

Abstract
During S-phase, minor DNA damage may be overcome by DNA damage tolerance (DDT) pathways that bypass such obstacles, postponing repair of the offending damage to complete the cell cycle and maintain cell survival. In translesion DNA synthesis (TLS), specialized DNA polymerases replicate the damaged DNA, allowing stringent DNA synthesis by a replicative polymerase to resume beyond the offending damage. Dysregulation of this DDT pathway in human cells leads to increased mutations rates that may contribute to the onset of cancer. Furthermore, TLS affords human cancer cells the ability to counteract chemotherapeutic agents that elicit cell death by damaging DNA in actively replicating cells. Currently, it is unclear how this critical pathway unfolds, in particular where and when TLS occurs on each template strand. Given the semi-discontinuous nature of DNA replication, it is likely that TLS on the leading and lagging strand templates is unique for each strand. Since the discovery of DDT in the late 1960’s, most studies on TLS in eukaryotes have focused on DNA lesions resulting from ultraviolet (UV) radiation exposure. In this review, we re-visit these and other related studies to dissect the step-by-step intricacies of this complex process, provide our current understanding of TLS on leading and lagging strand templates, and propose testable hypotheses to gain further insights.
Graphical abstract
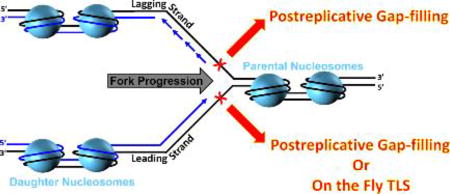
1. Introduction
The genomes of cellular organisms encode genetic information in strands of DNA that assemble into an antiparallel DNA double helix. Each time a cell divides, the genome must be faithfully copied and transferred to a daughter cell for genetic inheritance. The former relies on replicative DNA polymerases (pols) that read a template strand in the 3′ to 5′ direction and synthesize complementary DNA in the 5′ to 3′ direction4. In eukaryotes, DNA replication emanates from many replication origins (Ori) that are activated at different times over the course of an extended S-phase5. At each Ori, two replication forks are established (forming a replication bubble, Figure 1) and progress in opposite directions as the template strands of each fork are replicated in concert. Due to the antiparallel nature of the DNA double helix and the direction of DNA synthesis, each template strand is copied in a unique manner. The leading strand template is replicated continuously in the direction of replication fork progression while the nascent DNA on the lagging strand template is synthesized in short fragments (i.e., discontinuously) in the opposite direction. These short fragments, referred to as Okazaki fragments, are subsequently processed and ligated together to form a continuous, mature DNA strand4.
Figure 1.

Semi-discontinuous DNA replication from a replication bubble. Parental and nascent (daughter) DNAs are shown in black and blue, respectively, with arrows indicating the 5′ to 3′ direction of DNA synthesis. At each origin of replication (Ori, shown in green), two replication forks are established and progress in opposite directions (indicated by grey arrows) as the two template strands of each fork are replicated in concert. The leading strand templates are replicated continuously (solid line) in the direction of replication fork progression while the nascent DNA on the lagging strand templates is synthesized discontinuously (dashed line) in the opposite direction.
The replicative pols have very stringent polymerase domains as well as 3′ to 5′ exonuclease (“proofreading”) domains and, thus, cannot accommodate distortions to the native DNA sequence6–9 However, genomic DNA is continuously subjected to spontaneous damage from reactive metabolites and environmental mutagens. Prominent examples are modifications (lesions) to the native template bases that alter or eliminate their base pairing capability. Despite the protection provided by cellular DNA repair pathways, some lesions may evade detection and persist into S-phase10, 11. Consequently, DNA synthesis on an afflicted template strand abruptly stops upon encountering a lesion. Failure to restart often results in double-strand breaks which may lead to gross chromosomal rearrangements, cell-cycle arrest, and cell death10–13. Therefore, it is often more advantageous to circumvent such replicative arrests and postpone repair of the offending damage to complete the cell cycle and maintain cell survival11, 13, 14. Such a process, referred to as DNA damage tolerance (DDT), may be carried out by translesion DNA synthesis (TLS) where the replicative pol is exchanged for one or more TLS pols. With a more “open” pol active site and the lack of an associated proofreading activity, TLS pols can support stable, yet potentially erroneous, nucleotide incorporation opposite and beyond damaged templates (i.e. bypass), allowing DNA synthesis by the replicative pol to resume15.
Dysregulation of TLS in human cells leads to increased mutations rates that may contribute to the onset of cancer. Furthermore, TLS affords human cancer cells the ability to counteract chemotherapeutic agents that elicit cell death by damaging DNA in actively replicating cells16. Currently, it is unclear how this critical pathway unfolds, in particular where and when TLS occurs on each template strand. The most prevalent source of exogenous DNA damage is ultraviolet (UV) radiation provided by exposure to the sun. Since the discovery of DDT in the late 1960’s, most studies on TLS in eukaryotes have focused on DNA lesions resulting from UV radiation exposure. In this review, we re-visit these and other related studies to dissect the step-by-step intricacies of this complex process and provide our current understanding of TLS on leading and lagging strand templates. We begin with a brief overview of the eukaryotic replisome (Figure 2).
Figure 2.

A model of the eukaryotic replisome assembled at each replication fork. This model shown in cartoon form was generated from recent EM structures1, 2 and related studies (cited in main text). This model suggests that leading strand synthesis and at least the initiation of an Okazaki fragment occur on opposite sides of the helicase, necessitating an unexpected path for the templates as the dsDNA is unwound. The lagging strand template is sterically occluded from the central chamber of the MCM core and traverses the outside of the MCM core to reach pol α-primase. The leading strand template enters the CTD tier of the MCM core, traverses the central chamber or exits at an internal position, and then bends upward toward pol ε.
2. The eukaryotic replisome
At each replication fork, dsDNA is unwound by the replicative helicase, CMG (Cdc45, MCM proteins 2 – 7, and GINS), that translocates with a 3′ to 5′ polarity and, hence, tracks along the leading strand. This helicase is comprised of core proteins MCMs 2 – 7 (MCM core) and two accessory subunits, Cdc45 and GINS2, 17–19 The leading strand template enters through the C-terminal Domain (CTD) tier of the MCM core while the lagging strand template is sterically occluded2, 20. Upon unwinding of dsDNA, the single strand DNA (ssDNA) templates are immediately coated by the ssDNA binding protein, replication protein A (RPA), which binds to ssDNA with extremely high-affinity (< pM) at physiological ionic strength21. Binding of RPA protects ssDNA from cellular nucleases and prevents formation of alternative DNA structures22, 23. The bi-functional pol α-primase complex (pol α-primase) constitutively interacts with the CMG helicase through a Ctf4 bridge that connects it to GINS on the N-terminal half of the helicase complex. Ctf4 is a homotrimer with three identical binding sites; one site is occupied by GINS, one site is occupied by the DNA polymerase subunit of pol α-primase, and the last site is available to bind an unidentified third protein24. Pol α-primase is a four-subunit complex that contains both RNA primase and DNA polymerase activities in separate subunits and it synthesizes complementary RNA/DNA hybrid primers every 100 – 250 nucleotides on the exposed lagging strand template. In other words, re-priming is an innate property of discontinuous lagging strand DNA synthesis. The hybrid primers are comprised of 7–10 nucleotides of RNA followed by 10 – 12 nucleotides of DNA25–32. The switch from RNA to DNA occurs without dissociation of pol α-primase from DNA; the template is transferred internally from the primase active site to the DNA polymerase active site24.
The replicative pols anchor to ring-shaped sliding clamps to achieve the high degree of processivity required for efficient DNA replication. The highly-conserved toroidal structure of sliding clamps has a central cavity large enough to encircle double-stranded DNA (dsDNA) and slide freely along it. Thus, such an association tethers the pol to DNA, increasing the extent of continuous replication. The eukaryotic sliding clamp, PCNA, is a trimer of identical subunits aligned head-to-tail, forming a ring with two structurally distinct faces. The “C-terminal” or “front” face of the homotrimeric PCNA ring is a platform for interaction with pols33, 34. The complex of PCNA and a pol is often referred to as a pol holoenzyme. The closed circular structure of the PCNA ring necessitates an enzyme-catalyzed mechanism which not only opens it for assembly and closes it around DNA but targets it to sites where DNA synthesis is initiated and orients it correctly for replication. The eukaryotic clamp loader complex, replication factor C (RFC), utilizes ATP binding and hydrolysis to selectively load PCNA rings onto primer/template (P/T) junctions with recessed 3′ hydroxyl ends such that the “front” face of the ring is oriented towards the terminal 3′ hydroxyl end of the primer where DNA synthesis will initiate33. Thus, a PCNA ring is loaded onto each Okazaki fragment on the lagging strand template while a single PCNA ring may be utilized throughout leading strand replication of a given genomic segment. The eukaryotic replicative pols, δ and ε, then bind to the “front” face of PCNA encircling a P/T junction, completing formation of the replisome (Figure 2).
Extensive studies utilizing various experimental techniques indicate that the leading and lagging strand templates are primarily copied by pol ε and pol δ, respectively1, 31, 35–47. Recent biochemical and biophysical studies have revealed how this “division of labor” is established. Pol ε docks onto the C-terminal halves of MCMs 2 and 5 and contacts Cdc45 and GINS. Thus, pol ε and pol α are located on opposite sides of the CMG helicase. Pol δ is not stabilized on the leading strand by CMG and can be ejected by pol ε, even in the presence of PCNA31, 48, 49. Conversely, the affinity of pol ε for PCNA alone on the lagging strand is relatively weak such that pol ε can be ejected from PCNA by pol δ or RFC, even when the pol ε● PCNA complex is replicating DNA1, 31, 42, 43, 49–51. These unique interactions select and stabilize pol ε on the leading strand template with CMG and banish pol δ to the lagging strand where it utilizes the hybrid primers synthesized by pol α-primase1, 30, 31, 35–47. Altogether, these studies indicate that the lagging strand holoenzyme (pol δ●PCNA) does not contact the CMG helicase in an active replisome and, hence, does not travel with the replisome24, 31, 48, 49. In other words, Okazaki fragment (i.e., discontinuous) synthesis by pol δ on the lagging strand is not physically coupled to unwinding of the duplex DNA by the CMG helicase whereas continuous DNA synthesis by pol ε on the leading strand is.
3. Escaping detection: UV-induced DNA lesions in S-phase
There are two major UV-induced lesions (photodimers); cis-syn cyclobutane pyrimidine dimers (CPDs) account for ~67 – 83 % of the photoproducts and pyrimidine (6–4) pyrimidone photoproducts ((6–4) PPs) account for the rest (Figure 3)52. In eukaryotes, the main pathway for repairing these photodimers is nucleotide excision repair (NER)53 and it should be noted that NER is the only mechanism for repair of UV-induced photodimers in placental mammals, including humans54, 55. NER proceeds by two sub-pathways that differ primarily in the damage recognition step56. Global genome NER (GG-NER) can occur anywhere in the genome and is initiated by NER factors that recognize ssDNA regions within duplex DNA that is thermodynamically destabilized by a UV-induced photodimer. Alternatively, RNA polymerase II stalled at a UV-induced lesion may initiate transcription-coupled repair (TC-NER) of the offending damage in actively transcribed genes. Subsequent steps of both pathways funnel them to the “core” NER factors that complete the repair process53.
Figure 3.

The most prominent DNA lesions resulting from exposure to UV radiation. The numerical positions within the six-membered rings are indicated in blue. Exposure of adjacent pyrimidines, such as two thymidines, results in formation of (6–4) PPs and CPDs. The proportion of each is indicated. The only mechanism for repairing these photodimers within the human genome is the NER pathway, which proceeds by two sub-pathways: GG-NER and TC-NER. (6–4) PPs are efficiently and rapidly repaired by GG-NER while CPDs are repaired via TC-NER in a much slower fashion.
(6–4) PPs impose a significant distortion in duplex DNA and, hence, are efficiently recognized and rapidly repaired by the GG-NER machenery53. Indeed, Hela cells remove nearly all (6–4) PPs within 2–3 hours of UV radiation exposure56. However, CPDs do not dramatically destabilize duplex DNA and, thus, are less-efficiently recognized by GG-NER recognition factors. Rather, these lesions are repaired via TC-NER in a much slower fashion. In studies on human cells irradiated with UV, repair of CPD lesions was not observed until the repair of (6–4) PPs was nearly complete and only 10% of CPDs were removed within 2 – 3 hours after UV irradiation56, 57. Thus, the vast majority of (6–4) PPs are repaired prior to an encounter with a moving replisome while most CPD lesions persist into and throughout S-phase. However, both lesions can be accommodated, i.e., “tolerated,” during S-phase by TLS and this process absolutely requires the interaction of TLS pols with PCNA encircling the damaged DNA58–64.
Three TLS pols, η, ι, and ζ, provide alternative routes for TLS across a (6–4) PP. Pols η and ι function in an error-prone manner while pol ζ promotes high fidelity TLS58, 65. It should be noted that (6–4) PPs may also be accommodated in lower eukaryotes by an alternative, error-free DDT pathway that is independent of TLS pols. This pathway is prominent at low UV doses and readers are directed to a recent review (and references cited therein) for more information66. In contrast to TLS across (6–4) PPs, a single pol, pol η, carries out TLS across a CPD and does so with high fidelity (>90%). The magnitude of this feat is underscored by xeroderma pigmentosum variant (XPV), a human autosomal recessive genetic disorder in which the xpv gene encoding pol η is either mutated or deleted. In the absence of functional pol η, pols ζ, ι, and κ perform mutagenic TLS across UV-induced CPD lesions. Hence, patients afflicted with this disease are hypermutable to UV irradiation and suffer from sunlight sensitivity and extreme predisposition to sunlight-induced skin cancer67, 68.
4. When two worlds collide: The replisome encountering a UV-induced DNA lesion in S-phase
As the replisome approaches a UV-induced lesion, the CMG helicase will be the first to encounter the offending damage. CMG unwinds duplex DNA by a steric exclusion mechanism in which the leading strand template enters the central chamber of the MCM core and the lagging strand is sterically occluded1, 24. This suggests that DNA lesions on the excluded lagging strand do not stall/arrest the CMG helicase while lesions on the leading strand may. However, the CMG helicase does not “read” the leading strand template nor the phosphodiester backbone and translocates along the ssDNA template in a sequence-independent manner. Indeed, significant DNA lesions within a leading strand template, such as intrastrand crosslinks, do not impede CMG helicase movement. In fact, the CMG helicase can proceed past four, consecutive biotinylated thymidine nucleotides within a leading strand template20, 69. Thus, UV-induced lesions (Figure 3) are not envisioned to stall/arrest unwinding of duplex DNA by the CMG helicase. This has been directly demonstrated for the closely-related simian virus 40 (SV40) large T-antigen (T-Ag) helicase on single-origin plasmids containing a site-specific CPD in either template strand70. In contrast, the three pols within the eukaryotic replisome (α-primase, δ, and ε) cannot accommodate UV-induced lesions and DNA synthesis by these pols abruptly stops upon encountering such damage71–74. The impending consequences on semi-discontinuous DNA replication are unique for each pol and each is discussed below.
4.1. The Pol α-primase complex encountering a UV-induced lesion on a lagging strand template
During S-phase, pol α-primase synthesizes RNA/DNA hybrid primers on the leading strand templates at each replication origin but predominantly interacts with the lagging strand templates where hybrid primers are required for each Okazaki fragment. Hence, pol α-primase only “reads” a small fraction of a diploid genome (≤ 11% in humans)5. Furthermore, UV fluences comparable to what an individual experiences from one hour of mid-day sun only generate one lesion every 3 – 4 kilobases (kb) in somatic human cells, which equates to < 0.017% of the diploid genome incurring damage56, 75, 76. Altogether, this suggests that the likelihood of pol α-primase encountering a UV-induced lesion during S-phase is extremely rare. Unlike pols ε and δ, pol α-primase does not require a preexisting P/T junction to initiate or re-start DNA synthesis. Thus, in the rare event that pol α-primase encounters a UV-induced lesion, we envision that pol α-primase simply releases the lagging strand template and re-primes upstream (5′) of the UV-induced lesion, keeping pace with the progressing replisome. This leaves behind a ssDNA gap extending from the blocked 3′ hydroxyl terminus of the aborted primer to the 5′ terminus of the downstream primer. Hence, the burden of UV-lesions encountered by pol α-primase on a lagging strand template is passed onto a pol δ holoenzyme assembled on the blocked P/T junction or approaching from the nascent primer upstream of the offending damage (Figure 4). If the aborted primer is of sufficient length, RFC will load PCNA onto the P/T junction abutting the UV-induced lesion33, 77. However, a pol δ holoenzyme will not synthesize past a UV-induced lesion. If approaching from the upstream nascent primer, a pol δ holoenzyme can remove the aborted primer by strand-displacement synthesis but cannot replicate past the offending damage.
