

Abstract
There is no consensus of opinion on the origin of the large rate accelerations observed for enzyme-catalyzed hydride transfer. The interpretation of recent results from studies on hydride transfer reactions catalyzed by alcohol dehydrogenase (ADH) focus on the proposal that the effective barrier height is reduced by quantum-mechanical tunneling through the energy barrier. This interpretation contrasts sharply with the notion that enzymatic rate accelerations are obtained through direct stabilization of the transition state for the nonenzymatic reaction in water. The binding energy of the dianion of substrate for DHAP provides 11 kcal/mol stabilization of the transition state for the hydride transfer reaction catalyzed by glycerol-3-phosphate dehydrogenase (GPDH). We summarize evidence that the binding interactions between (GPDH) and dianion activators are utilized directly for stabilization of the transition state for enzyme-catalyzed hydride transfer. The possibility is considered, and then discounted, that these dianion binding interactions are utilized for the stabilization of a tunnel ready state (TRS) that enables efficient tunneling of the transferred hydride through the energy barrier, and underneath the energy maximum for the transition state. It is noted that the evidence to support the existence of a tunnel-ready state for the hydride transfer reactions catalyzed by ADH is ambiguous. We propose that the rate acceleration for ADH is due to the utilization of the binding energy of the cofactor in the stabilization of the transition state for enzyme-catalyzed hydride transfer.
Graphical Abstract
Introduction
Transition state theory has been defined as “a theory of the rates of elementary reactions which assumes a special type of equilibrium, having an equilibrium constant K‡, for conversion of the reactants to the transition state.”1 It follows from this definition that interactions, which stabilize an enzyme-bound transition state, will result in an increase in K‡ and an enzymatic rate acceleration.2 It is broadly accepted that enzymes operate to stabilize transition states, in order to achieve their rate accelerations;3, 4 but, there is dissatisfaction with the slow pace in closing real and imagined gaps in our understanding of enzyme catalysis, and with the slow progress towards the de novo design of proteins with enzyme-like catalytic activity.5 This discontent, and a sense of intellectual curiosity, has stimulated the development of models that emphasize the role of dynamics in enzymatic catalysis.5–7 Among these is the proposal that enzymatic catalysis of hydron transfer [L−, L• or L+] is promoted by protein dynamics, and proceeds mainly by tunneling of the transferred hydron through the energy barrier.5
We have used transition state theory in the interpretation of results from studies to characterize the effect of protein-dianion interactions on the kinetic parameters for enzyme-catalyzed reactions.8 Our first studies examined the isomerization reactions catalyzed by triosephosphate isomerase (TIM, Scheme 1A) and the decarboxylation reactions catalyzed by orotidine 5′-monophosphate decarboxylase (OMPDC, Scheme 1B), through unstable carbanion reaction intermediates. The activation barrier for formation of the transition states for nonenzymatic isomerization and decarboxylation is composed mainly of the thermodynamic barrier to formation of the respective carbanion intermediates, whose structures should closely resemble the corresponding transition state.9 There is a strong imperative to lower this activation barrier through the stabilization of the high-energy reaction intermediate (Figure 1); and, it is possible to rationalize the rate acceleration for these enzymes using transition state theory.10–13
Scheme 1.

Figure 1.

Reaction coordinate profiles which show: (1) The similarity between the activation barrier for nonenzymatic decarboxylation of OMP and the thermodynamic barrier for formation of the unstable vinyl carbanion reaction intermediate.24–26 (2) The similar effect of the protein catalyst on the activation barrier for nonenzymatic decarboxylation of OMP and the thermodynamic reaction barrier.
The modern reports of X-ray crystal structures of free and liganded forms of alcohol dehydrogenase (ADH) show that the cofactor NAD/NADH is poised at the enzyme active site to undergo the catalyzed hydride transfer reaction.14–19 The introduction of site-directed mutations of side chains at the active site of ADH result in falloffs in enzyme activity, which are consistent with the elimination of side-chains that provide stabilization of the transition state for hydride transfer.19–23 However, there is no consensus about the importance of transition state stabilization in this reaction, because of an alternate model, which emphasizes the role of quantum mechanical tunneling through the energy barrier. We summarize in this review the results of experiments that demonstrate a striking similarity in the stabilization of the transition states for the polar reactions catalyzed by OMPDC and TIM, and for the hydride transfer reaction catalyzed by glycerol-3-phosphate dehydrogenase (GPDH, Scheme 2) from interactions between the protein and the substrate phosphodianion, along with evidence against a significant contribution from quantum mechanical tunneling to the enzymatic rate acceleration for GPDH. These data provide support for the conclusion that catalysis by GPDH is due largely or entirely to stabilization of the transition state for hydride transfer by interactions with the protein catalyst. We suggest that this conclusion may also hold for hydride transfer reactions catalyzed by alcohol dehydrogenase.
Scheme 2.
Transition State Stabilization from Protein-Dianion Binding Interactions
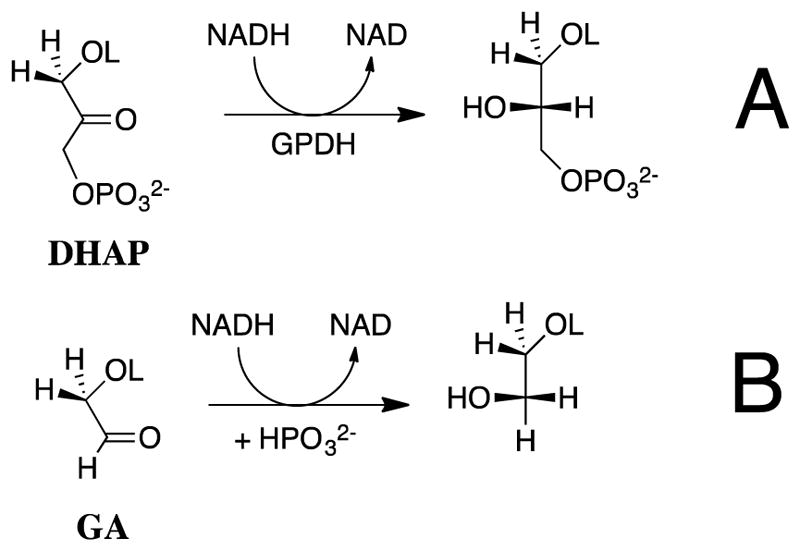
If transition state theory holds, then our results require that the transition state for GPDH-catalyzed reduction of glycolaldehyde (GA) is 11 kcal/mol higher in energy than that for reduction of the whole substrate dihydroxyacetone phosphate (DHAP, Scheme 2).27, 28 Figure 2 shows that the former transition state is stabilized 8 kcal/mol by the binding of phosphite dianion [HPi] at a standard state of 1.0 M, and an additional 3 kcal/mol by the connection [“connection energy”]29 of these substrate pieces (Figure 2).27, 28 By comparison, the transition states for OMPDC and TIM-catalyzed reactions of truncated substrates are 12 kcal/mol higher in energy than for the reaction of the corresponding whole substrate;30, 31 and, the transition states for reactions of truncated substrates are stabilized by 8 and 6 kcal/mol,30, 31 respectively, by the binding of phosphite dianion at a standard state of 1.0 M. Similar results have been obtained in studies on the reactions catalyzed by phosphoglucomutase32 and 1-deoxy-D-xylulose-5-phosphate reductoisomerase.33
Figure 2.

