

Abstract
In humans, PCNA sliding clamps encircling DNA coordinate various aspects of DNA metabolism throughout the cell cycle. A critical aspect of this is restricting PCNA to the vicinity of its DNA target site. For example, PCNA must be maintained at/near primer/template (P/T) junctions during DNA synthesis. With a diverse array of cellular factors implicated, many of which interact with PCNA, DNA, or both, it is unknown how this critical feat is achieved. Furthermore, current biochemical assays that examine the retention of PCNA near P/T junctions are inefficient, discontinuous, qualitative, and significantly deviate from physiologically-relevant conditions. To overcome these challenges and limitations, we recently developed a novel and convenient FRET assay that directly and continuously monitors the retention of human PCNA at a P/T junction. Here we describe in detail the design, methodology, interpretation, and limitations of this quantitative FRET assay using the single strand DNA (ssDNA) binding protein, SSB, from Escherichia coli as an example. This powerful tool is broadly applicable to any ssDNA-binding protein and may be utilized and/or expanded upon to dissect DNA metabolic pathways that are dependent upon PCNA.
In humans, PCNA sliding clamps encircling DNA coordinate various aspects of DNA metabolism throughout the cell cycle, ranging from DNA replication and chromatin assembly to DNA damage tolerance and DNA repair pathways5. The highly conserved toroidal structure of the PCNA sliding clamp has a central cavity large enough to encircle double-stranded DNA (dsDNA) and slide freely along it6. Accordingly, PCNA is highly-mobile on DNA and, in the absence of any blocks, will rapidly diffuse away from its site of action7, 8. Hence, a critical aspect of PCNA-dependent DNA metabolism is restricting PCNA to the vicinity of its DNA target site. During DNA synthesis, PCNA must be maintained at/near primer/template (P/T) junctions and prohibiting diffusion of PCNA along the adjacent ssDNA is central to this1. Such activity is imperative not only for unperturbed DNA replication but also for DNA damage tolerance (DDT) and DNA damage repair pathways where ssDNA regions are generated that are kilobases long. However, with a diverse array of cellular factors implicated in these pathways, many of which interact with PCNA, ssDNA, or both, it is unknown how diffusion of PCNA along ssDNA is restricted for each. Deciphering how this critical feat is achieved is limited by the availability of assays that examine the retention of PCNA at/near P/T junctions
Current biochemical assays utilize circular plasmids and radioactive-labeled PCNA9–11 or PCNA antibodies12 to monitor the retention of PCNA on DNA after a fixed incubation period. These discontinuous assays are very time-consuming and inefficient. Furthermore, these assays often generate qualitative data and significantly deviate from physiological-relevant conditions. For instance, after incubations at room temperature or 37°C, reaction mixtures are chilled on ice and then analyzed at 4°C10, 12. To overcome these challenges and limitations, we recently developed a novel and convenient FRET assay that directly and continuously monitors the retention of human PCNA at a P/T junction. Here we describe in detail the design, methodology, interpretation, and limitations of this quantitative FRET assay using the single strand DNA (ssDNA) binding protein, SSB, from Escherichia coli as an example. This powerful tool is broadly applicable to any ssDNA-binding protein and may be utilized and/or expanded upon to dissect DNA metabolic pathways that are dependent upon PCNA.
Materials
Oligonucleotides
Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA) and purified on 19:1 Acrylamide:Bis-Acrylamide denaturing polyacrylamide gels (see main text for percentages) using the SequaGel® UreaGel System (National Diagnostics, Atlanta, GA). Oligonucleotides were extracted from gels by a modified “Crush and Soak” technique as follows. First, the DNA band of interest was excised from the gel with a razor blade and crushed into a 50 mL falcon tube by pushing it through a 10 mL sterile syringe. TE Buffer (10 mM TrisHCl, pH 8.0, 1 mM EDTA) was then added to the crushed gel pieces, the solution was vortexed, flash frozen in liquid N2, and rocked overnight at room temperature. Gel pieces were removed by a Steriflip® vacuum-driven filtration system (Millipore, Billerica, MA) and the filtered solution was desalted with a C18 reverse phase column (Sep-pak, Waters, Milford, MA). Concentrations were determined from the absorbance at 260 nm using the calculated extinction coefficients. For annealing, the primer strands were mixed with a stoichiometric amount of the corresponding, complementary template strands in 1X Annealing Buffer (10 mM TrisHCl, pH 8.0, 100 mM NaCl, 1 mM EDTA), heated to 95 °C for 5 minutes, and allowed to slowly cool to room temperature.
Proteins
Recombinant human proteins were utilized for all experiments within this study. Wild-type and a mutant PCNA that can be site-specifically labeled with a Cy5 dye were purified as described previously 2. A truncated form of RFC (hRFCp140ΔN555) described previously was used in all reported studies and is referred to as simply RFC throughout the text2. RPA was expressed and purified from E. coli and the concentration was determined from the reported extinction coefficient as described13. Recombinant single-strand DNA-binding protein from Escherichia coli (referred to as SSB) was purchased from Promega (Madison, WI). All concentrations of SSB referred to herein refer to the concentration of the homotetramer.
Fluorescence microscopy
All measurements were obtained in a 200 μL quartz cuvette (240 μL total reaction volume) utilizing a Jobin Yvon fluoromax-4 fluorimeter.
