

Abstract
Advances in allogeneic hematopoietic cell transplantation for sickle cell disease have improved outcomes, but there is limited analysis of healthcare utilization in this setting. We hypothesized that, compared to late transplantation, early transplantation (at age <10 years) improves outcomes and decreases healthcare utilization. We performed a retrospective study of children transplanted for sickle cell disease in the USA during 2000–2013 using two large databases. Univariate and Cox models were used to estimate associations of demographics, sickle cell disease severity, and transplant-related variables with mortality and chronic graft-versus-host disease, while Wilcoxon, Kruskal-Wallis, or linear trend tests were applied for the estimates of healthcare utilization. Among 161 patients with a 2-year overall survival rate of 90% (95% confidence interval [CI] 85–95%) mortality was significantly higher in those who underwent late transplantation versus early (hazard ratio (HR) 21, 95% CI 2.8–160.8, P=0.003) and unrelated compared to matched sibling donor transplantation (HR 5.9, 95% CI 1.7–20.2, P=0.005). Chronic graftversus host disease was significantly more frequent among those translanted late (HR 1.9, 95% CI 1.0–3.5, P=0.034) and those who received an unrelated graft (HR 2.5, 95% CI 1.2–5.4; P=0.017). Merged data for 176 patients showed that the median total adjusted transplant cost per patient was $467,747 (range: $344,029–$799,219). Healthcare utilization was lower among recipients of matched sibling donor grafts and those with low severity disease compared to those with other types of donor and disease severity types (P<0.001 and P=0.022, respectively); no association was demonstrated with late transplantation (P=0.775). Among patients with 2-year pre- and post-transplant data (n=41), early transplantation was associated with significant reductions in admissions (P<0.001), length of stay (P<0.001), and cost (P=0.008). Early transplant outcomes need to be studied prospectively in young children without severe disease and an available matched sibling to provide conclusive evidence for the superiority of this approach. Reduced post-transplant healthcare utilization inpatient care indicates that transplantation may provide a sustained decrease in healthcare costs over time.
Introduction
Sickle cell disease (SCD) affects approximately 100,000 people in the United States of America (USA) with 2,000 new cases detected via newborn screening annually. There is a lack of clinical predictors to estimate overall outcomes of SCD-associated morbidities, including painful crises and organ dysfunction, which respond variably to medical management, have a devastating impact on quality of life, and can lead to premature death.1 As a result, many people with SCD are left with sequelae of the disease and its complications. Allogeneic hematopoi-etic cell transplantation (alloHCT) remains the only established curative option for these individuals.
Despite mounting evidence of rising alloHCT success rates over time, such that the 5-year disease-free survival in children with SCD is now 92%, many still regard alloHCT as an experimental therapy, only for patients with severe disease.2,3 The indications for alloHCT remain unclear for non-transplant providers when compared to the benefits of medical management.4 In addition, a recent retrospective study from Belgium suggested that patients with SCD managed medically with hydroxyurea may have a better survival than those treated with alloHCT.5
However, short-term improvements in outcome with medical therapy must be balanced against a disease with an unpredictable clinical course and substantial impact on healthcare utilization. USA individuals with SCD account for an estimated $1.6 billion per year in healthcare costs.6 SCD ranked fifth among the top ten diagnoses of hospital stays among Medicaid super-utilizers.7 The substantial healthcare utilization and cost of SCD-related morbidity suggests that a greater focus on curative approaches for this disease is needed.
AlloHCT, when successful, can be curative, but also carries the risks of death and substantial morbidity from chronic graft-versus-host disease (GvHD). In addition, the initial cost of alloHCT represents a significant financial burden of approximately $400,000 in the transplant year.8 This research investigates alloHCT for pediatric SCD using a comprehensive, systematic database analysis exploring patient-, disease-, and transplant-related variables that may reduce healthcare utilization over time while sustaining excellent clinical outcomes. The findings may provide transplant and non-transplant physicians with additional information to help choose between recommending medical therapy and alloHCT.
Methods
Data sources
Outcomes analysis
The Center for International Blood and Marrow Transplant Research (CIBMTR) database contains alloHCT data for recipients and their donors. Data are collected prior to and at various intervals post-alloHCT. Upon CIBMTR registration, a weighted randomization scheme selects a subset of patients for more detailed data collection in comprehensive research forms (CRF) which provide more specific transplant-related data (SCD complications, pre-transplant therapy, etc.) (Online Supplementary Figure A1).
Healthcare utilization analysis
CIBMTR data on all alloHCT recipients are submitted as transplant essential data (TED) (Online Supplementary Figure A2). TED forms record donor and recipient demographic, clinical, and transplant data but lack specific CRF data.