Diagram prepared with the assumption that transition state theory can be used to model the relative activation barriers to formation of transition states for GPDH-catalyzed reactions at a standard state of 1.0 M.27, 28 (1) Reduction of DHAP by NADH at the ternary complex [E•DHAP•NADH]‡. (2) Reduction of GA by NADH, activated by phosphite dianion, at the quaternary complex [E•GA•HPi•NADH]‡. (3) Reduction of GA by NADH at the ternary complex [E•GA•NADH]‡. The transition state stabilization obtained by interactions with the substrate dianion is equal to the 11 kcal/mol difference in the activation barriers for GPDH-catalyzed reduction of DHAP and GA. The 3 kcal/mole greater transition state stabilization from protein-dianion interactions at [E•DHAP•NADH]‡ compared with [E•GA•HPi•NADH]‡ is the advantage obtained from the connection of the pieces at the quaternary complex.
The disconnect between protein structures, obtained from the snapshots provided by X-ray crystallography, and enzyme function in phosphite dianion activation of truncated substrates, is striking. An examination of X-ray crystal structures for TIM,34–36 OMPDC,11, 37, 38 and GPDH39 over many years failed to predict that protein-dianion interactions might activate these enzymes for catalysis. The absence of direct stabilizing interactions between phosphite dianion and the putative transition states for these enzymatic reactions is worth noting; and, in the case of OMPDC the large 8–10 Å separation between the dianion binding site and the catalytic sites precludes strong direct stabilizing interactions.37, 40, 41 The observation of dianion activation of any one of these enzyme-catalyzed reactions is surprising, while the similarity in the kinetic parameters for dianion activation of the diverse set of reactions catalyzed by TIM, OMPDC and GPDH suggests that there exists a common, generalizable, mechanism for activation of a broad range of enzymatic reactions through protein dianion interactions.28
The X-ray crystal structures for TIM,34–36 OMPDC,11, 37, 38 and GPDH,39 each show the presence of a gripper loop near the respective enzyme active sites. The active site at each unliganded enzyme is accessible to substrate, but in each case substrate binding is accompanied by a protein conformational change, during which the flexible loop folds over the phosphodianion and sequesters the substrate, at a protein cage, from interaction with bulk solvent.3, 8, 42 The large difference between the structures of the open unliganded (EO) and the closed liganded (EC) forms of TIM, OMPDC and GPDH prompted the proposal that only EC is catalytically active (Scheme 3).3, 43 The specificity in the binding of phosphite dianion to EC results from interactions between the protein catalyst and phosphodianion. Strong dianion activation is observed when a small fraction of the enzyme exists in the active closed form (KC ≪ 1, Scheme 3), and the dianion binding energy is utilized to increase the fraction of enzyme in the active closed form. The barrier for conversion of EO to EC represents, minimally, the barriers to extrusion of protein bound waters to bulk solvent, and to freezing of large conformational motions of the flexible protein loops and of catalytic side chains at the ordered structure for the active protein-substrate cage.3, 8, 42, 44, 45
Scheme 3.
We have examined the effect of site-directed mutations of TIM,46–54 OMPDC,40, 41, 55–57 and GPDH58–60 on the activity of these enzymes for catalysis of the reactions of the whole substrate and the substrate in pieces. The results of these studies on the reactions of the substrate in pieces have been fully rationalized using Scheme 3. This work establishes Scheme 3 as a broadly generalizable model to rationalize the utilization of substrate binding interactions in enzyme activation.
Utilization of Dianion Binding Interactions for Stabilization of the Transition State for GPDH-Catalyzed Hydride Transfer
The kinetic parameters from Scheme 4 for phosphite dianion activation of GPDH for catalysis of the reduction of GA were used to construct the free energy diagram shown in Figure 3.28 This diagram shows: (i) That phosphite dianion and the transition state for enzyme-catalyzed reduction of GA (GA‡) bind specifically to the closed form of GPDH (EC, Scheme 3); and, (ii) That the −5.9 kcal/mol difference between the −1.6 kcal/mol free energy for binding of HPi to E′O and the −7.5 kcal/mol free energy for binding of HPi to the transition state complex to EC•GA‡ is the part of the intrinsic dianion binding energy that is utilized to drive the unfavorable conformational change from EO to EC.4
Scheme 4.

Figure 3.

Free energy diagram for GPDH-catalyzed reduction of glycolaldehyde (GA) by free GPDH, which exists mainly in the inactive open form (E′O, Scheme 3) and by GPDH that is saturated with phosphite dianion and exists in the active closed form (E′C•HPO32−). This diagram was constructed using kinetic parameters for Scheme 4 that were reported in Ref 28. The observed tighter binding of phosphite dianion to the transition state complex E′C•GA‡ to give EC•HPO32−•GA‡ (ΔGint = −7.5 kcal/mol) than to the free enzyme EO to give EC•HPO32− (ΔGobsd = −1.6 kcal/mol) is attributed the binding energy utilized to drive an unfavorable enzyme conformational change (ΔGC = +5.9 kcal/mol) that converts E′O to E′C (Scheme 3).