Results
Overview of the FRET Assay
The unique assays described herein enables the retention of human PCNA at a P/T junction to be directly and continuously monitored. These assays rely on FRET between Cy5 and Cy3 dyes. Specifically, Cy5-labeled PCNA can be excited through FRET from a Cy3-labeled P/T DNA substrate only when the two dyes are in close proximity (< ~10 nm). RFC loads PCNA onto a P/T junction such that the “front” face of the sliding clamp ring is oriented towards the P/T junction. The Cy5 FRET acceptor on PCNA is located on the “back” face of the sliding clamp ring and, hence, will face the Cy3 FRET donor at the duplex end of the P/T DNA substrate (Figure 1). Thus, an increase in the fluorescence emission intensity at 665 nm (Cy5 FRET acceptor fluorescence emission max, I665) and a concomitant decrease in the fluorescence emission intensity at 561 nm (Cy3 FRET donor fluorescence emission max, I561), i.e., a FRET signal, directly indicates that Cy5-PCNA is loaded onto a Cy3-labeled P/T DNA substrate.
Figure 1.
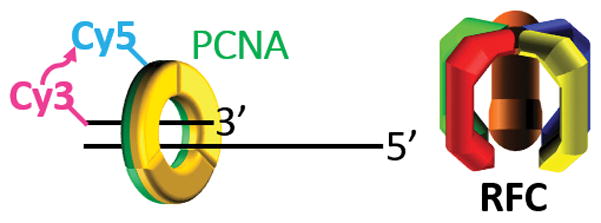
Schematic representation of a Cy5-labeled PCNA ring loaded onto a Cy3-labeled P/T DNA substrate by RFC. The front face (orange) of the PCNA ring faces the P/T junction. The Cy5 FRET acceptor is located on the back face (green) of the PCNA ring and faces the Cy3 FRET donor at the duplex end of the P/T DNA substrate.
Design of the FRET Pair
The FRET Acceptor: Cy5-labeled Human PCNA
The position of PCNA residue 107 within the homotrimeric PCNA clamp ring is structurally conserved among eukaryotes, solvent-exposed, and has been previously utilized in S. cerevisiae and human PCNA for labeling as a FRET acceptor through mutation to cysteine2, 14–16. Human PCNA contains six cysteine residues, only two (C27, C62) of which are surface-exposed2. This was confirmed by Ellman’s test (data not shown)17. Through site-directed mutagenesis, the surface-exposed cysteines of human PCNA were changed to methionine (C27M, C62M) and asparagine 107 was changed to cysteine (N107C)2. These mutations do not affect the interaction of PCNA with RFC or the ability of RFC to load PCNA onto DNA (Figure S1)2. This mutant PCNA (C27M/C62M/N107C) can be site-specifically labeled with a Cy5 FRET acceptor as follows.
Add TCEP (750 μM) to mutant PCNA (150 μM monomer) in labeling buffer (20 mM TrisHCl, pH 7.4, 0.2 M NaCl, 10% glycerol) and equilibrate mixture to room temperature.
Dissolve one Cy5 maleimide mono-reactive dye pack (1 mg, GE Healthcare) in 50 μL of anhydrous DMF and protect from light.
Add 12.5 μL of Cy5/DMF mixture (from step 2) to PCNA, cover mixture from light and rock at room temperature for 30 min.
Repeat Step 3 three more times (4 times total).
Incubate PCNA/Cy5 mixture at 4°C overnight with rocking
Separate Cy5-labeled PCNA from the free Cy5 dye by gravity size exclusion chromatography (SEC) using Sephadex G25 medium (GE Healthcare) prepared in labeling buffer (Column radius = 0.845 cm, Column height = 29 cm, Bed Volume = 65 mL).
Determine Cy5 labeling efficiency by absorbance according to the manufacturer’s protocol using ε280 = 13670 M−1cm−1 for PCNA.
The labeling efficiencies of three independent preparations were 38.3 +/− 1.19%, 35.5 +/− 0.713%, 38.8 +/−1.05% of PCNA monomers, indicating that each PCNA homotrimer has ~1.0 labeled monomer on average for the described procedure. It is not critical to attain this labeling efficiency as the number of Cy5 labels per PCNA homotrimer does not affect the interaction of PCNA with RFC or the ability of RFC to load PCNA onto DNA (Figure S1)2. Thus, for a given concentration of PCNA homotrimer, the labeling efficiency will only affect the maximal FRET observed in the experiments described below (see also Special Note below on labeling efficiency and FRET distance).
The FRET Donor: Cy3P/BioT DNA
The annealed DNA substrates utilized in the assay are comprised of primer and template strands that are each uniquely designed by considering the following (Figure 2);
Figure 2.

Cy3P/BioT DNA substrate for SSB. The duplex region is identical to that described previously for RPA1 and the size (29 bp) is in agreement with the requirements for assembly of a PCNA ring onto DNA by RFC2,3. The length (70 nt) of the 5′ polyT ssDNA overhang shown in red will accommodate only a single SSB molecule at 200 mM ionic strength and permits RFC-catalyzed loading of PCNA. When pre-bound to Neutravidin, the biotin attached to the 3′-end of the template strand prevents loaded PCNA from sliding off the duplex end. A Cy3 dye attached to the 5′end of the primer strand serves as the FRET donor.
The double-stranded DNA (dsDNA)/duplex region of the annealed P/T DNA substrate must accommodate the requirements for assembly of human PCNA onto DNA by human RFC2, 6. The complementary sequences of the primer and template strands determine the length of the dsDNA region. As such, the primer must be at least 25 nucleotides (nt) in length and the 3′ sequence of the template strand must be complementary to the full length of the primer strand.
-
The poly(dT) sequence of the template strand determines the length of the single-stranded DNA (ssDNA) region of the annealed P/T DNA substrate. This region is referred to herein as the 5′ poly(dT) overhang and must be long enough to accommodate only a single molecule of a given ssDNA-binding protein yet also permit RFC-catalyzed loading of PCNA onto DNA (see Special Note below). The poly(dT) sequence is recommended as it ensures proper annealing of the P/T DNA substrate and prohibits formation of secondary structures in the ssDNA region.
