

Abstract
Mitochondrial abnormalities impact the development of myofibrillar myopathies. Therefore, understanding the mechanisms underlying the removal of dysfunctional mitochondria from cells is of great importance toward understanding the molecular events involved in the genesis of cardiomyopathy. Earlier studies have ascribed a role for BAG3 in the development of cardiomyopathy in experimental animals leading to the identification of BAG3 mutations in patients with heart failure which may play a part in the onset of disease development and progression. BAG3 is co-chaperone of heat shock protein 70 (HSP70), which has been shown to modulate apoptosis and autophagy, in several cell models. In this study, we explore the potential role of BAG3 in mitochondrial quality control. We demonstrate that siRNA mediated suppression of BAG3 production in neonatal rat ventricular cardiomyocytes (NRVCs) significantly elevates the level of Parkin, a key component of mitophagy. We found that both BAG3 and Parkin are recruited to depolarized mitochondria and promote mitophagy. Suppression of BAG3 in NRVCs significantly reduces autophagy flux and eliminates expression of Tom20, an essential import receptor for mitochondria proteins, after induction of mitophagy. These observations suggest that BAG3 is critical for the maintenance of mitochondrial homeostasis under stress conditions, and disruptions in BAG3 expression impact cardiomyocyte function.
Keywords: BAG3, Parkin, Tom20, mitophagy, proteasome
INTRODUCTION
BAG3 is a member of the BAG family of proteins which functions as a regulator of the Hsp70/Hsc70 chaperone system (Rosati et al., 2011). In cardiac muscle, BAG3, through association with the sarcomeric Z-disk, maintains the integrity and contractility of heart muscle. Mutations in the BAG3 gene have been reported to cause the cardiac muscle disorder known as familial dilated cardiomyopathy (DCM) (Arimura et al., 2011). BAG3 is critical for survival; its deficiency in mice caused skeletal and cardiac tissue abnormalities and strong myofibrillar degeneration which led to massive induction of apoptosis followed by death after 4 weeks (Homma et al., 2006). Furthermore, reduction of BAG3 in adult mouse ventricular myocytes interfered with contraction and calcium signaling (Feldman et al., 2016). Recent studies have demonstrated enhanced expression of BAG3 and increased levels of its chaperonic activity along with heat shock proteins during cellular stress and aging, suggesting a crucial role for BAG3 in determining the cellular stress response (Behl et al., 2011). BAG3 is known to play an important role in intracellular protein homeostasis by participating either in protein folding and/or clearance of damaged proteins (Rodriguez et al., 2016). To this end, BAG3 functions along with other proteins such as SQSTM1/P62 (referred to as P62 from here on), HSP70, and LC3 in a process called macroautophagy in which misfolded or damaged proteins and organelles are sequestered within autophagosomes and targeted to lysosomes for degradation (Behl et al., 2011). In this process, BAG3 targets misfolded proteins to the aggresome for further degradation through interaction with the dynein motor (Gamerdinger et al., 2011). Moreover, BAG3 functions as an anti-apoptotic protein and inhibits apoptotic cell death through its interactions with Bcl-2 family proteins (Arimura et al., 2011; Liao et al., 2001).
Dysregulation of mitochondrial homeostasis plays an important role in the development of various types of disease in which mitochondrial abnormalities have been described, including cancer, neurodegeneration, and cardiomyopathies (Vincow et al., 2013; Chan et al., 2011; Maron et al., 2006). For example, damage to the mitochondrial respiratory chain leads to the uncontrolled generation of reactive oxygen species (ROS) which, by oxidizing cellular components, act as a stress signal and lead to caspase activation, thus triggering apoptotic signaling pathways (Chan et al., 2011). In support of this finding, abnormalities in mitochondrial distribution, morphology, and respiratory chain function have been reported in patients with myofibrillar myopathies. In addition, alteration of NADH, SDH or COX distribution is a hallmark feature of many myofibrillar disorders (Jackson et al., 2015). Parkin, encoded by the PARK2 gene, has been reported as an important regulator of mitochondrial quality control and mutations associated with Parkin are associated with the development of neurodegenerative diseases such as the autosomal recessive form of Parkinson’s disease (Yoshii et al., 2011). Parkin translocates from the cytosol to the mitochondria in response to mitochondrial membrane potential loss and targets damaged mitochondria for degradation and clearance via ubiquitination (Narendra et al., 2008). In this regard, recent studies have focused on utilizing Parkin overexpression to enhance the elimination of damaged mitochondria as well as understanding potential mechanisms of mitochondrial quality control and maintenance within cells (Yoshii et al., 2011; Narendra et al., 2008; Kim et al., 2013).
Previous studies concluded that Parkin deletion did not interfere with cardiomyocyte contractility or affect mitochondrial characteristics, while disruption of the mitochondrial fission protein, Drp1 (dynamin-related protein 1), led to both alterations of the cardiac phenotype and myocardial dysfunction (Cao et al., 2015). These observations, along with the role of mitochondrial abnormalities in the development of myofibrillar myopathies, led us to hypothesize that BAG3 may play an important role in the clearance of dysfunctional mitochondria. Thus, we investigated the role of BAG3 in the clearance of damaged mitochondria in response to treatment with the electron uncoupler, carbonyl cyanide m-chlorophenylhydrazone (CCCP). We observed an increase in Parkin expression in response to knockdown of BAG3 and identified the translocation of both Parkin and BAG3 to the depolarized mitochondria thus promoting its degradation. Although no direct interaction between BAG3 and Parkin was detected, BAG3 suppression significantly reduced the clearance of Tom20 after CCCP treatment, indicating that mitophagy is impaired in the absence of BAG3. To our knowledge, this is the first study reporting BAG3 as a key regulator of mitophagy induced by CCCP electron transport chain uncoupler.
