 to Regulate Five Radiation-Induced ERGs
The above results left us with 17 of 51 ERGs and 5 of 48 LRGs that do not exhibit strong dependence on any of the pathways we have evaluated. ROS are known to be additional sensors of IR exposure (Anuranjani and Bala, 2014). To examine the role of ROS in the response to IR, we pre-treated BMDMs with the ROS scavenger, N-acetyl-L-cysteine (NAC), for 1 hr prior to irradiation. The cells were then collected 0, 0.5, 1, 2, 6, and 24 hr after irradiation, followed by RNA-seq analysis. Only two ERGs, Slc7a11 and Chac1, exhibited strong sensitivity to NAC (<30% of maximum RPKM), even at a high dose (1 mM; Figure 5A–C). A third ERG, Trib3, just missed the 30% cut-off for classification as a NAC-sensitive gene (Figure 5B,C).
The transcription factor NRF2 is arguably the most compelling mediator of the anti-oxidant responses (Anuranjani and Bala, 2014). RNA-seq was therefore performed with WT and Nrf2−/− BMDMs collected 0, 0.5, 1, 2, 6, and 24 hrs after irradiation. The greatest decreases in radiation-induced transcription were observed with precisely the same three genes that exhibited the highest sensitivity to NAC: Slc7a11, Chac1, and Trib3 (Figure 5A–C). Two additional members of the Slc7a family, Slc7a1 (43% of WT) and Slc7a5 (34% of WT), exhibited transcript levels that were reduced to a more moderate extent in the Nrf2−/− cells (Figure 5A–C). These results confirm the importance of ROS signaling in the radiation response in BMDMs. The results also demonstrate that NRF2 is largely responsible for mediating the ROS response. However, the results show that ROS plays a dominant role in the activation of a surprisingly small number of potently induced genes. Notably, the 5 genes regulated by NRF2 encode functionally linked proteins (see Discussion).
A Critical Role for p53 in IR-Specific Inflammatory Gene Activation in Comparison to Lipid A-Induced Inflammation
A major goal is to understand how a transcriptional response is tailored to a stimulus, given that many stimuli induce common pathways such as the NF-κB, MAPK, and IRF pathways. With this in mind, we compared our RNA-seq data sets from irradiated BMDMs with data sets from BMDMs stimulated with Lipid A, which activates TLR4 (Tong et al. 2016). Forty-two of the 51 radiation-induced ERGs, and 45 of the 48 LRGs, were also induced by Lipid A (Figure 6A). The strong overlap is not surprising, as both ATM and TLR4 are potent inducers of NF-κB, MAPKs, and either IRF1 or IRF3. Nevertheless, a careful comparison of the responses to the two stimuli allowed us to gain critical insights into the stimulus-specificity of the radiation response.
Figure 6. Identification and Analysis of 12 IR-Specific Genes: Roles of NRF2 and p53 in IR Specificity.

(A) A dot plot displays the Log2 fold induction by Lipid A and IR for 42 commonly induced ERGs and 45 commonly induced LRGs.
(B) The graph displays the maximum fold induction of the 12 IR-specific genes by IR and Lipid A.
(C) A heat map shows the expression kinetics (percent of maximum expression) for the 12 IR-specific genes following treatment with IR (column 1–6) and Lipid A (column 7–11). Columns 12–15 highlight the dependence of each IR-specific ERG on ATM, MAPKs, NRF2 and ROS.
(D) The scatter plot at the left shows the maximum transcript levels for commonly induced ERGs in p53−/− (y-axis) and Atm−/− (x-axis) BMDMs. The two genes containing p53 binding sites within 1 kb of the TSS are highlighted in blue (see Figure 7). The center plot displays maximum transcript levels for IR-specific ERGs in p53−/− (y-axis) and Atm−/− (x-axis) BMDMs. NRF2-independent genes are highlighted in blue. The right plot displays maximum transcript levels for IFNAR-dependent LRGs, commonly induced IFNAR-independent LRGs, and IR-specific IFNAR-independent LRGs in p53−/− (y-axis) and Atm−/− (x-axis) BMDMs.