Special Note: the human PCNA clamp loader, replication factor C (RFC), binds to the P/T junction and adjacent ssDNA when loading PCNA onto DNA. Hence, this region of nucleic acid must be accessible to RFC2. This FRET assay was originally designed for human replication protein A (hRPA) and, hence, the ssDNA region was 33 nt in length. At physiological ionic strength (200 mM), hRPA binds to ssDNA with sub-pM affinity and low cooperatively18, 19. Under these conditions, the occluded binding site size is 30 +/− 2 nt and the interaction site size is 24 +/− 1 nt18, 19. The former is the length of ssDNA covered when hRPA binds and the latter is the length of ssDNA that directly interacts with hRPA. Furthermore, binding is not static. Rather, hRPA rapidly diffuses along ssDNA (D > 2800 +/− 200 nt2s−1 at 25°C for ssDNA less than 90 nts in length)20. Thus, on a Cy3P/BioT DNA substrate in which the 5′ poly(dT) overhang is 33 nt long, the P/T junction is accessible to RFC in the presence of RPA. Indeed, RFC-catalyzed loading of Cy5-PCNA onto this Cy3P/BioT DNA substrate was not affected by saturating hRPA20.
The primer strand serves as the FRET donor and, hence, is labeled with a 5′ terminal Cy3 dye.
Once loaded onto the annealed P/T DNA substrate, human PCNA may dissociate back into solution by diffusion along the ssDNA or the duplex region. To selectively monitor diffusion of human PCNA along the ssDNA adjacent to the P/T junction, a 3′-terminal biotin label may be attached to the template strand. When in complex with Neutravidin, biotin serves as a block that prevents loaded PCNA from sliding off the duplex end of the annealed P/T DNA substrate2.
Once designed and synthesized, the Cy3-labeled primer and biotin-labeled template should be purified via denaturing polyacrylamide gel electrophoresis (PAGE). The Cy3-labeled primer (a 29-mer) and biotin-labeled template (a 99-mer) strands depicted in Figure 2 were purified on 16% and 8% acrylamide monomer denaturing PAGE gels, respectively. Complete labeling of the primer strand with Cy3 can be verified using the extinction coefficients for the primer oligonucleotide sequence and the Cy3 label. Strand annealings should be carried out at a 1:1 ratio of Cy3-labeled primer to biotin-labeled template to ensure the poly(dT) sequence of the template strand is the only source of ssDNA. Herein, a Cy3-labeled primer annealed to a biotin-labeled template is referred to as a Cy3P/BioT DNA substrate.
The FRET assay described here was first developed for hRPA1 but is broadly applicable to any nonspecific ssDNA-binding protein, independent of binding mode and binding site size. In order to demonstrate this, we utilized the ssDNA-binding protein, SSB, from Escherichia coli as an example. SSB is a functional homolog of hRPA and is often utilized as an RPA surrogate for in vitro studies. SSB and RPA each bind ssDNA with extremely high affinity (in pM range) and rapidly diffuse along ssDNA. Both proteins display ionic strength-dependent transitions between ssDNA-binding modes that involve switches in the size of ssDNA occluded20. Furthermore, binding of either protein to ssDNA is non-cooperative at physiological ionic strength (200 mM)18, 19, 21–23. However, several key features distinguish these functional homologs. hRPA is a heterotrimer that binds ssDNA with an occluded site size of 30 +/−2 nt at physiological ionic strength18, 19. Furthermore, hRPA binds ssDNA in a relatively elongated manner, extending the bound ssDNA to within 10% of its full contour length 23–26. In contrast, SSB is homotetramer that binds ssDNA with an occluded site size of 65 nt at 200 mM ionic strength. In this binding mode, referred to as (SSB)65, the ssDNA is fully-wrapped around SSB, contacting each subunit within the homotetramer. This shortens the contour length of the bound ssDNA by more than 50%21, 25. Thus, SSB and RPA bind to ssDNA with contrasting binding modes (extended versus fully-wrapped) and binding site sizes (30 nt versus 65 nt).
Accordingly, a Cy3P/BioT DNA substrate was designed with a 70 nt long poly(dT) region that will accommodate only a single SSB molecule at 200 mM ionic strength yet permit RFC-catalyzed loading of PCNA onto DNA. The 3′ sequence of the template strand (29 nt) is complementary to the full length of the primer strand (29 nt). The primer and template strands are labeled with a 5′ Cy3 FRET donor and a 3′ biotin, respectively. The primer and template strands were annealed at a 1:1 ratio, yielding the Cy3P/BioT70 DNA substrate (Figure 2). The duplex region (29 bp) is identical to that described previously for RPA1 and is in agreement with the requirements for assembly of a PCNA ring onto DNA by RFC2, 3.
Qualitative assay to probe the retention of PCNA on a P/T DNA substrate by a ssDNA-binding protein
As an initial test of whether a given ssDNA-binding protein, such as SSB, retains Cy5-PCNA on a Cy3P/BioT DNA substrate, FRET can be qualitatively monitored in a stepwise manner (Figure 3A), as follows:
Figure 3.