MATERIALS AND METHODS
Cell Isolation and Culture Conditions
All experiments were performed under protocols approved by the Temple University Institutional Animal Care and Use Committee. Cardiomyocytes were isolated from 1–2 day old Sprague-Dawley rats (Charles River) as previously described (Gupta et al., 2016). Isolated cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Life Technologies) supplemented with 2% fetal bovine serum (FBS, Denville Scientific inc.) and 25 μg/mL Gentamicin (Life Technologies).
Adenoviral Transduction
Cardiomyocytes were transduced in a minimal volume of DMEM containing either Adeno-null (Vector Biolabs), as an internal control, or Adeno-siBAG3 (Vector Biolabs) with a multiplicity of infection of 2 at 37°C for 2 hr, then refed with medium containing 2% FBS and 25 μg/mL Gentamicin. 72 hr post-transduction, cells were subjected to other treatments.
Plasmids and Transfections
HEK cells were transfected using Lipofectamine 3000 transfection reagent (Life Technologies) according to the manufacturer’s instructions. Cells were incubated with Opti-MEM reduced-serum medium (Life Technologies) with YFP-Parkin (Addgene) with or without BAG3 siRNA (Dharmacon) for 2 hr, then fed with culture medium (DMEM, 10% FBS, 25 μg/mL Gentamicin) followed by overnight incubation. Medium then was replaced with culture medium and 48 hr post-transfection, cells were subjected to other treatments.
Mitochondrial Isolation
Mitochondrial enriched fractions were prepared using the sucrose gradient method. Cells were scraped in mitochondrial isolation buffer containing 250 mM sucrose, 10 mM Tris-HCl (pH 7.4), and 0.1 mM EGTA and homogenized using glass teflon homogenizers. Cells then were centrifuged at 4,000 rpm for 10 min to pellet nuclei and cell debris. Supernatant was transferred to a new tube and centrifuged at 14,000 rpm for 30 min to pellet mitochondria. Mitochondrial pellets were washed two times with mitochondrial isolation buffer and then lysed in RIPA lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) for further analysis.
Western Blotting
Cells were washed with PBS and then lysed in RIPA lysis buffer supplemented with 10% protease inhibitor cocktail (Sigma-Aldrich) and rotated at 4°C for 30 min. Insoluble material was separated by centrifugation at 14,000 rpm for 10 min and supernatants containing soluble proteins were used for further analysis. Protein concentration was determined using Bio-Rad protein assay reagent (Bio-Rad). Equal amounts of proteins were resolved by 10% or 12% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes (LI-COR, Inc., Lincoln, NE). Membranes were blocked in Odyssey blocking buffer (LI-COR) for 1 hr at room temperature (RT) and then incubated with primary (3 hr at RT) and secondary (1 hr at RT) antibodies followed by washing with PBST containing 0.5% Tween 20 (Amresco, 1 ×, 5 min) and PBS (3 ×, 5 min ea.). After staining with secondary antibodies, membranes were scanned with an Odyssey® CLx Imaging System (LI-COR, Inc., Lincoln, NE) The following primary antibodies were used for Western blotting: BAG3 (Proteintech, 10599-1-AP), SQSTM1/P62 (Cell Signaling Technology, 5114), Parkin (Abcam, ab77924), HSP70 (Santa Cruz Biotech, sc-1060), LC3 (Sigma, L8918), PINK1 (Abcam, ab23707), Tom20 (Abcam, ab199641), GAPDH (Santa Cruz, sc-32233), β-Actin (Santa Cruz, sc-47778), VCP (Santa Cruz, sc-20799), Beclin-1 (Cell Signaling Technology, 3738), Cox-2 (Santa Cruz, sc-1745), Cytochrome c (Cell Signaling Technology, 4272), BAX (Santa Cruz, sc-493) and BAD (Cell Signaling Technology, 9292).
RNA Isolation and Quantitative Reverse Transcription-PCR (qRT-PCR)
RNA was extracted from NRVCs using the RNeasy Mini Kit (Ambion) according to the manufacturer’s protocol. On column DNA digestion was performed using DNase I (QIAGEN) to decrease DNA contamination in the eluted RNA solution. 1 μg of the isolated RNA was used for reverse transcription and cDNA synthesis with M-MLV Reverse Transcriptase (Invitrogen). qRT-PCR was performed in 20 μL reaction volume with 10 μL SybrGreen master mix (Roche) and Primers. β-actin was used as a reference gene to normalize data. The following primers were used: Park2 FW: 5′-ACCCAACCTCAGACAAGGAC-3′; Park2 RV: 5′-AGACAGGGTTCCTGACATCC-3′; BAG3 FW: 5′-GGCCCTAAGGAAACTGCAT-3′; BAG3 RV: 5′-GGGAATGGGAATGTAACCTG-3′; HSP70 FW: 5′-GCTTCAGACCTCCCTTTGAG-3′; HSP70 RV: 5′-TCCAAGATGCTACGAAGTGG-3′; SQSTM1/P62 FW: 5′-GAGTCATGCTGCACTCCACT-3′; SQSTM1/P62 RV: 5′-TATCAGGCAGGAATGATGGA-3′.
Fluorescence Microscopy
Cells were washed with PBS, fixed with 4% paraformaldehyde (10 min at RT), and permeabilized using 0.5% Triton-X (10 min at RT). Cells then were masked with 0.1 M glycine (pH 3.5, 30 min at RT) followed by blocking (1% BSA, 0.1% Tween 20 in 1x PBS, 30 min at RT). Cells were probed with primary antibody (overnight at 4°C), rinsed in PBS and stained with Alexa Fluor® secondary antibody (Thermo Fisher Scientific) for 1 hr at RT. After mounting in VECTASHIELD hard set mounting medium with DAPI (Vector Laboratories), images were acquired using Leica fluorescent microscope. Tom20 (Abcam, ab56783), BAG3 (Proteintech, 10599-1-AP) and Parkin (Abcam, ab15954) were used as primary antibodies.