Figure 4 shows a representation of the X-ray crystal structure of the nonproductive complex between GPDH, DHAP and NAD+. The loop residues 292–297 that fold over DHAP when the substrate binds to GPDH are shown in green. The stabilizing interactions between the phosphodianion of DHAP and GPDH are due mainly or entirely to interactions with the guanidinium cation side chain of R269 and the amide side chain of N270; and, we propose that these side chains also interact with enzyme-bound phosphite dianion. The results of studies of the effect of R269A and N270A mutations on the GPDH-catalyzed reduction of the whole substrate DHAP and the substrate pieces GA + HPi by NADH show that the interactions of these side chains account for most, or all, of the 11 kcal/mol intrinsic dianion binding energy of the substrate DHAP (Figure 2).58, 60
Figure 4.
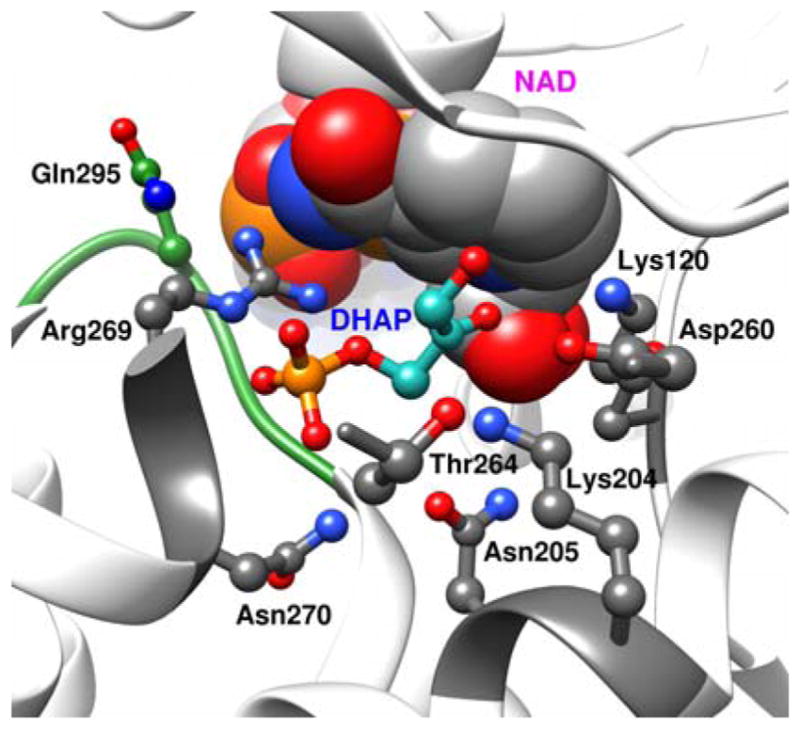
A rendering of the X-ray crystal structure of the nonproductive ternary complex between GPDH, DHAP and NAD+ (PDB entry 1WPQ), which shows: (a) The loop residues 292–297 (green) that fold over DHAP. (b) Arg269 and Asn270 that interact with the substrate phosphodianion. (c) The network of hydrogen-bonded side chains Asn270, Thr264, Asn205, Lys204 Asp260 and Lys120. Reprinted with permission from Ref 5. Copyright 2016 American Chemical Society.
R269A GPDH
The R269A mutation of GPDH results in a 110-fold increase in Km (2.8 kcal/mol effect) and a 41,000-fold decrease in kcat (6.3 kcal/mol effect). These effects sum to a 9.1 kcal/mol destabilization of the transition state for GPDH-catalyzed reduction of DHAP by NADH,60 which represents ca 80% of the total 11 kcal/mol dianion binding energy. The 6.3 kcal/mol effect of this mutation that is specifically expressed at the transition state for hydride transfer (kcat effect) is ca 80% of the 7.5 kcal/mol phosphite dianion binding energy (Figure 3) expressed at the transition state for GPDH-catalyzed reduction. The striking size of the apparent side-chain interaction (9.1 kcal/mol) suggests that the architecture of the active site of GPDH has evolved to optimize stabilizing interactions, at the transition state for hydride transfer, between the side chain cation from R269 and ligand negative charge q, which increases from −2 at the reactant to as large as −3 (Scheme 5) at the transition state. The high density of negative charge at the transition state is important, because the magnitude of Coloumbic interactions increases with the product of the square of the interacting charges. The difference between the 2.8 kcal/mol ground-state and the total 9.1 kcal/mol stabilization, shows that the stabilizing interactions develop mainly at the transition state. The strong phosphite dianion interactions also develop specifically at the transition state for GPDH-catalyzed reduction of GA (Figure 3), which is consistent with the conclusion that these interactions involve the side chain cation from R269.60
Scheme 5.

N270A GPDH
The N270A and R269A/N270A mutations of GPDH result in 5.6 and 11.5 kcal/mol destabilization, respectively of the transition state for GPDH-catalyzed reduction of DHAP by NADH. These results are summarized in Scheme 6, where the total effect of the R269A/N270A mutations on ΔG‡DHAP is similar to the 11 kcal/mol intrinsic dianion binding energy. We proposed that the 5.6 kcal/mol effect of the N270A mutation on ΔG‡DHAP for wildtype GPDH represents the loss of a 2.4 kcal/mol interaction between the amide side chain and the phosphodianion observed for the N270A mutation of R269A GPDH plus an additional ca. 3.2 kcal/mol weakening of the 9.1 kcal/mol interaction between the phosphodianion and the side chain of R269 (Scheme 6).58
Scheme 6.