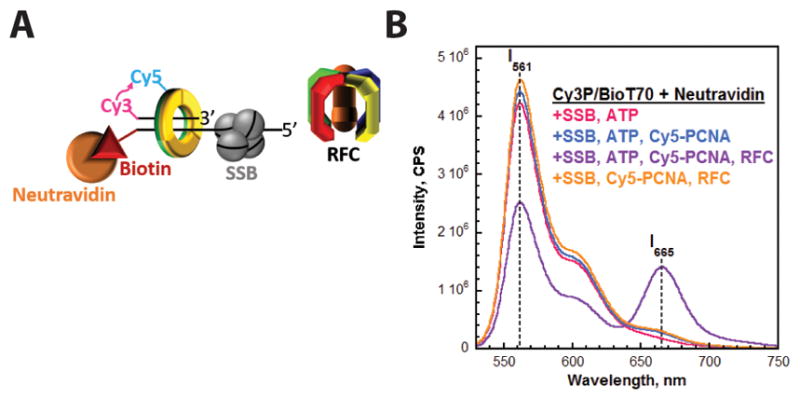
Monitoring the retention of PCNA on DNA through FRET. (A) Schematic representation of PCNA encircling a P/T junction bound by SSB. (B) Fluorescence emission spectra in the presence of SSB. Cy3P/BioT70 DNA (100 nM), Neutravidin (400 nM), ATP (1 mM), and SSB (200 nM) were pre-equilibrated at 25°C. Cy5-PCNA (110 nM homotrimer) and RFC (110 nM) were sequentially added, the solution was excited at 514 nm, and the fluorescence emission spectra was recorded from 530 to 750 nm. The fluorescence emission intensities at 665 nm (Cy5 FRET acceptor fluorescence emission max, I665) and 561 nm (Cy3 FRET donor fluorescence emission max, I561) are indicated. Cy5-PCNA can be excited through FRET from Cy3P/BioT70 only when the two dyes are in close proximity (<~10 nm). This is indicated by an increase in I665 and concomitant decrease in I561.
All experiments are performed at room temperature (23±2 °C) in 1X Tris buffer (25 mM TrisOAc, pH 7.7, 10 mM Mg(OAc)2, 125 mM KOAc) supplemented with 0.1 mg/mL BSA and 1 mM DTT and the final ionic strength is adjusted to physiological (200 mM) by the addition of appropriate amounts of KOAc.
All assay solutions contain a Cy3P/BioT DNA (100 nM Cy3P/BioT70), Neutravidin (400 nM), and excess of a ssDNA-binding protein (200 nM SSB). To ensure saturation of the ssDNA, the concentration of the respective ssDNA-binding protein should be at least 10-fold above its KD for ssDNA.
To this solution, ATP (1 mM), Cy5-PCNA (110 nM) and RFC (110 nM) are sequentially added. Cy5-PCNA is in slight excess (10%) of the Cy3P/BioT DNA substrate to maximize the FRET signal. RFC will not load more than one Cy5-labeled PCNA ring onto a P/T DNA substrate in which the duplex region is 25 to 29 bp1, 2.
Two minutes after the addition of RFC, the solution is excited at 514 nm (5 nm slit width) and the fluorescence emission spectra is recorded (5 nm slit width) from 530 to 750 nm.
For each condition described below, steps are 2 – 4 are repeated except certain reaction components are omitted from step 3.
As depicted in Figure 3B, a peak at 665 nm is not observed when both Cy5-PCNA and RFC are omitted (magenta trace). A very small, broad peak is observed at 665 nm when only RFC is omitted (blue trace) and this is due to direct excitation of Cy5-PCNA. When both RFC and Cy5-PCNA are present (purple trace), a sharp, intense peak is observed at 665 nm and the fluorescence peak at 561 nm is concomitantly and substantially decreased, indicative of FRET. RFC-catalyzed loading of PCNA absolutely requires ATP6. Indeed, the observed FRET signal disappears when ATP is removed (orange trace), confirming that Cy5-PCNA encircling the Cy3P/BioT70 DNA substrate is responsible for the observed FRET signal. Under these conditions, RFC immediately dissociates into solution after hydrolyzing ATP and releasing a closed Cy5-PCNA ring onto the duplex region of a Cy3P/BioT DNA substrate1. Hence, the only pathway for dissociation of Cy5-PCNA from a Cy3P/BioT DNA substrate in the presence of Neutravidin is diffusion of PCNA off the ssDNA end. Accordingly, a FRET signal will only be observed if a ssDNA-binding protein prohibits diffusion of PCNA along the ssDNA adjacent to the P/T junction. Altogether, these qualitative results indicate that SSB, like RPA, stabilizes/retains PCNA at a P/T junction by prohibiting diffusion of the sliding clamp ring along the adjacent ssDNA.
Quantitative assays to decipher how a ssDNA-binding protein retains PCNA on a P/T DNA substrate
A ssDNA-binding protein may prohibit diffusion of loaded PCNA along the ssDNA adjacent to a P/T junction by one of three pathways; 1) Hold RFC at a P/T junction with PCNA10, 12; 2) Concurrently bind PCNA and the adjacent ssDNA (or P/T junction), tethering loaded PCNA to a blocked P/T junction or; 3) Bind tightly to the ssDNA adjacent to a P/T junction and physically block diffusion of loaded PCNA along ssDNA. In pathway 3, diffusion of loaded PCNA is only prohibited on the ssDNA adjacent to a P/T junction whereas PCNA diffusion is prohibited in both directions in the pathways 1 and 2. Such behaviors can be exploited in quantitative, steady-state FRET assays to discern between these pathways for a given ssDNA-binding protein. Two such assays are described in detail below utilizing SSB as an example. At physiological ionic strength (200 mM), SSB binds to ssDNA with extremely high affinity (pM) in the fully-wrapped mode but does not bind RFC nor PCNA10, 21. Thus, one would hypothesize that SSB retains PCNA at a P/T junction via pathway 3.
Titrations of the steady-state FRET signal
All experiments are performed at room temperature in 1X Tris buffer supplemented with 0.1 mg/mL BSA and 1 mM DTT and the final ionic strength is adjusted to physiological (200 mM) by the addition of appropriate amounts of KOAc.