Figure 4.

Pol α-primase encountering a UV-induced lesion (
 ) on a lagging strand template. This model shown in cartoon form only displays pol α-primase on the lagging strand template for simplicity.
) on a lagging strand template. This model shown in cartoon form only displays pol α-primase on the lagging strand template for simplicity.
4.2. Pol ε encountering a UV-induced lesion on a leading strand template
Pol ε docks onto the CMG helicase within the replisome (Figure 2) and this interaction is maintained during replication fork progression, promoting continuous DNA synthesis on a leading strand template31, 43, 48. During unperturbed S-phase, replication forks progress with a mean fork velocity of ~1.6 kb/min in human78–80 and Saccharomyces cerevisiae (budding yeast)81, dramatically slower than dNTP insertion catalyzed by pol ε at 37°C (kpol > 500 s−1)82. This suggests that fork progression is driven by the relatively slow CMG helicase and, hence, leading strand DNA synthesis is rate-limited by unwinding of the DNA duplex. The drastic contrast in these enzymatic activities is critical as it ensures tight coupling of leading strand synthesis and DNA unwinding such that complementary dNTPs are inserted by pol ε as soon as template base pairs are available, limiting exposure of a leading strand template. Upon encountering a UV-induced lesion within a leading strand template, the coordinated activities of pol ε and the CMG helicase become “uncoupled”; DNA synthesis by pol ε abruptly stops (i.e., kpol = 0)71–74 while the CMG helicase and, hence, the replication fork progress onward, exposing long stretches of the leading strand template before eventually stalling downstream of the offending damage83–85. Herein, such events such events are simply referred to as “uncoupling”. During uncoupling, RPA coats the exposed leading strand template, which may be longer than 3 kilobases (kb) in S. cerevisiae and 20 kb in humans85, 86. This raises two critical issues for when a UV-induced lesion is encountered within a leading strand template; 1) Is the undamaged lagging strand template also exposed during uncoupling or does lagging strand DNA synthesis continue? 2) Does the progressing CMG helicase carry non-replicating pol ε along with it or is pol ε left behind at the UV-induced lesion? Each is discussed in detail below.
4.2.1 Lagging strand DNA synthesis after a UV-induced lesion is encountered on a leading strand template
Pol α-primase constitutively interacts with the CMG helicase during unperturbed S-phase and this interaction is maintained upon uncoupling24, 26, 28, 30–32, 87. Thus, DNA synthesis may continue on an undamaged lagging strand template after a UV-induced lesion is encountered within a leading strand template. Direct visual evidence of this was provided by electron microscopy (EM) studies on chromosomal replication forks obtained from S. cerevisiae. In untreated cells, both template strands were fully replicated up to the fork or a small ssDNA stretch (<400 nt) was observed on only one of the template strands, interpreted as the lagging strand. After UV irradiation, ssDNA stretches as long as 3 kb were observed on only one side of the fork while the opposing side was either fully-replicated up to the fork or contained a small ssDNA stretch (<400 nt). Furthermore, in essentially all replication bubbles containing extended ssDNA regions at both forks, the two regions were distributed in trans. These observations suggested an unrepaired UV lesion encountered in a leading strand template only blocks leading strand synthesis and the extended ssDNA regions (≤ 3 kb) were generated by uncoupling. Hence, lagging strand synthesis continues onward with the uncoupled, and progressing replication fork after a UV-induced lesion is encountered within the leading strand template. This agreed with later studies on extracts from Xenopus laevis (frog) that suggested that new RNA primers continue to be synthesized by pol α-primase on a lagging strand template after uncoupling and these primers are extended by pol δ88. Furthermore, the same conclusion had been reached in earlier studies on human cell free extracts that utilized various experimental techniques to thoroughly analyze the replication of single-origin plasmids containing a unique, site-specific CPD in the leading strand template70, 89–95. Altogether, these studies suggest that DNA synthesis by pol ε abruptly stops upon encountering a UV-induced lesion within a leading strand template but unwinding of the DNA duplex by the CMG helicase and DNA synthesis by pol δ on the undamaged lagging strand template continue onward.
4.2.2 Location of pol ε after a UV-induced lesion is encountered on a leading strand template
Pol ε maintains several contacts to the CMG helicase within an actively progressing replisome. Specifically, pol ε docks onto the C-terminal halves of MCMs 2 and 5 and contacts Cdc45 and GINS (Figure 2)31, 43, 48. These binding interactions may persist after a UV-induced lesion is encountered in a leading strand template. Indeed, free, i.e. non-replicating, pol ε binds to the CMG helicase with very high affinity (low nM) in solution such that CMG helicase and pol ε can be purified from budding yeast cells as a stable protein complex42, 49. Furthermore, a study on X. laevis egg extracts that analyzed chromatin binding of replication fork proteins suggested that when both template DNA strands are continuous, the CMG helicase remains intact upon uncoupling from leading strand DNA synthesis and maintains contact with pol ε as the replication fork progresses87. This raises three possibilities; 1) pol ε maintains contact with both the CMG helicase and the blocked P/T junction during uncoupling, forming an extensive, RPA-coated ssDNA loop; 2) pol ε releases the blocked P/T junction upon uncoupling and is carried away by the progressing CMG helicase or 3) a combination of both possibilities.
At physiological ionic strength, human RPA has an occluded binding site size of 30 +/−2 nt of ssDNA. Assuming a mean fork velocity of ~1.6 kb/min in human cells, this length of ssDNA is exposed within ~1 s of an uncoupling event78–80, 96, 97. When bound by RPA, a ssDNA sequence is stretched to within 10% of its full contour length and its bending rigidity is increased 2–3 fold98–100. Such an extended and stiffened conformation antagonizes loop formation and is expected to strain a P/T junction●pol ε●CMG helicase complex. Furthermore, RPA is ~240-fold more abundant than pol ε in human cells and binds to ssDNA with extremely high affinity (< pM), more than 3 orders of magnitude tighter than the affinity of non-replicating pol ε for a native P/T junction49, 101, 102. Thus, RPA effectively outcompetes non-replicating pol ε for ssDNA. Altogether, this suggests that pol ε is released from a blocked P/T after uncoupling. Supporting in vivo evidence was provided by studies that analyzed proteins on daughter DNAs in relation to progressing (active) or stalled replication forks.
In untreated human cells, pol ε and the CMG helicase are highly enriched near the elongating P/T junctions at active replication forks, indicating assembly and progression of the replisome103–105. Upon treatment with hydroxyurea (HU) or aphidocolin (Aph), both of which trigger uncoupling without damaging DNA, the abundance of the CMG helicase near blocked P/T junctions decreased over time while RPA and its interacting proteins were recruited, consistent with the increased ssDNA generated during uncoupling. Under these conditions, the abundance of pol ε near blocked P/T junctions also decreased with time, indicating that pol ε released the blocked P/T junction after fork uncoupling. Furthermore, pol ε and the CMG helicase decreased with similar kinetics, consistent with the CMG helicase maintaining contact with pol ε after uncoupling and carrying the polymerase away from blocked P/T junctions on leading strand templates83, 87, 103, 106, 107. These studies on human cells agree with previous studies on S. cerevisiae cells that tracked the movement of replication fork proteins along genomic DNA. When cells were synchronized in G1 phase of the cell cycle, MCM6, cdc45, GINS, and pol ε were all detected at an early replication origin, indicating assembly of the replisome. When cells were released into S-phase in the presence of HU, cdc45, GINS, and pol ε were all detected kb away from an early replication origin after increasing periods of time, suggesting that an intact CMG helicase carries pol ε away from the replication origin upon uncoupling from DNA synthesis. Altogether, the biochemical and cellular studies described in this section collectively suggest that the CMG helicase remains intact upon uncoupling from leading strand DNA synthesis and maintains contact with a non-replicating pol ε as the DNA is unwound. Pol ε may also maintain contact with the blocked P/T junction after uncoupling, forming a ssDNA loop. However, these complexes are expected to be short-lived (~1 s) due to the well-characterized properties of RPA described above. Released from a blocked P/T junction on the leading strand template, non-replicating pol ε is carried away downstream by the CMG helicase.
In contrast to pol ε, PCNA is likely to be left behind at the blocked P/T junction on a leading strand template. As discussed above in section 2, the affinity of pol ε for PCNA encircling a P/T junction is dramatically weak compared to pol δ (see also section 4.3 below) and, hence, the contribution of the sliding clamp to continuous DNA synthesis by pol ε is relatively minimal1, 31, 42, 43, 49–51. In fact, cellular pol ε was initially isolated from pol δ-containing fractions and referred to as “PCNA-independent pol δ” or “pol δ2” based on the marginal stimulation of its DNA synthesis activity by PCNA50. Thus, pol ε likely disengages from PCNA upon encountering a UV-induced lesion, akin to that described below for pol δ (section 4.3), and, hence, leaves PCNA behind at the blocked P/T junction. Recent in vivo studies revealed that enzyme-catalyzed unloading/recycling of PCNA from a P/T junction will not occur during S-phase until the primer is fully-extended and ligated to the downstream duplex region108. This suggests that a PCNA ring residing at a blocked P/T junction on a leading strand template persists as the replication fork progresses onward (Figure 5).
Figure 5.

Pol ε encountering a UV-induced lesion within a leading strand template. This model shown in cartoon form depicts discontinuous lagging strand DNA synthesis as a dashed line for simplicity. Arrows indicate the direction of DNA synthesis (5′ ➜ 3′). The heterohexameric MCM core is depicted as a barrel. (1) Upon encountering a UV-induced lesion (
) within a leading strand template, DNA synthesis by pol ε abruptly stops but the CMG helicase remains intact and continues unwinding DNA, exposing long stretches of the leading strand template. During such uncoupling, RPA coats the exposed leading strand template and DNA synthesis continues on the undamaged lagging strand template as the replication fork progresses. Furthermore, non-replicating pol ε maintains contact with the CMG helicase and is carried downstream of the offending lesion while PCNA is left behind at the blocked P/T junction. Pol ε may also maintain contact with the blocked P/T junction upon uncoupling, forming ssDNA loop. However, such complexes are expected to be short-lived due to RPA●ssDNA interactions. (2) Uncoupled from leading strand DNA synthesis, the CMG helicase and, hence, lagging strand DNA synthesis eventually stall downstream of the offending damage, halting progression of the replication fork.
4.3. Pol δ encountering a UV-induced lesion on a lagging strand template
Pol δ alone has dramatically low, if any, affinity for P/T DNA and must anchor to PCNA to efficiently replicate the lagging strand templates49, 51, 77, 109 . At a progressing replication fork, pol δ effectively captures a PCNA ring encircling a nascent P/T junction on the lagging strand and initiates DNA synthesis77, 109–113. A pol δ holoenzyme inserts dNTPs at least 3 orders of magnitude faster than pol δ dissociates from PCNA encircling DNA109, 113. Thus, anchoring to PCNA dramatically increases the processivity of pol δ-mediated synthesis on a lagging strand template. Recent studies on the human pol δ holoenzyme revealed that pol δ rapidly and passively dissociates into solution upon stalling, leaving PCNA behind on the DNA77, 109. This scenario mimics the encounter of a UV-induced lesion within a lagging strand template71. As described above, the pol δ holoenzyme does not contact the CMG helicase within an active replisome and, hence, is not physically coupled to unwinding of the DNA duplex31, 43, 48, 49. Thus, when a pol δ holoenzyme encounters a UV-induced lesion in a lagging strand template, DNA synthesis on the afflicted Okazaki fragment abruptly stops but the replisome may continue unimpeded. Indeed, extensive ssDNA regions (> 400 nt) leading up to a replication fork were never observed on both template strands in the aforementioned EM studies on chromosomal replication forks obtained from UV-irradiated S. cerevisiae85. This suggests that an unrepaired UV lesion encountered in a lagging strand template does not stall progression of the replication fork nor leading strand DNA synthesis. Rather, pol ε continues to replicate the undamaged leading strand template as the CMG helicase unwinds the duplex DNA and pol α-primase performs “scheduled” synthesis of RNA/DNA hybrid primers every 100 – 250 nt on the exposed lagging strand template (Figure 6). Accordingly, DNA synthesis by pol δ must also continue upstream of the offending damage to limit the exposure of the lagging strand template. Such behavior would generate small ssDNA gaps behind a progressing replication fork. This will be discussed in detail in section 5.2 below.
Figure 6.

Pol δ encountering a UV-induced lesion within a lagging strand template. For simplicity, this model shown in cartoon form depicts continuous leading strand DNA synthesis as a solid line and only displays PCNA, RPA and pol δ proteins on the lagging strand template. Arrows indicate the direction of DNA synthesis (5′ ➜ 3′). Upon encountering a UV-induced lesion (X) within a lagging strand template, pol δ rapidly and passively dissociates into solution, leaving PCNA behind on the DNA. Pol δ may reiteratively dissociate and re-bind to the PCNA encircling the blocked P/T junction but pol δ-mediated DNA synthesis will not resume on the afflicted Okazaki fragment until the lesion is bypassed. Meanwhile, pol ε continues to replicate the undamaged leading strand template as the CMG helicase unwinds the duplex DNA. In this schematic, “scheduled” re-priming of the lagging strand template by pol α-primase has not yet occurred.
Altogether, the aforementioned studies confirm the original model put forth from the seminal studies on UV-irradiated cells carried out decades before 89, 114–136. That is, DNA synthesis on both templates does not stop when a UV-induced lesion is encountered within either template. Rather, DNA synthesis continues on the undamaged template until it too reaches a photodimer.
5. The GPS for TLS: Correlation between re-priming the damaged template and TLS
Upon encountering a UV-induced lesion within a given template, resumption of DNA synthesis by the respective replicative pol requires a primer terminus annealed to an undamaged section of that template. This may be provided by one of two pathways. First, one or more TLS pols may insert dNTPs across from and beyond the offending DNA lesion such that the aborted primer terminus is extended to an undamaged section of the respective template. Alternatively, the damaged template may be re-primed, providing a nascent primer terminus on an undamaged section of the respective template and leaving behind a ssDNA gap containing the lesion. Hence, a re-priming event may provide a beacon for when and where TLS occurs. As each template is replicated and re-primed in a unique fashion, this suggests that TLS on leading and lagging strand templates is distinct. Each is discussed in detail below.
5.1. Solicited re-priming of a leading strand template, TLS, and fork re-start
A UV-induced lesion encountered within a leading strand template uncouples the activities of pol ε and the CMG helicase and eventually stalls progression of the replication fork downstream of the offending damage (Figure 5). Accordingly, progression of the replication fork can only be re-started (i.e., fork restart) when leading strand DNA synthesis and DNA unwinding are re-coupled and this requires a primer terminus suitable for extension by pol ε to be provided to the stalled CMG helicase. This may be achieved in one of two ways. First, the offending DNA lesion may be bypassed via TLS such that the aborted primer terminus is extended to an undamaged section of the leading strand template where a replicative pol may faithfully extend the primer to the stalled CMG helicase, allowing fork-restart. In this pathway, referred to as “on the fly” TLS, bypass of the UV-induced lesion is required for fork restart and leading strand DNA synthesis remains continuous (Figure 7)137. In other words, “on the fly” TLS allows leading strand DNA synthesis to continue without the formation of ssDNA gaps opposite DNA lesions. Alternatively, the damaged leading strand template may be re-primed downstream (3′) of the offending damage, providing a nascent primer terminus on an undamaged section of the leading strand template. This leaves behind a ssDNA gap extending from the blocked P/T junction to the 5′ end of the newly-synthesized primer downstream of the offending damage.
Figure 7.