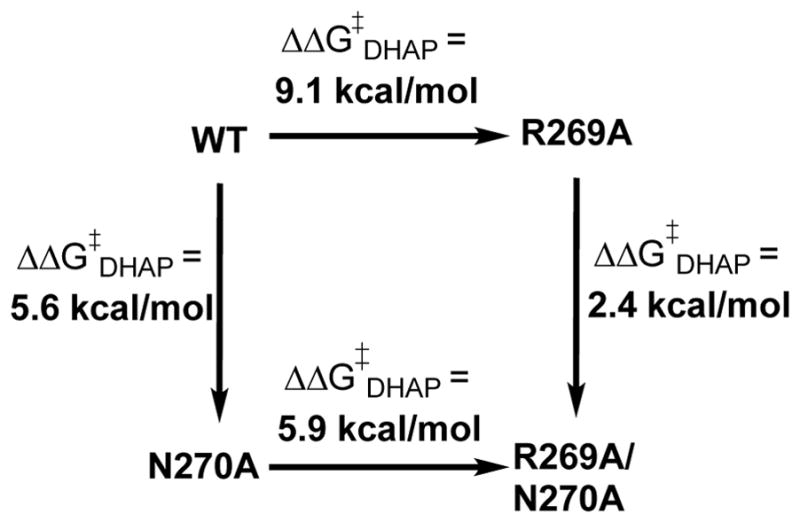
The N270A mutation results in two further surprising changes in the kinetic parameters for the reactions of the substrate pieces.58 (i) A 2.2 kcal/mol stabilization of the transition state for unactivated hydride transfer from NADH to GA, instead of the expected destabilization. (ii) A change in the effect of phosphite dianion on GPDH-catalyzed reduction of GA by NADH, from strongly activating for wildtype GPDH to inhibiting. These results are consistent with a reorganization in the structure of the active site for the N270A mutants, which alters the preferred mode of GA binding. We propose that phosphite dianion activation of wildtype GPDH-catalyzed reduction of GA occurs by the binding of the substrate pieces GA + HPi at a structured binding pocket, while the N270A mutation results in an erosion of the structure of this binding pocket, and changes in enzyme reactivity that have not yet been rationalized.
Optimization of a Protein-Dianion Interaction
Ionic interactions between the transition state for GPDH-catalyzed reduction of DHAP by NADH and the cationic side chain of R269 provide a 9 kcal/mol stabilization, which corresponds to 4 million fold rate effect.60 By comparison, the cationic side chains of K12 and R235 provide 8 and 5.8 kcal/mol stabilization, respectively of the transition states for the isomerization of D-glyceraldehyde 3-phosphate catalyzed by triosephosphate isomerase and for the decarboxylation of orotidine 5′-monophosphate (OMP) catalyzed by OMP decarboxylase (OMPDC).61, 62 These results show that the structures of TIM, OMPDC and GPDH have evolved to optimize transition state stabilization from the interaction between the phosphodianion and a side chain cation. Such interactions in water are weak. For example, the value of KGua = 0.18 M for breakdown of [OMP•Gua+], the ion pair between the OMP trianion and guanidine cation,62 corresponds to a binding energy of only 1.0 kcal/mol (Scheme 7A). Ion pairing interactions are stronger for a ligand bound to a protein catalyst, compared with aqueous solution, because the effective dielectric constants at the interior of proteins is much smaller than for water.63, 64
Scheme 7.

A large fraction of the stabilizing interactions associated with ion pair formation (Kd, Scheme 7) is utilized for the reduction in the translational and rotational entropy of the separate free ions.29, 65 The effect of a change in the molecularity on the free energy change associated with ion-pair formation has been examined in studies on rescue of enzyme activity, which is lost upon mutation of an essential cationic amino acid side chain, by small molecule analogs of the excised side chain (Schemes 7B and 7C). The K12G mutation of triosephosphate isomerase eliminates the interaction between the side chain cation and substrate phosphate group and results in a 500,000 fold reduction in kcat/Km. This corresponds to the loss of a 7.8 kcal/mol stabilizing interaction (Figure 5).61 The activity that is rescued by addition of 1.0 M NH4+ or by different RNH3+ has been determined, to give the values of -ΔGRNH3‡ listed in Figure 5. The apparent advantage of the covalent connection between the protein and the side chain cation, ΔGS‡, is then calculated as the difference between the overall 7.8 kcal/mole effect of the K12G mutation and -ΔGRNH3‡.61
Figure 5.

A comparison of -ΔGRNH3‡, the free energy associated with the binding of NH4+ or RNH3+ to the transition state for K12G mutant TIM-catalyzed isomerization of GAP, and the apparent transition state stabilization obtained from connecting these activators to the mutant enzyme, ΔGS‡. The sum of these terms is equal to the overall stabilization of the transition state for the wildtype TIM-catalyzed isomerization of GAP by interaction with the cationic side chain of Lys12. Reprinted with permission from Ref 61. Copyright 2010 American Chemical Society.
The decrease in ΔGS‡ from 6.3 to 4.4 kcal/mol observed as GRNH3‡ increases for activation by alkyl ammonium cations (Figure 5) reflects the contribution of hydrophobic protein•cation interactions towards reducing the price for formation of the transition state from free reactants.61 The minimum value of ΔGS‡ = 6.3 kcal/mol for activation by NH4+ provides an estimate for the intrinsic entropic advantage for reaction of wildtype TIM compared with the K12G•NH4+ pieces, where there is the minimal stabilization of the complex to the cationic piece.
The large 9.1 kcal/mol stabilizing interaction between the cationic side chain of R269 and the transition state for GPDH-catalyzed reduction of DHAP by NADH is reflective of strong protein-cation and protein-anion interactions, which favor the self-assembly of enzyme (R269A GPDH + Gua+) and substrate (GA + HPi) pieces at the active site of GPDH to form the catalytically active [R269AGPDH•GA•HPi•Gua+] complex (Figure 6A).59 The results of experiments to characterize the reactivity of this complex are summarized in Figure 6B. The R269A mutation results in a 9.1 kcal/mole reduction in the activation barrier for GPDH-catalyzed reduction of DHAP by NADH. The addition of 1.0 M Gua+ and 1.0 M HPi results in a 3.5 kcal/mol lower activation barrier than is observed for R269A GPDH catalyzed reduction of DHAP [−(ΔGact‡)Gua + HPi, Figure 6B]. The advantage obtained from connection of the enzyme and substrate pieces is (ΔG‡S)Gua + HPi = 5.6 kcal/mol (Figure 6B), or roughly 2.8 kcal/mol for each covalent connection. This is substantially smaller that the single connection energies of (4 – 6) kcal/mol from previous comparisons of the reactive of whole enzymes or substrates with the corresponding pieces.28, 59 This small advantage obtained from the covalent covalent connection of the enzyme and substrate pieces is consistent with a relatively strong stabilization of the complex to the pieces, as illustrated in Figure 5 for activation of the K12G mutant of TIM. We propose that the active site of GPDH has evolved specifically to optimize stabilizing electrostatic interactions between the side chain of R269 and the highly negatively charged transition state for GPDH-catalyzed reduction of DHAP (Scheme 5).
Figure 6.