The Pediatric Health Information System (PHIS; Children’s Hospital Association, Overland Park, KS, USA) records the corresponding inpatient healthcare utilization data. PHIS, a confidential database of 43 member hospitals in the USA (Online Supplementary Figure A3), has participating hospitals submit de-identified data with an encrypted medical record number for identification of readmissions at the same hospital. Institutional and patient-specific information, including patient’s age, date of service, visit codes, length of stay (LOS), adjusted costs, and daily billing data, are collected. The PHIS has been merged for similar research purposes including a number of recent scientific publications.9
Merging and validating datasets
Patients in the PHIS database who underwent alloHCT for SCD during the study period were identified utilizing International Classification of Diseases version 9 (ICD9) and alloHCT diagnosis-related group (DRG) codes (282.6 and 1803, respectively) as well as PHIS procedure codes. These patients were identified within the CIBMTR using a probabilistic algorithm. This process occurred under the guidance of the CIBMTR via the National Marrow Donor Program institutional review board.
TED data were merged with PHIS data to determine risk factors and clinical outcomes associated with healthcare utilization (Online Supplementary Figure A4). A target of 85% merge accuracy was set based on the available database population and previously published reports.9–11 Once linked, the merge accuracy was assessed by performing institutional level validation under an existing pilot institutional review board process.8 This validation confirmed 100% patient identification in this subset. SCD-related complications identified in the PHIS were validated against CRF data, where available, and showed concordance.
Determination of the severity of sickle cell disease
TED/PHIS variables were used to determine SCD severity. Younger patients (age <10 years) without disease sequelae were considered low risk. Younger patients with disease sequelae or older patients (age ≥10 years) without disease sequelae were considered moderate risk. Older patients with disease sequelae or patients of any age with stroke were considered high risk. Disease sequelae were defined as any episode of acute chest syndrome, and/or three or more vaso-occlusive crises requiring hospitalization in 1 year.12
Variables and outcomes
Outcomes analysis
The study population consisted of children 21 years or younger who had undergone alloHCT for SCD in the USA between 2000–2013 and for whom CRF data were available. The CRF provided information on clinical risk factors and outcomes including overall survival, graft failure, grade II–IV acute GvHD, chronic GvHD, and GvHD-related event free survival (GREFS). GREFS was defined as the survival free of graft failure, chronic GvHD, or death and was used to better assess the post-alloHCT morbidity and associations of clinical risk factors with outcomes.
Healthcare utilization analysis
The total adjusted cost reported to the PHIS is based on a fixed hospital-wide ratio of cost to charge adjusted by geographical location. Adjusted costs for each service unit or department (clinical, pharmacy, imaging, etc.) were reported using service-specific ratios of cost to charge. Charges in the PHIS database were adjusted for the wage and price index (published annually in the Federal Register) and reported from the hospital perspective. Total adjusted costs were determined for all inpatient admissions for each patient and include direct medical costs, excluding provider fees, incurred. Indirect costs, outpatient costs, and costs incurred at non-PHIS hospitals were not captured.
Adjusted cost data only were analyzed as the primary outcome of interest because charges and reimbursements vary across each institution and state. Patients without available adjusted cost data were excluded (n=7). Zero-dollar research or study-related costs, reflecting largely workup, medication, or laboratory-related account credits, were included in the analysis. Additional healthcare utilization outcomes included number of admissions and LOS. PHIS data were used for descriptive analyses of the selected cohort of patients throughout all the study periods.
Healthcare utilization for the initial alloHCT admission (conditioning to first recorded discharge) and alloHCT year (conditioning to day +365) was described and considered separately in the analyses. Likewise, the pre-alloHCT period (2 years preceding transplant through to the day of transplant conditioning) and the post-alloHCT period (2 years from day +366 onward) were analyzed separately (Online Supplementary Figure B).
Analysis of allogeneic hematopoietic cell transplant year
Factors influencing healthcare utilization during the alloHCT year were analyzed using the following TED clinical variables -age at transplant, gender, performance status, recipient cytomegalovirus status, income level, insurance, distance from center, SCD complications, donor type, graft source, conditioning regimens, and transplant year. A secondary analysis of disease severity and healthcare utilization was also performed.
Pre- and post-allogeneic hematopoietic cell transplantation comparison
To standardize costs for comparisons, the total adjusted cost per 30 hospital days was calculated for each patient with both pre- and post-alloHCT inpatient admissions and used as the primary healthcare utilization outcome. The change in an individual patient’s healthcare utilization pre- and post-alloHCT was compared. This change in healthcare utilization was also analyzed by disease severity.
Statistical analysis
Outcomes analysis
After documenting descriptive statistics, Cox proportional hazards modeling of CRF level data determined the impact of risk factors on alloHCT outcomes. Due to low event rates and small sample sizes, only bivariate analyses involving one explanatory variable at a time were performed. The cumulative incidences of acute GvHD and chronic GvHD were calculated using a competing risk framework.
Healthcare utilization analysis
TED/PHIS data were evaluated to identify whether demographic factors, SCD severity, or alloHCT variables correlate with healthcare utilization changes during the alloHCT year using Wilcoxon, Kruskal-Wallis, or linear trend tests. Healthcare utilization pre- and post-alloHCT was compared using the Wilcoxon signed rank test for continuous variables and the McNemar test for binary variables. Finally, the impact of SCD severity on healthcare utilization across these two time periods was examined by Poisson regression for number of visits and LOS. Total adjusted cost were analyzed by linear regression. All analyses were performed using SAS version 9.3 statistical software (Cary, NC, USA).