“On the fly” TLS on a leading strand template. An unrepaired UV-induced lesion (
) encountered in a leading strand template leads to stalling of the replication fork downstream of the lesion (as depicted in Figure 5). TLS on a leading strand template can occur by one of two pathways. In “on the fly” TLS, bypass of the UV-induced lesion occurs before progression of the replication fork has been re-started through a re-priming event, as follows: 1) One or more TLS pols bypass the offending DNA lesion, extending the aborted primer terminus to an undamaged section of the leading strand template. 2) Pol δ faithfully extends the primer to the stalled CMG helicase where 3) the bound pol ε rapidly replaces pol δ on the leading strand template, re-starting progression of the stalled replication fork. In this scenario, replication fork re-start requires TLS. As an alternative to “on the fly” TLS, UV-induced lesions within a leading strand template may be bypassed by postreplicative gap-filling (Figure 8).
Given the extensive unwinding of duplex DNA upon uncoupling at a UV-induced lesion, re-priming of a leading strand template may generate relatively large ssDNA gaps behind a progressing replication fork. In the seminal studies on UV-irradiated cells, estimates of the gap sizes ranged from 150 to 1250 nt117, 132. The former agreed with ssDNA gaps generated on a lagging strand template by interruption of an Okazaki fragment (see section 5.2 and Figure 9 below). The latter implied a leading strand template can be re-primed downstream of a UV-induced lesion. Visual confirmation was later provided by EM studies on chromosomal replication forks obtained from UV-irradiated S. cerevisiae where ssDNA gaps ranging from 200 to more than 1600 nts in length were observed. Importantly, in approximately half of the molecules containing multiple ssDNA gaps, discontinuities were observed on opposite sides of the same fork, confirming that re-priming events occur on both templates85. In this alternative pathway, fork-restart requires a re-priming event rather than TLS and leading strand DNA synthesis is no longer continuous. Bypass of the offending damage and “filling in” of the ssDNA gap occurs after replicative DNA synthesis has resumed on the afflicted template. Hence, TLS in this scenario is commonly referred to as “postreplicative gap-filling” (see Figure 8 below).
Figure 9.

Postreplicative gap-filling on a lagging strand template. DNA synthesis by pol δ abruptly stops upon encountering a UV-induced lesion (
) within a lagging strand template but the replisome and, hence, the replication fork continue unimpeded (as detailed in Figure 6). Bypass of the offending damage occurs by postreplicative gap-filling as follows. 1) Pol α-primase performs “scheduled” synthesis of an RNA/DNA hybrid primer upstream (5′) of the offending damage on the exposed lagging strand template. PCNA residing at the blocked P/T junction remains and a pol δ holoenzyme is assembled on the nascent P/T junction with a new PCNA ring, allowing lagging strand DNA synthesis to resume and continue upstream of the offending DNA lesion. This prevents excessive exposure of the lagging strand template and generates a postreplicative gap less than or equal to the size of an Okazaki fragment (~100 – 250 nt) extending from the blocked P/T junction to the 5′ terminus of the downstream Okazaki fragment. As the replisome progresses in the absence of TLS, the replication fork moves further and further ahead of the postreplicative gap. 2) Eventually, one or more TLS pols bypass the offending DNA lesion within the postreplicative gap, extending the aborted primer terminus to an undamaged section of the lagging strand template. 3) Pol δ then “fills in” the remainder of the postreplicative gap and the 5′ RNA end of the downstream duplex region is removed as in Okazaki fragment processing/metabolism3. 4) The resident PCNA is removed some time after the fully-extended primer terminus is ligated to the 5′ end of the downstream Okazaki fragment.
Figure 8.

Postreplicative gap-filling on a leading strand template. An unrepaired UV-induced lesion (
) encountered in a leading strand template leads to stalling of the replication fork downstream of the lesion (as depicted in Figure 5). In postreplicative gap-filling, bypass of the UV-induced lesion occurs after the replication fork has been re-started through a re-priming event, as follows. 1) PrimPol is recruited to the RPA-coated ssDNA downstream of a UV-induced lesion by a direct interaction with RPA. Once localized, PrimPol synthesizes a nascent primer at a random location downstream of the offending damage and a new PCNA ring is loaded onto the nascent 3′ hydroxyl terminus. This leaves behind an RPA-coated ssDNA gap (postreplicative gap) containing PCNA and the lesion. 2) Pol δ faithfully extends the nascent primer terminus to the stalled CMG helicase where 3) the bound pol ε rapidly replaces pol δ on the leading strand template, re-starting progression of the stalled replication fork. 4) Eventually, the offending DNA lesion within the postreplicative gap (shown) is bypassed by one or more TLS pols, extending the aborted primer terminus to an undamaged section of the leading strand template. 5) Pol δ “fills in” the remainder of the postreplicative gap and the 5′ RNA end of the downstream duplex region is removed as in Okazaki fragment processing/metabolism3. 6) The resident PCNA is removed some time after the fully-extended primer terminus is ligated to the 5′ end of the downstream daughter DNA.
On a leading strand template, a single primer is continuously extended by pol ε in the direction of replication fork progression. Thus, in contrast to the lagging strand template where re-priming is an innate property, re-priming events on a leading strand template are unprogrammed and must be solicited. Pol α is reloaded onto exposed leading strand templates and such events are required for activation of the ATR checkpoint at ssDNA gaps where a re-priming event has occurred138. Thus, it is tempting to speculate, as others have, that when a UV-induced lesion is encountered within a leading strand template, pol α mediates fork-restart by re-priming the leading strand template downstream of the offending damage88. However, this has yet to be demonstrated and additional work is required to garner support for this model. Meanwhile, recent studies have implicated a novel primase for re-priming leading strand templates, as discussed below.
5.1.1 Re-priming of a leading strand template by PrimPol
Primase-Polymerases (PrimPols) are a novel family within the archaeao-eukaryotic primase (AEP) superfamily that have been identified in most eukaryotes, including mammals. Notably, PrimPols are absent from D. melanogaster, S. pombe, S. cerevisiae, and C. elegans74, 139. Unlike most primases that are heterodimeric and utilize NTPs, PrimPols are monomers that prefer dNTPs for de novo nucleic acid synthesis. Furthermore, PrimPols can extend primers, even across UV-induced lesions, and both primase and DNA polymerase activities are catalyzed by the same active site73, 74, 140–142. In humans, the highly-conserved C-terminus of PrimPol is required for its interaction with RPA139, 143. Following treatment of human cells with either HU or Aph, PrimPol accumulated into nuclear foci on genomic DNA and such behavior required the initiation of DNA replication (i.e., entry into S-phase) and the RPA-binding domain of PrimPol73, 74, 139. Similar behavior was also observed for PrimPol after UV treatment and PrimPol foci colocalized with UV-induced lesions and PCNA73, 74. Survival assays revealed that depletion or knockout of PrimPol sensitizes cells to UV treatment and such behavior was non-epistatic with both pol η and pol ζ 74, 141, 144 . However, re-localization of PrimPol was not observed after DNA strand breaks were generated by ionizing radiation (IR) and PrimPol knockout cells are not sensitive to IR74. Together, this suggested that PrimPol functions in the tolerance of UV-induced lesions encountered during S-phase and its role in this process is RPA-dependent but nonredundant with that of TLS pols η and ζ . Hence, PrimPol may serve to re-prime leading strand templates downstream of UV-induced lesions. Such activity had been demonstrated in vitro with various obstructions that block DNA synthesis and, indeed, primers synthesized by PrimPol can be efficiently extended by pol ε141, 144, 145. Clever genetic assays validated this model in vivo.
Upon incorporation of a chain-terminating nucleotide analog (CTNA) at the 3′ terminus of a growing DNA chain, further extension is prevented and DNA synthesis can only resume from a nascent primer. PrimPol knockout cells are hypersensitive to killing by CTNA treatment and suppression of this phenotype by complementation absolutely required the primase activity of PrimPol144. The same behavior was also observed with UV treatment, suggesting that re-priming of damaged DNA templates by PrimPol is critical for the cellular tolerance of UV-induced lesions 74, 141, 144. Indeed, loss of PrimPol markedly increased the delay or blockage of replication fork progression in UV-irradiated cells and reversion of this phenotype was only achieved by complementation with PrimPol containing active primase activity73, 74, 142, 144. Altogether, this suggests that RPA directly recruits PrimPol to an exposed leading strand template where it re-primes downstream of a UV-induced lesion, allowing fork-restart. PrimPol activity is independent of PCNA and PrimPol does not interact with PCNA in vivo. Furthermore, an interaction between PrimPol and any constituent of the eukaryotic replisome (Figure 2) has yet to be reported in the literature. Hence, it is likely that re-priming by PrimPol occurs anywhere along the RPA-coated ssDNA. However, it remains to be seen how such a feat is achieved within the nuclear environment as PrimPol is very distributive on naked DNA, inserting up to 4 nucleotides within a single binding encounter139, 141, 142, and its primase activity is inhibited by RPA143.
Extensive studies on UV-irradiated cells suggest that both of the aforementioned pathways for fork-restart (“on the fly” TLS and postreplicative gap-filling) are operational in all eukaryotes but the propensity for each may vary depending on the nature of the damage (i.e., CPD versus (6–4) PP) and/or the organism76, 85, 137, 146–158. In each pathway, long stretches of the leading strand template may be exposed by uncoupling of pol ε and CMG activities85, 86. Given the properties of pol ε-mediated DNA synthesis discussed up to this point, this raises a critical issue for when a UV-induced lesion is encountered on a leading strand template; after a primer terminus suitable for extension by a replicative pol is provided downstream of a UV-induced lesion by either TLS or re-priming, which replicative pol (δ or ε) binds to the resident PCNA and faithfully extends the primer to the stalled CMG helicase or fills in the postreplicative gap? This is discussed in detail below.
5.1.2 Pol δ copies the undamaged ssDNA on each template strand during DDT
In cellular studies on X. laevis, only pol δ accumulated on replication-arrested chromatin in response to UV or Aph treatment while the level of chromatin-bound pol ε did not change88, 159. Furthermore, the level of pol δ on chromatin gradually decreased after release from Aph-induced fork arrest159. These observations are similar to those from later studies on human cells that analyzed proteins on daughter DNAs in relation to progressing (active) or stalled replication forks. In untreated human cells, both pol δ and pol ε were enriched near elongating P/T junctions at active replication forks, consistent with assembly and progression of the replisome. Upon treatment with HU, only pol δ was enriched near the blocked P/T junctions behind stalled replication forks104. Considering these observations, we propose that the undamaged ssDNA exposed during DDT is predominantly replicated by pol δ. Indeed, genetic experiments on S. cerevisiae revealed that only pol δ is required for DDT of UV-induced lesions while pol ε has a minor role, if any160. Recent in vitro studies have also provided staunch supporting evidence for the proposed model.
Both modes of TLS absolutely require the interaction of pols with PCNA encircling DNA. The affinity of pol ε alone for PCNA encircling a P/T junction is dramatically weak compared to pol δ such that pol δ outcompetes pol ε for PCNA and even ejects pol ε from PCNA encircling a P/T DNA substrate, even when the pol ε● PCNA complex is replicating the DNA1, 31, 42, 43, 49–51. Thus, the contrasting binding affinities of the replicative pols for PCNA encircling a P/T junction drive the selection of pol δ for replicating the extensive stretches of ssDNA during DDT. Altogether, the independent biochemical and cellular studies described in this section provide strong evidence that the undamaged ssDNA exposed during DDT is predominantly replicated by pol δ while pol ε plays a minor role, if any. Perhaps emerging technologies, such as HydEn-seq, that map DNA synthesis by the replicative pols to specific template strands within cells will be exploited in the future to definitively confirm this47.
In the event pol δ faithfully extends a primer to a stalled CMG helicase during DDT, the bound pol ε rapidly replaces pol δ on the leading strand template, re-starting progression of the stalled replication fork. This exchange is driven by re-assembly of an active replisome (Figure 2) and may be promoted further by “collision release” of pol δ from PCNA upon colliding with the stall CMG helicase1, 30, 31, 35–51. Considering these various pol exchange events, we propose that TLS on a leading strand template may occur by either “on the fly” TLS (Figure 7) or postreplicative gap-filling (Figure 8) as described in the indicated figures.
5.2. Scheduled re-priming of a lagging strand template and TLS
Upon encountering a UV-induced lesion within a lagging strand template, pol δ rapidly dissociates into solution, leaving PCNA behind on the DNA109. However, leading strand DNA synthesis and “scheduled” re-priming of the lagging strand template continue onward with the progressing replication fork (Figure 6). The rate of replication fork progression in human and S. cerevisiae cells is ~1.6 kb/min78, 79, 81, indicating that a ssDNA template for an Okazaki fragment (100 – 250 nt) is exposed and primed every 3.8 – 9.4 s on average. During this time frame, pol δ may reiteratively dissociate and re-bind to the PCNA encircling the blocked P/T junction but pol δ-mediated DNA synthesis cannot resume on the afflicted Okazaki fragment until the lesion is bypassed via TLS. Hence, bypass of the UV-induced lesion and the subsequent resumption of pol δ-mediated synthesis on the afflicted Okazaki fragment has a very narrow window of opportunity (< 10 s) before the lagging strand template undergoes scheduled re-priming upstream (5′) of the offending damage. However, TLS pols are not “on standby” for such rapid events. TLS pols were not identified near actively progressing replication forks in human cells, unlike all other components of the replisome, suggesting that TLS pols do not travel with progressing replication forks103, 104 . Indeed, pol η is distributed uniformly throughout the nucleus during unperturbed S-phase in human cells, in contrast to PCNA, which is concentrated into intranuclear foci at sites of DNA replication161. Furthermore, the protein levels of TLS pols ζ and η in human and S. cerevisiae cells do not fluctuate significantly during the cell cycle nor in response to UV irradiation151, 162–164. Collectively, this suggests that TLS on a UV-damaged Okazaki fragment most likely occurs after pol δ-mediated DNA synthesis has initiated from the nascent hybrid primer upstream (5′) of the offending damage.
According to the aforementioned model, an RPA-coated ssDNA gap less than or equal to the size of an Okazaki fragment is generated behind a progressing replication fork, extending from the UV-induced lesion to the 5′ terminus of the downstream Okazaki fragment. The seminal studies on UV-irradiated eukaryotic cells initially suggested this 114–118, 120–130, 132, 133 and staunch supporting evidence was provided by extensive studies on the SV40 model system which hijacks the replication machinery of the infected mammalian host cell for viral DNA replication115, 116, 134, 136, 165–174. In particular, studies that utilized human cell free extracts and various experimental techniques to thoroughly analyze the replication of SV40-based plasmids containing a single Ori and a unique, site-specific CPD demonstrated that a UV lesion encountered in the lagging strand template only blocks synthesis of the Okazaki fragment containing the lesion; both leading and lagging strand synthesis continue onward with the progressing replication fork, essentially unaffected70, 89–95. This was later confirmed by EM studies on chromosomal replication forks obtained from S. cerevisiae. In the absence of UV irradiation, ssDNA gaps were not detected. After UV irradiation, 30 – 40% of the replication forks contained one or more ssDNA gaps, most of which (>70%) were smaller than 400 nt. Interestingly, most (>55%) ssDNA gaps were observed more than 2.5 kb from the replication fork and as far away as 15 – 20 kb. Furthermore, the percentage of DNA molecules containing ssDNA gaps only marginally increased (by 17%) when TLS was abolished, indicating that bypass of UV-induced lesions does not dictate the frequency of ssDNA gaps left behind on a lagging strand template85. Rather, such gaps are generated by the relatively fast re-priming of lagging strand templates compared to TLS and these discontinuities are not immediately filled in behind a progressing replication fork.
As mentioned above, enzyme-catalyzed unloading/recycling of PCNA from a P/T junction will not occur until the primer is fully-extended and ligated to the downstream duplex region108. This suggests that PCNA residing at a blocked P/T junction on a lagging strand template persists as the replication fork progresses onward and, hence, pol δ holoenzymes are rapidly re-assembled with new PCNA rings on the nascent hybrid primers upstream of the offending damage. Considering this, we propose that TLS on a lagging strand template occurs by postreplicative gap-filling as described in Figure 9. Hence, “on the fly” TLS is limited to DNA lesions encountered within a leading strand template.