Specifically, even though Lipid A was a more potent inducer of most genes that were induced in common by Lipid A and IR (Figure 6A), 9 IR-induced ERGs and 3 IR-induced LRGs exhibited little or no induction in response to Lipid A (Figure 6B). Each of these 12 genes was induced less than 3-fold by Lipid A and its induction magnitude by radiation exceeded its Lipid A induction magnitude by at least 3-fold (Figure 6B). Notably, 4 of the IR-specific ERGs were from the group of 5 NRF2-dependent genes (Figure 6C, top); the fifth NRF2-dependent gene, Slc7a11, was strongly induced by Lipid A, presumably via an NRF2-independent mechanism (data not shown). All 5 of the remaining IR-specific ERGs were included in the “unknown” gene cluster for which no dependencies were identified (see Figure 5A). The 3 IR-specific LRGs were from the small group of 5 IFNAR-independent LRGs.
During a search for factors that may regulate these 8 IR-specific genes (5 ERGs and 3 LRGs), the transcription factor p53 emerged as a candidate due to evidence from ChIP-seq experiments in MEFs that p53 can bind in close proximity to a few of these genes (Kenzelmann Broz et al., 2013). p53 is activated by DNA damage via ATM, with a critical role of activated p53 in blocking cell cycle progression at the G1-S checkpoint by up-regulation of cell cycle check point inhibitor, p21 (Vousden and Prives, 2009).
To evaluate a possible role for p53 in the radiation response in BMDMs, WT and p53−/− BMDMs were examined by RNA-seq, with cells collected 0, 0.5, 1, 2, 6, and 24 hrs after irradiation. Strong p53 dependence (<30% of WT RPKM in p53−/− cells) was observed with 4 of the 5 IR-specific ERGs that did not exhibit NRF2 dependence (Figure 6D, middle), as well as 2 of the 3 IR-specific LRGs (Figure 6D, right)). Interestingly, only 2 of the other genes from the full set of 99 radiation-induced genes, Cdkn1a (p21) and Mdm2, exhibited p53 dependence (Figure 6D, left). These two genes, which play central roles in p53’s functions as an inhibitor of the G1-S transition and in negative feedback regulation, were also induced by Lipid A (Tong et al., 2016).
Taken together, p53 and NRF2 explain the induction of 10 of the 12 IR-specific genes. Notably, weak ATM-dependence was observed at most of the p53-dependent genes, suggesting that ATM and another kinase may act with partial redundancy in irradiated cells to activate p53, consistent with previous evidence that ATM can phosphorylate and activate p53 (Banin et al., 1998; Canman et al., 1998)
To evaluate further the p53 dependence of this small group of genes, ChIP-seq was performed with antibodies directed against active p53 phosphorylated on serine 15 (p-p53). BMDMs were treated with a crosslinking agent 0, 0.5, 1, 2, and 6 hrs post-irradiation, followed by ChIP-seq analysis. The number of called peaks increased significantly by the 0.5-hr time point and remained high at the 1, 2, and 6-hr time points (Figure S5B). Approximately 35% of peaks were within 1 kb of an annotated TSS (Figure S5D), and 5,395 of the peaks were observed in at least 3 of the 4 samples between 0.5 and 6 hr (Figure S5B, right panel). We focused on these 5,395 reproducible peaks for the subsequent analysis.
An examination of the 99 radiation-induced genes revealed that only 12 exhibited called p-p53 peaks within 1 kb of their TSS; 6 of these peaks exhibited average scores of 69 or more, whereas the other 6 exhibited average scores of 16 or less (Figure 7B). The 6 peaks with the strongest scores were located within 1 kb of the TSSs for the 6 ERGs that exhibited strong p53 dependence, including the 4 p53-dependent, IR-specific ERGs (Fam212b, Slc19a2, Phlda3, and Sesn2) and the 2 p53-dependent ERGs induced by both IR and Lipid A (Mdm2 and Cdkn1a) (Figure 7B; see also Figure 6D). Although strong p-p53 binding by ChIP-seq was a strong predictor of p53 dependence, a strong motif score within 1 kb of the TSS was a poor predictor of both binding and p53 dependence (Figure 7C); most of the strong binding events coincided with strong motifs, but binding was not observed at most other promoters with similarly strong motifs. Notably, binding within 1 kb of the TSS was not observed at either of the p53-dependent LRGs (Figure 7B), suggesting that these genes are either indirect targets of p53 or that p-p53 binds distant enhancers for these genes.