Results
Outcomes analysis
Demographics
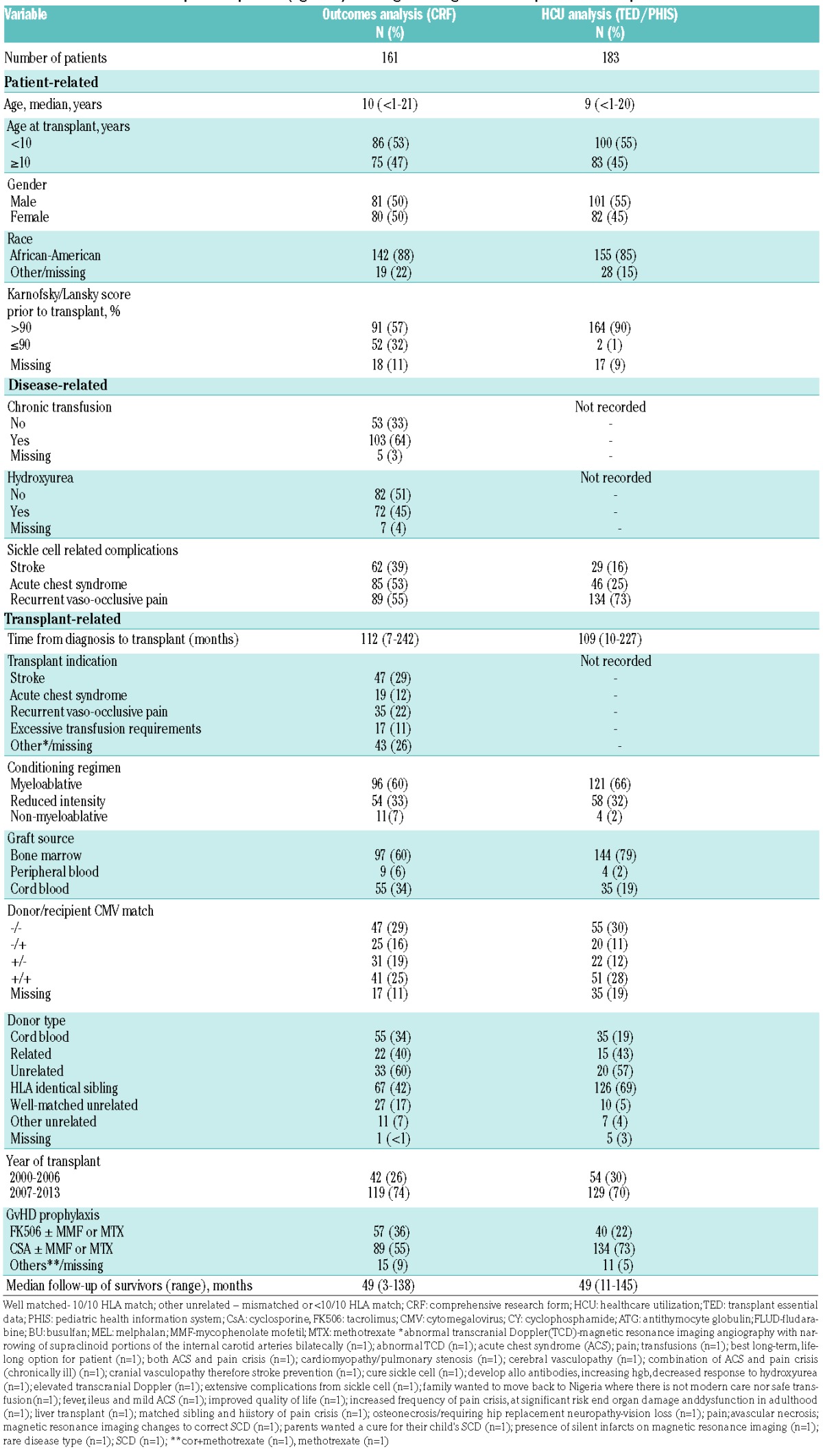
CRF data were available for 161 patients with a median age of 10 years (range <1–21) of whom 50% were female (Table 1; Online Supplementary Table A). The majority (84%) of patients had the HbSS genotype. The most commonly documented transplant indication was stroke (29%) followed by recurrent vaso-occlusive crises (22%). However, 39% had stroke as a documented SCD complication, 55% vaso-occlusive crises, and 53% acute chest syndrome. The majority of patients reported use of medical therapy, hydroxyurea (45%) and chronic transfusions (64%). The most common source of a graft for transplantation was a matched sibling donor (MSD) (42%) and the majority of patients received myeloablative conditioning (60%).
Table 1.
Characteristics of USA pediatric patients (age ≤21) receiving first allogeneic hematopoietic cell transplant for sickle cell disease.