All assay solutions contain a Cy3P/BioT DNA (100 nM Cy3P/BioT70), Neutravidin (400 nM), and ATP (1 mM). The Cy3-labeled primer and biotin-labeled template are annealed at a 1:1 ratio and SSB has an occluded binding site size of 65 nt at physiological ionic strength (200 mM)21. Hence, the concentration of SSB binding sites is identical to the concentration of the Cy3P/BioT70 DNA substrate (100 nM).
To this solution, a ssDNA-binding protein (0 – 180 nM SSB), Cy5-PCNA (110 nM), and RFC (110 nM) are sequentially added. RFC and PCNA are present in slight excess (10%) to ensure saturation of the DNA. See Special Notes below on the concentration of ssDNA-binding protein and the observed FRET values.
Two minutes after the addition of RFC (t = 2 min), the solution is excited at 514 nm and the fluorescence emission spectra is recorded from 530 to 750 nm. For a given concentration of ssDNA-binding protein, the FRET magnitude must be reproducibly quantified from the recorded spectra. As described above, FRET is indicated by an increase in I665 and a concomitant decrease in I561. Hence, the FRET magnitude, F, is equal to the ratio of I665 to I561 (F = I665/I561). This method is more reproducible than monitoring the absolute values for I561 and/or I665 as these values fluctuate significantly between replicates due to cuvette handling.
Spectra are recorded every minute until a constant FRET magnitude is maintained for at least two minutes. Within this plateau region, the reaction has reached equilibrium where PCNA loading and dissociation are equal and a net change in FRET is no longer observed. In other words, the concentration of the Cy5-PCNA·Cy3P/BioT DNA complex remains constant and, hence, the observed FRET represents the steady-state FRET.
Data points (at least two) within the plateau region are averaged to obtain the final steady-state FRET value
-
For each concentration of ssDNA-binding protein, steps 2 – 6 are carried out in triplicate and the average steady-state FRET (and standard deviation) is calculated. The concentration of ssDNA-binding protein should be increased until the FRET is saturated.
Special Note: At physiological ionic strength (200 mM), SSB binds to ssDNA with extremely high affinity (KD ~ pM)21. Thus, binding of SSB is stoichiometric at 100 nM Cy3P/BioT70 (~103 higher than the KD of SSB for ssDNA). Under these conditions, FRET should increase linearly until the concentrations of the ssDNA-binding protein and the Cy3P/BioT DNA substrate (i.e., ssDNA binding sites) are equal. At this point, referred to as the “breakpoint,” the FRET plateaus and remains constant thereafter, indicating that the Cy3P/BioT70 DNA substrate is saturated with Cy5-PCNA1. On the other hand, if the KD of the respective ssDNA-binding protein is comparable to the concentration of ssDNA binding sites (100 nM), high concentrations of the ssDNA-binding protein are required to achieve saturation (~10-fold higher than its KD). Under these conditions, FRET should increase with ssDNA-binding protein and display hyperbolic behavior, indicative of equilibrium binding.
Special Note: If the labeled PCNA homotrimer contains more than one Cy5 FRET acceptor (i.e., labeling efficiency > 33.33%), the maximal FRET values observed cannot be used to accurately determine the absolute distance between the FRET dyes. When multiple Cy5 FRET acceptors are available for each Cy3 FRET donor, the probability of energy transfer and, hence, FRET increases. Hence, the inflated FRET values would underestimate the distance between the FRET dyes under these conditions27.
The average steady-state FRET is plotted versus the concentration of the single-strand DNA binding protein.
As observed in Figure 4, the FRET increased linearly with SSB concentration up to ~100 nM, indicative of stoichiometric (SSB)65 binding, and plateaued thereafter. At the breakpoint where the Cy3P/BioT70 DNA substrate (100 nM) is saturated with Cy5-PCNA, the concentration of SSB (113 nM) is roughly equal to the concentration SSB binding sites in the assay (100 nM). Altogether, this suggests that tight binding of a single SSB molecule to the ssDNA region of the Cy3P/BioT70 DNA substrate stabilizes a Cy5-PCNA ring at the P/T junction by prohibiting diffusion of the sliding clamp ring along the adjacent ssDNA. Such behavior is consistent with all three pathways described above. These pathways can then be discerned by repeating steps 1 thru 8 with the following exceptions. In step 2, maintain the concentration of ssDNA-binding protein at a constant, saturating level (180 nM SSB). In step 3, utilize a range of RFC concentrations (27.5 nM – 110 nM).
Figure 4.
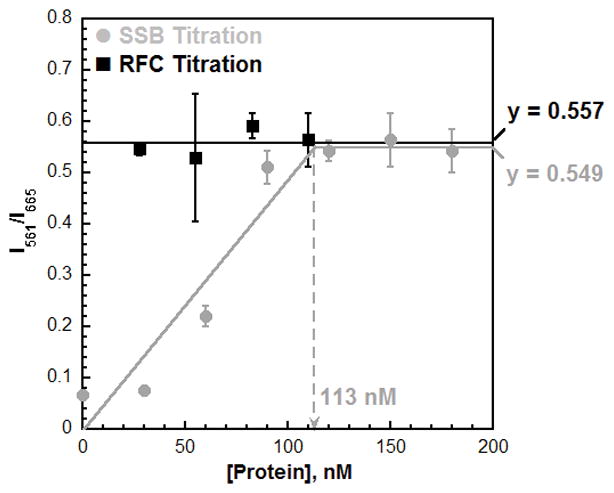
Titrations of the steady-state FRET. The Cy3P/BioT70 DNA substrate (100 nM with 400 nM Neutravidin), ATP (1 mM), and Cy5-PCNA were held constant (110 nM homotrimer) and either saturated with RFC (110 nM) and titrated with SSB (0 – 180 nM) (
 ) or saturated with SSB (180 nM) and titrated with RFC (30 – 110 nM) (·). Results are plotted versus the concentration of the respective titrant. When SSB was titrated, the FRET signal increased linearly with SSB concentration up to 90 nM. Data points within this concentration range (0 – 90 nM SSB) were fit to a linear equation. After 90 nM, the FRET plateaus and remains constant with SSB concentration. Data points within this concentration range (120 – 180 nM) were fit to a flat line. The two lines intersect at the “breakpoint” where the Cy3P/BioT70 DNA substrate is saturated with Cy5-PCNA. When RFC was titrated, the FRET signal remained constant at a level (0.557) equivalent to that observed at saturating concentrations of RFC and SSB (0.549). These FRET values are identical to those observed for RPA (Table S1). This suggests that Cy5-PCNA encircling a P/T junction is in the same FRET state when the adjacent ssDNA is bound by either RPA or SSB.