(A) Representation of the structure of the nonproductive complex between R269A GPDH, NAD+, Gua+, GA and HPi. generated from the X-ray crystal structure of the ternary complex between wildtype enzyme, NAD+ and DHAP (PDB entry 1WPQ). (B) A comparison between the effect of the R269A mutation on the activation barrier to wildtype GPDH-catalyzed reduction of DHAP (ΔGR269A‡ − ΔGWT‡ = 9.1 kcal/mol) and of the relative activation barriers for R269A GPDH catalyzed reduction of DHAP and for GA activated by 1.0 M Gua+ and 1.0 M HPi ([−(ΔGact‡)Gua + HPi). The advantage obtained from connection of the enzyme and substrate pieces is (ΔG‡S)Gua + HPi = 5.6 kcal/mol.
Beyond Pauling’s Model?
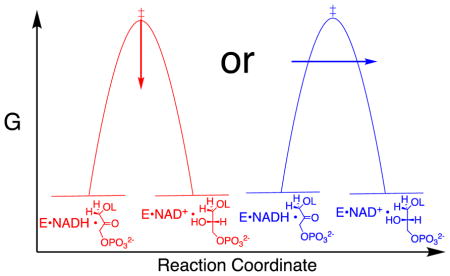
The results from the studies on GPDH, summarized above, show that the interaction between the side chain cation of R269 and the substrate phosphodianion are essential for the observation of robust catalysis. We have proposed that this ion pairing interaction provides a 2.8 kcal/mol stabilization of the Michaelis complex to substrate, and a much stronger 9.1 kcal/mol stabilization of the transition state for GPDH-catalyzed hydride transfer from NADH to GPDH. We now consider the possibility that these binding interactions are utilized in the stabilization of a “tunneling ready state” (TRS), which is proposed to constitute a special case of the more general transition state.66 The TRS has been defined as “the ensemble of states from which H tunneling occurs and like the general case of TS is a saddle point along a coordinate that represents the motions that bring the system from the ground state to the TRS”.67
The two models for enzymatic catalysis through a classical transition state and through a TRS, invoke different roles for the protein. The first model envisions the protein as participating directly in transition state stabilization. By contrast, the existence of a TRS implies that classical transition state stabilization alone is not sufficient to obtain the enzymatic rate acceleration. In this case, the energetic cost to organization of the protein into the TRS must be recovered, with dividends, as a rate enhancement for enzyme-catalyzed hydride transfer: this would arise from the advantage for tunneling through the reaction barrier, as compared with passage over the classical transition state.
It is important to separate the question of whether there is quantum mechanical tunneling during enzyme-catalyzed hydride transfer, from the question of whether this tunneling results in a large rate enhancement, compared to a reaction that passes over the energy barrier. The first question was addressed in computational studies, which provide evidence for tunneling of the transferred hydron, but only as the position of the hydron draws very close to that for the semi-classical transition state. The tunneling that occurs near the top of the energy barrier results in only a small decrease in the “effective” barrier height. The calculations give kinetic isotope effects on enzyme-catalyzed hydride transfer that are well below the semiclassical limit for a reaction that passes over the energy barrier, consistent a minor role for tunneling,68–70 so that the observation of primary deuterium kinetic isotope effects in the semi-classical range does not exclude a reaction that occurs with tunneling.71, 72
With respect to the second question, tunneling will only make a large contribution to an enzymatic rate enhancement when the energetic cost of organization of the protein into the TRS is recovered, so that the apparent reaction barrier is lower than the sum of the barrier for the reaction that passes through the transition state plus the barrier to formation of the TRS. If the effective barrier to a reaction with tunneling is lower than for formation of a semiclassical transition state, then the hydron must tunnel though the surface at an energy substantially below that for the transition state. Enzyme-catalyzed hydride transfers, which occur mainly by tunneling, should result in primary kinetic deuterium isotope effects (1°DKIE) that exceed the semi classical limit of ca. 7, because the longer QM wavelength for -H compared with -D gives rise to a greater uncertainty in the position of the light isotope. This favors tunneling of -H as the width of the energy barrier approaches that for the transferred -H.73
Kinetic Isotope Effect on GPDH-catalyzed Hydride Transfer
The following primary deuterium kinetic isotope effects were determined for GPDH-catalyzed reduction of DHAP by NADL [L = H, D] by fitting the data to the ordered reaction mechanism shown in Scheme 8,74, 75 where A = NADH and B = DHAP: D(V/KA) = 1.2; D(V/KB) = 1.5; DV = 1.5. 78 These 1°DKIEs are smaller than the intrinsic 1°DKIEs expected for enzyme-catalyzed hydride transfer to the carbonyl group when hydride transfer is fully rate determining.68–70 This is because the barriers to isotope-independent substrate binding and product release steps, compared with isotope dependent hydride transfer, have been equalized by evolutionary pressures to optimize the efficiency of protein catalysis.76, 77
Scheme 8.

The tight binding to GPDH ((Km)DHAP = 50 μM)28 favors slow release of the sticky substrate DHAP and partially rate determining substrate binding, which suppresses the 1°DKIE. By contrast, the weak binding of the pieces GA (Kd = 5 mM) and HPi (KX = 70 mM, Scheme 9) to GPDH favors fast and reversible ligand binding, and the observation of intrinsic 1°DKIEs on hydride transfer.
Scheme 9.
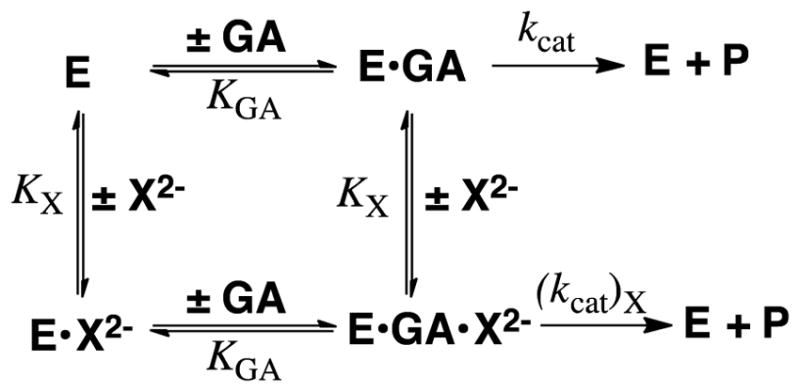
We determined the kinetic parameters (Scheme 9) for GPDH-catalyzed dianion activated reduction of GA by NADH and NADD and the 1°DKIE reported in Figure 7 on enzyme-catalyzed hydride transfer from NADL to GA.78 The large dissociation constants KGA for GA (6 mM) and KX for X2− (17–105 mM) require that ligand binding to GPDH be fast and reversible, so that kcat/KGA and kcat are controlled by the same rate determining hydride transfer. This is supported by the observation of similar 1°DKIE on these kinetic parameters (Figure 7).78 The strongest evidence that the 1°DKIEs from Figure 7 are intrinsic is the observation that they are independent of the reactivity of the dianion activator. If binding of phosphite dianion were partly rate determining for this fast reaction, because dianion disassociation is slow compared with hydride transfer, then the change to more weakly binding dianions should result in faster dianion disassociation and an increase in the primary 1°DKIE, but this is not observed.