Transplant outcomes
The 2-year overall survival was 90% [95% confidence interval (CI): 85–95%]: 96% (95% CI: 89–100%) for cord blood transplant (CBT), 94% (95% CI: 86–98%) for MSD transplants, and 74% (95% CI: 54–90%) for transplants from well-matched unrelated donors (MUD) (P=0.002) (Online Supplementary Table B and Online Supplementary Figure C). All 16 deaths occurred among children with pre-alloHCT complications of SCD and were due to organ failure (37.5%), infections (25%), GvHD (6.2%), and other/unknown causes (31.2%) (Online Supplementary Table C). The majority of deaths (62.5%) occurred during the alloHCT year; six patients died after day +365 (2 from organ failure, 1 from infection, 1 from sickle cell-associated vasculopathy, and 2 from a missing/other unspecified cause).
The cumulative incidence of acute GvHD at day 100 was 14% (95% CI: 9–20%), and chronic GvHD developed in 31% (95% CI: 23–38%) at 2 years. Of those with chronic GvHD, 64% had extensive disease with over half having a MSD (n=9) or MUD (n=11). The 2-year GREFS was 64% (95% CI: 56–71%).
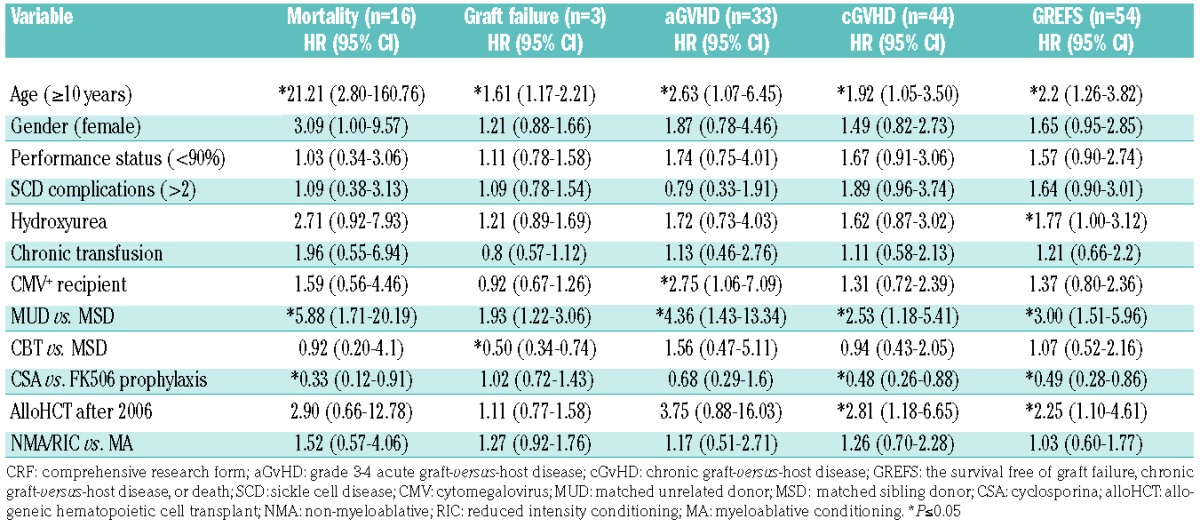
Bivariate analysis
Age ≥10 years and use of a MUD showed significant negative associations with outcomes (Table 2). The use of cyclosporine-A prophylaxis and year of alloHCT exhibited significant associations with chronic GvHD-related outcomes (Table 2).
Table 2.
Cox regression model of outcomes with patient- and transplant-related variables as reported in CRF data (n=161).
Healthcare utilization analysis
Demographics
Combined TED/PHIS data were available for 183 patients with a median age of 9 years (range: <1–20) of whom 45% were female (Table 1). With regards to SCD complications, 73% had vaso-occlusive crises, 25% had acute chest syndrome, and 16% had a stroke prior to alloHCT. This translated into 19.7% with low severity disease, 36.6% with moderate severity, and 43.7% with high severity disease (Online Supplementary Table D). The majority of patients received MSD alloHCT (69%) and a myeloablative regimen (66%). Complete cost data were available for 176 of the 183 patients.
Allogeneic hematopoietic cell transplantation admission
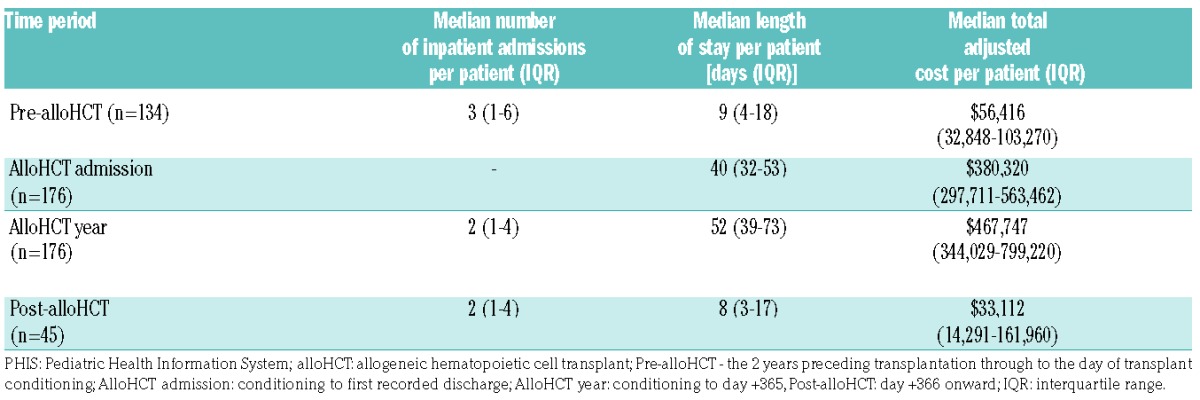
The median total adjusted cost per patient was $380,320 [interquartile range (IQR): $297,710–$563,462] with a median LOS of 39.5 days (IQR: 31–53) (Table 3). The highest costs were associated with room and nursing charges followed by pharmacy costs (Online Supplementary Figure D).