) or saturated with SSB (180 nM) and titrated with RFC (30 – 110 nM) (·). Results are plotted versus the concentration of the respective titrant. When SSB was titrated, the FRET signal increased linearly with SSB concentration up to 90 nM. Data points within this concentration range (0 – 90 nM SSB) were fit to a linear equation. After 90 nM, the FRET plateaus and remains constant with SSB concentration. Data points within this concentration range (120 – 180 nM) were fit to a flat line. The two lines intersect at the “breakpoint” where the Cy3P/BioT70 DNA substrate is saturated with Cy5-PCNA. When RFC was titrated, the FRET signal remained constant at a level (0.557) equivalent to that observed at saturating concentrations of RFC and SSB (0.549). These FRET values are identical to those observed for RPA (Table S1). This suggests that Cy5-PCNA encircling a P/T junction is in the same FRET state when the adjacent ssDNA is bound by either RPA or SSB.
In pathway 1 described above, a ssDNA-binding protein holds/traps RFC at a P/T junction with PCNA10, 12. On a Cy3P/BioT DNA substrate in which diffusion of Cy5-PCNA off the duplex end is prohibited by a biotin·Neutravidin complex, trapped RFC prevents Cy5-PCNA from sliding off the ssDNA end. Accordingly, RFC will not turnover (i.e., act catalytically) and, hence, a stoichiometric amount of RFC is required to load all Cy5-PCNA. On the other hand, if RFC is released into solution after loading Cy5-PCNA and the ssDNA-binding protein alone prevents Cy5-PCNA from sliding off the ssDNA end (by either pathway 2 or 3), RFC will act catalytically. As observed in Figure 4, the steady-state FRET was independent of RFC concentration and remained constant at a level (0.557) equivalent to that observed at saturating concentrations of RFC and SSB (0.549). This indicates that limiting concentrations of RFC can load all Cy5-PCNA onto the Cy3P/BioT70 DNA substrate in the presence of SSB. Thus, RFC is released into solution after loading Cy5-PCNA and SSB alone prevents Cy5-PCNA from sliding off the ssDNA end of the Cy3P/BiotT70 DNA substrate. Altogether, these steady-state FRET titrations effectively eliminate pathway 1 for SSB.
Effect of Neutravidin on the Steady-State FRET
In pathway 3 described above, diffusion of loaded PCNA is only prohibited on the ssDNA adjacent to a P/T junction whereas PCNA diffusion is prohibited in both directions in the pathways 1 and 2. Thus, for a given ssDNA-binding protein, pathway 3 can be distinguished on a Cy3P/BioT DNA substrate by characterizing the effect of neutravidin on the steady state FRET. To do so, the steady state FRET is calculated exactly as described above (Steps 1 – 8) for various conditions and the results are directly compared. The assay solution for each reaction condition is detailed below using SSB as an example.
Control Reaction 1 (CR1): Cy3P/BioT DNA (100 nM Cy3P/BioT70), Neutravidin (400 nM), ATP (1 mM), saturating ssDNA-binding protein (200 nM SSB), Cy5-PCNA (110 nM homotrimer) and RFC (110 nM). Under this condition, SSB stabilizes all loaded Cy5-PCNA on the Cy3P/BioT70 DNA substrate and a robust steady-state FRET is observed (CR1 in Figure 5). The observed value represents the FRET maximum.
Control Reaction 2 (CR2): The assay solution is identical to “Control Reaction 1” described above except RFC is omitted. Under this condition, Cy5-PCNA remains in solution and, hence, the observed FRET represents the FRET minimum from direct excitation of Cy5-PCNA (CR2 in Figure 5).
Control Reaction 3 (CR3). The assay solution is identical to “Control Reaction 1” described above except the ssDNA-binding protein of interest is omitted. Under this condition, loaded Cy5-PCNA immediately slides off the ssDNA end of the Cy3P/BioT70 DNA substrate in the absence of SSB and, hence, the observed FRET is identical to that observed for Control 2 above (CR3 in Figure 5). This confirms that binding of SSB to the ssDNA region of the Cy3P/BioT70 DNA substrate is required to stabilize Cy5-PCNA at the P/T junction.
Reaction (R). The assay solution is identical to “Control Reaction 1” described above except neutravidin is omitted. Under this condition, diffusion of Cy5-PCNA off the ssDNA end of the Cy3P/BioT DNA substrate is prohibited by the respective ssDNA-binding protein and, hence, the only pathway for dissociation of Cy5-PCNA is diffusion off the duplex end.
Figure 5.