Figure 7. A Precise Correlation between Strong p53 Binding within 1 kb of the TSS and Strong p53 Dependence of IR-Induced Genes.

(A) The heat maps summarize the expression kinetics of the 99 IR-induced genes in BMDMs treated with IR (columns 1–6) and Lipid A (columns 7–11). The impacts of the various perturbations examined in this study are also shown (columns 12–17). The genes are separated into distinct classes on the basis of their specific dependencies. All values represent averages from 2 independent RNA-seq experiments performed with different BMDM preparations; each experiment with mutant BMDMs was performed with a parallel and full time-course with WT BMDMs.
(B) The scatter plot shows the maximum transcript level for each of the 99 IR-induced genes in p53−/− BMDMs (y-axis), along with the p-p53 ChIP-seq peak score within 1 kb of the TSS for each gene. Note that the 6 ERGs exhibiting the strongest p53-dependence are the only genes exhibiting exceptionally strong p-p53 ChIP-seq peaks within 1 kb of the TSS. The 2 p53-dependent LRGs lack p-p53 binding sites within 1 kb of the TSS
(C) The scatter plot displays the strongest p53 binding site motif score within 1 kb of the TSS for each of the 99 IR-induced genes, along with the p-p53 ChIP-seq peak score within 1 kb of the TSS for each gene. See also Figures S5–S7.
The perfect correlation between the 6 p53-dependent ERGs and the 6 p-p53 ChIP-seq peaks with scores of 69 or greater was unexpected. Genome-wide, only 132 of the 5,495 reproducible ChIP-seq peaks exhibited average scores of 69 or greater, with only 48 of these 132 peaks within 1 kb of a TSS (Figure S5C,D). Another atypical yet common property was observed with the 6 p53-dependent ERGs: in each case, the p-p53 ChIP-seq peak was located downstream rather than upstream of the TSS, between +102 and +836, with the binding site either in the first intron of the gene or within the 5’ untranslated leader sequence (Figure S6, left and middle panels). Thus, the 6 genes that are most strongly activated by IR in a p53-dependent manner share common properties - unusually strong ChIP-seq peaks, with the peaks downstream of the TSS but within 1 kb.
DISCUSSION
Although a large number of sensors, signaling pathways, and transcription factors have been implicated in the response to IR, little information has been available regarding the degree to which each contributes to the transcriptional output. A more general deficiency exists in our knowledge of how each environmental insult elicits a tailored, stimulus-specific response. We found that ATM is a dominant regulator of approximately half of the ERGs, presumably via the activation of NF-κB and other inducible factors. ATM plays a dual role in opposing a tonic IFN response driven by STING, DNA-PK, and IRF3, and in activating IFN responses following IR treatment via IRF1. ROS activation of NRF2 serves as another important IR sensing pathway, although this pathway plays a dominant, non-redundant role at only a small number of potently induced genes. p53 is a dominant regulator of another small set of potently induced genes. The p53 target genes exhibited weak ATM dependence, suggesting that ATM and another kinase act with partial redundancy to induce p53. Notably, the NRF2 and p53 target genes account for almost all genes found to be induced in an IR-specific manner in comparison to lipid A.
A key feature of our approach, which allowed us to provide such precise connections between radiation-induced genes and their dominant regulators, was the focus on genes induced by at least 4-fold. A long-term goal is to understand the regulation of all genes that are induced by IR in a statistically significant manner, which includes many genes induced between 1.5- and 4-fold. However, efforts to mechanistically evaluate all statistically induced genes are fraught with challenges that currently are difficult to overcome in a meaningful way. In particular, when analyzing loss-of-function data, it is necessary to consider the continuum of effects that is typically observed. Induced genes rarely exhibit complete dependence on a particular pathway, generally leaving residual induction in mutant strains. For our studies, we considered a factor to be a “dominant” regulator of a gene if gene induction in a mutant strain was reduced to less than 30% of the induction level observed in WT cells. This approach is imperfect and further refinements will be required in the future. However, the challenge would have been greatly amplified if we had attempted to evaluate genes induced by only 1.5 or 2-fold. For example, when a gene is induced by only 2-fold, it is extremely difficult to evaluate with confidence the degree of dependence of the gene’s induction on a mutant factor? It therefore is preferable to focus only on the most potently induced genes, and to then build on the resulting framework in future studies.