ATP Assay
Intracellular ATP was measured using the ATP determination kit (Molecular Probes) according to the manufacturer’s protocol. Mitochondria were isolated as described above and lysed in RIPA buffer. Standard reaction solutions containing 8.9 mL dH2O, 0.5 mL 20× reaction buffer, 0.1 mL 0.1M DTT, 0.5 mL of 10 mM D-luciferin and 2.5 uL firefly luciferase (5 mg/mL) were prepared. 10 μL of the lysed mitochondria was added to 90 μL of the standard reaction solution and the luminescence was immediately read using a luminometer (Femtomaster FB 12 luminometer, Zylux).
Cell Death Assays
NRVCs were plated in 96-well plates at a density of 10,000 cells/well. Cells were transduced with either Ad-null or Ad-siBAG3 for 72 hr followed by treatment with different concentrations of CCCP. Cell death was measured using the SYTOX Green cell death assay (Invitrogen. Cells were incubated with SYTOX Green solution for 15 minutes at 37°C followed by measurement of fluorescent emission at excitation/emission maxima of 504/523 nm to detect cells with damaged plasma membranes (SYTOX Green DNA intercalation) vs. live cells (impermeable to SYTOX Green) using a spectrophotometer.
Statistical Analysis
Student’s t-test was used to determine the statistical significance of differences between two pairs of data. P <0.05 was considered as statistically significant.
RESULTS
BAG3 knockdown decreases autophagy flux and BAG3 translocates to the mitochondria upon depolarization
In order to assess the role of BAG3 in the regulation of autophagy, cells were treated with either CCCP (20 μM, 6 hr) or bafilomycin A1 (Baf A1, 50 nM, 6 hr) and autophagic activity was evaluated by measuring LC3 I cleavage, an indicator of autophagy, using an anti-LC3 antibody to detect LC3 II on the autophagosomal outer membrane (Pankiv et al., 2007). CCCP was utilized to uncouple the proton gradient and interfere with the electron transport chain across mitochondrial membrane, leading to loss of the mitochondrial membrane potential. CCCP treatment activated mitophagy and increased LC3 II levels, but autophagy flux was reduced in NRVCs with lower levels of BAG3 compared to control cells. Unexpectedly, cotreatment with CCCP and bafilomycin A1 led to a decrease in both LC3 I and LC3 II levels; suggesting that the mitophagy pathway may be inhibited when mitochondrial damage is induced and the lysosome is blocked. Moreover, reductions in LC3 I and LC3 II under these conditions were significantly higher in cells with reduced BAG3 levels compared to control cells. In order to ensure the inhibition of autophagy, the autophagy receptor, P62, was examined and results revealed a significant reduction in P62 levels after bafilomycin A1 and CCCP treatment in BAG3 knock-down cells; indicating an important role for BAG3 in regulating mitophagy (Fig. 1A). Since longer stress conditions may result in the complete degradation of autophagy proteins, we examined cells treated with CCCP for a shorter time period (4 hr) and mitochondrial enriched fractions were then isolated by sucrose gradient separation. Results revealed that BAG3 levels did not change in the whole cell lysates (Fig. 1B). By isolating mitochondria, we found that BAG3 translocated to the mitochondrial fraction in response to the membrane potential loss (Fig. 1C). Interestingly, BAG3’s binding partner, HSP70, did not translocate to mitochondria. Parkin translocation from the cytosol to the depolarized mitochondria was observed, as has been reported by others (Narendra et al., 2008) and BAG3 knock-down significantly increased Parkin recruitment. P62, Beclinl and valosin-containing protein (VCP) also translocated to the mitochondria after depolarization, while their degradation was not observed in the whole cell extracts (Fig. 1B and C).
Figure 1. BAG3 regulates autophagy and is recruited to mitochondria upon depolarization.
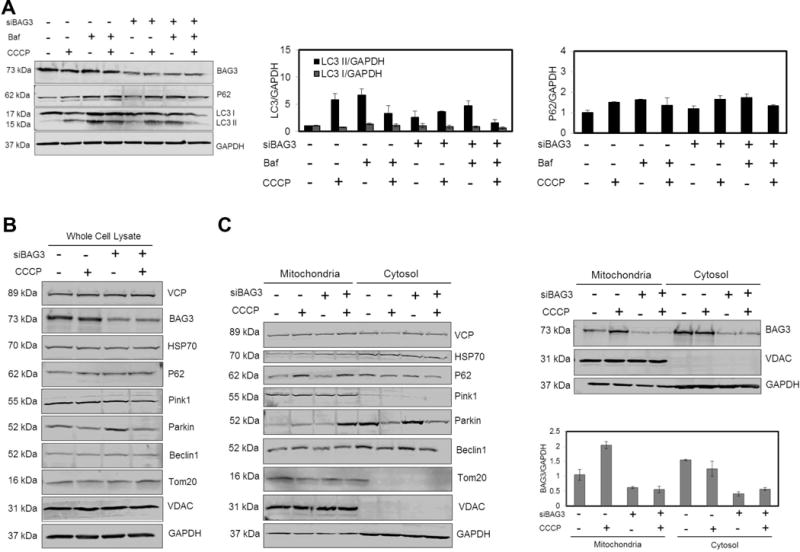
(A) NRVCs were treated with CCCP (20 μM, 6 hr) in the absence or presence of Bafilomycin A1 (Baf, 50 nM, 6 hr) and autophagy markers LC3 and P62 were evaluated using Western blot analysis. CCCP treatment increased autophagy marker, LC3 II, and BAG3 knockdown cells had lower autophagy flux compared to control cells. (B) The protein expression analysis of the whole cell extract after treatment with CCCP (20 μM, 4 hr). (C) NRVCs were treated with CCCP (20 μM, 4 hr) and then mitochondrial fraction was isolated. Mitochondrial and cytosolic fractions were analyzed using Western blot analysis. BAG3 and Parkin translocated to mitochondria upon depolarization and BAG3 knock-down increased the level of Parkin translocated to depolarized mitochondria. VDAC and GAPDH were used as markers of mitochondria and cytosol, respectively (data were normalized with respect to GAPDH).
BAG3 promotes mitochondrial degradation through the autophagy-lysosome pathway when the proteasome is inhibited
Upregulation of BAG3 expression upon proteasome inhibition has been previously reported and BAG3 is believed to mediate the initiation of the autophagy-lysosomal degradation pathway (Liu et al., 2013). We further investigated the mechanism through which BAG3 promotes mitochondrial clearance by inhibiting proteasome activity using MG132 (5 μM for 12 hr). LC3 II levels increased upon blocking of proteasome activity suggesting the activation of autophagy in the absence of the proteasome degradation pathway. Furthermore, proteasome inhibition led to an increase in BAG3, HSP70, and P62 protein levels (Fig. 2A). When cells were treated with both MG132 and CCCP for 12 hr, BAG3, HSP70, and P62 proteins returned to their basal levels. The ratio of a given protein level to actin level as an internal control is presented in Fig. S1. Moreover, qRT-PCR data showed that mRNA level of these proteins increased when cells were treated with either CCCP or MG132 (Fig. 2B). Collectively, these data suggest that BAG3, HSP70, and P62 proteins mediate the disposal of damaged mitochondria and their levels are maintained within the normal range under stress conditions. Western blot analysis indicated the translocation of VCP and Beclin1 to depolarized mitochondria, and levels degraded at 12 hr after CCCP treatment.
Figure 2. BAG3 mediates clearance of defected mitochondria through autophagy-lysosome when proteasome is inhibited.
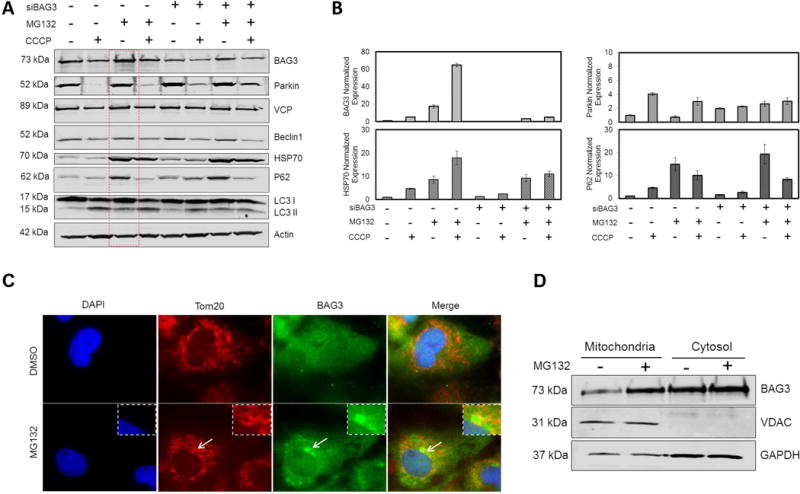
NRVCs were treated with CCCP (20 μM) in the presence or absence of MG132 (5 μM). 12 hr post-treatment, cells were analyzed with (A) Western blot for protein analysis or (B) qRT-PCR for mRNA expression analysis. (C) NRVCs were treated with MG132 (20 μM, 5 hr) and BAG3 and Tom20 localizations were visualized with a fluorescence microscope. (D) Mitochondrial and cytosolic fractions were analyzed for BAG3 translocations after proteasome inhibition (20 μM, 5 hr). By inhibiting proteasome, LC3 II level increased, suggesting activation of autophagy-lysosome degradation pathway in the absence of proteasome-ubiquitin pathway. Both mRNA and protein expressions of BAG3, HSP70 and P62 increased by inhibiting proteasome; while Parkin level remained unchanged.
Fluorescence microscopy showed the localization of BAG3 to perinuclear aggresomes when proteasomal activity was inhibited. Interestingly, these aggresomes were surrounded by a Tom20 network (Fig. 2C). BAG3 levels in both mitochondria and cytosol fractions increased after proteasome inhibition (Fig. 2D).
BAG3 knockdown increases Parkin levels and impairs clearance of depolarized mitochondria
Fluorescence microscopy showed that 2 hr CCCP (20 μM) treatment caused fragmentation of the mitochondrial network in that Parkin translocated from the cytosol to the fragmented Tom20 network (Fig. 3A). Parkin degradation was also observed along with degradation of damaged mitochondria as seen by Western blotting of lysates from NRVCs treated with 20 μM CCCP for 6 hr. Interestingly, reduction of BAG3 levels in NRVCs led to a significant increase in endogenous Parkin levels (Fig. 3B). Numerous studies have utilized cellular systems that overexpress Parkin for studying the mitochondrial degradation pathway. In light of these observations, we investigated the effect of BAG3 on the expression of exogenous Parkin in the rat cardiac moyoblast cell line, H9c2. Flow cytometry analysis showed that BAG3 knock-down significantly enhanced exogenous Parkin expressed as a YFP-tagged fusion protein (Fig. 3C and S2). These observations indicate that BAG3 regulates both endogenous and exogenous expression of Parkin, suggesting that BAG3 may function upstream of Parkin in the mitochondrial clearance pathway.