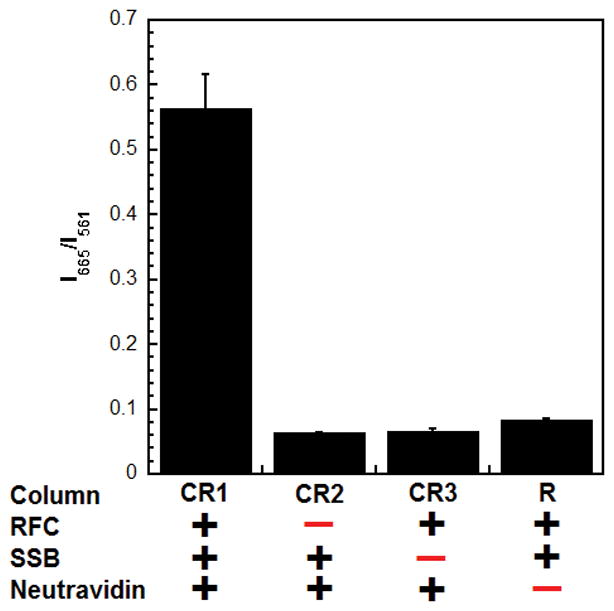
Characterization of the steady state FRET signal. Cy5-PCNA was assembled onto the Cy3P/BioT70 DNA substrate with various components omitted and the FRET signal (I665/I561) was measured.
For the “Reaction” condition, a steady-state FRET will only be observed if the respective ssDNA-binding protein indirectly (pathway 1) or directly (pathway 2) “holds” Cy5-PCNA at the P/T junction. However, FRET was not observed for SSB when Neutravidin was omitted (R in Figure 5). This effectively eliminates pathways 1 and 2 for SSB and confirms that the Neutravidin/biotin complex alone prohibits Cy5-PCNA from diffusing off the duplex end of the Cy3P/BioT70 substrate. Altogether, results from the various FRET assays described here indicate that SSB maintains human PCNA at a P/T junction by pathway 3 exclusively; SSB binds tightly to the ssDNA adjacent to a P/T junction and restricts PCNA to the upstream duplex region by physically blocking diffusion of the sliding clamp along ssDNA.
Limitations
The FRET assays described here are broadly applicable to any protein that binds to ssDNA in a sequence-independent manner (i.e., nonspecifically). However, care should be taken when analyzing ssDNA-binding proteins that also bind P/T junctions and/or PCNA as these proteins may interfere with RFC-catalyzed loading of PCNA onto DNA. In the presence of ATP, human RFC binds to free PCNA (in solution) with very tight affinity (0.2 nM)28, 29. Under the conditions of the assays (110 nM RFC, 110 nM Cy5-PCNA), even a ssDNA-binding protein with tight affinity for PCNA would require very high concentrations to effectively compete with RFC for binding to PCNA. For instance, an IC50 of 6.3 μM is expected for a ssDNA-binding protein with a KD for PCNA of 20 nM (Figure 6A). Thus, it is unlikely that a ssDNA-binding protein will interfere with PCNA loading by competing with RFC for binding to free PCNA in solution (i.e., competitive inhibition). Nonetheless, this should be verified through stringent controls if FRET is not observed or the observed FRET decreases in the titration assays described above. In contrast, ssDNA-binding proteins with specificity for P/T junctions should be carefully considered. An RFC·PCNA complex binds tightly to a P/T junction (KD ~ 5 – 10 nM)30, 31. Hence, a ssDNA-binding protein with a comparable affinity (KD < 50 nM) may effectively compete with an RFC·PCNA complex at modest concentrations (< 1 μM) (Figure 6B). For an uncharacterized ssDNA-binding protein, stringent controls should be performed to rule out inhibition of PCNA loading by competing with RFC·PCNA complexes for binding to P/T junctions.
Figure 6.
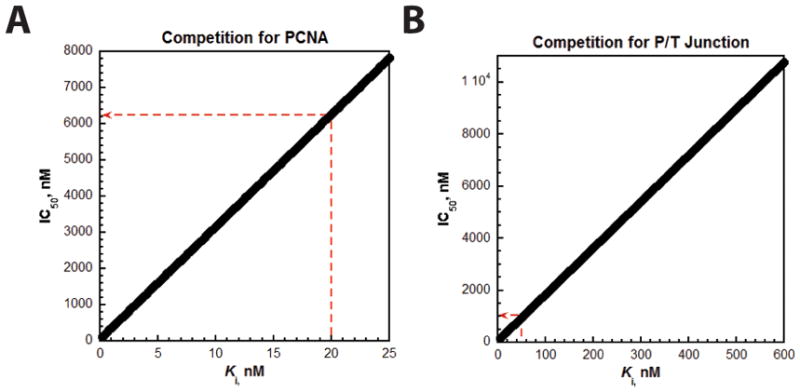
Competitive inhibition of PCNA loading. A ssDNA-binding protein may competitively inhibit RFC-catalyzed loading of PCNA by binding to PCNA (panel A) or a P/T junction (panel B) with an affinity described by Ki(inhibition constant). The IC50 is the concentration of the ssDNA-binding protein that decreases PCNA loading by 50% and is related to Ki by the exact solution provided by Munson and Rodbard4. (A) Competitive inhibition of PCNA loading by binding to PCNA. The IC50’s were calculated based on the binding affinity (KD) of RFC for PCNA (0.2 nM) and plotted versus Ki. The IC50 for a Ki of 20 nM is indicated. (B) Competitive inhibition of PCNA loading by binding to a P/T junction. The IC50’s were calculated based on the binding affinity (KD) of an RFC·PCNA complex for a P/T junction (5 nM) and plotted versus Ki. The IC50 for a Ki of 50 nM is indicated.
Supplementary Material
RPA and SSB stabilize Cy5-PCNA in the same FRET state at a P/T junction.
Cy5-labeling efficiency does not affect the interaction of PCNA with RFC.
Acknowledgments
Funding Source Statement. This work was supported by NIH Grant GM13306 (S.J.B.).