The importance of these decisions is readily apparent from the studies of ROS/NRF2 and p53. Using stringent criteria, we identified a limited but clear set of IR-specific genes. The ROS/NRF2 and p53 pathways accounted for the induction of almost all of these genes, suggesting that they are the most important pathways that account for the specificity of the IR response. Remarkably, every potently induced p53 target gene contained an unusually strong p53 binding site, with the binding site located downstream of the TSS. Thus, although p53 binds thousands of genomic sites, the stability and location of the binding event may be important for potent regulation by p53. An examination of the RNA-seq and ChIP-seq data sets revealed other likely p53 target genes that are induced more weakly but with apparent p53 dependence (data not shown). These genes sometimes but not always contain strong p53 binding sites downstream and within 1kb of the TSS.
One important goal is to elucidate the logic through which molecular mechanisms of gene regulation are connected to biology. Do specific sensors, signaling pathways, and transcription factors activate genes with related biological functions and with a clear underlying logic? Strikingly, 4 of the 5 NRF-2 dependent genes – Slc7a1, Slc7a5, Slc7a11, and Chac1 – encode proteins that play important roles in the homeostasis of cellular glutathione and free radicals (Figure S7A), and two of them – Chac1 and Trib3 – encode proteins that regulate apoptosis. Thus, the potent activation of these 5 genes by NRF-2 may represent a coordinated and logical biological response to the generation of ROS by IR.
Most of the genes identified here as p53 targets have previously been described. Of interest, only 2 of the 6 strongly induced p53 target genes - Cdkn1a and Mdm2 – were also strongly activated by lipid A, presumably in a p53-independent manner. The other 4 exhibited strong dependence on p53 and were among the small group of IR-specific genes, suggesting that they may serve as high-confidence biomarkers for p53 activation. Literature search-based gene ontology analyses classified the 6 p53 target genes into multiple categories: inhibitor of cell growth and proliferation (Sesn2, Phlda3), vitamin-B1/Thiamin homeostasis (Slc19a2), pro-apoptotic/ tumor suppressor (Sesn2, Phlda3, Slc19a2), cell-cycle arrest (Cdkn1a), and negative regulation of p53 activity (Mdm2) (data not shown). Cdkn1a (Cyclin dependent kinase inhibitor/p21) is widely studied as an important mediator of cell cycle arrest induced by p53 (Deng et al., 1995; Gartel and Radhakrishnan, 2005), and Mdm2 is an ubiquitin ligase and a component of p53 auto-regulatory feedback network that binds to the transactivation domain of p53 to facilitates degradation and nuclear export (Chène, 2003).
Sestrin2 contributes to the tumor suppression function of p53 by negatively regulating the mTOR pathway (Budanov and Karin, 2008). Phlda3 also contributes to the tumor suppression and pro-apoptotic functions of p53 by inhibiting the Akt pathway (Kawase et al., 2009). Fam212b (Family of sequence homology 212 member b) is a poorly understood, p53-regulated gene (Allen et al., 2014), and Slc19a2 (Thiamine transporter, THTR-1) plays an important role in thiamin homeostasis (Lo et al., 2001).
One of the most surprising findings to emerge from this study was the strong dependence of Ifnb1 expression and the Type I IFN response on IRF1. Although the IFN response induced by IR was previously suggested to be activated by STING via IRF3, STING and IRF3 played an important role only in the tonic IFN response. Much remains to be learned about the complex regulation of the tonic IFN response, and also about the mechanism by which IR activates IRF1 in an ATM-dependent manner. Importantly, our findings suggest that IRF1 may be an attractive target for efforts to suppress potentially damaging IFN responses in irradiated cells.