6. Critical aspects of TLS in eukaryotes
UV-induced lesions that persist into S-phase can be accommodated by TLS and this DDT pathway absolutely requires the interaction of TLS pols with PCNA encircling the damaged DNA58–64. As discussed above, extensive stretches of ssDNA are generated when UV-induced lesions are encountered by the replisome, especially within the leading strand template, and the aforementioned models for TLS provide options for when these abnormalities are resolved. Recent studies indicate that postreplicative gaps generated in response to UV irradiation can persist throughout S-phase and into G2/M147–149, 151. Furthermore, PCNA remains associated with these postreplicative gaps, even after bulk DNA synthesis is completed147. Thus, critical aspects of eukaryotic TLS on either template strand are maintaining PCNA at blocked P/T junctions and permitting access of TLS pols and related factors to postreplicative gaps throughout S-phase. Each is discussed below.
6.1. Dynamics of PCNA on UV-damaged DNA
Retention of PCNA at a blocked P/T junction may be promoted during DDT by; 1) inhibiting the unloading of PCNA from DNA; 2) prohibiting diffusion of PCNA along DNA; 3) promoting the loading of PCNA onto DNA or; 4) a combination of one or more of these possibilities (Figure 10). As mentioned above, enzyme-catalyzed unloading of PCNA from a P/T junction will not occur until the primer is fully-extended and ligated to the downstream duplex region108. A PCNA ring may rapidly diffuse along the newly-synthesized dsDNA behind a progressing replication fork175. However, chromatin assembly is rapidly initiated on this nascent duplex DNA by the deposition of nucleosomes within 250 base pairs of a progressing replication fork (see section 6.2 below)176, 177. Furthermore, high-affinity transcription factors essential for the establishment of chromatin structure also re-bind to nascent DNA duplexes immediately after passage of a replication fork178. Thus, diffusion of PCNA along the dsDNA adjacent to blocked P/T junctions is likely restricted during S-phase by physical blocks. However, PCNA may diffuse along the adjacent ssDNA which varies greatly in length and may be as long as 20 kb in human cells85, 86. If PCNA can vacate a blocked P/T junction in this manner, new PCNA rings are not envisioned to be continuously re-loaded as PCNA levels are limiting179 and the RFC clamp loader travels with progressing replication forks during S-phase104, 179. Thus, preventing diffusion of PCNA along ssDNA may be critical to maintain PCNA at blocked P/T junctions for ensuing TLS.
Figure 10.

Dynamics of PCNA on a UV-damaged template. During DDT, the retention of PCNA at a blocked P/T junction abutting a UV-induced lesion (
) is governed by three activities; 1) Enzymatic loading of PCNA onto the P/T junction; 2) Enzymatic unloading of PCNA from the P/T junction and; 3) diffusion of PCNA along either the nascent dsDNA or the adjacent ssDNA. If PCNA can vacate a blocked P/T junction, limiting PCNA rings will not be continuously re-loaded. However, diffusion of PCNA along DNA and enzyme-catalyzed unloading PCNA from DNA are prohibited during TLS, promoting retention of PCNA at blocked P/T junctions during S-phase.
Dramatically increasing the frictional drag of PCNA by tethering a large object to the sliding clamp ring only marginally decreases the diffusion coefficient of PCNA on DNA175. Hence, a large protein simply binding to PCNA encircling a blocked P/T junction will not prohibit diffusion of PCNA along the adjacent ssDNA180. Human RPA binds to ssDNA with extremely high affinity (<pM) at physiological ionic strength and might prohibit diffusion of PCNA along ssDNA by serving as a physical block21, 181. The effect of RPA on the occupancy of various P/T DNA substrates was recently monitored by utilizing a unique Cy3-Cy5 FRET pair that directly reports on the retention of human PCNA on DNA110. Results from various kinetic assays indicated that RPA binds sufficiently tight to ssDNA adjacent to a P/T junction such that it restricts PCNA to the upstream duplex region by physically blocking diffusion of PCNA along ssDNA180.
6.2. Access to postreplicative gaps during S-phase
Genomic DNA is packaged into chromatin within the nucleus of a eukaryotic cell. The fundamental unit of chromatin is the nucleosome which consists of dsDNA wrapped around a histone octamer comprised of one (H3-H4)2 tetramer and two H2A-H2B dimers182. During S-phase, nucleosomes ahead of a replication fork (comprised of parental histones) must be disassembled for the replisome to gain access to the parental DNA. Immediately following passage of a replication fork, histone chaperones deposit parental and newly-synthesized histones onto the nascent daughter DNA, re-forming nucleosomes182, 183. This process is tightly-coupled to on-going DNA replication and occurs within 250 – 600 base pairs of a progressing replication fork (Figure 11A)176, 177. Upon assembly onto nascent DNA, nucleosomes are immediately repositioned into an evenly (regularly) spaced arrangement by ATP-dependent chromatin remodeling enzymes184. Nucleosomal DNA is then further compacted into higher-order structures, culminating with the formation of chromatin where access to genomic DNA is limited185.
Figure 11.

Accessibility of postreplicative gaps. (A) Chromatin dynamics during S-phase. On the lagging strand template, Okazaki fragments yet to be ligated are indicated by dashed lines. During S-phase, nucleosomes ahead of a replication fork are disassembled as the replication fork progresses. Immediately following passage of a replication fork, nucleosomes are re-formed on the nascent daughter DNA by various histone chaperones. B) Monoubiquitination of PCNA and CAF-1 binding. Surface of the human PCNA ring generated with VMD (PDB 1AXC). Coloring scheme is indicated. Each PCNA monomer consists of two independent domains joined by an interdomain connecting loop (IDCL). Three PCNA monomers arrange in a head-to-tail manner, resulting in a ring with structurally distinct faces. The “front” face contains the IDCLs and interacts with pols. Following UV irradiation, single ubiquitin moieties are covalently attached to PCNA rings at the conserved lysine residue 164 (K164). C) Ubiquitin docking site 2. Shown in cartoon form is a side profile of a PCNA subunit-subunit interface. Coloring scheme is identical to panel B. At the interface, the N-terminal domain from one PCNA monomer interacts with the C-terminal domain from the adjacent monomer. At ubiquitin docking site 2, a ubiquitin moiety conjugated to K164 interacts with loop J (indicated) and residues within the subunit-subunit interface. (D) Selective inhibition of CAF-1 activity at postreplicative gaps. Upon formation of postreplicative gaps within either template, chromatin assembly continues in the wake of a progressing replication fork. PCNA is retained at a postreplicative gap and is monoubiquitinated by Rad6/Rad18. We propose that the ubiquitin moieties dock onto the PCNA ring at ubiquitin docking site 2, sheltering/disrupting the CAF-1 binding sites on PCNA. This precludes CAF-1 binding to PCNA, selectively inhibiting nucleosome assembly at/near a postreplicative gap until the gap is filled.
Postreplicative gaps create an intriguing paradox for this process. Chromatin assembly must resume in the wake of restarted replication forks in order for the cell to continue progression through S-phase186. However, postreplicative gaps may persist into G2/M and, hence, must remain accessible throughout S-phase147–149, 151. Upon filling in and sealing of the gap, chromatin must be fully-restored on the nascent DNA to complete the cell cycle187. Thus, chromatin assembly at/near postreplicative gaps must be selectively and dynamically regulated during S-phase, but how? Given these demands, a reversible post-translational modification (PTM) is quite befitting of the task. In response to agents that halt progression of replication forks, such as UV radiation, Rad6/Rad18 catalyzes the attachment of single ubiquitin moieties (i.e. monoubiquitination) to PCNA rings encircling damaged DNA. This PTM is unique to eukaryotic TLS and the ubiquitin attachment site on PCNA (K164) has been fully conserved throughout evolution (Supplemental Figure 1)34. PCNA monoubiquitination is imperative for replication through UV-induced lesions34 and extensive in vivo studies collectively suggest that this PTM is restricted to PCNA residing at postreplicative gaps and is required for postreplicative gap-filling130, 137, 146, 147, 150, 151, 153, 156, 188. In particular, the ubiquitin moieties conjugated to PCNA after exposing human cells to UV are not removed until the bulk of DNA synthesis (i.e., S-phase) has been completed and, hence, postreplicative gaps have been filled150, 189. In the ensuing sections we propose that PCNA monoubiquitination selectively governs chromatin assembly at/near a postreplicative gap until the gap is filled. We begin with a brief overview of monoubiquitinated PCNA and TLS.
6.2.1 Monoubiquitinated PCNA: A Brief Overview
Most studies on the role of monoubiquitinated PCNA during TLS have focused on TLS pols belonging to the Y-family of DNA pols. Members of this family, which includes pol η, contain at least one ubiquitin-binding domain (UBD) in addition to their PCNA-binding domains15. Following UV irradiation, PCNA encircling damaged DNA is monoubiquitinated during S-phase and pol η co-localizes with PCNA in replication factories (foci) on damaged DNA. Both activities are imperative for replication through UV-induced lesions34, 190–192. The seminal in vivo studies proposed that the ubiquitin moieties attached to PCNA may serve to selectively recruit TLS pols via their UBDs to sites of DNA damage where they may also displace a blocked replicative pol190–192. However, the authors pointed out that the studies did not provide evidence for a direct interaction between the UBD of pol η and a ubiquitin moiety conjugated to PCNA191. Indeed, pol η co-localized with PCNA in replication foci during unperturbed S-phase in human cells and this activity required the UBD of pol η even though PCNA monoubiquitination was absent192. Thus, one cannot extrapolate these in vivo findings to conclude that a direct interaction between pol η and a ubiquitin moiety conjugated to PCNA is required or even occurs during DDT190–192. Furthermore, this model posits that TLS pols bind tighter to monoubiquitinated PCNA than to native PCNA due to additive binding domains193, which is counterintuitive to TLS and inconsistent with the remarkably low error rates observed in human cells after UV exposure65, 194. At postreplicative gaps, which may be kilobases long and persist into G2/M, a tighter interaction with monoubiquitinated PCNA would selectively promote error-prone DNA synthesis by Y-family TLS pols downstream of a UV-induced lesion. Finally, three of the seven human TLS pols lack a UBD16. In particular, pol ζ, a B-family TLS pol implicated in TLS following UV exposure (see above), does not contain a UBD yet assembly of pol ζ into replication foci at UV-induced lesions requires PCNA monoubiquitination and is independent of Rev1, a Y-family TLS pol that also recruits other TLS pols195, 196. Together, this argues strongly against a “universal” and direct role of monoubiquitinated PCNA recruiting TLS pols to sites of DNA damage. In follow up studies on these seminal in vivo reports, it was observed that accumulation of pol η into replication foci after UV irradiation was independent of PCNA monoubiquitination197, 198 and did not require the UBD of pol η199. Rather, monoubiquitinated PCNA and the UBD of pol η independently retained localized pol η within replication foci, increasing its residence time. Independent reports arrived at the same conclusion but challenged the requirement of the UBD of pol η for TLS following UV irradiation59, 60. Altogether, these cellular studies suggest that the ubiquitin moieties conjugated to PCNA following UV irradiation do not serve to directly recruit pol η to PCNA encircling damaged DNA. Biochemical studies have since confirmed this.
In studies on isolated binding domains of human pol η, the affinity of a PCNA-binding domain for PCNA was more than 190-fold tighter than the affinity of its UBD for ubiquitin, arguing against a selectively enhanced affinity for monoubiquitinated PCNA thru additive binding domains200, 201. Ensuing studies on the wild-type, full-length protein confirmed that binding of human pol η to PCNA is independent of PCNA monoubiquitination; human pol η binds to PCNA and monoubiquitinated PCNA with identical affinities77. Accordingly, exchange of pol δ for pol η at a P/T junction (i.e. polymerase switching) and the ensuing TLS by pol η across a UV-induced CPD are both independent of PCNA monoubiquitination77, 109. Thus, direct binding of pol η to the ubiquitin moieties conjugated to PCNA, if it occurs, is dispensable for pol η-mediated TLS across a UV-induced CPD lesion, in agreement with the aforementioned in vivo studies59, 60, 77, 109, 197–201. Furthermore, monoubiquitination of PCNA encircling DNA has no effect on the formation, stability, or processivity of the pol δ holoenzyme77, 109. Similar behaviors are expected for pol ε holoenzymes on leading strand templates as the ubiquitin moieties attached to PCNA do not alter the conformation of the sliding clamp ring 202 nor do they shelter the region of PCNA that interacts with DNA pols 203, 204. Collectively, this argues against PCNA monoubiquitination indirectly promoting TLS by destabilizing the replicative pol holoenzymes. Importantly, this also indicates that the ubiquitin moieties attached to PCNA do not need to be removed after TLS so that DNA synthesis by the replicative pols can resume, in agreement with the extended lifetime of monoubiquitinated PCNA in UV-irradiated human cells150, 189. Altogether, these recent cellular and biochemical studies suggest that monoubiquitinated PCNA serves an indirect role in postreplicative gap-filling that persists until the gap is filled.
6.2.2 A Proposal: Monoubiquitination of PCNA inhibits CAF1-mediated nucleosome assembly
Chromatin assembly factor 1 (CAF-1) is unique among histone chaperones in that it has no apparent affinity for either single- or double-stranded DNA and is instead recruited by a direct interaction with PCNA encircling DNA. Once localized, CAF-1 deposits a (H3-H4)2 tetramer onto DNA, initiating nucleosome assembly, and this activity is required following exposure to UV irradiation182, 183, 205–208. However, CAF-1 can efficiently trigger bidirectional propagation of nucleosomal arrays from a ssDNA gap in the absence of DNA synthesis and, indeed, ssDNA gaps can be enveloped by histones and “nucleosome-like” particles209–215. In vivo, such activity may impede postreplicative gap-filling by limiting access of TLS pols and other associated factors to the gap and, hence, must be prevented until the gap is filled. On the other hand, chromatin must be fully-restored on the nascent DNA upon filling in and sealing of the gap to complete the cell cycle187. Thus, CAF-1 activity must be selectively and dynamically regulated at/near postreplicative gaps.
PCNA remains associated with postreplicative gaps generated by UV irradiation, even after bulk DNA synthesis is completed, and such retention is independent of PCNA monoubiquitination by Rad6/Rad18147. In a recent study on human cells, accumulation of pol η into replication foci after UV irradiation was independent of PCNA monoubiquitination but monoubiquitinated PCNA increased the residence time of pol η within the foci. In other words, monoubiquitination of PCNA transiently immobilized pol η within replication foci. The same behavior was observed after treatment with a DNA intercalating agent, DRAQ5, that temporarily disrupts chromatin structure, resulting in the release of histones and exposure of genomic DNA. However, DRAQ5 does not elicit PCNA monoubiquitination nor activate a DNA damage checkpoint, suggesting that transient opening of chromatin promotes access of pol η to UV-damaged DNA197. Thus, we propose the single ubiquitin moieties conjugated to PCNA after DNA damage facilitate the temporal exposure of DNA at/near postreplicative gaps by regulating CAF-1 activity.
PCNA residues (R61, D63) that are required for CAF-1-mediated nucleosome assembly are fully-conserved (Supplemental Figure 1) and located in a distinct region far removed from the region of PCNA that interacts with replication proteins (Figure 11B–C)206. Indeed, PCNA interacts with components of the replication machinery and CAF-1 independently208. In recent modeling studies, the ubiquitin moieties conjugated to PCNA at K164 spent most of the time (65 – 80%) docked onto the sliding clamp ring at two discrete locations (Figure 11B). In both locations, the surface of the ubiquitin moieties that interacted with PCNA was the canonical hydrophobic surface centered on conserved residues L8, I44, and V70, in agreement with a recent NMR study on human, monoubiquitinated PCNA193, 203, 204, 216. 25 – 30% of the time, ubiquitin interacted with the back face of the PCNA ring (ubiquitin docking position 1). This position was first identified in the crystal structure of monoubiquitinated PCNA from budding yeast and is not likely to be biologically relevant as strains harboring mutations within ubiquitin docking site 1 display wild-type resistance to UV irradiation and are not growth defective193, 203, 204, 208. 40 – 50% of the time, ubiquitin was docked onto the side of the PCNA ring at the subunit-subunit interface (ubiquitin docking site 2), intimately close to PCNA residues (R61, D63) required for CAF-1 binding203, 204. In this preferred orientation (ubiquitin docking site 2), the ubiquitin moieties interact with residues at the subunit-subunit interfaces of the PCNA ring, particularly loop J (Figure 11C)203, 204.
Budding yeast strains harboring a E104A/D105A mutation in loop J do not display defects in cell growth or viability under normal conditions. Indeed, the E104A/D105A mutation has no effect on the ability of PCNA to stimulate the activities of DNA replication proteins, pol δ in particular208, 217. However, E104A/D105A strains are grossly defective in postreplicative gap-filling following UV irradiation and, hence, are drastically more sensitive to killing by UV compared to wild-type cells207, 208. Furthermore, the enhanced UV sensitivity of the E104A/D105A mutant was not elevated further when either rad6 or rad18 were deleted. Altogether, this suggests that the E104A/D105A mutation in ubiquitin docking site 2 specifically impairs a Rad6/Rad18 dependent process (i.e. postreplicative gap-filling) in a manner that is independent of the interactions between PCNA and the DNA replication machinery217. Considering these observations, we propose that the ubiquitin moieties conjugated to PCNA at a postreplicative gap preferentially dock onto the sliding clamp ring at ubiquitin docking site 2, sheltering or disrupting the CAF-1 binding sites on PCNA (Figure 11D). This precludes CAF-1 binding to PCNA and, hence, selectively inhibits nucleosome assembly at/near a postreplicative gap until the gap is filled. Future studies are underway to test this proposal.
6.2.3 Maintenance of monoubiquitinated PCNA throughout S-phase
As mentioned above, postreplicative gaps may persist into G2/M phase of the cell cycle and, hence, must remain accessible throughout S-phase147–149, 151. This is particularly relevant in human cells where postreplicative gaps generated by UV irradiation at the G1/S border disappear primarily in G2 phase when the bulk of chromosomal replication has been completed, ~16 – 24 hours after irradiation 151. According to the proposed model (Figure 11D), monoubiquitinated PCNA must be generated and maintained at postreplicative gaps for an extensive period of time. Indeed, this PTM is upregulated in human cells following UV exposure and remains elevated until the bulk of DNA synthesis (i.e., S-phase) has been completed and, hence, the postreplicative gaps have been filled150. A similar trajectory was observed in genetic studies on budding yeast146. As described below, this feat is achieved on one hand by promoting PCNA monoubiquitination and on the other hand by inhibiting the removal of this PTM.
The signal for PCNA monoubiquitination is the build-up and persistence of RPA-coated ssDNA downstream of blocked P/T junctions during S-phase. Specifically, the Rad6/Rad18 complex is recruited to RPA-coated ssDNA where it catalyzes monoubiquitination of PCNA encircling the blocked P/T junction upstream34. The upregulation of Rad6/Rad18 activity during DDT has been extensively reviewed34. Given the model proposed in Figure 11D, it is relevant to re-iterate that ATP-dependent chromatin remodeling enzymes enhance PCNA monoubiquitination and this may ensure inhibition of CAF-1 activity. Recent in vivo studies from human and S. cerevisiae indicate that the RSC (remodels the structure of chromatin) and NuRD (nucleosome remodeling deacetylase) complexes are required for normal PCNA ubiquitination in response to UV irradiation. This suggests these remodelers direct Rad6/Rad18 to PCNA encircling blocked P/T junctions by increasing the accessibility of DNA surrounding UV-induced lesions, perhaps by repositioning/removing nucleosomes deposited by CAF-1 prior to PCNA monoubiquitination.
USP1 is a member of the ubiquitin-specific protease (USP) family of deubiquitinating enzymes (DUBs) and is responsible for removing ubiquitin from PCNA218. The USP1 protein is degraded during G1 phase, rapidly accumulates as the cell transitions into S-phase, and remains elevated until the late stages of mitosis 219–226. The high levels of USP1 during unperturbed S-phase allow any unwarranted ubiquitination of PCNA that may occur during normal DNA replication to be counteracted. However, upon exposure to DNA-damaging agents that block progression of the replication fork, the activity of USP1 must be down-regulated to permit the buildup of monoubiquitinated PCNA on DNA227. Interestingly, the pathway by which this is achieved is unique to the DNA-damaging agent itself150, 189, 227–231. In response to UV irradiation, transcription of the USP1 gene is shut off and the USP1 protein undergoes internal autocleavage by its own protease activity on a sequence immediately after a well-conserved C-terminal glycine-glycine region221, 227, 230. The auto-generated C-terminal fragment (Gln-Usp1Ct) is inactive and bears an N-terminal glutamine that targets it for ubiquitin-dependent degradation via the Arg/N-end rule pathway232. Together, this significantly decreases the cellular concentration of active USP1 protein for an extended period of time following UV exposure, permitting the buildup and maintenance of monoubiquitinated PCNA at postreplicative gaps150, 221, 226, 227, 230. Such regulation is essential for replication through UV-induced lesions150, 227.
7. Conclusions and perspectives
Beginning in the mid 1960’s, many independent groups began investigating how eukaryotic cells replicate genomic DNA following exposure to DNA-damaging agents, UV radiation in particular. These pioneering studies collectively demonstrated that eukaryotes can perform DNA synthesis across UV-induced lesions and such activity is imperative for cell survival114, 116–118, 120–130, 188, 233–244. Substantial efforts in the ensuing years aimed to temporally and spatially define how this complex process unfolds in vivo. In this review, we thoroughly analyzed these and other related studies to dissect the step-by-step intricacies of this complex process and provide our current understanding of TLS on leading and lagging strand templates. The major points are summarized below.
There are two major UV-induced lesions. CPDs are more prominent and most persist into and throughout S-phase. The less-prominent (6–4) PPs are likely repaired prior to an encounter with a moving replisome (Figure 3). Both lesions can be accommodated, i.e., “tolerated,” during S-phase by TLS.
UV-induced lesions do not to stall/arrest unwinding of duplex DNA by the CMG helicase at the lesion site.
In the extremely rare event that pol α-primase encounters a UV-induced lesion on a lagging strand template, pol α-primase simply releases the lagging strand template and re-primes upstream (5′) of the UV-induced lesion, keeping pace with the progressing replisome. This leaves behind a ssDNA gap extending from the blocked 3′ hydroxyl terminus of the aborted primer to the 5′ terminus of the downstream primer and passes the burden of the UV-lesion onto pol δ (Figure 4).
When a UV-induced lesion is encountered within a leading strand template, the coordinated activities of pol ε and the CMG helicase become “uncoupled”; DNA synthesis by pol ε abruptly stops while the CMG helicase progresses onward, exposing long stretches of the leading strand template before eventually stalling downstream of the offending damage. During such uncoupling, DNA synthesis continues on the undamaged lagging strand template as the CMG helicase continues to unwind duplex DNA and RPA coats the exposed leading strand template (Figure 5). The stalled replication fork may re-started by either “on the fly” TLS (Figure 7) or postreplicative gap-filling (Figure 8).
When a UV-induced lesion is encountered within a lagging strand template, only DNA synthesis on the damaged Okazaki fragment stops; The CMG helicase and leading strand DNA synthesis continue unimpeded and lagging strand DNA synthesis resumes upstream of the offending damage through a “scheduled” re-priming event (Figures 6 and 9). The damage is later bypassed by postreplicative gap-filling behind the progressing replication fork.
“On the fly” TLS occurs only on leading strand templates while postreplicative gap-filling may occur on either template strand.
During DDT, PCNA is retained at blocked P/T junctions by concerted endeavors (Figure 10); 1) enzymatic unloading of PCNA from the P/T junction is prohibited; 2) Physical blocks such as nucleosomes restrict diffusion of PCNA along the dsDNA adjacent to the blocked P/T junction and; 3) RPA binds tight to the ssDNA adjacent to a blocked P/T junction, restricting PCNA to the upstream duplex by physically blocking diffusion of PCNA along ssDNA.
In response to agents that halt progression of replication forks, Rad6/Rad18 catalyzes the attachment of single ubiquitin moieties (i.e. monoubiquitination) to PCNA rings encircling damage DNA. This PTM is generated and maintained throughout S-phase and into G2/M by the synergistic upregulation of Rad6/Rad18 activity and inhibition of the USP1 protease that removes ubiquitin from PCNA.
PCNA monoubiquitination is restricted to PCNA residing at postreplicative gaps and is required for postreplicative gap-filling. This PTM is imperative for replication through UV-induced lesions but its functional role is currently unknown. We propose that the ubiquitin moieties conjugated to PCNA at a postreplicative gap dock onto the sliding clamp ring, sheltering or disrupting the CAF-1 binding sites on PCNA (Figure 11). This precludes CAF-1 binding to PCNA and, hence, selectively inhibits chromatin assembly at/near a postreplicative gap until the gap is filled.
TLS across the two major UV-induced lesions, CPDs and (6–4)PPs, is predominantly error-free in vivo and is carried out by pols η and ζ, respectively. However, as studies in the field have progressed, additional TLS pols have been identified, further complicating the matter. Currently, at least seven TLS pols are utilized in human cells to combat the onslaught of diverse DNA-damaging agents16, 34. With an “open” DNA polymerase active site, these pols can accommodate multiple DNA lesions, including CPDs and (6–4)PPs, albeit with varying fidelities15. Hence, the appropriate TLS pol (or combination of TLS pols) must be selected to maximize the fidelity of DNA synthesis following exposure to UV radiation. Following UV irradiation of human cells, pols η, ζ, κ, and ι, accumulate into replication foci on damaged DNA34, 196. In particular, Rad6/Rad18 may physically deliver pol η●pol ι complexes to UV-irradiated DNA in a process driven by damaged-induced PTMs to the involved proteins34, 245–247. Furthermore, UV irradiation elicits monoubiquitination of PCNA by Rad6/Rad18 and phosphorylation of pol η, both of which are imperative for pol η-mediated TLS in vivo34, 198. However, Polη has an eightfold preference for mis-insertion at (6–4)PPs and pols ζ, ι, and κ all perform mutagenic TLS across CPDs in human cells lacking functional pol η 58, 65, 67, 68. Thus, DNA-damaging agents and the cellular response they trigger may generically target TLS pols to damaged DNA but selection of the appropriate TLS pol(s) and, hence, the fidelity of DNA damage bypass is ultimately governed by the DNA lesion itself and perhaps the surrounding sequence. In other words, kinetics and thermodynamics drive selection of TLS pols during DDT and future biochemical studies are required to define this critical process.
Supplementary Material
Figure S1. Conservation of PCNA
Acknowledgments
We would like to thank Dr. Michelle Spiering, PhD of the Benkovic lab for critical reading of this manuscript.
Biographies
Mark Hedglin was born in Clarks Summit, PA and received his B.A. in Biochemistry from Ithaca College. While at Ithaca College, he performed research in the lab of Dr. Scott Ulrich, focusing on the rational design of isozyme-specific histone deacetylases (HDAC) inhibitors. He was also a member of the football team and was a two-time District I Academic All-American and an ESPN Academic All-American. He obtained his Ph.D. in Chemical Biology at the University of Michigan under the supervision of Dr. Patrick J. O’Brien. His graduate studies focused on elucidating the searching mechanism of a human DNA repair enzyme. In 2010, his interests shifted from DNA damage repair to DNA damage tolerance and he joined the laboratory of Dr. Stephen J. Benkovic in the Department of Chemistry at the Pennsylvania State University where he was the recipient of the Ruth L. Kirschstein National Research Service Award from National Cancer Institute of the NIH. His postdoctoral studies focus on human DNA replication and DNA damage Tolerance.
Stephen J. Benkovic was born in Orange, NJ. He received his B.S. degree in Chemistry and A.B. degree in English Literature from Lehigh University, and his Ph.D. in Organic Chemistry from Cornell University. After a period as a postdoctoral research associate at the University of California, Santa Barbara, he joined the Chemistry Department at Penn State University in 1965 and became a Full Professor of Chemistry in 1970, followed by recognitions as an Evan Pugh Professor of Chemistry, and in 1988 the holder of the Eberly Chair in Chemistry. Benkovic’s work has spanned many diverse fields and topics including DNA replication, dynamic protein motion in catalysis, de novo purine biosynthesis, cellular communication, and boron therapeutics. His work has been recognized by many awards and fellowships including the NIH Career Development Award, the Pfizer Award in Enzyme Chemistry, the Benjamin Franklin Medal in Life Science, the National Medal of Science, and the National Academy of Science Award in Chemical Sciences. In addition, he has been elected to memberships in the American Academy of Arts and Sciences, the National Academy of Sciences, the Institute of Medicine, and the American Philosophical Society. One of Benkovic’s recent interests is human DNA damage tolerance, particularly translesion DNA synthesis.
References
- 1.Sun J, Shi Y, Georgescu RE, Yuan Z, Chait BT, Li H, O’Donnell ME. The architecture of a eukaryotic replisome. Nat Struct Mol Biol. 2015;22:976–982. doi: 10.1038/nsmb.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costa A, Renault L, Swuec P, Petojevic T, Pesavento JJ, Ilves I, MacLellan-Gibson K, Fleck RA, Botchan MR, Berger JM. DNA binding polarity, dimerization, and ATPase ring remodeling in the CMG helicase of the eukaryotic replisome. Elife. 2014;3:e03273. doi: 10.7554/eLife.03273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balakrishnan L, Bambara RA. Okazaki fragment metabolism. Cold Spring Harb Perspect Biol. 2013;5:185–196. doi: 10.1101/cshperspect.a010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lujan SA, Williams JS, Kunkel TA. DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol. 2016;26:640–654. doi: 10.1016/j.tcb.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leonard AC, Mechali M. DNA replication origins. Cold Spring Harb Perspect Biol. 2013;5:a010116. doi: 10.1101/cshperspect.a010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nature reviews. 11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol. 13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Washington MT, Carlson KD, Freudenthal BD, Pryor JM. Variations on a theme: eukaryotic Y-family DNA polymerases. Biochimica et biophysica acta. 1804:1113–1123. doi: 10.1016/j.bbapap.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annual review of biochemistry. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 10.Lehmann AR. Replication of damaged DNA by translesion synthesis in human cells. FEBS letters. 2005;579:873–876. doi: 10.1016/j.febslet.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 11.Chun AC, Jin DY. Ubiquitin-dependent regulation of translesion polymerases. Biochemical Society transactions. 38:110–115. doi: 10.1042/BST0380110. [DOI] [PubMed] [Google Scholar]
- 12.Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, Green CM. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA repair. 2007;6:891–899. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Andersen PL, Xu F, Xiao W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell research. 2008;18:162–173. doi: 10.1038/cr.2007.114. [DOI] [PubMed] [Google Scholar]
- 14.Friedberg EC. Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol. 2005;6:943–953. doi: 10.1038/nrm1781. [DOI] [PubMed] [Google Scholar]
- 15.Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nature reviews. Molecular cell biology. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soultanas P. Loading mechanisms of ring helicases at replication origins. Molecular microbiology. 84:6–16. doi: 10.1111/j.1365-2958.2012.08012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boos D, Frigola J, Diffley JF. Activation of the replicative DNA helicase: breaking up is hard to do. Current opinion in cell biology. 24:423–430. doi: 10.1016/j.ceb.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 19.McGlynn P. Helicases at the replication fork. Advances in experimental medicine and biology. 2013;767:97–121. doi: 10.1007/978-1-4614-5037-5_5. [DOI] [PubMed] [Google Scholar]
- 20.Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, Walter JC. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146:931–941. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen B, Sokoloski J, Galletto R, Elson EL, Wold MS, Lohman TM. Diffusion of human replication protein A along single-stranded DNA. J Mol Biol. 2014;426:3246–3261. doi: 10.1016/j.jmb.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balakrishnan L, Bambara RA. Eukaryotic lagging strand DNA replication employs a multi-pathway mechanism that protects genome integrity. The Journal of biological chemistry. 286:6865–6870. doi: 10.1074/jbc.R110.209502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annual review of biochemistry. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 24.Zhang D, O’Donnell M. The Eukaryotic Replication Machine. The Enzymes. 2016;39:191–229. doi: 10.1016/bs.enz.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Frick DN, Richardson CC. DNA primases. Annual review of biochemistry. 2001;70:39–80. doi: 10.1146/annurev.biochem.70.1.39. [DOI] [PubMed] [Google Scholar]
- 26.Zhou B, Arnett DR, Yu X, Brewster A, Sowd GA, Xie CL, Vila S, Gai D, Fanning E, Chen XS. Structural basis for the interaction of a hexameric replicative helicase with the regulatory subunit of human DNA polymerase alpha-primase. The Journal of biological chemistry. 287:26854–26866. doi: 10.1074/jbc.M112.363655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuchta RD, Stengel G. Mechanism and evolution of DNA primases. Biochimica et biophysica acta. 1804:1180–1189. doi: 10.1016/j.bbapap.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel SS, Pandey M, Nandakumar D. Dynamic coupling between the motors of DNA replication: hexameric helicase, DNA polymerase, and primase. Current opinion in chemical biology. 15:595–605. doi: 10.1016/j.cbpa.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng L, Shen B. Okazaki fragment maturation: nucleases take centre stage. Journal of molecular cell biology. 3:23–30. doi: 10.1093/jmcb/mjq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, Ivanova ME, Kilkenny ML, Renault L, Kjaer S, Matak-Vinkovic D, Labib K, Costa A, Pellegrini L. A Ctf4 trimer couples the CMG helicase to DNA polymerase alpha in the eukaryotic replisome. Nature. 2014;510:293–297. doi: 10.1038/nature13234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Georgescu RE, Schauer GD, Yao NY, Langston LD, Yurieva O, Zhang D, Finkelstein J, O’Donnell ME. Reconstitution of a eukaryotic replisome reveals suppression mechanisms that define leading/lagging strand operation. eLife. 2015;4:e04988. doi: 10.7554/eLife.04988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.You Z, De Falco M, Kamada K, Pisani FM, Masai H. The mini-chromosome maintenance (Mcm) complexes interact with DNA polymerase alpha-primase and stimulate its ability to synthesize RNA primers. PloS one. 2013;8:e72408. doi: 10.1371/journal.pone.0072408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hedglin M, Kumar R, Benkovic SJ. Replication clamps and clamp loaders. Cold Spring Harb Perspect Biol. 2013;5:a010165. doi: 10.1101/cshperspect.a010165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hedglin M, Benkovic SJ. Regulation of Rad6/Rad18 Activity During DNA Damage Tolerance. Annual review of biophysics. 2015;44:207–228. doi: 10.1146/annurev-biophys-060414-033841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shcherbakova PV, Pavlov YI. 3’-->5’ exonucleases of DNA polymerases epsilon and delta correct base analog induced DNA replication errors on opposite DNA strands in Saccharomyces cerevisiae. Genetics. 1996;142:717–726. doi: 10.1093/genetics/142.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karthikeyan R, Vonarx EJ, Straffon AF, Simon M, Faye G, Kunz BA. Evidence from mutational specificity studies that yeast DNA polymerases delta and epsilon replicate different DNA strands at an intracellular replication fork. Journal of molecular biology. 2000;299:405–419. doi: 10.1006/jmbi.2000.3744. [DOI] [PubMed] [Google Scholar]
- 37.Fukui T, Yamauchi K, Muroya T, Akiyama M, Maki H, Sugino A, Waga S. Distinct roles of DNA polymerases delta and epsilon at the replication fork in Xenopus egg extracts. Genes to cells : devoted to molecular & cellular mechanisms. 2004;9:179–191. doi: 10.1111/j.1356-9597.2004.00716.x. [DOI] [PubMed] [Google Scholar]
- 38.Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM. Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication. Genes & development. 2004;18:2764–2773. doi: 10.1101/gad.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, Suzuki M, Kunkel TA. Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Current biology : CB. 2006;16:202–207. doi: 10.1016/j.cub.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Molecular cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langston LD, Zhang D, Yurieva O, Georgescu RE, Finkelstein J, Yao NY, Indiani C, O’Donnell ME. CMG helicase and DNA polymerase epsilon form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:15390–15395. doi: 10.1073/pnas.1418334111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J, Agarwal T, O’Donnell ME. Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nature structural & molecular biology. 2014;21:664–670. doi: 10.1038/nsmb.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu C, Gan H, Han J, Zhou ZX, Jia S, Chabes A, Farrugia G, Ordog T, Zhang Z. Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Molecular cell. 2014;56:551–563. doi: 10.1016/j.molcel.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyabe I, Kunkel TA, Carr AM. The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS genetics. 2011;7:e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reijns MA, Kemp H, Ding J, de Proce SM, Jackson AP, Taylor MS. Lagging-strand replication shapes the mutational landscape of the genome. Nature. 2015;518:502–506. doi: 10.1038/nature14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clausen AR, Lujan SA, Burkholder AB, Orebaugh CD, Williams JS, Clausen MF, Malc EP, Mieczkowski PA, Fargo DC, Smith DJ, Kunkel TA. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nature structural & molecular biology. 2015;22:185–191. doi: 10.1038/nsmb.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sengupta S, van Deursen F, de Piccoli G, Labib K. Dpb2 integrates the leading-strand DNA polymerase into the eukaryotic replisome. Current biology : CB. 2013;23:543–552. doi: 10.1016/j.cub.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 49.Schauer GD, O’Donnell ME. Quality control mechanisms exclude incorrect polymerases from the eukaryotic replication fork. Proc Natl Acad Sci U S A. 2017;114:675–680. doi: 10.1073/pnas.1619748114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burgers PM, Bambara RA, Campbell JL, Chang LM, Downey KM, Hubscher U, Lee MY, Linn SM, So AG, Spadari S. Revised nomenclature for eukaryotic DNA polymerases. European journal of biochemistry / FEBS. 1990;191:617–618. doi: 10.1111/j.1432-1033.1990.tb19165.x. [DOI] [PubMed] [Google Scholar]
- 51.Chilkova O, Stenlund P, Isoz I, Stith CM, Grabowski P, Lundstrom EB, Burgers PM, Johansson E. The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic acids research. 2007;35:6588–6597. doi: 10.1093/nar/gkm741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beukers R, Eker AP, Lohman PH. 50 years thymine dimer. DNA repair. 2008;7:530–543. doi: 10.1016/j.dnarep.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 53.Scharer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harbor perspectives in biology. 2013;5:a012609. doi: 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lucas-Lledo JI, Lynch M. Evolution of mutation rates: phylogenomic analysis of the photolyase/cryptochrome family. Mol Biol Evol. 2009;26:1143–1153. doi: 10.1093/molbev/msp029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCready SJ, Osman F, Yasui A. Repair of UV damage in the fission yeast Schizosaccharomyces pombe. Mutat Res. 2000;451:197–210. doi: 10.1016/s0027-5107(00)00050-6. [DOI] [PubMed] [Google Scholar]
- 56.Kemp MG, Sancar A. DNA excision repair: where do all the dimers go? Cell Cycle. 2012;11:2997–3002. doi: 10.4161/cc.21126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu J, Adar S, Selby CP, Lieb JD, Sancar A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes & development. 2015;29:948–960. doi: 10.1101/gad.261271.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shriber P, Leitner-Dagan Y, Geacintov N, Paz-Elizur T, Livneh Z. DNA sequence context greatly affects the accuracy of bypass across an ultraviolet light 6–4 photoproduct in mammalian cells. Mutation research. 2015;780:71–76. doi: 10.1016/j.mrfmmm.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Acharya N, Yoon JH, Gali H, Unk I, Haracska L, Johnson RE, Hurwitz J, Prakash L, Prakash S. Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase eta in translesion DNA synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17724–17729. doi: 10.1073/pnas.0809844105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Acharya N, Yoon JH, Hurwitz J, Prakash L, Prakash S. DNA polymerase eta lacking the ubiquitin-binding domain promotes replicative lesion bypass in humans cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10401–10405. doi: 10.1073/pnas.1005492107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Makarova AV, Stodola JL, Burgers PM. A four-subunit DNA polymerase zeta complex containing Pol delta accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic acids research. 2012;40:11618–11626. doi: 10.1093/nar/gks948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Northam MR, Garg P, Baitin DM, Burgers PM, Shcherbakova PV. A novel function of DNA polymerase zeta regulated by PCNA. The EMBO journal. 2006;25:4316–4325. doi: 10.1038/sj.emboj.7601320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haracska L, Johnson RE, Unk I, Phillips BB, Hurwitz J, Prakash L, Prakash S. Targeting of human DNA polymerase iota to the replication machinery via interaction with PCNA. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14256–14261. doi: 10.1073/pnas.261560798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vidal AE, Kannouche P, Podust VN, Yang W, Lehmann AR, Woodgate R. Proliferating cell nuclear antigen-dependent coordination of the biological functions of human DNA polymerase iota. The Journal of biological chemistry. 2004;279:48360–48368. doi: 10.1074/jbc.M406511200. [DOI] [PubMed] [Google Scholar]
- 65.Yoon JH, Prakash L, Prakash S. Error-free replicative bypass of (6–4) photoproducts by DNA polymerase zeta in mouse and human cells. Genes & development. 2010;24:123–128. doi: 10.1101/gad.1872810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Livneh Z, Cohen IS, Paz-Elizur T, Davidovsky D, Carmi D, Swain U, Mirlas-Neisberg N. High-resolution genomic assays provide insight into the division of labor between TLS and HDR in mammalian replication of damaged DNA. DNA Repair (Amst) 2016;44:59–67. doi: 10.1016/j.dnarep.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 67.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 68.Ziv O, Geacintov N, Nakajima S, Yasui A, Livneh Z. DNA polymerase zeta cooperates with polymerases kappa and iota in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11552–11557. doi: 10.1073/pnas.0812548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Veaute X, Mari-Giglia G, Lawrence CW, Sarasin A. UV lesions located on the leading strand inhibit DNA replication but do not inhibit SV40 T-antigen helicase activity. Mutation research. 2000;459:19–28. doi: 10.1016/s0921-8777(99)00052-x. [DOI] [PubMed] [Google Scholar]
- 71.McCulloch SD, Kokoska RJ, Chilkova O, Welch CM, Johansson E, Burgers PM, Kunkel TA. Enzymatic switching for efficient and accurate translesion DNA replication. Nucleic acids research. 2004;32:4665–4675. doi: 10.1093/nar/gkh777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kusumoto R, Masutani C, Shimmyo S, Iwai S, Hanaoka F. DNA binding properties of human DNA polymerase eta: implications for fidelity and polymerase switching of translesion synthesis. Genes to cells : devoted to molecular & cellular mechanisms. 2004;9:1139–1150. doi: 10.1111/j.1365-2443.2004.00797.x. [DOI] [PubMed] [Google Scholar]
- 73.Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L, Mendez J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol. 2013;20:1383–1389. doi: 10.1038/nsmb.2719. [DOI] [PubMed] [Google Scholar]
- 74.Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD, Doherty AJ. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell. 2013;52:566–573. doi: 10.1016/j.molcel.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams JI, Cleaver JE. Excision repair of ultraviolet damage in mammalian cells. Evidence for two steps in the excision of pyrimidine dimers. Biophysical journal. 1978;22:265–279. doi: 10.1016/S0006-3495(78)85488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Elvers I, Johansson F, Groth P, Erixon K, Helleday T. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic acids research. 2011;39:7049–7057. doi: 10.1093/nar/gkr420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hedglin M, Pandey B, Benkovic SJ. Characterization of human translesion DNA synthesis across a UV-induced DNA lesion. Elife. 2016;5:e19788. doi: 10.7554/eLife.19788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Conti C, Sacca B, Herrick J, Lalou C, Pommier Y, Bensimon A. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Molecular biology of the cell. 2007;18:3059–3067. doi: 10.1091/mbc.E06-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Terret ME, Sherwood R, Rahman S, Qin J, Jallepalli PV. Cohesin acetylation speeds the replication fork. Nature. 2009;462:231–234. doi: 10.1038/nature08550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. The Journal of cell biology. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sekedat MD, Fenyo D, Rogers RS, Tackett AJ, Aitchison JD, Chait BT. GINS motion reveals replication fork progression is remarkably uniform throughout the yeast genome. Mol Syst Biol. 2010;6:353. doi: 10.1038/msb.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zahurancik WJ, Klein SJ, Suo Z. Kinetic mechanism of DNA polymerization catalyzed by human DNA polymerase epsilon. Biochemistry. 2013;52:7041–7049. doi: 10.1021/bi400803v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes & development. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang DJ, Lupardus PJ, Cimprich KA. Monoubiquitination of proliferating cell nuclear antigen induced by stalled replication requires uncoupling of DNA polymerase and mini-chromosome maintenance helicase activities. The Journal of biological chemistry. 2006;281:32081–32088. doi: 10.1074/jbc.M606799200. [DOI] [PubMed] [Google Scholar]
- 85.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Molecular cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 86.Lonn U, Lonn S. Extensive regions of single-stranded DNA in aphidicolin-treated melanoma cells. Biochemistry. 1988;27:566–570. doi: 10.1021/bi00402a010. [DOI] [PubMed] [Google Scholar]
- 87.Hashimoto Y, Puddu F, Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nature structural & molecular biology. 2011;19:17–24. doi: 10.1038/nsmb.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Van C, Yan S, Michael WM, Waga S, Cimprich KA. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J Cell Biol. 2010;189:233–246. doi: 10.1083/jcb.200909105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Svoboda DL, Vos JM. Differential replication of a single, UV-induced lesion in the leading or lagging strand by a human cell extract: fork uncoupling or gap formation. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:11975–11979. doi: 10.1073/pnas.92.26.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Svoboda DL, Briley LP, Vos JM. Defective bypass replication of a leading strand cyclobutane thymine dimer in xeroderma pigmentosum variant cell extracts. Cancer research. 1998;58:2445–2448. [PubMed] [Google Scholar]
- 91.Cordeiro-Stone M, Zaritskaya LS, Price LK, Kaufmann WK. Replication fork bypass of a pyrimidine dimer blocking leading strand DNA synthesis. The Journal of biological chemistry. 1997;272:13945–13954. doi: 10.1074/jbc.272.21.13945. [DOI] [PubMed] [Google Scholar]
- 92.Cordeiro-Stone M, Makhov AM, Zaritskaya LS, Griffith JD. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. Journal of molecular biology. 1999;289:1207–1218. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 93.Cordeiro-Stone M, Nikolaishvili-Feinberg N. Asymmetry of DNA replication and translesion synthesis of UV-induced thymine dimers. Mutation research. 2002;510:91–106. doi: 10.1016/s0027-5107(02)00255-5. [DOI] [PubMed] [Google Scholar]
- 94.Nikolaishvili-Feinberg N, Cordeiro-Stone M. Bypass replication in vitro of UV-induced photoproducts blocking leading or lagging strand synthesis. Biochemistry. 2001;40:15215–15223. doi: 10.1021/bi011474t. [DOI] [PubMed] [Google Scholar]
- 95.Carty MP, Lawrence CW, Dixon K. Complete replication of plasmid DNA containing a single UV-induced lesion in human cell extracts. The Journal of biological chemistry. 1996;271:9637–9647. doi: 10.1074/jbc.271.16.9637. [DOI] [PubMed] [Google Scholar]
- 96.Kim C, Snyder RO, Wold MS. Binding properties of replication protein A from human and yeast cells. Mol Cell Biol. 1992;12:3050–3059. doi: 10.1128/mcb.12.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim C, Paulus BF, Wold MS. Interactions of human replication protein A with oligonucleotides. Biochemistry. 1994;33:14197–14206. doi: 10.1021/bi00251a031. [DOI] [PubMed] [Google Scholar]
- 98.Gibb B, Silverstein TD, Finkelstein IJ, Greene EC. Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Anal Chem. 2012;84:7607–7612. doi: 10.1021/ac302117z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gibb B, Ye LF, Gergoudis SC, Kwon Y, Niu H, Sung P, Greene EC. Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014;9:e87922. doi: 10.1371/journal.pone.0087922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen J, Le S, Basu A, Chazin WJ, Yan J. Mechanochemical regulations of RPA’s binding to ssDNA. Sci Rep. 2015;5:9296. doi: 10.1038/srep09296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumaran S, Kozlov AG, Lohman TM. Saccharomyces cerevisiae replication protein A binds to single-stranded DNA in multiple salt-dependent modes. Biochemistry. 2006;45:11958–11973. doi: 10.1021/bi060994r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell. 2015;59:998–1010. doi: 10.1016/j.molcel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. The Journal of biological chemistry. 2013;288:31458–31467. doi: 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, Remeseiro S, Rodrigo-Perez S, Rojas AM, Mendez J, Munoz J, Fernandez-Capetillo O. A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 2013;3:1105–1116. doi: 10.1016/j.celrep.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nitani N, Yadani C, Yabuuchi H, Masukata H, Nakagawa T. Mcm4 C-terminal domain of MCM helicase prevents excessive formation of single-stranded DNA at stalled replication forks. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12973–12978. doi: 10.1073/pnas.0805307105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bruck I, Kaplan DL. Cdc45 protein-single-stranded DNA interaction is important for stalling the helicase during replication stress. The Journal of biological chemistry. 2013;288:7550–7563. doi: 10.1074/jbc.M112.440941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kubota T, Katou Y, Nakato R, Shirahige K, Donaldson AD. Replication-Coupled PCNA Unloading by the Elg1 Complex Occurs Genome-wide and Requires Okazaki Fragment Ligation. Cell reports. 2015;12:774–787. doi: 10.1016/j.celrep.2015.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hedglin M, Pandey B, Benkovic SJ. Stability of the human polymerase delta holoenzyme and its implications in lagging strand DNA synthesis. Proc Natl Acad Sci U S A. 2016;113:E1777–1786. doi: 10.1073/pnas.1523653113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hedglin M, Perumal SK, Hu Z, Benkovic S. Stepwise assembly of the human replicative polymerase holoenzyme. eLife. 2013;2:e00278. doi: 10.7554/eLife.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kumar R, Nashine VC, Mishra PP, Benkovic SJ, Lee TH. Stepwise loading of yeast clamp revealed by ensemble and single-molecule studies. Proc Natl Acad Sci U S A. 2010;107:19736–19741. doi: 10.1073/pnas.1014139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou Y, Meng X, Zhang S, Lee EY, Lee MY. Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PLoS One. 2012;7:e39156. doi: 10.1371/journal.pone.0039156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Langston LD, O’Donnell M. DNA polymerase delta is highly processive with proliferating cell nuclear antigen and undergoes collision release upon completing DNA. J Biol Chem. 2008;283:29522–29531. doi: 10.1074/jbc.M804488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Edenberg HJ. Inhibition of DNA replication by ultraviolet light. Biophysical journal. 1976;16:849–860. doi: 10.1016/S0006-3495(76)85735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Edenberg HJ. Inhibition of simian virus 40 DNA replication by ultraviolet light. Virology. 1983;128:298–309. doi: 10.1016/0042-6822(83)90257-x. [DOI] [PubMed] [Google Scholar]
- 116.Sarasin AR, Hanawalt PC. Replication of ultraviolet-irradiated simian virus 40 in monkey kidney cells. Journal of molecular biology. 1980;138:299–319. doi: 10.1016/0022-2836(80)90288-0. [DOI] [PubMed] [Google Scholar]
- 117.Lehmann AR. Postreplication repair of DNA in ultraviolet-irradiated mammalian cells. J Mol Biol. 1972;66:319–337. doi: 10.1016/0022-2836(72)90418-4. [DOI] [PubMed] [Google Scholar]
- 118.Lehmann AR. Post-replication repair of DNA in ultraviolet-irradiated mammalian cells. No gaps in DNA synthesized late after ultraviolet irradiation. European journal of biochemistry / FEBS. 1972;31:438–445. doi: 10.1111/j.1432-1033.1972.tb02550.x. [DOI] [PubMed] [Google Scholar]
- 119.Lehmann AR, Stevens S. Postreplication Repair of DNA in Chick Cells - Studies Using Photoreactivation. Biochimica et biophysica acta. 1975;402:179–187. doi: 10.1016/0005-2787(75)90037-4. [DOI] [PubMed] [Google Scholar]
- 120.Lehmann AR, Kirk-Bell S, Arlett CF, Paterson MC, Lohman PH, de Weerd-Kastelein EA, Bootsma D. Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proceedings of the National Academy of Sciences of the United States of America. 1975;72:219–223. doi: 10.1073/pnas.72.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fujiwara Y. Characteristics of DNA synthesis following ultraviolet light irradiation in mouse L cells. Postreplication repair. Experimental cell research. 1972;75:485–489. doi: 10.1016/0014-4827(72)90456-9. [DOI] [PubMed] [Google Scholar]
- 122.Fujiwara Y, Kondo T. Caffeine-sensitive repair of ultraviolet light-damaged DNA of mouse L cells. Biochem Biophys Res Commun. 1972;47:557–564. doi: 10.1016/0006-291x(72)90915-1. [DOI] [PubMed] [Google Scholar]
- 123.Buhl SN, Stillman RM, Setlow RB, Regan JD. DNA chain elongation and joining in normal human and xeroderma pigmentosum cells after ultraviolet irradiation. Biophysical journal. 1972;12:1183–1191. doi: 10.1016/S0006-3495(72)86154-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Buhl SN, Setlow RB, Regan JD. DNA repair in Potorous tridactylus. Biophysical journal. 1974;14:791–803. doi: 10.1016/S0006-3495(74)85949-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Doniger J. DNA replication in ultraviolet light irradiated Chinese hamster cells: the nature of replicon inhibition and post-replication repair. Journal of molecular biology. 1978;120:433–446. doi: 10.1016/0022-2836(78)90429-1. [DOI] [PubMed] [Google Scholar]
- 126.Cleaver JE, Thomas GH. Single strand interruptions in DNA and the effects of caffeine in Chinese hamster cells irradiated with ultraviolet light. Biochemical and biophysical research communications. 1969;36:203–208. doi: 10.1016/0006-291x(69)90315-5. [DOI] [PubMed] [Google Scholar]
- 127.Meyn RE, Humphrey RM. Deoxyribonucleic acid synthesis in ultraviolet-light-irradiated Chinese hamster cells. Biophysical journal. 1971;11:295–301. doi: 10.1016/S0006-3495(71)86215-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rauth AM, Tammemagi M, Hunter G. Nascent DNA synthesis in ultraviolet light-irradiated mouse, human and Chinese hamster cells. Biophysical journal. 1974;14:209–220. doi: 10.1016/S0006-3495(74)85908-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Park SD, Cleaver JE. Postreplication repair: questions of its definition and possible alteration in xeroderma pigmentosum cell strains. Proc Natl Acad Sci U S A. 1979;76:3927–3931. doi: 10.1073/pnas.76.8.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Prakash L. Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Molecular & general genetics : MGG. 1981;184:471–478. doi: 10.1007/BF00352525. [DOI] [PubMed] [Google Scholar]
- 131.Meneghini R. Gaps in DNA synthesized by ultraviolet light-irradiated WI38 human cells. Biochimica et biophysica acta. 1976;425:419–427. doi: 10.1016/0005-2787(76)90006-x. [DOI] [PubMed] [Google Scholar]
- 132.Meneghini R, Cordeiro-Stone M, Schumacher RI. Size and frequency of gaps in newly synthesized DNA of xeroderma pigmentosum human cells irradiated with ultraviolet light. Biophysical journal. 1981;33:81–92. doi: 10.1016/S0006-3495(81)84873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Boyer JC, Kaufmann WK, Brylawski BP, Cordeiro-Stone M. Defective postreplication repair in xeroderma pigmentosum variant fibroblasts. Cancer research. 1990;50:2593–2598. [PubMed] [Google Scholar]
- 134.Mezzina M, Menck CF, Courtin P, Sarasin A. Replication of simian virus 40 DNA after UV irradiation: evidence of growing fork blockage and single-stranded gaps in daughter strands. Journal of virology. 1988;62:4249–4258. doi: 10.1128/jvi.62.11.4249-4258.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cordeiro-Stone M, Schumacher RI, Meneghini R. Structure of the replication fork in ultraviolet light-irradiated human cells. Biophysical journal. 1979;27:287–300. doi: 10.1016/S0006-3495(79)85218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Berger CA, Edenberg HJ. Pyrimidine dimers block simian virus 40 replication forks. Mol Cell Biol. 1986;6:3443–3450. doi: 10.1128/mcb.6.10.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Molecular cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 138.Yan S, Michael WM. TopBP1 and DNA polymerase alpha-mediated recruitment of the 9-1-1 complex to stalled replication forks: implications for a replication restart-based mechanism for ATR checkpoint activation. Cell Cycle. 2009;8:2877–2884. doi: 10.4161/cc.8.18.9485. [DOI] [PubMed] [Google Scholar]
- 139.Wan L, Lou J, Xia Y, Su B, Liu T, Cui J, Sun Y, Lou H, Huang J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013;14:1104–1112. doi: 10.1038/embor.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rechkoblit O, Gupta YK, Malik R, Rajashankar KR, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structure and mechanism of human PrimPol, a DNA polymerase with primase activity. Sci Adv. 2016;2:e1601317. doi: 10.1126/sciadv.1601317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ, Blanco L. PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell. 2013;52:541–553. doi: 10.1016/j.molcel.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Keen BA, Jozwiakowski SK, Bailey LJ, Bianchi J, Doherty AJ. Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Res. 2014;42:5830–5845. doi: 10.1093/nar/gku214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Guilliam TA, Jozwiakowski SK, Ehlinger A, Barnes RP, Rudd SG, Bailey LJ, Skehel JM, Eckert KA, Chazin WJ, Doherty AJ. Human PrimPol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res. 2015;43:1056–1068. doi: 10.1093/nar/gku1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kobayashi K, Guilliam TA, Tsuda M, Yamamoto J, Bailey LJ, Iwai S, Takeda S, Doherty AJ, Hirota K. Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle. 2016;15:1997–2008. doi: 10.1080/15384101.2016.1191711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ, Sale JE, Doherty AJ. PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol Cell. 2016;61:161–169. doi: 10.1016/j.molcel.2015.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010;141:255–267. doi: 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 147.Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–955. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Callegari AJ, Clark E, Pneuman A, Kelly TJ. Postreplication gaps at UV lesions are signals for checkpoint activation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8219–8224. doi: 10.1073/pnas.1003449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Callegari AJ, Kelly TJ. UV irradiation induces a postreplication DNA damage checkpoint. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15877–15882. doi: 10.1073/pnas.0607343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Niimi A, Brown S, Sabbioneda S, Kannouche PL, Scott A, Yasui A, Green CM, Lehmann AR. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc Natl Acad Sci U S A. 2008;105:16125–16130. doi: 10.1073/pnas.0802727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Diamant N, Hendel A, Vered I, Carell T, Reissner T, de Wind N, Geacinov N, Livneh Z. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic acids research. 2012;40:170–180. doi: 10.1093/nar/gkr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Gali H, Hendel A, Johansson F, Erixon K, Livneh Z, Mullenders LH, Haracska L, de Wind N. Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol Cell Biol. 2009;29:3113–3123. doi: 10.1128/MCB.00071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hendel A, Krijger PH, Diamant N, Goren Z, Langerak P, Kim J, Reissner T, Lee KY, Geacintov NE, Carell T, Myung K, Tateishi S, D’Andrea A, Jacobs H, Livneh Z. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011;7:e1002262. doi: 10.1371/journal.pgen.1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, Menck CF. Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res. 2016;44:5717–5731. doi: 10.1093/nar/gkw280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Quinet A, Vessoni AT, Rocha CR, Gottifredi V, Biard D, Sarasin A, Menck CF, Stary A. Gap-filling and bypass at the replication fork are both active mechanisms for tolerance of low-dose ultraviolet-induced DNA damage in the human genome. DNA repair. 2014;14:27–38. doi: 10.1016/j.dnarep.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 156.Temviriyanukul P, van Hees-Stuivenberg S, Delbos F, Jacobs H, de Wind N, Jansen JG. Temporally distinct translesion synthesis pathways for ultraviolet light-induced photoproducts in the mammalian genome. DNA repair. 2012;11:550–558. doi: 10.1016/j.dnarep.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 157.Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Verspuy J, Gali H, Haracska L, de Wind N. Mammalian polymerase zeta is essential for post-replication repair of UV-induced DNA lesions. DNA repair. 2009;8:1444–1451. doi: 10.1016/j.dnarep.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 158.Despras E, Daboussi F, Hyrien O, Marheineke K, Kannouche PL. ATR/Chk1 pathway is essential for resumption of DNA synthesis and cell survival in UV-irradiated XP variant cells. Hum Mol Genet. 2010;19:1690–1701. doi: 10.1093/hmg/ddq046. [DOI] [PubMed] [Google Scholar]
- 159.Sasakawa N, Fukui T, Waga S. Accumulation of FFA-1, the Xenopus homolog of Werner helicase, and DNA polymerase delta on chromatin in response to replication fork arrest. Journal of biochemistry. 2006;140:95–103. doi: 10.1093/jb/mvj130. [DOI] [PubMed] [Google Scholar]
- 160.Torres-Ramos CA, Prakash S, Prakash L. Requirement of yeast DNA polymerase delta in post-replicational repair of UV-damaged DNA. The Journal of biological chemistry. 1997;272:25445–25448. doi: 10.1074/jbc.272.41.25445. [DOI] [PubMed] [Google Scholar]
- 161.Kannouche P, Broughton BC, Volker M, Hanaoka F, Mullenders LH, Lehmann AR. Domain structure, localization, and function of DNA polymerase eta, defective in xeroderma pigmentosum variant cells. Genes & development. 2001;15:158–172. doi: 10.1101/gad.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci U S A. 2006;103:8971–8976. doi: 10.1073/pnas.0510167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pabla R, Rozario D, Siede W. Regulation of Saccharomyces cerevisiae DNA polymerase eta transcript and protein. Radiat Environ Biophys. 2008;47:157–168. doi: 10.1007/s00411-007-0132-1. [DOI] [PubMed] [Google Scholar]
- 164.Gong J, Siede W. SBF transcription factor complex positively regulates UV mutagenesis in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2009;379:1009–1014. doi: 10.1016/j.bbrc.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Williams JI, Cleaver JE. Perturbations in simian virus 40 DNA synthesis by ultraviolet light. Mutation research. 1978;52:301–311. doi: 10.1016/0027-5107(78)90169-0. [DOI] [PubMed] [Google Scholar]
- 166.Stacks PC, White JH, Dixon K. Accommodation of pyrimidine dimers during replication of UV-damaged simian virus 40 DNA. Mol Cell Biol. 1983;3:1403–1411. doi: 10.1128/mcb.3.8.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Clark JM, Hanawalt PC. Replicative intermediates in UV-irradiated simian virus 40. Mutation research. 1984;132:1–14. doi: 10.1016/0167-8817(84)90061-0. [DOI] [PubMed] [Google Scholar]