Figure 3. BAG3 regulates Parkin expression and BAG3 knock down impairs mitopahgy in cardiomyocytes.
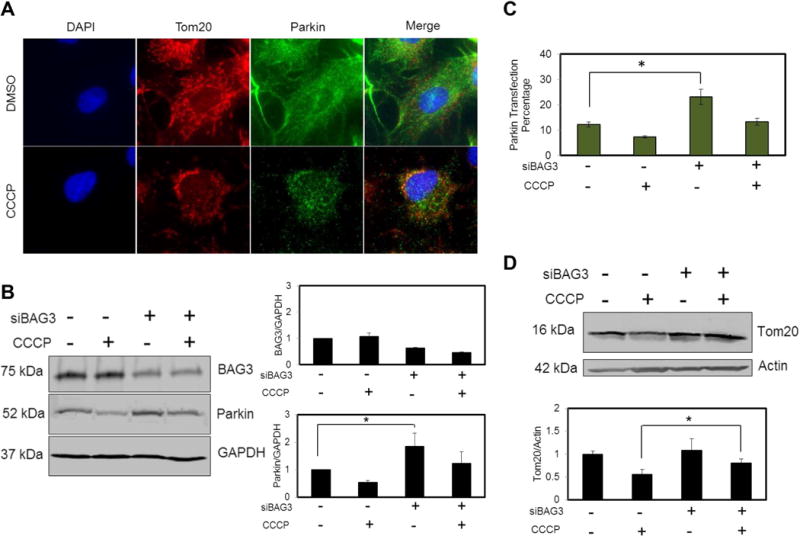
(A) CCCP (20 μM, 2 hr) treatment caused the fragmentation of Tom20 network and Parkin was recruited to the depolarized mitochondria. (B) Parkin degraded along with the mitochondrial degradation after CCCP treatment (20 μM, 6 hr). Parkin endogenous level increased as a result of BAG3 knock down. (C) H9c2 cells were transfected with YFP-Parkin with or without BAG3 siRNA. 48 hr after transfection, cells were treated with DMSO or CCCP (20 μM, 4 hr) and fluorescent signal was detected using flow cytometry analysis. Cells with BAG3 supression showed higher levels of Parkin expressions. (D) Knock down of BAG3 significantly inhibited the clearance of Tom20 after CCCP treatment (20 μM, 12 hr).
In another experiment, we investigated the effect of BAG3 on the mitochondrial outer membrane protein, Tom20. Our results showed that BAG3 knock-down significantly decreased the clearance of damaged Tom20 in NRVCs treated with 20 μM of CCCP for 12 hr (Fig. 3D).
BAG3 knock-down increases sensitivity to apoptosis
Three days after transduction, NRVCs were treated with 20 μM CCCP for 4 hr and then mitochondrial fractions were isolated to analyze ATP level. Intracellular ATP levels were decreased upon treatment of cells with CCCP. Although knock-down of BAG3 only changed ATP levels after CCCP treatment, NRVCs knocked-down for BAG3 had higher ATP levels compared to control cells. Here, BAG3 expression was measured in cytosolic protein extracts (Fig. 4A). Western blot data also showed that levels of the mitochondrial-encoded Cox-2 protein in the whole cell protein lysates increased after CCCP treatment (Fig. 4B).
Figure 4. Cellular death and bioenergetics assays after CCCP treatment.
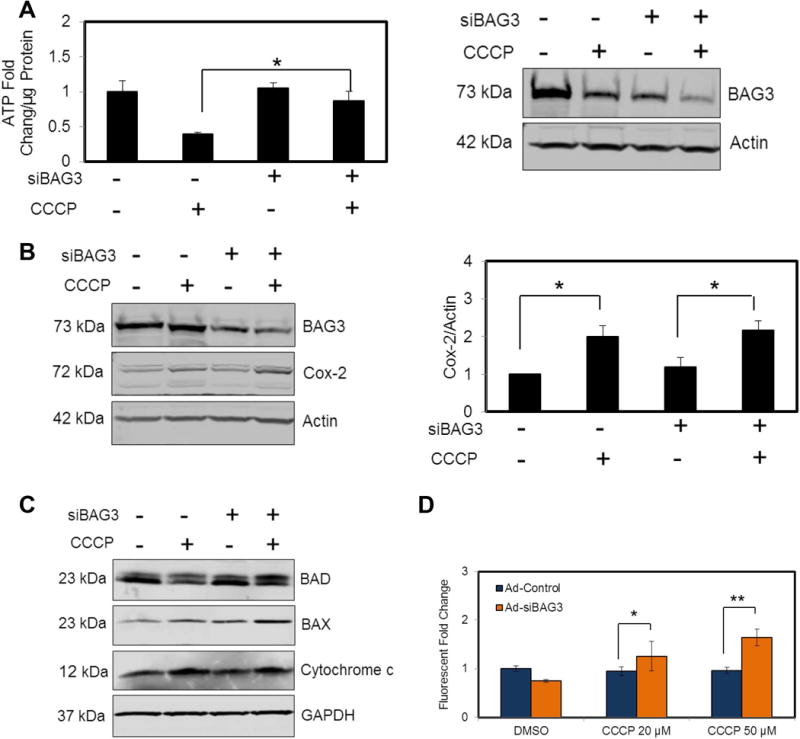
(A) Mitochondrial fractions were isolated and intracellular ATP level was measured. CCCP treatment led to the decrease in ATP level. BAG3 knock-down cells showed a higher ATP level compared to the control cells treated with CCCP, suggesting the impairment of mitochondrial clearance in the absence of BAG3. (B) Cox-2 protein level increased after CCCP treatment. (C) The expression of pro-apoptotic proteins after CCCP treatment was analyzed using Western blot analysis. (D) NRVCs were treated with CCCP (20 μM or 50 μM) and cell death was analyzed using SYTOX Green cell death assay. BAG3 knock-down cells showed more sensitivity to mitochondrial stress condition. *P<0.05; **P<0.01.