Table 3.
Inpatient healthcare utilization analysis as reported over time in the PHIS.
Allogeneic hematopoietic cell transplantation year
The median total adjusted cost per patient was $467,747 (IQR: $344,029–$799,219) with a median LOS of 52 days (IQR: 38–72) (Table 3). Again, the highest costs were associated with room and nursing charges followed by pharmacy costs (Online Supplementary Figure D).
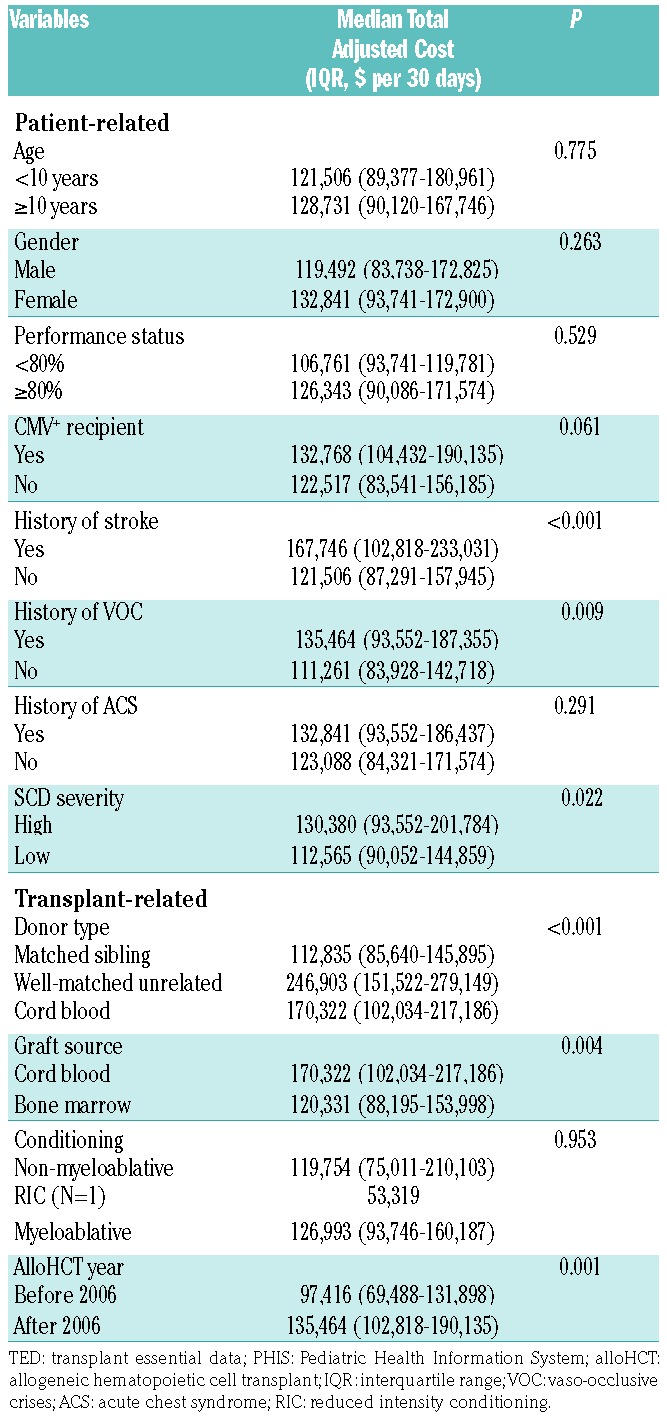
During the alloHCT year, age ≥10 years and total adjusted cost per 30 days were not significantly associated (P=0.775) (Table 4). Total adjusted cost was also not associated with income level, insurance type, or distance from transplant center (P=0.417, 0.918, and 0.253, respectively). The total adjusted cost per 30 days was lower for MSD transplants than for CBT or MUD transplants (P<0.001). CBT was associated with a higher total adjusted cost per 30 days compared to bone marrow transplants (P=0.004). Increased total adjusted cost per 30 days were associated with prior stroke (P=0.004) and vaso-occlusive crises (P=0.009) but not acute chest syndrome (P=0.291). Overall, total adjusted cost per 30 days increased with SCD severity (P=0.022).
Table 4.
Comparison of inpatient total adjusted costs per 30 days by TED/PHIS patient and transplant variables during the alloHCT year (n=176).
Pre- and post-allogeneic hematopoietic cell transplantation
Two-year inpatient healthcare utilization data were available for 134 pre-alloHCT patients and 45 post-alloHCT. The median total adjusted cost per patient was $56,416 (IQR: $32,848–$103,270) pre-alloHCT; this decreased to $33,112 (IQR: $14,291–$161,959) post-alloHCT (Table 3). The highest adjusted costs during these periods were associated with room and nursing charges followed by laboratory costs (Online Supplementary Figure D).
Only 41 patients were admitted to the hospital during both pre- and post-transplant periods: healthcare utilization was significantly reduced with total adjusted cost per 30 days decreasing from $9,393 (IQR: $4,595–$31,291) to $1,873 (IQR: $571–$6,504) (P=0.008) (Figure 1). Of these, 30 patients had high severity disease and had a significant reduction in healthcare utilization (admissions P<0.001, LOS P<0.001, cost P=0.002) (Online Supplementary Table E).
Figure 1.

Comparison of 2-year pre- and post-allogeneic hematopoietic cell transplant inpatient healthcare utilization per 30 days as reported in the Pediatric Health Information System (n=41). Pre-alloHCT: the 2 years preceding transplant through to the day of transplant conditioning; Post-alloHCT: day +366 onward; alloHCT: hematopoietic cell transplant; PHIS: Pediatric Health Information System: visits: inpatient admissions; LOS: length of stay.
Discussion
Substantial advances in alloHCT for SCD have been made in the past two decades; nevertheless, the eligibility of patients, especially those without severe disease, remains controversial. This study provides additional insight into eligibility as alloHCT outcomes were favorably linked to age and donor type suggesting that early alloHCT, before the age of 10 years, and MSD alloHCT have optimal outcomes with the latter also showing a significant healthcare utilization advantage. These outcomes are in keeping with a recent report from an international study of adult and pediatric MSD recipients describing lower event-free survival rates with increasing age at transplantation and support the recent expert panel recommendation of early alloHCT, prior to the onset of SCD complications, for children with SCD and an available MSD.13,14
MSD alloHCT is not, however, a viable option for many patients, as fewer than 25% will have a suitable HLA-matched donor, necessitating MUD alloHCT for severe disease. Among patients undergoing MUD alloHCT, higher GvHD risk could be hypothesized as the etiology of poorer outcomes and increased healthcare utilization of older recipients.13,15–17 Although the GvHD-related mortality rate in this study is lower than that in published reports, the low percentage of deaths after day +365 may mitigate the mortality associated with chronic GvHD. However, the 26% mortality among the MUD alloHCT patients remains well over the 5% mortality threshold accepted by most parents and adolescents for the cure of SCD, as recently published.18 Efforts are still needed to elucidate more precisely the cause of death (unknown cause in 31%) and transplant-related mortality, especially among patients who died after the alloHCT year, in order to improve transplant procedures to prevent these complications. Our findings support the current practice of restricting MUD alloHCT to individuals with severe disease.19
The results of the analysis of donor type were consistent with published reports and indicated that the overall survival of patients treated with CBT is similar to that of recipients of MSD bone marrow; however, healthcare utilization was higher with CBT.20 Many of the drawbacks of CBT have been associated with insufficient cell dose. Ongoing research into cord blood expansion may mitigate this limitation and the associated healthcare utilization by reducing the delay in engraftment.21–23 However, this cohort of both related and unrelated CBT showed no statistical difference in GREFS or other GvHD outcomes, with a lower cost compared to MUD, suggesting that more analysis is needed to determine the optimal donor type if a MSD is unavailable.
Donor type and SCD severity had a significant impact on both outcomes and healthcare utilization, such that patients with high severity disease and MUD had poorer outcomes and increased healthcare utilization. The correlation of healthcare utilization and disease severity, not age, is unclear but may suggest that healthcare utilization is linked to management of persistent SCD complications after alloHCT. Although some end-organ disease is reversible after alloHCT, complications of chronic lung disease and pain can persist in the first year post-alloHCT.24 These complications correlate with increased healthcare utilization in individuals with SCD.25 In addition, severe disease remains an indication for MUD alloHCT research trials suggesting this as a possible confounder in healthcare utilization analysis. MUD outcomes associated with degree of HLA matching, supportive care measures, and infection must also be considered in more detail.16 Understanding and mitigating risk factors associated with poor outcomes and increased healthcare utilization following MUD alloHCT is needed because improvements in unrelated alloHCT clinical outcomes have the potential to have the greatest clinical and financial impact.
The healthcare utilization analysis also described variations over time with a subset of patients having a significant reduction in healthcare utilization pre- and post-alloHCT. However, the current sample size and/or 2-year time period may not be sufficient to document a change in inpatient healthcare utilization for the entire population of patients. Donor type variations (e.g., CBT) and disease severity within our small sample size likely have a role; more robust analysis is ongoing to understand this phenomenon better. In addition, outpatient healthcare utilization was not described which may account for the substantial reduction of data available for pre- and post-alloHCT analysis. However, previous publications on healthcare utilization in this population indicated that outpatient costs remained flat pre- and post-alloHCT.8
Limitations
This is a retrospective study and analysis is therefore limited to variables and data collected by the CIBMTR at the time of alloHCT. This limitation is somewhat mitigated by using multiple sources of data (PHIS and CIBMTR, both TED and CRF forms) to increase sample size and data availability or quality. Retrospective studies also do not allow control of exposures (pre-alloHCT treatment, conditioning, etc.) which may influence outcomes. Bivariate and multivariate analyses can elucidate cofounders; however, due to sample size and low event rates, multivariate analysis was not performed in this study. However, univariate analysis was used to document the impact of these exposures on outcomes.
The retrospective nature of this database study also does not allow for comparison to controls with SCD who have not been transplanted or incorporation of prospective metrics including quality of life in the analysis. Previous studies have documented the quality of life improvements gained after alloHCT for SCD.26,27 Other studies have described significant quality of life differences between non-transplant interventions such as chronic transfusion and hydroxyurea.1,28 Outcomes and cost of care for children have been well documented, with a recent analysis of management with hydroxyurea based on Medicaid claims data suggesting that lifetime costs of care may be influenced by the age of hydroxyurea initiation.5,29 These data largely demonstrate the substantial lifetime costs associated with caring for individuals with SCD and the need to compare costs across the various available treatment options.6 Collectively these findings suggest a multicenter case control study incorporating quality of life is needed to truly understand the full impact of alloHCT on outcomes and healthcare utilization. Prospective studies, including STRIDE2 (NCT02766465), are underway to fill this gap in knowledge of alloHCT for SCD.
The study includes a population that is heterogeneous for donor type, graft source, and conditioning regimen. Analysis of this complex group reflects the current clinical paradigm and its potential to influence healthcare utilization; however, diversity does introduce confounders. A recent study showed that conditioning regimen does not influence outcomes, and graft source only influences overall survival, while age and year of transplant influence both overall and event-free survival.13 Survival outcomes of this cohort are similar to this and other previously published estimates; these findings suggest that the heterogeneity of our cohort had limited influence on the outcomes analysis.
The study focuses on a pediatric population which is unique in that children are largely less affected by the disease than their adult counterparts and alloHCT has quite a different long-term impact. Certainly, efforts to offer early MSD alloHCT will have a significant effect on adult care; in the meantime, a large number of adults live with SCD. Therefore, efforts to better understand the cost of “late alloHCT” are needed. Specifically, the combination of favorable outcomes of CBT and advances in cord blood expansion technology may make this a more viable option for adults.21–23 Although, this study excluded data on haploidentical transplants, promising clinical outcomes to date suggest this is as another viable means of expanding the donor pool for adults and children with SCD.24 Ultimately, as the field advances with changes in conditioning regimens, modified donor source options, and the advent of gene therapy, ongoing analysis of outcomes and cost will be needed.
Studies of alloHCT for SCD are also limited by the absence of sufficient data on late effects. The risk of impaired fertility and long-term quality of life are not well described. Many studies have documented the impact of alloHCT on sperm production and ovarian failure in this population, but the actual fertility risk remains unclear particularly in light of the use of newer reduced intensity regimens that are less gonadotoxic.3,17,30 However, a recent study of patients’ and parents’ attitudes toward alloHCT documented that 56% were willing to accept infertility18 suggesting the potential for cure may far outweigh the risk of this complication. At the same time, another recent study of alloHCT not only reported the impact on fertility but also on sexual function and other patient-reported outcomes. In a cohort of individuals studied at least 10 years after transplantation for malignant disease, reports of sexual problems, restrictions in social function, memory and attention concerns, denial of life and health insurance were significant.31 Future studies should determine late outcomes after transplantation for SCD, collecting data not only on end-organ complications but also patient-reported outcomes including organ function and quality of life evaluations for both alloHCT recipients and controls.
Finally, this study involved a USA population and its findings cannot be easily extrapolated to a global population. However, recent publications have documented the impact of alloHCT throughout the developing world. AlloHCT outcomes in India and Mexico have been described as nearly equivalent to those in the developed world but are performed at substantially lower costs.32,33 International experience suggests that further study of USA transplant approaches to learn potential cost efficiencies from the global experience will make alloHCT more viable for SCD and other diseases throughout the world.
Conclusion
Performing analyses of both clinical and financial outcomes is challenging because of the lack of a single, exhaustive data source. The data merging process is invaluable in that it successfully utilizes existing datasets and combines them to create the largest and most accurate surrogate for assessment in a comprehensive manner. This provides proof of principle for this methodology and type of analysis and builds a foundation for future research in the field. Specifically, the superior clinical outcomes among children <10 years old lay the basis for prospective studies among low-risk SCD patients for whom a MSD is available. In addition, alloHCT, although costly, can provide a sustained decrease in healthcare utilization for patients over time. These results and future studies will provide guidance and insight into health policy determinations for the optimal use of this curative therapy for children with SCD.
Supplementary Material
Acknowledgments
Special acknowledgments to our patients and families. The numerous mentors and advisors who helped develop and support this project. Dr. Arnold was supported in part by a Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Program award. Dr. Aplenc was supported by 1R01CA166581.
CIBMTR Support List: the CIBMTR is supported primarily by Public Health Service Grant/Cooperative Agreement 5U24-CA076518 from the National Cancer Institute (NCI), the National Heart, Lung and Blood Institute (NHLBI) and the National Institute of Allergy and Infectious Diseases (NIAID); a Grant/Cooperative Agreement 5U10HL069294 from NHLBI and NCI; a contract HHSH250201200016C with Health Resources and Services Administration (HRSA/DHHS); two Grants N00014-15-1-0848 and N00014-16-1-2020 from the Office of Naval Research; and grants from *Actinium Pharmaceuticals, Inc.; Alexion; *Amgen, Inc.; Anonymous donation to the Medical College of Wisconsin; Astellas Pharma US; AstraZeneca; Atara Biotherapeutics, Inc.; Be the Match Foundation; *Bluebird Bio, Inc.; *Bristol Myers Squibb Oncology; *Celgene Corporation; Cellular Dynamics International, Inc.; Cerus Corporation; *Chimerix, Inc.; Fred Hutchinson Cancer Research Center; Gamida Cell Ltd.; Genentech, Inc.; Genzyme Corporation; Gilead Sciences, Inc.; Health Research, Inc. Roswell Park Cancer Institute; HistoGenetics, Inc.; Incyte Corporation; Janssen Scientific Affairs, LLC; *Jazz Pharmaceuticals, Inc.; Jeff Gordon Children’s Foundation; The Leukemia & Lymphoma Society; Medac, GmbH; MedImmune; The Medical College of Wisconsin; *Merck & Co, Inc.; *Mesoblast; MesoScale Diagnostics, Inc.; *Miltenyi Biotec, Inc.; National Marrow Donor Program; Neovii Biotech NA, Inc.; Novartis Pharmaceuticals Corporation; Onyx Pharmaceuticals; Optum Healthcare Solutions, Inc.; Otsuka America Pharmaceutical, Inc.; Otsuka Pharmaceutical Co, Ltd. – Japan; PCORI; Perkin Elmer, Inc.; Pfizer, Inc; *Sanofi US; *Seattle Genetics; *Spectrum Pharmaceuticals, Inc.; St. Baldrick’s Foundation; *Sunesis Pharmaceuticals, Inc.; Swedish Orphan Biovitrum, Inc.; Takeda Oncology; Telomere Diagnostics, Inc.; University of Minnesota; and *Wellpoint, Inc. The views expressed in this article do not reflect the official policy or position of the National Institute of Health, the Department of the Navy, the Department of Defense, Health Resources and Services Administration (HRSA) or any other agency of the U.S. Government.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/11/1823
Corporate Members.
References
- 1.Panepinto JA, Bonner M. Health-related quality of life in sickle cell disease: past, present, and future. Pediatr Blood Cancer. 2012;59(2):377–385. [DOI] [PubMed] [Google Scholar]
- 2.Gluckman E. Allogeneic transplantation strategies including haploidentical transplantation in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2013;2013:370–376. [DOI] [PubMed] [Google Scholar]
- 3.Walters MC, De Castro LM, Sullivan KM, et al. Indications and results of HLA-identical sibling hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2016;22(2):207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mikles B, Bhatia M, Oyeku SO, Green NS. Hematology provider perspectives on hematopoietic stem cell transplantation for pediatric sickle cell disease. Blood. 2012;120(21):4276–4276.23175658 [Google Scholar]
- 5.Lê PQ, Gulbis B, Dedeken L, et al. Survival among children and adults with sickle cell disease in Belgium: benefit from hydroxyurea treatment. Pediat Blood Cancer. 2015;62(11):1956–1961. [DOI] [PubMed] [Google Scholar]
- 6.Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84(6):323–327. [DOI] [PubMed] [Google Scholar]
- 7.Jiang HJ, Weiss AJ, Barrett ML, Sheng M. Characteristics of hospital stays for super-utilizers by payer, 2012: Statistical Brief #190. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville (MD) 2015. [Google Scholar]
- 8.Arnold SD, Jin Z, Sands S, Bhatia M, Kung AL, Satwani P. Allogeneic hematopoietic cell transplantation for children with sickle cell disease is beneficial and cost-effective: a single-center analysis. Biol Blood Marrow Transplant. 2015;21(7):1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berry JG, Hall DE, Kuo DZ, et al. Hospital utilization and characteristics of patients experiencing recurrent readmissions within children’s hospitals. JAMA. 2011;305(7):682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Hall M, Fisher BT, et al. Merging Children’s Oncology Group data with an external administrative database using indirect patient identifiers: a report from the Children’s Oncology Group. PLoS One. 2015;10(11):e0143480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aplenc R, Fisher BT, Huang YS, et al. Merging of the National Cancer Institute-funded cooperative oncology group data with an administrative data source to develop a more effective platform for clinical trial analysis and comparative effectiveness research: a report from the Children’s Oncology Group. Pharmacoepidemiol Drug Saf. 2012;21(Suppl 2):37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shenoy S. Has stem cell transplantation come of age in the treatment of sickle cell disease? Bone Marrow Transplant. 2007;40(9):813–821. [DOI] [PubMed] [Google Scholar]
- 13.Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2016;129(11):1548–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99(5):811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SJ, Klar N, