

Abstract
Balance of labile methyl groups (choline, methionine, betaine, and folate) is important for normal liver function. Quantitatively, a significant use of labile methyl groups is in production of phosphatidylcholines (PCs), which are ligands for the nuclear receptor LRH-1. We studied the role of LRH-1 in methyl-pool homeostasis and determined its metabolic effects using the methionine and choline deficient diet (MCD), which depletes methyl groups and results in a deleterious decrease in PC to phosphatidylethanolamine (PE) ratio. We found that MCD-fed liver-specific LRH-1 knockout mice (Lrh-1−/−) do not show the expected decreased methyl-pool and PC/PE ratio and are resistant to the hepatitis and fibrosis normally induced by the diet. Adaptive responses observed in WT mice on MCD diet are also observed in Lrh-1−/− mice on a normal diet. This includes reduced expression of the highly active methyltransferase Gnmt and the biliary phospholipid floppase Mdr2/Abcb4, resulting in reduced consumption of methyl groups and biliary PC secretion. In vitro studies confirm that Gnmt and Mdr2 are primary LRH-1 target genes. Additional similarities between hepatic gene expression profiles in MCD-fed WT and untreated Lrh-1−/− mice suggest that methyl-pool deficiency decreases LRH-1 activity, and this was confirmed by in vitro functional results in cells maintained in MCD media. We conclude that LRH-1 is a novel transcriptional regulator of methyl-pool balance. When the methyl-pool is depleted, decreased LRH-1 transactivation suppresses expression of key genes to minimize loss of labile methyl groups.
Keywords: methionine-choline deficient diet, non-alcoholic fatty liver disease, nuclear receptors, LRH-1, Mdr2, Gnmt
Introduction
Balance of labile methyl groups (choline, betaine, methionine, and folate) and a constant supply of methyl-donors (S-adenosylmethionine) are important for liver function (1, 2). Patients with chronic liver disease such as liver cirrhosis, alcoholic and non-alcoholic fatty liver disease (NAFLD) or hepatocellular carcinoma often have reduced levels of methyl-donors (3–5). In addition, diets deficient in labile methyl groups or genetical manipulation to decrease generation of methyl-donors result in liver injury in both humans and animal models, ranging from fatty liver to steatohepatitis and hepatocellular carcinoma (2, 6, 7). Conversely, dietary supplementation with methyl groups and -donors improves metabolic liver disease and function, which has been the basis for several clinical trials investigating the clinical effects of methyl-donors in humans with chronic liver disease (1). However, excess of methyl-donors can also lead to liver injury (1, 2). Thus, hepatic methyl-donor levels need to be maintained within a certain range, and either deficiency or excess can lead to abnormal liver function.
S-adenosylmethionine (SAM) is the principal methyl-donor for more than 200 methylation reactions. SAM is generated from methionine via the enzyme methionine-adenosyltransferase (Mat), which exists as multiple isoforms (1, 2). Methyltransferases (MTs) transfer methyl groups from SAM to several different substrates including DNA, RNA, proteins, phosphatidylethanolamine (PE), glycine and guanidinoacetate. For regeneration of SAM, input of labile methyl groups from either the choline-betaine pathway or from the folate cycle are required. The three most abundant MTs are glycine-n-methyltransferase (Gnmt), guanidinoacetate-n-methyltransferase (Gamt) and phosphatidylethanolamine-n-methyltransferase (Pemt), which generate sarcosine, creatine and distinct phosphatidylcholines (PCs) (8), respectively (1, 2). Gnmt is particularly important as it acts as a cellular buffer for maintaining constant cellular SAM levels (1, 2). Besides its best known role in transmethylation, SAM is linked to cysteine and glutathione biosynthesis via the transulfuration pathway, and is critically involved in polyamine biosynthesis and radical chemical reactions (1). The regulation of SAM homeostasis is thought to be primarily based on enzyme activation and inhibition by intermediates of SAM metabolism (2). Little is known on the potential impact of transcriptional regulation in SAM homeostasis.
Quantitatively, a significant use of labile methyl groups in liver is generation of PCs in transmethylation reactions using the Pemt pathway (9). Like the beneficial effects of supplementation of labile methyl groups, PC supplementation shows beneficial `lipotropic' effects on metabolic liver disease, as first described by Charles Best in the 1930's (10). These observations suggest that at least some of the lipotropic effects of labile methyl groups may be exerted by the action of PCs. 75 years later, PCs were identified as ligands for the nuclear receptor LRH-1/NR5A2 (11, 12) and the lipotropic effects of certain PCs have been shown to clearly depend on the presence of LRH-1 (12). However, it is not known whether changes in the methyl-pool affect LRH-1 signaling and if LRH-1 contributes to methyl-pool homeostasis.
To test the potential role of LRH-1 in methyl pool and PC metabolism, we employed the methionine-choline deficient diet (MCD) model, which markedly alters SAM homeostasis and PC pools and results in metabolic liver injury resembling non-alcoholic steatohepatitis (NASH) (7, 13, 14). We found that LRH-1 is a direct transcriptional regulator of methyl-donor homeostasis and biliary PC secretion as well as a regulatory target to maintain appropriate methyl pool levels.
Materials and Methods
For a more detailed description of methods used please see the supplementary material and methods section.
Animal studies and diets
Generation of Lrh-1 liver specific knockout (Lrh-1−/−) mice has been previously described (15). Age-matched floxed Lrh-1f/f littermates served as wildtype (WT) controls. Methionine choline deficient diet (MCD, TD.90262) and the corresponding amino acid control diet (chow, TD.94149) containing 350g/kg choline dihydrogen citrate and 8.2g/kg methionine were custom made by Harlan Laboratories, Madison, WI, USA. 8–12 weeks old male WT and Lrh-1−/− mice were fed either chow diet or MCD diet for 2 (acute effects on injury, SAM and PC metabolism) or 8 weeks (prolonged effects on fibrosis). Methods were approved by Baylor College of Medicine's Institutional Animal Care and Use Committee.