Figure 7.

The 1°DKIEs determined for dianion activated reduction of GA catalyzed by GPDH that were determined from the fit of kinetic data to the mechanism shown in Scheme 9.78 Essentially the same disassociation constants KX and KGA were obtained from the fit of data for the reactions of NADH and NADD, so that the 1°DKIE on the kinetic parameters kcat, kcat/KGA and kcat/KGAKX for the different dianion activated reactions are equal to the quoted values of kH/kD.
Cutting substrates into pieces has little effect on the structure of the transition state for reactions catalyzed by triosephosphate isomerase51, 52 and orotidine 5′-monophosphate decarboxylase.40 We likewise propose that the transition states for GPDH-catalyzed reactions of the whole substrate DHAP and the pieces GA + X2− are similar, and that the intrinsic 1°DKIE on the reaction of the whole substrate is similar to the values of kH/kD. reported for the pieces in Figure 7.
The magnitude of these intrinsic 1°DKIE for GPDH-catalyzed reductions of GA by NADL (2.7 ± 0.3, Figure 7) is marginally smaller than the computed value of ≈ 3.6 for the intrinsic 1°DKIE on hydride transfer catalyzed by alcohol dehydrogenase.69 The intrinsic 1°DKIE of ca 2.7 provides strong evidence for the conclusion that the isotope effect is due largely to the change in the difference in zero-point energy for -H compared with -D, on moving from reactants to the transition state, and that there is only a small contribution from tunneling to this 1°DKIE.
We conclude that there is no evidence that the difference in the QM wavelengths of -H and -D provide a substantial advantage to the reaction of H, as expected for the tunneling of -H and -D underneath the reaction barrier. Such tunneling has been proposed to give rise to 1°DKIEs of kH/kD > 80 for the C–H cleavage catalyzed by soybean lipoxygenase at room temperature (Scheme 10).79–82 The observation of 1°DKIE in the semi-classical range does not rule out the existence of tunneling during GPDH catalyzed hydride transfer. However, the small 1°DKIEs observed for the dianion-activated reactions (Figure 7) provide strong evidence that such tunneling does not provide a rate advantage, compared with the reaction that passes over the barrier.
Scheme 10.

Alcohol Dehydrogenase
The 1°DKIE reported for alcohol dehydrogenase catalyzed reduction of aldehydes and ketones, or oxidation of alcohols range from 1.2 – 6.4.19, 83–87 The relatively small isotope effects on the physiological reactions of ethanol/acetaldehyde may be suppressed because hydride transfer is not fully rate-determining.84, 85 However, the isotope effects determined for the slower reactions of nonphysiological substrates are not sufficiently large to provide evidence for quantum-mechanical tunneling.19, 83, 84, 87 We suggest that the barriers to these enzymatic reactions are determined largely by the barrier to formation of the transition state for a semi-classical hydride transfer reaction, and that there is no large reduction in this barrier from quantum mechanical tunneling. The ambiguity associated with other evidence presented to support the proposal that protein-substrate interactions enhance tunneling during alcohol dehydrogenase-catalyzed hydride transfer has been discussed in detail by Truhlar.88 The binding energy of the small ethanol/acetaldehyde substrate for ADH is almost certainly insufficient to account for the enyzmatic rate acceleration, so that the binding energy of NAD+/NADH cofactor must make a significant contribution to this acceleration. We propose that a significant fraction of this cofactor binding energy is utilized to activate ADH for catalysis of hydride transfer, in a manner similar to that described above for dianion activation of GPDH for catalysis. We are considering experiments with the NAD+ cofactor in pieces, similar to those with DHAP in pieces described above for GHPD
Summary and Concluding Remarks
The notion that chemical catalysts achieve their rate acceleration through transition state stabilization is part of the fabric of the chemical literature. Enzymes are special in the sense that the transition state stabilizations required to rationalize their rate accelerations are much larger than for man-made catalysts. However, to the best of our knowledge, there is no rate acceleration for any enzymatic reaction that exceeds the interactions possible between the putative transition state and the protein catalyst.
We find that protein-dianion interactions make a large contribution to the rate acceleration for GPDH-catalyzed reduction of DHAP, and we present here arguments that these interactions represent direct stabilization of the reaction transition state. The 1°DKIE determined for these reactions are consistent with a reaction whose rate is controlled largely by the barrier to formation of the reaction transition state.
It might seem sensible for the sake of avoiding futile controversy, to suspend judgement about the role of tunneling in enzymatic reactions, until coming to grips with the experimental evidence published to support the proposal this role. However, this evidence is difficult to evaluate. We note that such suspension of judgement may slow progress towards the development of a consensus; and, that the resulting stagnation may prove more damaging than heated discussion. Such discussion often generates ideas for experiments that provide the results necessary for the development of a consensus. We hope that this article spurs discussion and the development of creative experimental protocol to distinguish between these two models to rationalize the rate acceleration for enzyme-catalyzed hydride transfer.
Acknowledgments
We are grateful to the US National Institutes of Health Grants GM116921 and GM039754 for generous support of our work.
References
- 1.IUPAC, Compendium of Chemical Terminology. 2. International Union of Pure and Applied Chemistry; Hoboken, NJ: 1997. [Google Scholar]
- 2.Pauling L. Nature. 1948;161:707–709. doi: 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- 3.Amyes TL, Richard JP. Biochemistry. 2013;52:2021–2035. doi: 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jencks WP. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 5.Nagel ZD, Klinman JP. Nat Chem Biol. 2009;5:543–550. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- 6.Schramm VL. Ann Rev Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammes-Schiffer S, Benkovic SJ. Ann Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 8.Amyes TL, Malabanan MM, Zhai X, Reyes AC, Richard JP. Prot Eng Des & Sel. 2017;30:157–165. doi: 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammond GS. J Am Chem Soc. 1955;77:334–338. [Google Scholar]
- 10.Richard JP. J Am Chem Soc. 1984;106:4926–4936. [Google Scholar]
- 11.Wu N, Mo Y, Gao J, Pai EF. Proc Natl Acad Sci U S A. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warshel A, Strajbl M, Villa J, Florian J. Biochemistry. 2000;39:14728–14738. doi: 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]
- 13.Vardi-Kilshtain A, Doron D, Major DT. Biochemistry. 2013;52:4382–4390. doi: 10.1021/bi400190v. [DOI] [PubMed] [Google Scholar]
- 14.Plapp BV, Charlier HA, Jr, Ramaswamy S. Arch Biochem Biophys. 2016;591:35–42. doi: 10.1016/j.abb.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 15.Meijers R, Cedergren-Zeppezauer E. Chem-Biol Int. 2009;178:24–28. doi: 10.1016/j.cbi.2008.10.043. [DOI] [PubMed] [Google Scholar]
- 16.Adolph HW, Zwart P, Meijers R, Hubatsch I, Kiefer M, Lamzin V, Cedergren-Zeppezauer E. Biochemistry. 2000;39:12885–12897. doi: 10.1021/bi001376s. [DOI] [PubMed] [Google Scholar]
- 17.Yahashiri A, Rubach JK, Plapp BV. Biochemistry. 2014;53:881–894. doi: 10.1021/bi401583f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plapp BV, Ramaswamy S. Biochemistry. 2012;51:4035–4048. doi: 10.1021/bi300378n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramaswamy S, Park DH, Plapp BV. Biochemistry. 1999;38:13951–13959. doi: 10.1021/bi991731i. [DOI] [PubMed] [Google Scholar]
- 20.Herdendorf TJ, Plapp BV. Chem-Biol Int. 2011;191:42–47. doi: 10.1016/j.cbi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LeBrun LA, Park DH, Ramaswamy S, Plapp BV. Biochemistry. 2004;43:3014–3026. doi: 10.1021/bi036103m. [DOI] [PubMed] [Google Scholar]
- 22.Bahnson BJB, Park DHD, Kim KK, Plapp BVB, Klinman JPJ. Biochemistry. 1993;32:5503–5507. doi: 10.1021/bi00072a003. [DOI] [PubMed] [Google Scholar]
- 23.Kulkarni YS, Liao Q, Petrović D, Krüger DM, Strodel B, Amyes TL, Richard JP, Kamerlin SCL. J Am Chem Soc. 2017;139:10514–10525. doi: 10.1021/jacs.7b05576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goryanova B, Amyes TL, Gerlt JA, Richard JP. J Am Chem Soc. 2011;133:6545–6548. doi: 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsang WY, Wood BM, Wong FM, Wu W, Gerlt JA, Amyes TL, Richard JP. J Am Chem Soc. 2012;134:14580–14594. doi: 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsang WY, Amyes TL, Richard JP. Biochemistry. 2008;47:4575–4582. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reyes AC, Zhai X, Morgan KT, Reinhardt CJ, Amyes TL, Richard JP. J Am Chem Soc. 2015;137:1372–1382. doi: 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jencks WP. Proc Nat Acad Sci. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amyes TL, O’Donoghue AC, Richard JP. J Am Chem Soc. 2001;123:11325–11326. doi: 10.1021/ja016754a. [DOI] [PubMed] [Google Scholar]
- 31.Amyes TL, Richard JP, Tait JJ. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 32.Ray WJ, Jr, Long JW, Owens JD. Biochemistry. 1976;15:4006–4017. doi: 10.1021/bi00663a015. [DOI] [PubMed] [Google Scholar]
- 33.Kholodar SA, Allen CL, Gulick AM, Murkin AS. J Am Chem Soc. 2015;137:2748–2756. doi: 10.1021/ja512911f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alber TC, Davenport RC, Jr, Giammona DA, Lolis E, Petsko GA, Ringe D. Cold Spring Harbor Symp Quant Biol. 1987;52:603–613. doi: 10.1101/sqb.1987.052.01.069. [DOI] [PubMed] [Google Scholar]
- 35.Lolis E, Petsko GA. Biochemistry. 1990;29:6619–6625. doi: 10.1021/bi00480a010. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Sugio S, Komives EA, Liu KD, Knowles JR, Petsko GA, Ringe D. Biochemistry. 1994;33:2830–2837. doi: 10.1021/bi00176a012. [DOI] [PubMed] [Google Scholar]
- 37.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc Natl Acad Sci U S A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Appleby TC, Kinsland C, Begley TP, Ealick SE. Proc Nat Acad Sci. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ou X, Ji C, Han X, Zhao X, Li X, Mao Y, Wong LL, Bartlam M, Rao Z. J Mol Biol. 2006;357:858–869. doi: 10.1016/j.jmb.2005.12.074. [DOI] [PubMed] [Google Scholar]
- 40.Goldman LM, Amyes TL, Goryanova B, Gerlt JA, Richard JP. J Am Chem Soc. 2014;136:10156–10165. doi: 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amyes TL, Ming SA, Goldman LM, Wood BM, Desai BJ, Gerlt JA, Richard JP. Biochemistry. 2012;51:4630–4632. doi: 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malabanan MM, Amyes TL, Richard JP. Curr Op Struct Biol. 2010;20:702–710. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Go MK, Amyes TL, Richard JP. Biochemistry. 2009;48:5769–5778. doi: 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richard JP, Zhai X, Malabanan MM. Bioorg Chem. 2014;57:206–212. doi: 10.1016/j.bioorg.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richard JP, Amyes TL, Goryanova B, Zhai X. Curr Op Chem Biol. 2014;21:1–10. doi: 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malabanan MM, Amyes TL, Richard JP. J Am Chem Soc. 2011;133:16428–16431. doi: 10.1021/ja208019p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malabanan MM, Go MK, Amyes TL, Richard JP. Biochemistry. 2011;50:5767–5779. doi: 10.1021/bi2005416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malabanan MM, Koudelka AP, Amyes TL, Richard JP. J Am Chem Soc. 2012;134:10286–10298. doi: 10.1021/ja303695u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malabanan MM, Nitsch-Velasquez L, Amyes TL, Richard JP. J Am Chem Soc. 2013;135:5978–5981. doi: 10.1021/ja401504w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richard JP, Amyes TL, Malabanan MM, Zhai X, Kim KJ, Reinhardt CJ, Wierenga RK, Drake EJ, Gulick AM. Biochemistry. 2016;55:3036–3047. doi: 10.1021/acs.biochem.6b00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhai X, Amyes TL, Richard JP. J Am Chem Soc. 2014;136:4145–4148. doi: 10.1021/ja501103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhai X, Amyes TL, Richard JP. J Am Chem Soc. 2015;137:15185–15197. doi: 10.1021/jacs.5b09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhai X, Amyes TL, Wierenga RK, Loria JP, Richard JP. Biochemistry. 2013;52:5928–5940. doi: 10.1021/bi401019h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhai X, Go MK, Donoghue AC, Amyes TL, Pegan SD, Wang Y, Loria JP, Mesecar AD, Richard JP. Biochemistry. 2014;53:3486–3501. doi: 10.1021/bi500458t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Desai BJ, Wood BM, Fedorov AA, Fedorov EV, Goryanova B, Amyes TL, Richard JP, Almo SC, Gerlt JA. Biochemistry. 2012;51:8665–8678. doi: 10.1021/bi301188k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goryanova B, Goldman LM, Amyes TL, Gerlt JA, Richard JP. Biochemistry. 2013;52:7500–7511. doi: 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goryanova B, Spong K, Amyes TL, Richard JP. Biochemistry. 2013;52:537–546. doi: 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reyes AC, Amyes TL, Richard JP. Biochemistry. 2016;55:1429–1432. doi: 10.1021/acs.biochem.6b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reyes AC, Amyes TL, Richard JP. J Am Chem Soc. 2016;138:15251–15259. doi: 10.1021/jacs.6b09936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reyes AC, Koudelka AP, Amyes TL, Richard JP. J Am Chem Soc. 2015;137:5312–5315. doi: 10.1021/jacs.5b02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Go MK, Amyes TL, Richard JP. J Am Chem Soc. 2010;132:13525–13532. doi: 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barnett SA, Amyes TL, Wood MB, Gerlt JA, Richard JP. Biochemistry. 2010;49:824–826. doi: 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kukic P, Farrell D, McIntosh LP, García-Moreno EB, Jensen KS, Toleikis Z, Teilum K, Nielsen JE. J Am Chem Soc. 2013;135:16968–16976. doi: 10.1021/ja406995j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karp DA, Gittis AG, Stahley MR, Fitch CA, Stites WE, García-Moreno B. Biophys J. 2007;92:2041–2053. doi: 10.1529/biophysj.106.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Page MI, Jencks WP. Proc Nat Acad Sci U S. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pudney CR, Hay S, Sutcliffe MJ, Scrutton NS. J Am Chem Soc. 2006;128:14053–14058. doi: 10.1021/ja0614619. [DOI] [PubMed] [Google Scholar]
- 67.Roston D, Kohen A. Proc Nat Acad Sci. 2010;107:9572–9577. doi: 10.1073/pnas.1000931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Truhlar DG, Gao J, Alhambra C, Garcia-Viloca M, Corchado J, Sánchez ML, Villà J. Acc Chem Res. 2002;35:341–349. doi: 10.1021/ar0100226. [DOI] [PubMed] [Google Scholar]
- 69.Alhambra C, Corchado JC, Sánchez ML, Gao J, Truhlar DG. J Am Chem Soc. 2000;122:8197–8203. [Google Scholar]
- 70.Billeter SR, Webb SP, Agarwal PK, Iordanov T, Hammes-Schiffer S. J Am Chem Soc. 2001;123:11262–11272. doi: 10.1021/ja011384b. [DOI] [PubMed] [Google Scholar]
- 71.Tsang WY, Richard JP. J Am Chem Soc. 2009;131:13952–13962. doi: 10.1021/ja905080e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wong KY, Richard JP, Gao J. J Am Chem Soc. 2009;131:13963–13971. doi: 10.1021/ja905081x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Caldin EF. Chem Rev. 1969;69:135–156. [Google Scholar]
- 74.Argyrou A, Blanchard JS. Biochemistry. 2004;43:4375–4384. doi: 10.1021/bi049974k. [DOI] [PubMed] [Google Scholar]
- 75.Cook PF, Cleland WW. Biochemistry. 1981;20:1790–1796. doi: 10.1021/bi00510a013. [DOI] [PubMed] [Google Scholar]
- 76.Knowles JR, Albery WJ. Acc Chem Res. 1977;10:105–111. [Google Scholar]
- 77.Albery WJ, Knowles JR. Biochemistry. 1976;15:5631–5640. doi: 10.1021/bi00670a032. [DOI] [PubMed] [Google Scholar]
- 78.Reyes AC, Amyes TL, Richard JP. J Am Chem Soc. 2016;138:14526–14529. doi: 10.1021/jacs.6b07028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jonsson T, Glickman MH, Sun S, Klinman JP. J Am Chem Soc. 1996;118:10319–10320. [Google Scholar]
- 80.Sharma SC, Klinman JP. Biochemistry. 2015;54:5447–5456. doi: 10.1021/acs.biochem.5b00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carr CAM, Klinman JP. Biochemistry. 2014;53:2212–2214. doi: 10.1021/bi500070q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Knapp MJ, Rickert K, Klinman JP. J Am Chem Soc. 2002;124:3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 83.Klinman JP. Biochemistry. 1976;15:2018–2026. doi: 10.1021/bi00654a032. [DOI] [PubMed] [Google Scholar]
- 84.Klinman JP. J Biol Chem. 1972;247:7977–7987. [PubMed] [Google Scholar]
- 85.Bush K, Shiner VJ, Mahler HR. Biochemistry. 1973;12:4802–4805. doi: 10.1021/bi00747a037. [DOI] [PubMed] [Google Scholar]
- 86.Shore JD, Gutfreund H. Biochemistry. 1970;9:4655–4659. doi: 10.1021/bi00826a006. [DOI] [PubMed] [Google Scholar]
- 87.Cook PF, Cleland WW. Biochemistry. 1981;20:1805–1816. doi: 10.1021/bi00510a015. [DOI] [PubMed] [Google Scholar]
- 88.Truhlar DG. J Phys Org Chem. 2010;23:660–676. [Google Scholar]