In order to investigate the role of BAG3 in mitochondria-associated apoptosis, NRVCs were treated with 20 μM CCCP for 6 hr and the expressions of pro-apoptotic proteins such as Bcl-2-associated death protein (BAD) and Bcl-2-associated X protein (BAX) were analyzed by Western blot. BAD and BAX are involved in the initiation of apoptosis and their upregulation is associated with increased cell death (Brunelle et al., 2009). Cytochrome c levels were elevated after CCCP treatment. BAX and BAD levels were upregulated after CCCP treatment in whole cell extracts from NRVCs knocked-down for BAG3, indicating that BAG3 knock-down sensitizes the cells to mitochondrial stress conditions (Fig. 4C).
In another experiment, cell death was measured after 6 hr treatment of NRVCs with increasing concentrations of CCCP. Results showed that BAG3 knock-down significantly increased cell death as measured by SYTOX Green when mitochondrial stress was applied (Fig. 4D).
DISCUSSION
The results presented herein ascribe a role for BAG3 in mitophagy and demonstrate translocation of BAG3 along with Parkin to mitochondria upon depolarization. Evidently, BAG3 facilitates the clearance of damaged Tom20 and its down regulation increases the level of Parkin translocation to the mitochondria after CCCP treatment. It was previously reported that BAG3 knock-down increased the amount of mitochondria in HeLa cells (Rodriguez et al., 2016). The accumulation of dysfunctional mitochondria within cells has been implicated in the development of neurodegenerative disorders such as Parkinson’s disease. In this regard, mitophagy functions as a protective response and selectively targets damaged mitochondria for degradation (Matsuda et al., 2015). Upon mitochondrial depolarization, Parkin as an E3 ubiquitin ligase ubiquitinates the damaged mitochondria in a PINK1-dependent manner (Vincow et al., 2013). The ubiquitin tag is then recognized by the mitophagy machinery and the selected mitochondria are targeted for lysosomal degradation. This model was further refined by Lazarou et al., (2015) reporting that PINK1 phosphorylates ubiquitin thus activating Parkin recruitment, and Parkin triggers recruitment of autophagy receptors by adding ubiquitin chains to the mitochondrial outer membrane proteins. Moreover, the recruitment of the autophagy receptors, NDP52 and Optineurin (OPTN), by PINK1 were found to be independent of Parkin, as this was observed in HeLa cells lacking Parkin (Lazarou et al., 2015). Moreover, in vivo studies showed that even though mitochondrial fragmentation was observed in respiratory chain-deficient dopaminergic neurons, Parkin did not translocate to the defective mitochondria and the absence of Parkin did not alter the clearance of dysfunctional mitochondria (Sterky et al., 2011). These observations are highly consistent with our findings indicating that BAG3 may function upstream of Parkin in mitochondrial clearance.
The two main pathways through which intracellular degradation is carried out to maintain protein quality control are the ubiquitin-proteasome and the autophagy-lysosome pathways (Pankiv et al., 2007). Parkin has been reported to mediate the proteasomal degradation of mitochondrial outer membrane proteins (Chan et al., 2011; Yoshii et al., 2011). In mouse embryonic fibroblasts overexpressing Parkin, the degradation of mitochondrial outer membrane proteins happened through Parkin-mediated proteasomal pathway while the degradation of inner membrane and matrix proteins relied mainly on mitophagy (Yoshii et al., 2011). We found that Parkin levels did not increase by blocking proteasome activity, suggesting the possibility of a dual role for Parkin in both ubiquitin-proteasome and autophagy-lysosome pathways.
BAG3 has been reported to play an important role in regulating autophagy through aggresome targeting. In this pathway, BAG3 transports HSP70-bound substrates to aggresomes residing in the perinuclear area by interacting with microtubules. The cargo is then directed to lysosomes through the autophagy process for degradation (Gamerdinger et al., 2011). It has been reported that the HSP70 chaperon protein functions in protein quality control through recognition of misfolded proteins (Behl et al., 2011). BAG3 binds to the ATPase domain of HSP70 through its BAG domain and modulates its chaperonic activity (Liu et al., 2013; Arimura et al., 2011; Behl et al., 2011). Although BAG3 translocates to depolarized mitochondria, no such translocation was observed for HSP70. These results suggest that BAG3 may function upstream of HSP70 in the recognition of cargo in the mitophagy process.
Our results indicate that the autophagy receptor, P62, translocated to the mitochondria upon membrane potential loss. SQSTM1 has been reported to play a protective role under conditions of proteasome inhibition (Milan et al., 2015). P62 functions as a cargo receptor and binds to both ubiquitinated and non-ubiquitinated substrates and triggers their autophagic clearance (Pankiv et al., 2007; Watanabe et al., 2011; Behl et al., 2011).
Beclin 1 is the mammalian homolog of yeast Vp30/Atg6 and is essential for autophagosome formation. Upon blockage of the ubiquitin-proteasome pathway in HepG2 cells, BAG3 promoted autophagy initiation; which was not suppressed by silencing of Beclin 1, indicating a non-canonical autophagy initiation (Liu et al., 2013). VCP functions as a segregase in Parkin-mediated mitophagy and extracts autophagy components from the ubiquitinated proteins. VCP mutations in mouse embryonic fibroblasts overexpressing Parkin impaired mitophagy (Kim et al., 2013). Our results showed that both Beclin 1 and VCP translocated to the depolarized mitochondria and their degradation in whole cell lysate was observed upon treatment of the cells with CCCP.
Our data indicate that CCCP treatment causes ATP depletion in cardiomyocytes. CCCP interferes with mitochondrial ATP synthesis by damaging the electron transport chain and causes uncontrolled oxygen consumption. The mitochondrial encoded protein, Cox-2, which plays a role in electron transport, increases to maintain ATP homeostasis. Cells with reduced BAG3 expression indicated higher ATP levels compared to those maintaining basal BAG3 levels when they were treated with CCCP, suggesting less clearance of mitochondrial proteins. HeLa cells overexpressing Parkin showed faster clearance of depolarized mitochondria and less cell viability when they were cultured in galactose media compared to cells with endogenous Parkin level; indicating that more mitochondrial clearance caused a lower ATP concentration and led to cell death when glycolysis was inhibited (Narendra et al., 2008).
Accelerated cell death in cardiomyocytes is accompanied with the development of degenerative cardiovascular diseases (Brunelle et al., 2009). BAG3 functions as an anti-apoptotic protein through interaction with Bcl2 (Knezevic et al., 2015). Apoptotic cell death significantly increased in neonatal rat cardiomyocytes with BAG3 mutations (Arimura et al., 2011). High levels of BAG3 were found in pancreatic tumor cells compared to normal pancreas tissue which promoted malignancy by functioning as an anti-apoptotic protein (Liao et al., 2001). BAX and BAD belong to the Bcl2 protein family and function as pro-apoptotic proteins. Higher levels of BAX in pancreatic cancer cells were associated with longer survivability of pancreatic cancer patients (Liao et al., 2001). Cytochrome c levels significantly increased in response to CCCP treatment. BAX and BAD showed higher levels in BAG3 knock-down cells compared to the control cells after CCCP treatment. Increased levels of these pro-apoptotic proteins in the cells with reduced BAG3 levels makes cells more susceptible to death when mitochondrial stress is induced. Moreover, when the autophagic machinery is not working properly to clear damaged mitochondria, cells are exposed to the toxic effects of free radicals and oxidative stress which trigger death signaling. Collectively, these results indicate that BAG3 plays a vital role in mitochondrial quality control within cardiomyocytes and its reduction impairs the clearance of damaged mitochondria (Fig. 5).
Figure 5. Schematic diagram of Tom20 degradation in cardiomyocytes.
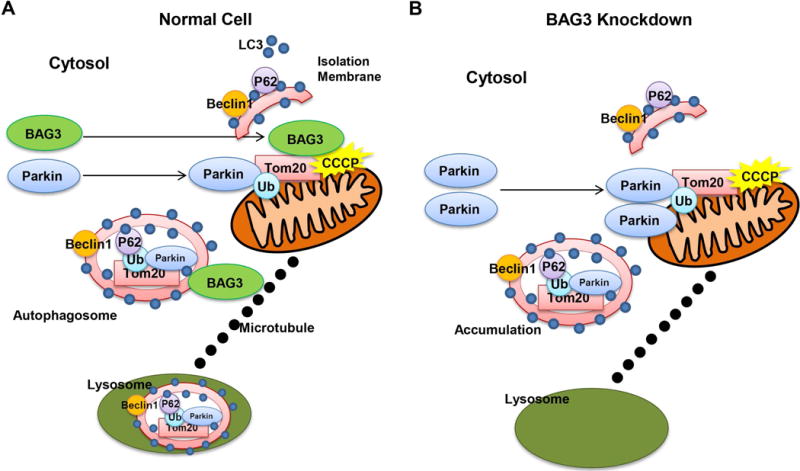
(A) BAG3 and Parkin translocate from cytosol to mitochondria upon depolarization. Parkin functions as an E3 ubiquitin ligase and ubiquitinates Tom20. Tom20 is then sequestered within autophagosome and BAG3 through interactions with microtubule, targets the cargo for lysosomal degradation. (B) BAG3 knockdown increases Parkin expression and more Parkin translocates from cytosol to depolarized mitochondria. Autophagy flux decreases and Tom20 clearance is impaired when BAG3 is not present, suggesting that BAG3 is one of the key regulators of mitophagy in cardiomyocytes.
In summary, the present study uses NRVCs to demonstrate a new pathway for the clearance of dysfunctional mitochondria in which BAG3 regulates both endogenous and exogenous expression of Parkin. BAG3 and Parkin are both recruited to the mitochondria upon depolarization. BAG3 knock-down reduced autophagy flux and impaired the clearance of defective mitochondria which led to the higher levels of toxicity within the cells and subsequent cell death. Proteasome inhibition suggested BAG3’s crucial role in activating the autophagy process along with other autophagy proteins such as HSP70 and P62. However, further mechanistic studies are needed to determine the mechanism through which BAG3 and Parkin function, whether Parkin ubiquitination signaling is essential for BAG3 recruitment, and whether BAG3 works upstream of Parkin. Moreover, the role of BAG3 as a key factor in mitophagy regulation in adult cardiomyocytes remains to be demonstrated. Taken together, our observations demonstrate that BAG3 may be a promising target for the treatment of heart failure and other myofbrillar myopathies.
Supplementary Material
Figure S1. Protein level normalized to Actin level after proteasome inhibition. NRVCs were treated with MG132 (5 μM, 12hr) in the presence or absence of CCCP (20 μM, 12 hr). Protein levels of (A) BAG3, (B) Parkin, (C) VCP, (D) Beclin 1, (E) HSP70, (F) P62 and (G) LC3 II are normalized to Actin levels.
Figure S2. BAG3 increases exogenous Parkin expression. H9c2 cells were transfected with fluorescent Parkin plasmid with or without BAG3 siRNA and fluorescent signals were detected using flow cytometry in (A) Parkin overexpression (B) Parkin overexpression and CCCP treatment, (C) Parkin overexpression and BAG3 knockdown, and (D) Parkin overexpression, BAG3 knockdown and CCCP treatment conditions.
Acknowledgments
We thank past and present members of the Department of Neuroscience and Center for Neurovirology for their insightful discussion and sharing of ideas and reagents. This work was made possible by grants awarded by NIH to KK (P30MH092177) and KK, JYC, AMF (R01HL123093)
Contract grant sponsor: P30MH092177 (Khalili)
Contract grant sponsor: R01HL123093 (Khalili, Cheung, Feldman)
Footnotes
Conflict of Interests
KK is a board member, scientific advisor, and holds equity in Excision Biotherapeutics, a biotech start-up who has licensed the viral gene editing technology from Temple University for commercial development and clinical trials.
References
- Arimura T, Ishikawa T, Nunoda S, Kawai S, Kimura A. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum Mutat. 2011;32:1481–1491. doi: 10.1002/humu.21603. [DOI] [PubMed] [Google Scholar]
- Behl C. BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy. 2011;7:795–798. doi: 10.4161/auto.7.7.15844. [DOI] [PubMed] [Google Scholar]
- Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao DJ, Lavandero S, Hill JA. Parkin Gone Wild Unbridled Ubiquitination. Circ Res. 2015;117:311–313. doi: 10.1161/CIRCRESAHA.115.307022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman AM, Gordon J, Wang J, Song J, Zhang XQ, Myers VD, Tilley DG, Gao E, Hoffman NE, Tomar D, Madesh M. BAG3 regulates contractility and Ca 2+ homeostasis in adult mouse ventricular myocytes. J Mol Cell Cardiol. 2016;92:10–20. doi: 10.1016/j.yjmcc.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011;12:149–156. doi: 10.1038/embor.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta MK, Tahrir FG, Knezevic T, White MK, Gordon J, Cheung JY, Khalili K, Feldman AM. GRP78 Interacting Partner Bag5 Responds to ER Stress and Protects Cardiomyocytes from ER Stress-Induced Apoptosis. J Cell Biochem. 2016 doi: 10.1002/jcb.25481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma S, Iwasaki M, Shelton GD, Engvall E, Reed JC, Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. 2006;169:761–773. doi: 10.2353/ajpath.2006.060250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S, Schaefer J, Meinhardt M, Reichmann H. Mitochondrial abnormalities in the myofibrillar myopathies. Eur J Neurol. 2015;22:1429–1435. doi: 10.1111/ene.12814. [DOI] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron. 2013;78:65–80. doi: 10.1016/j.neuron.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic T, Myers VD, Gordon J, Tilley DG, Sharp IIITE, Wang J, Khalili K, Cheung JY, Feldman AM. BAG3: a new player in the heart failure paradigm. Heart Fail Rev. 2015;20:423–434. doi: 10.1007/s10741-015-9487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Q, Ozawa F, Friess H, Zimmermann A, Takayama S, Reed JC, Kleeff J, Büchler MW. The anti-apoptotic protein BAG-3 is overexpressed in pancreatic cancer and induced by heat stress in pancreatic cancer cell lines. FEBS Lett. 2001;503:151–157. doi: 10.1016/s0014-5793(01)02728-4. [DOI] [PubMed] [Google Scholar]
- Liu BQ, Du ZX, Zong ZH, Li C, Li N, Zhang Q, Kong DH, Wang HQ. BAG3-dependent noncanonical autophagy induced by proteasome inhibition in HepG2 cells. Autophagy. 2013;9:905–916. doi: 10.4161/auto.24292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Amett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies an American heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Tanaka K. Cell biology: Tagged tags engage disposal. Nature. 2015;524:294–295. doi: 10.1038/nature15199. [DOI] [PubMed] [Google Scholar]
- Milan E, Perini T, Resnati M, Orfanelli U, Oliva L, Raimondi A, Cascio P, Bachi A, Marcatti M, Ciceri F, Cenci S. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy. 2015;11:1161–1178. doi: 10.1080/15548627.2015.1052928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Rodríguez AE, López-Crisosto C, Pena-Oyarzún D, Salas D, Parra V, Quiroga C, Morawe T, Chiong M, Behl C, Lavandero S. BAG3 regulates total MAP1LC3B protein levels through a translational but not transcriptional mechanism. Autophagy. 2016;12:287–296. doi: 10.1080/15548627.2015.1124225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati A, Graziano V, De Laurenzi V, Pascale M, Turco MC. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011;2:e141. doi: 10.1038/cddis.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L, Larsson NG. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci. 2011;108:12937–12942. doi: 10.1073/pnas.1103295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci. 2011;124:2692–2701. doi: 10.1242/jcs.081232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Protein level normalized to Actin level after proteasome inhibition. NRVCs were treated with MG132 (5 μM, 12hr) in the presence or absence of CCCP (20 μM, 12 hr). Protein levels of (A) BAG3, (B) Parkin, (C) VCP, (D) Beclin 1, (E) HSP70, (F) P62 and (G) LC3 II are normalized to Actin levels.
Figure S2. BAG3 increases exogenous Parkin expression. H9c2 cells were transfected with fluorescent Parkin plasmid with or without BAG3 siRNA and fluorescent signals were detected using flow cytometry in (A) Parkin overexpression (B) Parkin overexpression and CCCP treatment, (C) Parkin overexpression and BAG3 knockdown, and (D) Parkin overexpression, BAG3 knockdown and CCCP treatment conditions.