- 168.White JH, Dixon K. Gap filling and not replication fork progression is the rate-limiting step in the replication of UV-damaged simian virus 40 DNA. Mol Cell Biol. 1984;4:1286–1292. doi: 10.1128/mcb.4.7.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bowden GT, Giesselbach B, Fusenig NE. Postreplication repair of DNA in ultraviolet light-irradiated normal and malignancy transformed mouse epidermal cell cultures. Cancer research. 1978;38:2709–2718. [PubMed] [Google Scholar]
- 170.Thomas DC, Kunkel TA. Replication of UV-irradiated DNA in human cell extracts: evidence for mutagenic bypass of pyrimidine dimers. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7744–7748. doi: 10.1073/pnas.90.16.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Carty MP, Hauser J, Levine AS, Dixon K. Replication and mutagenesis of UV-damaged DNA templates in human and monkey cell extracts. Mol Cell Biol. 1993;13:533–542. doi: 10.1128/mcb.13.1.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Thomas DC, Nguyen DC, Piegorsch WW, Kunkel TA. Relative probability of mutagenic translesion synthesis on the leading and lagging strands during replication of UV-irradiated DNA in a human cell extract. Biochemistry. 1993;32:11476–11482. doi: 10.1021/bi00094a002. [DOI] [PubMed] [Google Scholar]
- 173.McGregor WG, Wei D, Maher VM, McCormick JJ. Abnormal, error-prone bypass of photoproducts by xeroderma pigmentosum variant cell extracts results in extreme strand bias for the kinds of mutations induced by UV light. Mol Cell Biol. 1999;19:147–154. doi: 10.1128/mcb.19.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gough G, Wood RD. Inhibition of in vitro SV40 DNA replication by ultraviolet light. Mutation research. 1989;227:193–197. doi: 10.1016/0165-7992(89)90045-6. [DOI] [PubMed] [Google Scholar]
- 175.Kochaniak AB, Habuchi S, Loparo JJ, Chang DJ, Cimprich KA, Walter JC, van Oijen AM. Proliferating cell nuclear antigen uses two distinct modes to move along DNA. The Journal of biological chemistry. 2009;284:17700–17710. doi: 10.1074/jbc.M109.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shibahara K, Stillman B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell. 1999;96:575–585. doi: 10.1016/s0092-8674(00)80661-3. [DOI] [PubMed] [Google Scholar]
- 177.Fennessy RT, Owen-Hughes T. Establishment of a promoter-based chromatin architecture on recently replicated DNA can accommodate variable inter-nucleosome spacing. Nucleic Acids Res. 2016;44:7189–7203. doi: 10.1093/nar/gkw331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–438. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Morris GF, Mathews MB. Regulation of proliferating cell nuclear antigen during the cell cycle. The Journal of biological chemistry. 1989;264:13856–13864. [PubMed] [Google Scholar]
- 180.Hedglin M, Benkovic SJ. Replication Protein A Prohibits Diffusion of the PCNA Sliding Clamp along Single-Stranded DNA. Biochemistry. 2017:1824–1835. doi: 10.1021/acs.biochem.6b01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tinker RL, Kassavetis GA, Geiduschek EP. Detecting the ability of viral, bacterial and eukaryotic replication proteins to track along DNA. EMBO J. 1994;13:5330–5337. doi: 10.1002/j.1460-2075.1994.tb06867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Li Q, Burgess R, Zhang Z. All roads lead to chromatin: Multiple pathways for histone deposition. Biochim Biophys Acta. 2012;1819:238–246. doi: 10.1016/j.bbagrm.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Corpet A, Almouzni G. Making copies of chromatin: the challenge of nucleosomal organization and epigenetic information. Trends Cell Biol. 2009;19:29–41. doi: 10.1016/j.tcb.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 184.Yadav T, Whitehouse I. Replication-Coupled Nucleosome Assembly and Positioning by ATP-Dependent Chromatin-Remodeling Enzymes. Cell Reports. 2016;15:715–723. doi: 10.1016/j.celrep.2016.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Svikovic S, Sale JE. The Effects of Replication Stress on S Phase Histone Management and Epigenetic Memory. J Mol Biol. 2016 doi: 10.1016/j.jmb.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 186.Ransom M, Dennehey BK, Tyler JK. Chaperoning histones during DNA replication and repair. Cell. 2010;140:183–195. doi: 10.1016/j.cell.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hoek M, Stillman B. Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc Natl Acad Sci U S A. 2003;100:12183–12188. doi: 10.1073/pnas.1635158100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.di Caprio L, Cox BS. DNA synthesis in UV-irradiated yeast. Mutat Res. 1981;82:69–85. doi: 10.1016/0027-5107(81)90139-1. [DOI] [PubMed] [Google Scholar]
- 189.Brown S, Niimi A, Lehmann AR. Ubiquitination and deubiquitination of PCNA in response to stalling of the replication fork. Cell cycle (Georgetown, Tex. 2009;8:689–692. doi: 10.4161/cc.8.5.7707. [DOI] [PubMed] [Google Scholar]
- 190.Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 191.Sabbioneda S, Green CM, Bienko M, Kannouche P, Dikic I, Lehmann AR. Ubiquitin-binding motif of human DNA polymerase eta is required for correct localization. Proc Natl Acad Sci U S A. 2009;106:E20. doi: 10.1073/pnas.0812744106. author reply E21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- 193.Freudenthal BD, Gakhar L, Ramaswamy S, Washington MT. Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nat Struct Mol Biol. 2010;17:479–484. doi: 10.1038/nsmb.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yoon JH, Prakash L, Prakash S. Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc Natl Acad Sci U S A. 2009;106:18219–18224. doi: 10.1073/pnas.0910121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Makarova AV, Burgers PM. Eukaryotic DNA polymerase zeta. DNA Repair (Amst) 2015;29:47–55. doi: 10.1016/j.dnarep.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yoon JH, Park J, Conde J, Wakamiya M, Prakash L, Prakash S. Rev1 promotes replication through UV lesions in conjunction with DNA polymerases eta, iota, and kappa but not DNA polymerase zeta. Genes Dev. 2015;29:2588–2602. doi: 10.1101/gad.272229.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sabbioneda S, Gourdin AM, Green CM, Zotter A, Giglia-Mari G, Houtsmuller A, Vermeulen W, Lehmann AR. Effect of proliferating cell nuclear antigen ubiquitination and chromatin structure on the dynamic properties of the Y-family DNA polymerases. Mol Biol Cell. 2008;19:5193–5202. doi: 10.1091/mbc.E08-07-0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gohler T, Sabbioneda S, Green CM, Lehmann AR. ATR-mediated phosphorylation of DNA polymerase eta is needed for efficient recovery from UV damage. J Cell Biol. 2011;192:219–227. doi: 10.1083/jcb.201008076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Despras E, Delrieu N, Garandeau C, Ahmed-Seghir S, Kannouche PL. Regulation of the specialized DNA polymerase eta: revisiting the biological relevance of its PCNA- and ubiquitin-binding motifs. Environ Mol Mutagen. 2012;53:752–765. doi: 10.1002/em.21741. [DOI] [PubMed] [Google Scholar]
- 200.Hishiki A, Hashimoto H, Hanafusa T, Kamei K, Ohashi E, Shimizu T, Ohmori H, Sato M. Structural basis for novel interactions between human translesion synthesis polymerases and proliferating cell nuclear antigen. J Biol Chem. 2009;284:10552–10560. doi: 10.1074/jbc.M809745200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bomar MG, Pai MT, Tzeng SR, Li SS, Zhou P. Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase eta. EMBO Rep. 2007;8:247–251. doi: 10.1038/sj.embor.7400901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dieckman LM, Freudenthal BD, Washington MT. PCNA structure and function: insights from structures of PCNA complexes and post-translationally modified PCNA. Subcell Biochem. 2012;62:281–299. doi: 10.1007/978-94-007-4572-8_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Tsutakawa SE, Yan C, Xu X, Weinacht CP, Freudenthal BD, Yang K, Zhuang Z, Washington MT, Tainer JA, Ivanov I. Structurally distinct ubiquitin- and sumo-modified PCNA: implications for their distinct roles in the DNA damage response. Structure. 2015;23:724–733. doi: 10.1016/j.str.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tsutakawa SE, Van Wynsberghe AW, Freudenthal BD, Weinacht CP, Gakhar L, Washington MT, Zhuang Z, Tainer JA, Ivanov I. Solution X-ray scattering combined with computational modeling reveals multiple conformations of covalently bound ubiquitin on PCNA. Proc Natl Acad Sci U S A. 2011;108:17672–17677. doi: 10.1073/pnas.1110480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Smith S, Stillman B. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell. 1989;58:15–25. doi: 10.1016/0092-8674(89)90398-x. [DOI] [PubMed] [Google Scholar]
- 206.Zhang Z, Shibahara K, Stillman B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature. 2000;408:221–225. doi: 10.1038/35041601. [DOI] [PubMed] [Google Scholar]
- 207.Kaufman PD, Kobayashi R, Stillman B. Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 1997;11:345–357. doi: 10.1101/gad.11.3.345. [DOI] [PubMed] [Google Scholar]
- 208.Ayyagari R, Impellizzeri KJ, Yoder BL, Gary SL, Burgers PM. A mutational analysis of the yeast proliferating cell nuclear antigen indicates distinct roles in DNA replication and DNA repair. Mol Cell Biol. 1995;15:4420–4429. doi: 10.1128/mcb.15.8.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Martini E, Roche DM, Marheineke K, Verreault A, Almouzni G. Recruitment of phosphorylated chromatin assembly factor 1 to chromatin after UV irradiation of human cells. J Cell Biol. 1998;143:563–575. doi: 10.1083/jcb.143.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gaillard PH, Moggs JG, Roche DM, Quivy JP, Becker PB, Wood RD, Almouzni G. Initiation and bidirectional propagation of chromatin assembly from a target site for nucleotide excision repair. EMBO J. 1997;16:6281–6289. doi: 10.1093/emboj/16.20.6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Moggs JG, Grandi P, Quivy JP, Jonsson ZO, Hubscher U, Becker PB, Almouzni G. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol Cell Biol. 2000;20:1206–1218. doi: 10.1128/mcb.20.4.1206-1218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Palter KB, Alberts BM. The use of DNA-cellulose for analyzing histone-DNA interactions. Discovery of nucleosome-like histone binding to single-stranded DNA. J Biol Chem. 1979;254:11160–11169. [PubMed] [Google Scholar]
- 213.Palter KB, Foe VE, Alberts BM. Evidence for the formation of nucleosome-like histone complexes on single-stranded DNA. Cell. 1979;18:451–467. doi: 10.1016/0092-8674(79)90064-3. [DOI] [PubMed] [Google Scholar]
- 214.Zofall M, Persinger J, Bartholomew B. Functional role of extranucleosomal DNA and the entry site of the nucleosome in chromatin remodeling by ISW2. Mol Cell Biol. 2004;24:10047–10057. doi: 10.1128/MCB.24.22.10047-10057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Almouzni G, Clark DJ, Mechali M, Wolffe AP. Chromatin assembly on replicating DNA in vitro. Nucleic Acids Res. 1990;18:5767–5774. doi: 10.1093/nar/18.19.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hibbert RG, Sixma TK. Intrinsic flexibility of ubiquitin on proliferating cell nuclear antigen (PCNA) in translesion synthesis. J Biol Chem. 2012;287:39216–39223. doi: 10.1074/jbc.M112.389890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Torres-Ramos CA, Yoder BL, Burgers PM, Prakash S, Prakash L. Requirement of proliferating cell nuclear antigen in RAD6-dependent postreplicational DNA repair. Proc Natl Acad Sci U S A. 1996;93:9676–9681. doi: 10.1073/pnas.93.18.9676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Villamil MA, Chen J, Liang Q, Zhuang Z. A noncanonical cysteine protease USP1 is activated through active site modulation by USP1-associated factor 1. Biochemistry. 2012;51:2829–2839. doi: 10.1021/bi3000512. [DOI] [PubMed] [Google Scholar]
- 219.Cotto-Rios XM, Jones MJ, Busino L, Pagano M, Huang TT. APC/CCdh1-dependent proteolysis of USP1 regulates the response to UV-mediated DNA damage. The Journal of cell biology. 2011;194:177–186. doi: 10.1083/jcb.201101062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Cotto-Rios XM, Jones MJ, Huang TT. Insights into phosphorylation-dependent mechanisms regulating USP1 protein stability during the cell cycle. Cell cycle (Georgetown, Tex. 10:4009–4016. doi: 10.4161/cc.10.23.18501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Molecular cell. 2005;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 222.Jaspersen SL, Charles JF, Morgan DO. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol. 1999;9:227–236. doi: 10.1016/s0960-9822(99)80111-0. [DOI] [PubMed] [Google Scholar]
- 223.Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science (New York, N.Y. 1998;282:1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
- 224.Villamil MA, Liang Q, Chen J, Choi YS, Hou S, Lee KH, Zhuang Z. Serine phosphorylation is critical for the activation of ubiquitin-specific protease 1 and its interaction with WD40-repeat protein UAF1. Biochemistry. 2012;51:9112–9123. doi: 10.1021/bi300845s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Cotto-Rios XM, Jones MJ, Busino L, Pagano M, Huang TT. APC/CCdh1-dependent proteolysis of USP1 regulates the response to UV-mediated DNA damage. The Journal of cell biology. 194:177–186. doi: 10.1083/jcb.201101062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D’Andrea AD. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Molecular cell. 2007;28:786–797. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 227.Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D’Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nature cell biology. 2006;8:339–347. doi: 10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- 228.Cotto-Rios XM, Bekes M, Chapman J, Ueberheide B, Huang TT. Deubiquitinases as a signaling target of oxidative stress. Cell reports. 2012;2:1475–1484. doi: 10.1016/j.celrep.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lee JG, Baek K, Soetandyo N, Ye Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nature communications. 4:1568. doi: 10.1038/ncomms2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Cotto-Rios XM, Bekes M, Chapman J, Ueberheide B, Huang TT. Deubiquitinases as a signaling target of oxidative stress. Cell reports. 2:1475–1484. doi: 10.1016/j.celrep.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular signalling. 24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Piatkov KI, Colnaghi L, Bekes M, Varshavsky A, Huang TT. The auto-generated fragment of the usp1 deubiquitylase is a physiological substrate of the N-end rule pathway. Molecular cell. 2012;48:926–933. doi: 10.1016/j.molcel.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lehmann AR, Stevens S. Postreplication repair of DNA in chick cells: studies using photoreactivation. Biochim Biophys Acta. 1975;402:179–187. doi: 10.1016/0005-2787(75)90037-4. [DOI] [PubMed] [Google Scholar]
- 234.Chiu SF, Rauth AM. Nascent DNA synthesis in ultraviolet light-irradiated mouse L cells. Biochim Biophys Acta. 1972;259:164–174. doi: 10.1016/0005-2787(72)90056-1. [DOI] [PubMed] [Google Scholar]
- 235.Buhl SN, Setlow RB, Regan JD. Recovery of the ability to synthesize DNA in segments of normal size at long times after ultraviolet irradiation of human cells. Biophys J. 1973;13:1265–1275. doi: 10.1016/S0006-3495(73)86061-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Barnett SW, Landaw EM, Dixon K. Test of models for replication of simian virus 40 DNA following ultraviolet irradiation. Biophys J. 1984;46:307–321. doi: 10.1016/S0006-3495(84)84027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Dahle D, Griffiths TD, Carpenter JG. Inhibition and recovery of DNA synthesis in UV-irradiated Chinese hamster V-79 cells. Photochem Photobiol. 1980;32:157–165. doi: 10.1111/j.1751-1097.1980.tb04003.x. [DOI] [PubMed] [Google Scholar]
- 238.D’Ambrosio SM, Setlow RB. Enhancement of postreplication repair in Chinese hamster cells. Proc Natl Acad Sci U S A. 1976;73:2396–2400. doi: 10.1073/pnas.73.7.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Meyn RE, Vizard DL, Hewitt RR, Humphrey RM. The fate of pyrimidine dimers in the DNA of ultraviolet-irradiated Chinese hamster cells. Photochem Photobiol. 1974;20:221–226. doi: 10.1111/j.1751-1097.1974.tb06570.x. [DOI] [PubMed] [Google Scholar]
- 240.Fujiwara Y, Kondo T. Postreplication repair of ultraviolet damage to DNA in xeroderma pigmentosum, other human and mouse cells in culture. J Radiat Res. 1974;15:81–89. doi: 10.1269/jrr.15.81. [DOI] [PubMed] [Google Scholar]
- 241.Fujiwara Y. Postreplication repair of ultraviolet damage to DNA, DNA-chain elongation, and effects of metabolic inhibitors in mouse L cells. Biophys J. 1975;15:403–415. doi: 10.1016/S0006-3495(75)85826-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kaufmann WK, Cleaver JE. Mechanisms of inhibition of DNA replication by ultraviolet light in normal human and xeroderma pigmentosum fibroblasts. J Mol Biol. 1981;149:171–187. doi: 10.1016/0022-2836(81)90297-7. [DOI] [PubMed] [Google Scholar]
- 243.Waters R, Yagi H, Jerina DM, Regan JD. Postreplication repair of DNA in human fibroblasts after UV irradiation or treatment with metabolites of benzo(a)pyrene. Chem Biol Interact. 1978;20:289–297. doi: 10.1016/0009-2797(78)90107-2. [DOI] [PubMed] [Google Scholar]
- 244.Rude JM, Friedberg EC. Semi-conservative deoxyribonucleic acid synthesis in unirradiated and ultraviolet-irradiated xeroderma pigmentosum and normal human skin fibroblasts. Mutat Res. 1977;42:433–442. doi: 10.1016/s0027-5107(77)80047-x. [DOI] [PubMed] [Google Scholar]
- 245.McIntyre J, Vidal AE, McLenigan MP, Bomar MG, Curti E, McDonald JP, Plosky BS, Ohashi E, Woodgate R. Ubiquitin mediates the physical and functional interaction between human DNA polymerases eta and iota. Nucleic Acids Res. 2013;41:1649–1660. doi: 10.1093/nar/gks1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yuasa MS, Masutani C, Hirano A, Cohn MA, Yamaizumi M, Nakatani Y, Hanaoka F. A human DNA polymerase eta complex containing Rad18, Rad6 and Rev1; proteomic analysis and targeting of the complex to the chromatin-bound fraction of cells undergoing replication fork arrest. Genes Cells. 2006;11:731–744. doi: 10.1111/j.1365-2443.2006.00974.x. [DOI] [PubMed] [Google Scholar]
- 247.Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–3896. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Conservation of PCNA