To conclude, although many questions remain to be answered about the regulation of the 99 potently induced genes that were the focus of this study, as well as the large number of genes that are induced more weakly by IR, the study provides a strong foundation for an understanding of the IR response. One important direction for the future will be to add an additional layer of complexity by determining how the response is altered when cell cycle checkpoints are activated. It will also be important to examine the variability of the response in other mouse cell types, in human cells, and in more complex physiological settings. Finally, in addition to the dominant regulators identified in this study, the expression of each gene may be influenced to a lesser extent by additional sensors, signaling pathways, and transcription factors. By continuing to build on this foundation, a full understanding of the radiation response can eventually be obtained.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen Smale (smale@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
WT C57BL/6, Atm−/−, StingGt/Gt, Scid, Irf1−/−, and Trp53−/− mice were obtained from Jackson Laboratory. Myd88−/−Trif−/− bones were obtained from G. Barton (UC Berkeley). Ifnar−/− and Irf3−/−, mice were obtained from G. Cheng (UCLA). Nrf2−/− mice were maintained in our own colony at UCLA. All the mutant strains used in this study were backcrossed to a C57BL/6 background as stated by Jackson inventory and the donating investigators. Catalog numbers and specific sources of the mice are provided in the Key Resources Table. Mice were maintained and bred in UCLA’s mouse housing rooms that are certified to breed and house immunocompromised and conventional mice from approved vendors. These rooms were supplied with autoclaved cages and irradiated feed and excluded detectable pathogens. The mice were monitored by the UCLA Division of Laboratory Animal Medicine.
Bone Marrow-Derived Macrophages
BMDMs were prepared from 6–10 week-old male mice as described previously (Ramirez-Carrozzi et al., 2009; Bhatt et al., 2012). Briefly, mice were euthanized by placing in a CO2 chamber followed by cervical dislocation. The euthanized mice were then sterilized by spraying with 70% ethanol. To isolate bones, all legs of the mice were pinned with palms down and stretched. An incision was made in the skin near the abdomen and the skin was then cut along the hind leg to expose the muscle. Muscle tissue was removed from the femur and tibia bones and the femur was dislocated or cut from the hip joint. The femur and tibia were separated and cleaned, and the ends were cut, followed by insertion of a needle (25 gauge, 5/8 in) attached to 10 ml syringe. Cold PBS (phosphate-buffered saline; 10 ml per mouse) was passed through one end of the bones with the syringe to flush out the bone marrow clumps from another end into a petri dish. Clumps were dissociated with the same syringe (without the needle) by drawing and flushing the clumps into the petri dish 3–4 times. The single cell suspension was passed through a 70 µM cell strainer in a 15 ml falcon tube. Cells were pelleted at 1300 rpm (~1000 g) for 10 min. The cell pellet was suspended in 1 ml RBC lysis buffer (Sigma, #R7757) and incubated at room temperature for 5 min, then diluted with 9 ml cold PBS. After centrifugation at 1300 rpm (~1000g) for 10 min, the cell pellet was suspended in 10 ml BMDM media (20% FBS, 10% CMG condition medium [containing M-CSF], 1x Pen-Strep [Gibco, #15140-122], 1x L-Glutamine [Gibco, #25030-081], 0.5 mM sodium pyruvate [Gibco, #11360-070] in Dubecco’s Modified Eagle Medium). 10 µl was used for cell counting using a hemocytometer. Cells were diluted to 0.65 million/ml and were dispensed 2 ml per well in 6 well plates. Cells were incubated at 37ºC in a CO2 incubator to allow differentiation of bone marrow monocytes into BMDMs. BMDM medium was changed on day 4 and experiments were performed on day 6.
METHOD DETAILS
Irradiation
The BMDMs were irradiated on day 6 with 2, 6, or 20 Gy of IR (for 6 Gy, ~1.6 Gy per min with Gulmay X-ray unit). RNA was harvested in Tri reagent (Molecular Research Center Inc, #TR118). For inhibitor studies, BMDMs were preincubated for 1 hr with ROS scavenger NAC (100 µM or 1mM, Sigma), ERK inhibitor PD0325901 (5 µM, Sigma), or p38 inhibitor BIRB0796 (5 µM, AXON Medchem).
RNA-seq
Total RNA was prepared using RNAeasy kit (Qiagen). Strand-specific libraries were generated using 400 ng of total RNA using the deoxyuridine triphosphate (dUTP) method (Levin et al., 2010) and TruSeq stranded RNA Sample Preparation Kit (Illumina). cDNA libraries were single-end sequenced (50bp) on Illumina HiSeq 2000 and eq 4000 machines.
ChIP-seq
ChIP-seq for IRF-1 (Cell Signaling #8478S), IRF-3 (Santa Cruz # sc-9082 X) and Phospho-(ser-15)-p53 (Cell Signaling # 9284S) was performed as described (Barish et al., 2010), with optimization for each antibody. ChIP-seq libraries were prepared using the Kapa Hyper Prep Kit (Kapa Biosystems), followed by sequencing (Illumina HiS eq 4000).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Seq Analysis
Reads were aligned to the mouse genome (NCBI37/mm9 build) with TopHat v1.5 by allowing reads to be aligned once with up to two mismatches per read. SeqMonk (Babraham Bioinformatics) was used to quantify against the exons of Refseq genes and RPKM values for genes were calculated as described (Mortazavi et al., 2008). Briefly, RPKM equals the number of reads mapped to exons of a gene divided by the exon length in kb times the total number of mapped reads in the data set (in millions). All RPKMs used for the analysis represent averages of two or three independent biological replicates, except the 24-hr time point from Irf3−/− cells (only one RNA-seq experiment was performed with this time point, as qPCR validated the absence of an impact of the IRF3 deficiency). Each mutant or chemically inhibited sample was analyzed simultaneously with a WT/uninhibited sample treated in parallel. A gene was initially included in the analysis if it met all of the following criteria from three WT replicates: The maximum average RPKM reached 3 at any of the time points, the fold induction level reached 4-fold, and the induced expression level was consistently different from the basal level (P<0.01), as determined by the DESeq package in R Bioconductor (Anders and Huber, 2010). Heat maps were generated by transforming RPKM into percentage considering peak RPKM as 100%.
To determine the impact of a perturbation on the expression of a gene, the maximum RPKM in the mutant or chemically inhibited samples was converted to a percentage after considering the WT maximum RPKM to be 100%.
ChIP-seq Analysis
Reads were aligned to the mouse genome (NCBI37/mm9 build) with Bowtie2. Uniquely mapped reads were used for peak calling and peaks were further annotated to nearby genes using HOMER (Heinz et al., 2010). Peaks were called if the normalized signal was enriched more than 4 fold over input with a poison P value lower than 1e-4 and false discovery rate of 0.01. Homer (makeUCSCfile command line) was used to generate bedgraph files of uniquely mapped reads for UCSC genome browser visualization. For p-p53 ChIP-seq analysis, peaks from four replicates were compared and only those with peaks in at least 3 replicates within a 100 bp window (mergePeaks function of homer) were selected. All peaks were annotated to Refseq genes based on the closest TSS using Homer; peaks were further corrected upon manual inspection of genome browser tracks of selected genes. To represent peak distributions, Venn diagrams were created using jvenn tool (Bardou et al., 2014).
Motif Analysis
Position weight matrices (PWMs) for p53 from a Transfac motif database were used to report the best matching p53 motif within 1 kb of the TSS by Pscan (Zambelli et al., 2009); the strength of individual motif was represented by a numerical value scale of 0–100.
DATA AND SOFTWARE AVAILABILITY
The accession number for RNA-seq and ChIP-seq data reported in this paper is GEO: GSE100963.
Supplementary Material
Highlights.