Cell Culture of AML-12 and C3HepG2 cells
Murine AML-12 and human C3A/HepG2 cells were kept in regular DMEM/F-12 media or MCD media (see above). All experiments using cell lines were run in triplicates and reproduced at least in two independent experiments.
Serum biochemistry
See supplementary material and methods section.
RNA isolation and RT-qPCR
See supplementary material and methods section.
Hydroxyproline assay
See supplementary material and methods section.
Lipidomics
See supplementary material and methods section.
Liquid Chromatography- Mass spectrometry
See supplementary material and methods section.
Isolation and determination of hepatic triglycerides, cholesterol and non-esterified fatty acids
See supplementary material and methods section.
H&E and Oil Red O staining
See supplementary material and methods section.
Bile flow and biliary phospholipid / bile acid measurement
See supplementary material and methods section.
Protein isolation and immunoblotting
See supplementary material and methods section. The following antibodies and conditions were used: Gnmt (AP1076b; 1:500; Abgent, San Diego, CA), β-Actin (sc-1616 HRP; 1:3000; Santa Cruz Biotechnology, Dallas, TX).
Microarray analysis
Microarray analysis was performed as described previously (15). Pooled total RNA from liver tissue of chow and MCD-fed WT and Lrh-1−/− mice (n=3 per group) was reverse transcribed and hybridized to the Illumina mouseRefseq-8v2 Expression BeadChip using standard protocols (Illumina, San Diego, CA). Chips were run in duplicates. Genes upregulated >0.80log2 (>1.74-fold) and downregulated >log2-0.80 (<0.57-fold) were regarded as significant and further analyzed.
Determination of LRH-1 binding sites
For genome-wide binding of LRH-1, we obtained a BED file from the lab of Tim Osborne (16) and annotated distance to TSS using Galaxy / Cistrome (www.cistrome.org).
siRNA knockdown of LRH-1 and DLPC treatment
See supplementary material and methods section.
Gnmt promoter and LRH-1 reporter uciferase assay and site directed mutagenesis
See supplementary material and methods section.
Chromatin immunoprecipitation (ChIP) for mouse and human LRH-1 binding sites
See supplementary material and methods section. Livers of WT and LRH-1−/− mice fed with either chow or 2 weeks of MCD diet (3 per group) and pooled tissue from human liver wedge biopsies, which served as normal control samples in a previous study (17), were processed for ChIP experiments using the ChIP-IT® High Sensitivity kit (Active Motif, Inc., Carlsbad, CA) and an anti-LRH-1 antibody (R&D Systems, Abingdon, UK, #PP-H2325-00) according the protocol. Primers for ChIP PCR are provides in suppl. table 2.
Statistical analysis
Numbers of mice or replicates for each group used in experiments are indicated in the figure legends. For statistical analysis, analysis of variance with Bonferroni posttesting (experiments with animals and primary hepatocytes) and Student t test (cell line experiments) was used (Sigmastat statistic program; Jandel Scientific, San Rafael, CA). A p value of less than .05 was considered significant. For statistical analysis of microarray experiments hypergeometric testing relative to the total numbers of transcripts and genes on the Illumina mouseRefseq-8v2 Expression BeadChip (see above) was performed using R software. Error bars represent means ± standard deviation.
Results
Lrh-1−/− mice are resistant to hepatitis and liver fibrosis induced by methyl-pool alterations
LRH-1 regulates diverse aspects of liver metabolism (12, 18, 19). Although, its natural ligand has not yet been identified, phospholipids (PL), in particular PCs are regarded as likely candidates (11, 12, 20). About 30% of total hepatic PC production is dependent on methyl pools via the Pemt pathway (9). The association between methyl-pool and PC production (21), along with PCs as potential LRH-1 ligands, suggests that LRH-1 may be involved in regulating methyl pool metabolism and/or that methyl pool alterations affect LRH-1 signaling. To explore this, we stressed WT and liver specific Lrh-1−/− mice with a diet completely depleted of methionine and choline, which are essential to feed and maintain methyl-pool cycling (Figure1a). MCD diet typically results in severe liver damage consisting of hepatic lipid droplet accumulation and liver inflammation at the early stage as well as liver fibrosis in later disease stages. Although there are important differences, particularly in insulin sensitivity, the phenotype of MCD-fed mice resembles non-alcoholic steatohepatitis (NASH) in humans (7).
Figure 1. Lrh-1−/− mice are resistant to hepatitis and liver fibrosis induced by methyl-pool alterations.

A. Schematic of methyl-group cycling. SAM, S-adenosylmethionine; Gnmt, glycine-n-methyltransferase; Gamt, guanidinoacetate-n-methyltransferase; Pemt, phosphatidylethanolamine-n-methyltransferase; phosphatidylcholine, PC; SAH, S-adenosylhomocysteine; MCD, Methionine-choline deficient diet; Mdr2/Abcb4, multidrug-resistance protein 2; PE, phosphatidylethanolamine; THF, tetrahydrofolate; B,C. Wildtype (WT) and liver specific LRH-1 knockout (Lrh-1−/−) mice were fed MCD diet for 2 weeks and markers of liver injury (ALT serum levels (B)) and inflammation (hepatic TNFα and Icam mRNA (C)) were measured. n=7-9 mice per group. D. Wildtype (WT) and liver specific LRH-1 knockout (Lrh-1−/−) mice were fed MCD diet for 8 weeks and markers of fibrosis (hepatic Col1a1 mRNA and hydroxyproline content) were determined. n=4 mice per group. * p<0.05, chow vs. MCD; # p<0.05, WT vs. Lrh-1−/− . Error bars represent means ± standard deviation.