Abbreviations and Textual Footnotes
- P/T DNA
primer/template DNA
- PCNA
proliferating cell nuclear antigen
- RFC
replication factor C
- RPA
replication protein A
- FRET
Forster Resonance Energy Transfer
- ssDNA
single-stranded DNA
- dsDNA
double-stranded DNA
- SSB
Escherichia coli single-strand DNA-binding protein
References
- 1.Hedglin M, Benkovic SJ. Biochemistry. 2017;56:1824–1835. doi: 10.1021/acs.biochem.6b01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hedglin M, Perumal SK, Hu Z, Benkovic S. eLife. 2013;2:e00278. doi: 10.7554/eLife.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Baranovskiy AG, Tahirov TH, Pavlov YI. J Biol Chem. 2014;289:22021–22034. doi: 10.1074/jbc.M114.570333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munson PJ, Rodbard D. J Recept Res. 1988;8:533–546. doi: 10.3109/10799898809049010. [DOI] [PubMed] [Google Scholar]
- 5.Choe KN, Moldovan GL. Mol Cell. 2017;65:380–392. doi: 10.1016/j.molcel.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hedglin M, Kumar R, Benkovic SJ. Cold Spring Harbor Perspect Biol. 2013;5:a010165. doi: 10.1101/cshperspect.a010165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kochaniak AB, Habuchi S, Loparo JJ, Chang DJ, Cimprich KA, Walter JC, van Oijen AM. J Biol Chem. 2009;284:17700–17710. doi: 10.1074/jbc.M109.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tinker RL, Kassavetis GA, Geiduschek EP. EMBO J. 1994;13:5330–5337. doi: 10.1002/j.1460-2075.1994.tb06867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao N, Turner J, Kelman Z, Stukenberg PT, Dean F, Shechter D, Pan ZQ, Hurwitz J, O’Donnell M. Genes Cells. 1996;1:101–113. doi: 10.1046/j.1365-2443.1996.07007.x. [DOI] [PubMed] [Google Scholar]
- 10.Yuzhakov A, Kelman Z, Hurwitz J, O’Donnell M. EMBO J. 1999;18:6189–6199. doi: 10.1093/emboj/18.21.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao N, Hurwitz J, O’Donnell M. J Biol Chem. 2000;275:1421–1432. doi: 10.1074/jbc.275.2.1421. [DOI] [PubMed] [Google Scholar]
- 12.Waga S, Stillman B. Mol Cell Biol. 1998;18:4177–4187. doi: 10.1128/mcb.18.7.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binz SK, Dickson AM, Haring SJ, Wold MS. Methods Enzymol. 2006;409:11–38. doi: 10.1016/S0076-6879(05)09002-6. [DOI] [PubMed] [Google Scholar]
- 14.Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, Kuriyan J. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 15.Kumar R, Nashine VC, Mishra PP, Benkovic SJ, Lee TH. Proc Natl Acad Sci USA. 2010;107:19736–19741. doi: 10.1073/pnas.1014139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhuang Z, Yoder BL, Burgers PM, Benkovic SJ. Proc Natl Acad Sci USA. 2006;103:2546–2551. doi: 10.1073/pnas.0511263103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riener CK, Kada G, Gruber HJ. Anal Bioanal Chem. 2002;373:266–276. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]
- 18.Kim C, Paulus BF, Wold MS. Biochemistry. 1994;33:14197–14206. doi: 10.1021/bi00251a031. [DOI] [PubMed] [Google Scholar]
- 19.Kim C, Snyder RO, Wold MS. Mol Cell Biol. 1992;12:3050–3059. doi: 10.1128/mcb.12.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen B, Sokoloski J, Galletto R, Elson EL, Wold MS, Lohman TM. J Mol Biol. 2014;426:3246–3261. doi: 10.1016/j.jmb.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waldman VM, Weiland E, Kozlov AG, Lohman TM. Nucleic Acids Res. 2016;44:4317–4329. doi: 10.1093/nar/gkw262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim C, Wold MS. Biochemistry. 1995;34:2058–2064. doi: 10.1021/bi00006a028. [DOI] [PubMed] [Google Scholar]
- 23.Sibenaller ZA, Sorensen BR, Wold MS. Biochemistry. 1998;37:12496–12506. doi: 10.1021/bi981110+. [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Le S, Basu A, Chazin WJ, Yan J. Sci Rep. 2015;5:9296. doi: 10.1038/srep09296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Treuner K, Ramsperger U, Knippers R. J Mol Biol. 1996;259:104–112. doi: 10.1006/jmbi.1996.0305. [DOI] [PubMed] [Google Scholar]
- 26.Blackwell LJ, Borowiec JA, Mastrangelo IA. Mol Cell Biol. 1996;16:4798–4807. doi: 10.1128/mcb.16.9.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shrestha D, Jenei A, Nagy P, Vereb G, Szollosi J. Int J Mol Sci. 2015;16:6718–6756. doi: 10.3390/ijms16046718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiomi Y, Usukura J, Masamura Y, Takeyasu K, Nakayama Y, Obuse C, Yoshikawa H, Tsurimoto T. Proc Natl Acad Sci USA. 2000;97:14127–14132. doi: 10.1073/pnas.97.26.14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang G, Gibbs E, Kelman Z, O’Donnell M, Hurwitz J. Proc Natl Acad Sci USA. 1999;96:1869–1874. doi: 10.1073/pnas.96.5.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schauer GD, O’Donnell ME. Proc Natl Acad Sci USA. 2017;114:675–680. doi: 10.1073/pnas.1619748114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hingorani MM, Coman MM. J Biol Chem. 2002;277:47213–47224. doi: 10.1074/jbc.M206764200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RPA and SSB stabilize Cy5-PCNA in the same FRET state at a P/T junction.
Cy5-labeling efficiency does not affect the interaction of PCNA with RFC.