Most IR-induced genes are dominantly regulated by one of a small number of sensors
IR and ATM activate an IFN response via an IRF1-dependent, STING-independent pathway
The tonic IFN response is activated by STING, DNA-PK, and IRF3, but limited by ATM
NRF2 activated by ROS and p53 largely explain the selectivity of the response to IR
Acknowledgments
We thank C. Garcia for maintaining mouse colonies, X. Liu for help with ChIP-seq optimization, L. Smale for designing the model figures, and the UCLA Broad Stem Cell Research Center Core for sequencing. This work was supported by NIH grants R01GM086372 (S.T.S.), K08AR066545 (P.O.S.), K08AR062593 (P.J.K.), and U19AI067769 (W.H.M).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
P.K.P. designed and performed experiments, analyzed the data, and wrote the manuscript. A.J.T. provide RNA-seq data sets from lipid A-stimulated BMDMs. P.O.S., P.J.K., K.S.I., and W.H.M. provided valuable guidance with the design and interpretation of experiments, with the computational analysis, and/or with preparation of the manuscript. S.T.S. designed experiments and wrote the manuscript.
References
- Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, Sullivan KD, Galbraith MD, Luo X, Kraus WL, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. ELife. 2014;3:e02200. doi: 10.7554/eLife.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anuranjani, null, Bala M. Concerted action of Nrf2-ARE pathway, MRN complex, HMGB1 and inflammatory cytokines - implication in modification of radiation damage. Redox Biol. 2014;2:832–846. doi: 10.1016/j.redox.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Bardou P, Mariette J, Escudié F, Djemiel C, Klopp C. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics. 2014;15:293. doi: 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, Dent AL, Tangirala RK, Evans RM. Bcl-6 and NF-κB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24:2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoletti-Stella A, Mariani E, Kurelac I, Maresca A, Caratozzolo MF, Iommarini L, Carelli V, Eusebi LH, Guido A, Cenacchi G, et al. Gamma rays induce a p53-independent mitochondrial biogenesis that is counter-regulated by HIF1α. Cell Death Dis. 2013;4:e663. doi: 10.1038/cddis.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585:1625–1639. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candéias SM, Testard I. The many interactions between the innate immune system and the response to radiation. Cancer Lett. 2015;368:173–178. doi: 10.1016/j.canlet.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chène P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat. Rev. Cancer. 2003;3:102–109. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Dong X, Luo M, Huang G, Zhang J, Tong F, Cheng Y, Cai Q, Dong J, Wu G, Cheng J. Relationship between irradiation-induced neuro-inflammatory environments and impaired cognitive function in the developing brain of mice. Int. J. Radiat. Biol. 2015;91:224–239. doi: 10.3109/09553002.2014.988895. [DOI] [PubMed] [Google Scholar]
- Gannon HS, Woda BA, Jones SN. ATM phosphorylation of Mdm2 Ser394 regulates the amplitude and duration of the DNA damage response in mice. Cancer Cell. 2012;21:668–679. doi: 10.1016/j.ccr.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J. and Galaxy Team. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Messina NL, Clarke CJP, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36:166–174. doi: 10.1016/j.immuni.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves AM, Johnston CJ, Misra RS, Williams JP, Finkelstein JN. Whole-Lung Irradiation Results in Pulmonary Macrophage Alterations that are Subpopulation and Strain Specific. Radiat. Res. 2015;184:639–649. doi: 10.1667/RR14178.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42:332–343. doi: 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslet L, Bay C, Nepper-Christensen S. Acute radiation syndrome (ARS) - treatment of the reduced host defense. Int. J. Gen. Med. 2012;5:105–115. doi: 10.2147/IJGM.S22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer MJ, Raulli R, DiCarlo-Cohen AL, Esker J, Hrdina C, Maidment BW, Moyer B, Rios C, Macchiarini F, Prasanna PG, et al. UNITED STATES DEPARTMENT OF HEALTH AND HUMAN SERVICES BIODOSIMETRY AND RADIOLOGICAL/NUCLEAR MEDICAL COUNTERMEASURE PROGRAMS. Radiat. Prot. Dosimetry. 2016;171:85–98. doi: 10.1093/rpd/ncw226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase T, Ohki R, Shibata T, Tsutsumi S, Kamimura N, Inazawa J, Ohta T, Ichikawa H, Aburatani H, Tashiro F, et al. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell. 2009;136:535–550. doi: 10.1016/j.cell.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–1031. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool J, Hamdi M, Cornelissen-Steijger P, van der Eb AJ, Terleth C, van Dam H. Induction of ATF3 by ionizing radiation is mediated via a signaling pathway that includes ATM, Nibrin1, stress-induced MAPkinases and ATF-2. Oncogene. 2003;22:4235–4242. doi: 10.1038/sj.onc.1206611. [DOI] [PubMed] [Google Scholar]
- Kozlov SV, Waardenberg AJ, Engholm-Keller K, Arthur JW, Graham ME, Lavin M. Reactive Oxygen Species (ROS)-Activated ATM-Dependent Phosphorylation of Cytoplasmic Substrates Identified by Large-Scale Phosphoproteomics Screen. Mol. Cell. Proteomics MCP. 2016;15:1032–1047. doi: 10.1074/mcp.M115.055723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Dimtchev A, Lavin MF, Dritschilo A, Jung M. A novel ionizing radiation-induced signaling pathway that activates the transcription factor NF-kappaB. Oncogene. 1998;17:1821–1826. doi: 10.1038/sj.onc.1202088. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. and 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu Y-T, Grishin NV, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- Lorimore SA, Coates PJ, Scobie GE, Milne G, Wright EG. Inflammatory-type responses after exposure to ionizing radiation in vivo: a mechanism for radiation-induced bystander effects? Oncogene. 2001;20:7085–7095. doi: 10.1038/sj.onc.1204903. [DOI] [PubMed] [Google Scholar]
- Mboko WP, Mounce BC, Wood BM, Kulinski JM, Corbett JA, Tarakanova VL. Coordinate regulation of DNA damage and type I interferon responses imposes an antiviral state that attenuates mouse gammaherpesvirus type 68 replication in primary macrophages. J. Virol. 2012;86:6899–6912. doi: 10.1128/JVI.07119-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Rashi-Elkeles S, Elkon R, Shavit S, Lerenthal Y, Linhart C, Kupershtein A, Amariglio N, Rechavi G, Shamir R, Shiloh Y. Transcriptional modulation induced by ionizing radiation: p53 remains a central player. Mol. Oncol. 2011;5:336–348. doi: 10.1016/j.molonc.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen EM, Day R, Singh VK. New approaches to radiation protection. Front. Oncol. 2014;4:381. doi: 10.3389/fonc.2014.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo I, Lee S, Kwak M-K. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxid. Med. Cell. Longev. 2016;2016:2428153. doi: 10.1155/2016/2428153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salkowski CA, Neta R, Wynn TA, Strassmann G, van Rooijen N, Vogel SN. Effect of liposome-mediated macrophage depletion on LPS-induced cytokine gene expression and radioprotection. J. Immunol. Baltim. Md 1950. 1995;155:3168–3179. [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- Sugihara T, Murano H, Nakamura M, Ichinohe K, Tanaka K. Activation of interferon-stimulated genes by gamma-ray irradiation independently of the ataxia telangiectasia mutated-p53 pathway. Mol. Cancer Res. MCR. 2011;9:476–484. doi: 10.1158/1541-7786.MCR-10-0358. [DOI] [PubMed] [Google Scholar]
- Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, Kidd VJ, Mak TW, Ingram AJ. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J. Biol. Chem. 2002;277:12710–12717. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- Victor CT, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Williams JP, Brown SL, Georges GE, Hauer-Jensen M, Hill RP, Huser AK, Kirsch DG, Macvittie TJ, Mason KA, Medhora MM, et al. Animal models for medical countermeasures to radiation exposure. Radiat. Res. 2010;173:557–578. doi: 10.1667/RR1880.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z-H, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015;32:48–53. doi: 10.1016/j.coi.2014.12.012. [DOI] [PubMed] [Google Scholar]
- Yoo SS, Jorgensen TJ, Kennedy AR, Boice JD, Shapiro A, Hu TC-C, Moyer BR, Grace MB, Kelloff GJ, Fenech M, et al. Mitigating the risk of radiation-induced cancers: limitations and paradigms in drug development. J. Radiol. Prot. Off. J. Soc. Radiol. Prot. 2014;34:R25–52. doi: 10.1088/0952-4746/34/2/R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli F, Pesole G, Pavesi G. Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res. 2009;37:W247–52. doi: 10.1093/nar/gkp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.