2 weeks of MCD-diet induced macrovesicular steatosis in both WT and liver specific Lrh-1−/− mice to comparable amounts. There was no significant difference on macroscopic appearance (not shown), histological evaluation, Oil-Red-O staining or by quantitative measurements of hepatic triglyceride, cholesterol and free fatty acid levels (Suppl. 1a). Decline in body weight (p=0.054) and liver to body weight ratio were also similar between genotypes (Suppl. 1b) and food intake did not differ. However, the expected increase in serum levels of ALT, AST and LDH, all biochemical markers of liver injury, observed in WT mice was absent in Lrh-1−/− mice (Figure 1b and Suppl. 1c). mRNA levels of TNFα and Icam, indicators of liver inflammation, were also increased only in MCD WT mice (Figure 1c). To further test if differences in liver injury at early stages translate into differences in fibrotic response at later injury stages, we treated another set of mice with MCD diet for 8 weeks. mRNA levels of collagen1a1 as well as direct determination of the fibrotic component hydroxyproline in liver tissue showed increased fibrosis only in MCD-fed WT but not MCD-fed Lrh-1−/− mice (Figure 1d).
Taken together, liver specific Lrh-1−/− mice show markedly diminished liver injury in response to methyl pool depletion.
Lrh-1−/− mice maintain normal phospholipid composition and methyl pool when methyl-pool metabolism is altered
MCD diet significantly reduces PC-pools and decreases PC/PE ratio, which is critical to maintain membrane integrity (22, 23). We performed a comprehensive hepatic lipidomic analysis to evaluate whether differences in PL content or composition may account for differences in hepatic injury between MCD-fed WT and Lrh-1−/− mice. Total PC content was significantly decreased after MCD diet in WT and, to a lesser but not significant different degree, also in Lrh-1−/− mice (Figure 2a). Hepatic PC/PE ratio was markedly decreased in MCD WT mice and this response was significantly blunted in MCD Lrh-1−/− mice (Figure 2a). The most pronounced differences were observed for PC 18:0/18:2 classes (Suppl. Figure 2a). Lipidomics analysis also revealed that Lyso-PC levels, which are an indirect marker of cell stress, inflammation and injury (24) were only significantly elevated in MCD-fed WT mice, independently confirming differences in injury between genotypes (Suppl. 2b).
Figure 2. Lrh-1−/− mice maintain normal phospholipid composition when methyl-pool metabolism is altered.
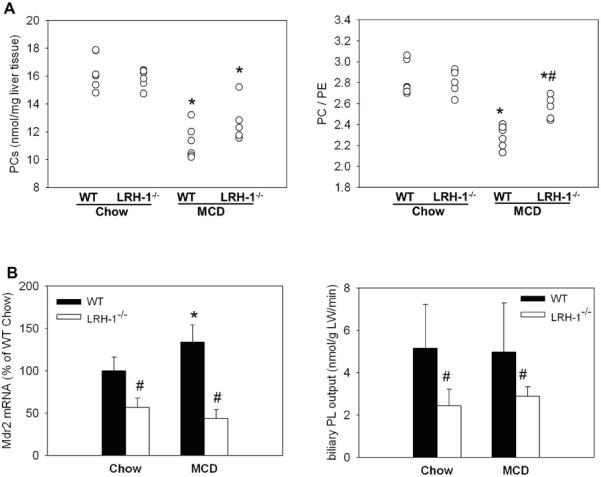
Wildtype (WT) and liver specific LRH-1 knockout (Lrh-1−/−) mice were fed MCD diet for 2 weeks. A. Comprehensive mass-spectrometry based lipidomics analysis of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) species. n=5-6 mice per group. B. Mdr2 mRNA levels (n=7-9 mice per group) and biliary phospholipid (PL) output (n=4-6 mice per group). * p<0.05, chow vs. MCD; # p<0.05, WT vs. Lrh-1−/−. Error bars represent means ± standard deviation.
A critical determinant of PC/PE ratio is multidrug resistance protein Mdr2/Abcb4, which shuttles PL from hepatocytes into bile. Various studies have reported that reduction of Mdr2 can stabilize PC/PE ratio and diminish injury resulting from MCD or depleted methyl-pools (22, 23). Chow- and MCD-fed Lrh-1−/− mice exhibited only 57% and 44% of Mdr2 mRNA expression compared to their WT littermates (Figure 2b). Functionally, reduced Mdr2 expression resulted in significantly lower biliary PL output in Lrh-1−/− mice than in WT animals (Figure 2b). This suggests that reduced expression and function of Mdr2 in Lrh-1−/− mice may contribute to reduced injury in MCD-fed Lrh-1−/− mice by stabilizing the critical PC/PE ratio.
MCD diet also significantly reduces methyl donors, particularly SAM. Imbalance in SAM, in particular reduced SAM levels, is a hallmark of several liver diseases including NASH (1). We performed another comprehensive metabolomic analysis of the methyl-pool to determine if additional differences in methyl-pool metabolites may also contribute to differences in injury (Suppl 3a). Levels of betaine, a direct product of choline, were robustly decreased to 26% and 32% in both WT and Lrh-1−/− mice after 2 weeks of MCD diet (Suppl. 3b). SAM levels and SAM to S-adenosylhomocysteine (SAH) ratio as an indicator of cellular methylation capacity were markedly decreased in MCD-fed WT livers after 2 weeks. In contrast, LRH-1−/− mice showed higher SAM and SAM/SAH starting levels (not significant) and maintained significantly higher SAM levels and SAM/SAH ratio after 2 weeks of MCD feeding. Of note, SAM levels and SAM/SAH ratio of MCD-fed LRH-1−/− mice were comparable to levels of normal WT mice (Figure 3a).
Figure 3. Lrh-1−/− mice maintain methyl-pools when methyl-pool metabolism is altered.
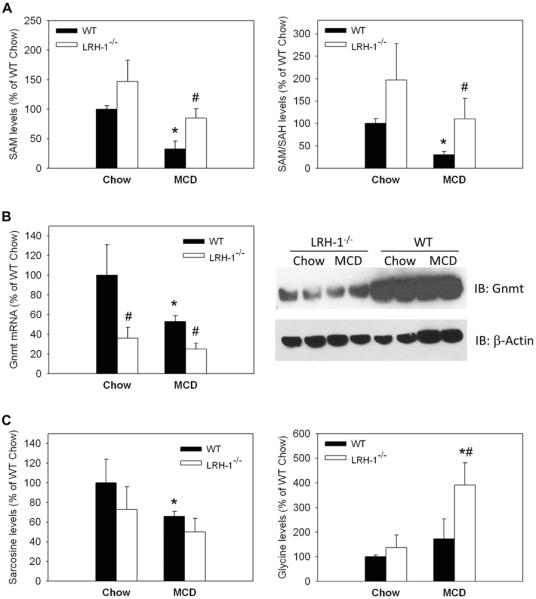
Wildtype (WT) and liver specific LRH-1 knockout (Lrh-1−/−) mice were fed MCD diet for 2 weeks. A. Mass-spectrometry based analysis of S-adenosylhomocysteine (SAM) and SAM / S-adenosylhomocysteine (SAH) ratio as readout for transmethylation capacity. n=3-4 mice per group. B. mRNA levels (n=7-9 mice per group) and immunoblot analysis (4 mice per group were pooled and blotted in duplicates) of the main hepatic methyltransferase Gnmt. C. Gnmt catalyzes the transmethylation of glycine into sarcosine. Mass-spectrometry based analysis of sarcosine and glycine. Sarcosine represents both sarcosine and alanine isomeric compounds. n=3-4 mice per group. * p<0.05, chow vs. MCD; # p<0.05, WT vs. Lrh-1−/−. Error bars represent means ± standard deviation.
LRH-1 is a transcriptional regulator of key enzymes of the methyl-pool cycle
A number of genes involved in methyl-pool metabolism were decreased in chow- or MCD-fed Lrh-1−/− mice (Suppl. 4). In particular, Gnmt was significantly underexpressed in Lrh-1−/− mice at both the mRNA and protein levels (Figure 3b). Gnmt is the most abundant hepatic methyltransferase and thereby a critical determinant of SAM usage. Metabolically, reduced Gnmt expression in Lrh-1−/− mice resulted in a trend towards reduction of its product sarcosine and accumulation of its starting metabolite glycine (Figure 3c and Suppl. 3a). Thus, reduced expression of Gnmt may contribute to reduced injury in MCD-fed Lrh-1−/− mice by maintaining SAM pools and methylation capacity.
We next evaluated whether the observed reduction of key genes of PC and methyl-pool metabolism, in particular Mdr2 and Gnmt, is a direct result of loss of LRH-1. siRNA-mediated knockdown of LRH-1 in the murine hepatocyte derived AML12 cell line resulted in significant reduction of Mdr2 and Gnmt mRNA (28% and 21% of si-scramble treated control cells, respectively) (Figure 4a). Conversely, treatment of AML12 cells with the specific LRH-1 ligand DLPC resulted in significant increase of Mdr2 and Gnmt mRNA levels (238% and 470% of vehicle treated control cells, respectively) (Figure 4b). Direct LRH-1 binding to the Gnmt promoter was significantly enriched 1.5-fold in LRH-1 ChIP-PCR experiments, with significantly less binding in WT MCD and no binding in LRH-1−/− mice (Figure 4c). In line with the ChIP-PCR results, cotranfection assays using the minimal −658kb promoter of Gnmt containing the LRH-1 binding site showed dose dependent LRH-1 transactivation. Specific mutation of the binding site at Gnmt −143 and −68 abolished LRH-1 responsiveness (Figure 4d). For the potential LRH-1 binding sites within the −1kb Mdr2 promoter region we could only detect modest binding by ChIP-PCR, and also only modest LRH-1 transactivation when using the −763 Mdr2 promoter region (not shown). We therefore conclude that the regulation of murine Mdr2 by LRH-1 may be indirect, with additional factors (e.g. bile acids) required, or that binding sites outside the screened minimal promoter are involved. Importantly, we did find direct binding of LRH-1 to the human GNMT and MDR3 promoter regions (Figure 4e), indicating that LRH-1 may also be an important regulator of human methyl-pool metabolism.
Figure 4. LRH-1 is a transcriptional regulator of key enzymes of the methyl-pool cycle.

A. AML-12 cells were treated with scrambled siRNA or a pool of 3 different Lrh-1 siRNAs for 24h. The classical transcriptional Lrh-1 target Cyp8b1 is significantly reduced in line with reduction of mRNA levels of Mdr2 and Gnmt. B. AML-12 cells were treated with vehicle or the LRH-1 ligand dilauroyl-sn-glycero-3-phosphocholine (DLPC) for 24h. mRNA levels of the classical Lrh-1 target gene Cyp8b1 are significantly increased in line with an induction of Mdr2 and Gnmt mRNA. n=triplicates. * p<0.05, vehicle vs. DLPC; # p<0.05, si-scrambled vs. si-Lrh-1. Error bars represent means ± standard deviation. C. Wildtype (WT) and liver specific LRH-1 knockout (Lrh-1−/−) mice were fed MCD diet for 2 weeks. LRH-1 ChIP-PCR for LRH-1 binding sites in the promoters of Cyp7a1, Cyp8b1 and Gnmt; n=3 mice per group. D. Luciferase assay using the minimal promoter of Gnmt (−661 to TSS) unmutated (WT Gnmt) or after mutation of the ChIPed binding sites at −143 (mutated Gnmt). E. LRH-1 ChIP-PCR from pooled liver tissue from normal control livers biopsies for classical LRH-1 target genes (left panel) and for human GNMT binding sites (right panel, black bars) and MDR3 binding sites (right panel, white bars). nc, negative control.
Analysis of a published murine LRH-1 ChIP-Seq dataset (16) showed LRH-1 binding sites at around −238 in the promoter of the Mdr2 gene and −79 in the Gnmt gene promoter (Table 1). Further in silico analysis of promoter regions of the most essential genes involved in methyl-pool metabolism (27 genes as referenced in recent extensive reviews (1, 2) as there is no separate gene ontology category for methyl-pool metabolism) revealed LRH-1 binding sites within 10kb of the transcription starting site for 74.1% of these methyl-pool cycle genes compared to 27.7% LRH-1 binding sites for the entire gene pool (p=6.57e–7 by hypergeometric-testing) (Table 1). Taken together, these results demonstrate that LRH-1 directly regulates multiple genes critically involved in PC and methyl-pool metabolism, suggesting a more general role for LRH-1 in regulating and maintaining methyl-pool metabolism.
Table 1.
Genes involved in methyl-pool/C1 metabolism with LRH-1 binding sites.
| Gene | TSS2pCenter | Gene | TSS2pCenter |
|---|---|---|---|
| Ada | - | Mthfd1 | −6 |
| Adk | - | Mthfd2 | +8330 |
| Ahcy | −4191 | Mthfr | −28|+2084 |
| Amd2 | −1145|+132 | Mthfs | - |
| Bhmt | −138 | Mtr | - |
| Bhmt2 | −18 | Mtr | - |
| Cbs | - | Pemt | −8721|9507 |
| Chdh | −3363|−9427|+13|+2876|+4206|+8940 | Sgms1 | −721|+1079 |
| Cth | +411 | Smpd1 | −66 |
| Dhfr | - | Srm | −201 |
| Gamt | −58|−5241|+3822|+9964 | Tyms | −124 |
| Gnmt | −79|−4161|+6930 | ||
| Mars1 | −40 | Nr0b2 | −209|−4217|−9950 |
| Mars2 | −54 | Cyp8b1 | −591−3536 |
| Mat1a | −7795 | Mdr2 | −238|−4727|−8665 |
| Mat2a | +5799 |
Note. The LRH-1 ChIP-Seq dataset published by Chong et al. (16) was used to screen for LRH-1 binding sites within 10kb of the transcription start site. Since there is no gene ontology category for C1 metabolism we used genes regarded to play key roles in C1 metabolism according to recent comprehensive reviews (1, 2). TSS2pCenter, transcription start site to center of peak.
Methyl pool depletion induces an LRH-1 antagonistic profile
To critically test the similarity of chow-fed Lrh-1−/− mice to MCD-fed WT, we compared their mRNA microarray signatures. 117 genes that changed in WT livers upon MCD-feeding (ΔMCD; 26.3% of all genes that changed upon MCD-feeding) were also changed in hepatic Lrh-1−/− mice (ΔLRH-1; 32.6% of all genes that changed in hepatic Lrh-1−/− mice compared to WT mice) yielding a highly significant overlap (p=1.17e–103). Genes that were upregulated by MCD diet (ΔMCD up; 68 out of 286, 23.7%) were likely to be upregulated in Lrh-1−/− mice (ΔLRH-1 up; 68 out of 201, 33.8%) and genes that were downregulated by MCD diet (ΔMCD down; 39 out of 159, 24.5%) were likely to be downregulated in Lrh-1−/− mice (ΔLRH-1 down; 39 out of 158, 24.7%) (Figure 5a). 61% of genes that were downregulated in Lrh-1−/− mice have LRH-1 binding sites within 10kb of the transcription start (p=1.31e–18) and this is shared by 48% of genes downregulated by MCD diet (p=3.36e–08) (not shown). Thus, MCD diet induces a transcriptional profile that is highly comparable to that of Lrh-1−/− mice.
Figure 5. Methyl-pool depletion induces an LRH-1 antagonistic profile.

A. mRNA microarray analysis for chow-fed WT and Lrh-1−/− as well as MCD-fed WT and Lrh-1−/− mice (3 mice per group were pooled and run in duplicates). Genes upregulated >0.80log2 (>1.74-fold) and downregulated >log2–0.80 (<0.57-fold) were further analyzed. ΔMCD = deregulated genes of WT chow vs. WT MCD; ΔLKO = deregulated genes of WT chow vs. Lrh-1−/− chow; B,C. C3HepG2 cells were transfected with a LRH-1 luciferase reporter, β-galactosidase and either LRH-1 construct or a corresponding empty vector (ev) and incubated for the indicated time points or concentrations with regular DMEM/F-12 media or MCD media. MCD media results in a time dependent decrease in LRH-1 luciferase reporter activity (B). After 12h incubation LRH-1 reporter activity shows a trend for reduced activity when 2/3 of regular media are substituted by MCD media and is significantly reduced when full MCD media is used (C). n=triplicates. D. C3HepG2 cells were incubated for 12h with regular control media or MCD media. mRNA levels of the classical LRH-1 target genes CYP8B1 and CYP7A1 as well as GNMT and GAMT are significantly downregulated. n=triplicates. * p<0.05, control media vs MCD media. Error bars represent means ± standard deviation.
To directly assess the impact of methyl-pool depletion on LRH-1 signaling we carried out transient transfections with an LRH-1 responsive luciferase reporter. MCD-media induced a striking time- and concentration-dependent decline in LRH-1 luciferase activity (Figure 5b and 5c). Importantly, MCD-media did not significantly affect viability and did not decrease transactivation by CAR as an example of another nuclear receptor (Suppl. Figure 4a and 4b). Moreover, MCD diet significantly reduced endogenous mRNA levels of classical LRH-1 target genes (i.e. CYP8b1, CYP7A1) and major methyl-pool metabolic genes (i.e. GNMT and GAMT) in C3AHepG2 cells (Figure 5d). Overall, these experiments indicate that methyl-pool depletion reduces LRH-1 signaling and that this in turn reduces transcription of several methyltransferases, the major methyl pool consuming enzymes.
Discussion
Homeostasis of methyl-donors is important for liver physiology, and is thought to be maintained via enzyme activation and inhibition by methyl-pool metabolites. Here, we describe a new level of transcriptional regulation by the nuclear receptor LRH-1. LRH-1 directly regulates expression of the most abundant methyltransferase, Gnmt, which balances SAM levels within a critical range. LRH-1 also regulates expression of the biliary phospholipid export floppase Mdr2, which channels biliary PC loss. When methyl pools are low, like in methionine and choline depleted states, LRH-1 signaling is down regulated, reducing SAM breakdown by suppression of Gnmt and biliary PC loss by suppression of Mdr2. Under harsh conditions of methionine and choline depletion, mice with genetic loss of LRH-1 are therefore resistant to detrimental effects of the MCD diet (Suppl. Figure 5).
MCD diet results in significant liver injury resembling non-alcoholic steatohepatitis (NASH) (7, 13). The resulting fat accumulation, which comprises a relatively benign component of injury, has been linked to the lack of choline (14, 25). The severe inflammatory/hepatitis aspect of injury, which leads to fibrosis and eventually cirrhosis, has been attributed rather to the lack of methionine and subsequent pronounced reduction of SAM and glutathione levels (14, 25). It is well known that the deleterious phenotype of MCD diet can completely be rescued by supplementing MCD-fed animals with SAM, highlighting its central role (14). The underlying hepatoprotective mechanisms of SAM include improved membrane fluidity, decreased tumor necrosis factor alpha expression, suppression of collagen synthesis by hepatic stellate cells, rise in mitochondrial GSH levels, change in DNA methylation, inactivation of CYP2E1 and protection against apoptosis (1, 26). We found that MCD-fed Lrh-1−/− mice have indistinguishable macrosteatosis compared to WT counterparts, yet are completely protected from hepatitis. This suggests that the higher SAM levels observed in Lrh-1−/− mice are an important part of the protective effect.
The three most abundant hepatic methyltransferases are Gnmt, Gamt and Pemt. We observed that both Gnmt and Gamt are significantly reduced in Lrh-1−/− mice under baseline chow fed conditions. The contribution of Gamt to overall SAM homeostasis is low (27), but Gnmt, comprising 1–3% of hepatic cytosolic protein, is critical for maintaining constant SAM levels (1). Gnmt knockout mice show markedly increased SAM, SAM/SAH levels and hyper-methylation capacity (28, 29). Gnmt knockout mice also exhibit fatty liver that is attributed to rerouting SAM into triglyceride synthesis via a novel pathway based on Pemt and PC breakdown, which may contribute to steatosis in MCD-fed Lrh-1−/− mice (21). When Gnmt knockout mice are fed a diet deficient in methionine SAM levels decrease/normalize and they are protected from steatotic liver injury (21). Our results suggest that LRH-1-dependent reduced expression of Gnmt may constitute a mechanism to maintain SAM levels and hepatic integrity in conditions when methyl-donors are scarce.
In response to the MCD, WT mice counteract further methyl-donor usage by downregulation of Gnmt and, at least transcriptionally, Gamt. This adaptive response of WT mice is similar to the basal state of Lrh-1−/− mice. More broadly, the global gene expression profile of MCD-fed WT animals indicates striking overlap with that of chow-fed Lrh-1−/− mice. A simple interpretation of this is that methionine-choline depletion may either deplete an endogenous LRH-1 agonist, or actively increase an endogenous LRH-1 antagonist. In either case, LRH-1 would be acting as both an active sensor and modulator of the methyl pool. Alternatively, LRH-1 activity may also be modulated post-translationally (30) independent of ligand by methyl-pool responsive pathways.
The importance of Mdr2 for PC-pool homeostasis is evident from the observation that feeding Pemt knockout mice a choline-depleted diet causes hyperacute liver failure within 3 days (22). These mice entirely lack the ability to generate PC from either the choline-dependent classical PC biosynthesis pathway or the alternative SAM-dependent Pemt pathway (9). However, the lethal phenotype is rescued by knockout of Mdr2, the “phospholipid floppase” transporter required for PC secretion into bile (22). These striking results are explained by the fact that the amount of PC in a mouse's normal daily biliary secretion is equivalent to the total pool of PC in its liver (31). In contrast to the very efficient recycling of bile acids, less than half of the biliary PC is returned to the liver (31), resulting in a significant net loss of methyl groups as well as fatty acids in the acyl side chains. Homozygous Mdr2 knockout mice almost completely lack biliary PC output and heterozygous Mdr2 knockout mice show a 50% decrease (32, 33). When heterozygous Mdr2 knockout mice were challenged with the MCD diet, they also showed significantly less liver injury (23). The inability to generate PC decreases the PC/PE ratio, which is important to maintain membrane fluidity and integrity. Choline-starved Pemt knockout mice experience a dramatic decrease in their PC/PE ratio, which is reversed when Mdr2 is knocked out (22). We found that Mdr2 transcripts are downregulated by 50% in Lrh-1−/− mice, and this was associated with a significant decrease in biliary phospholipid output. We also observed a significantly higher PC/PE ratio in MCD-fed Lrh-1−/− mice. This suggests that LRH-1- dependent reduction of Mdr2 is an additional component of the observed hepatoprotective response to methyl-pool deprived states.
PC synthesis by the Pemt pathway preferentially generates PC species rich in polyunsaturated fatty acids such as PC with (20:4) and PC with (22:6) (8, 34). However, the most pronounced differences in our lipidomic approach were in PC(18:0/18:2) classes, which are mainly synthesized by the CDP-choline pathway. The specific ligand of LRH-1, dilauryl-PC (DLPC), PC(12:0/12:0), is also not synthesized by the PEMT pathway. This argues against the Pemt pathway having an important role.
In contrast to the adaptive downregulation of the methyltransferases Gnmt and Gamt in MCD fed WT mice, we observed a modest increase in Mdr2 expression. The underlying molecular mechanism is not clear, but we speculate that increased bile acid levels upon MCD-feeding (35) may increase Mdr2 expression via the well-established effects of the bile acid receptor FXR on Mdr2 expression (36). Consistent with this, serum bile acid levels were modestly but significantly higher in MCD-fed WT mice (Suppl. figure 6a), which was paralleled by induction of adaptive hepatobiliary transporters (i.e. predominantly Mrp4 and Ostβ) (Suppl. figure 6b). Elevated serum bile acid levels may be secondary to steatohepatitis in MCD-fed WT mice (35) and therefore wold be expected to remain normal in MCD-fed Lrh-1−/− mice.
Although our results show a protective role of LRH-1 loss in the MCD model of NASH, the effects are specific to the this model and cannot be transferred to other models of NASH or NASH-induced fibrosis. These results also do not imply that LRH-1 ligands would aggravate NASH or NASH-fibrosis since it has clearly been shown that LRH-1 agonism improves stateatosis and insulin resistance in models of steatotic liver injury (12) and inhibits acute phase and inflammatory responses in the liver (37).
Taken together, we report a novel role for LRH-1 in transcriptional control of key genes of PC and methyl-pool metabolism. Methyl-pool depletion results in an LRH-1 antagonistic response which promotes maintenance of methyl-pools. Lastly, LRH-1 antagonists may allow adaption to methyl-pool depleted states and may represent a potential therapeutic direction for human liver diseases.
Supplementary Material
Acknowledgements
The current work was supported by the Metabolomics Core at Baylor College of Medicine, Houston, TX funded by the Cancer Prevention Research Institute of Texas (CPRIT)-RP120092 (to Arun Sreekumar and Nagireddy Putluri); MW and DDM were supported by an NIDDK NURSA (U19 DK62434) Cooperative Bridging Project grant. MW was supported by an Erwin Schrödinger Fellowship from the Austrian Science Fund FWF (J3119). SC was sponsored by the Program in Developmental Biology, Baylor College of Medicine, Houston, Texas 77030, USA.
Abbreviations
- Gamt
guanidinoacetate-n-methyltransferase
- Gnmt
glycine-n-methyltransferase
- LRH-1/NR5A2
liver receptor homologue-1
- MCD
methionine-choline depleted
- Mdr2/Abcb4
multidrug-resistance protein 2
- Mat
methionine adenosyltransferase
- MT
methyltransferase
- NAFLD
non-alcoholic liver disease
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- Pemt
phosphoethanolamine-n-methyltransferase
References
- 1.Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515–1542. doi: 10.1152/physrev.00047.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mato JM, Martinez-Chantar ML, Lu SC. Methionine metabolism and liver disease. Annu Rev Nutr. 2008;28:273–293. doi: 10.1146/annurev.nutr.28.061807.155438. [DOI] [PubMed] [Google Scholar]
- 3.Duce AM, Ortiz P, Cabrero C, Mato JM. S-adenosyl-L-methionine synthetase and phospholipid methyltransferase are inhibited in human cirrhosis. Hepatology. 1988;8:65–68. doi: 10.1002/hep.1840080113. [DOI] [PubMed] [Google Scholar]
- 4.Lu SC, Martinez-Chantar ML, Mato JM. Methionine adenosyltransferase and S-adenosylmethionine in alcoholic liver disease. J Gastroenterol Hepatol. 2006;21(Suppl 3):S61–64. doi: 10.1111/j.1440-1746.2006.04575.x. [DOI] [PubMed] [Google Scholar]
- 5.Kalhan SC, Edmison J, Marczewski S, Dasarathy S, Gruca LL, Bennett C, et al. Methionine and protein metabolism in non-alcoholic steatohepatitis: evidence for lower rate of transmethylation of methionine. Clin Sci (Lond) 2011;121:179–189. doi: 10.1042/CS20110060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeisel SH. Choline: critical role during fetal development and dietary requirements in adults. Annu Rev Nutr. 2006;26:229–250. doi: 10.1146/annurev.nutr.26.061505.111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 8.Watkins SM, Zhu X, Zeisel SH. Phosphatidylethanolamine-N-methyltransferase activity and dietary choline regulate liver-plasma lipid flux and essential fatty acid metabolism in mice. J Nutr. 2003;133:3386–3391. doi: 10.1093/jn/133.11.3386. [DOI] [PubMed] [Google Scholar]
- 9.Vance DE. Phospholipid methylation in mammals: from biochemistry to physiological function. Biochim Biophys Acta. 2014;1838:1477–1487. doi: 10.1016/j.bbamem.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Best CH, Huntsman ME. The effects of the components of lecithine upon deposition of fat in the liver. J Physiol. 1932;75:405–412. doi: 10.1113/jphysiol.1932.sp002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ortlund EA, Lee Y, Solomon IH, Hager JM, Safi R, Choi Y, et al. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat Struct Mol Biol. 2005;12:357–363. doi: 10.1038/nsmb910. [DOI] [PubMed] [Google Scholar]
- 12.Lee JM, Lee YK, Mamrosh JL, Busby SA, Griffin PR, Pathak MC, et al. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature. 2011;474:506–510. doi: 10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 14.Caballero F, Fernandez A, Matias N, Martinez L, Fucho R, Elena M, et al. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S-adenosyl-L-methionine and glutathione. J Biol Chem. 2010;285:18528–18536. doi: 10.1074/jbc.M109.099333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mamrosh JL, Lee JM, Wagner M, Stambrook PJ, Whitby RJ, Sifers RN, et al. Nuclear receptor LRH-1/NR5A2 is required and targetable for liver endoplasmic reticulum stress resolution. Elife. 2014;3:e01694. doi: 10.7554/eLife.01694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chong HK, Biesinger J, Seo YK, Xie X, Osborne TF. Genome-wide analysis of hepatic LRH-1 reveals a promoter binding preference and suggests a role in regulating genes of lipid metabolism in concert with FXR. BMC Genomics. 2012;13:51. doi: 10.1186/1471-2164-13-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marschall HU, Wagner M, Zollner G, Fickert P, Diczfalusy U, Gumhold J, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129:476–485. doi: 10.1016/j.gastro.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Lee YK, Moore DD. Liver receptor homolog-1, an emerging metabolic modulator. Front Biosci. 2008;13:5950–5958. doi: 10.2741/3128. [DOI] [PubMed] [Google Scholar]
- 19.Oosterveer MH, Mataki C, Yamamoto H, Harach T, Moullan N, van Dijk TH, et al. LRH-1-dependent glucose sensing determines intermediary metabolism in liver. J Clin Invest. 2012;122:2817–2826. doi: 10.1172/JCI62368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forman BM. Are those phospholipids in your pocket? Cell Metab. 2005;1:153–155. doi: 10.1016/j.cmet.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Una M, Varela-Rey M, Cano A, Fernandez-Ares L, Beraza N, Aurrekoetxea I, et al. Excess S-adenosylmethionine reroutes phosphatidylethanolamine towards phosphatidylcholine and triglyceride synthesis. Hepatology. 2013;58:1296–1305. doi: 10.1002/hep.26399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z, Agellon LB, Allen TM, Umeda M, Jewell L, Mason A, et al. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006;3:321–331. doi: 10.1016/j.cmet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 23.Igolnikov AC, Green RM. Mice heterozygous for the Mdr2 gene demonstrate decreased PEMT activity and diminished steatohepatitis on the MCD diet. J Hepatol. 2006;44:586–592. doi: 10.1016/j.jhep.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 24.Schmitz G, Ruebsaamen K. Metabolism and atherogenic disease association of lysophosphatidylcholine. Atherosclerosis. 2010;208:10–18. doi: 10.1016/j.atherosclerosis.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 25.Macfarlane DP, Zou X, Andrew R, Morton NM, Livingstone DE, Aucott RL, et al. Metabolic pathways promoting intrahepatic fatty acid accumulation in methionine and choline deficiency: implications for the pathogenesis of steatohepatitis. Am J Physiol Endocrinol Metab. 2011;300:E402–409. doi: 10.1152/ajpendo.00331.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]
- 27.Mudd SH, Brosnan JT, Brosnan ME, Jacobs RL, Stabler SP, Allen RH, et al. Methyl balance and transmethylation fluxes in humans. Am J Clin Nutr. 2007;85:19–25. doi: 10.1093/ajcn/85.1.19. [DOI] [PubMed] [Google Scholar]
- 28.Luka Z, Capdevila A, Mato JM, Wagner C. A glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic Res. 2006;15:393–397. doi: 10.1007/s11248-006-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, Martinez N, Varela M, Luka Z, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stein S, Oosterveer MH, Mataki C, Xu P, Lemos V, Havinga R, et al. SUMOylation-dependent LRH-1/PROX1 interaction promotes atherosclerosis by decreasing hepatic reverse cholesterol transport. Cell Metab. 2014;20:603–613. doi: 10.1016/j.cmet.2014.07.023. [DOI] [PubMed] [Google Scholar]
- 31.Li Z, Vance DE. Phosphatidylcholine and choline homeostasis. J Lipid Res. 2008;49:1187–1194. doi: 10.1194/jlr.R700019-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–481. doi: 10.1053/j.gastro.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 33.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 34.Pynn CJ, Henderson NG, Clark H, Koster G, Bernhard W, Postle AD. Specificity and rate of human and mouse liver and plasma phosphatidylcholine synthesis analyzed in vivo. J Lipid Res. 2011;52:399–407. doi: 10.1194/jlr.D011916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka N, Matsubara T, Krausz KW, Patterson AD, Gonzalez FJ. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology. 2012;56:118–129. doi: 10.1002/hep.25630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moschetta A, Bookout AL, Mangelsdorf DJ. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat Med. 2004;10:1352–1358. doi: 10.1038/nm1138. [DOI] [PubMed] [Google Scholar]
- 37.Venteclef N, Smith JC, Goodwin B, Delerive P. Liver receptor homolog 1 is a negative regulator of the hepatic acute-phase response. Mol Cell Biol. 2006;26:6799–6807. doi: 10.1128/MCB.00579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.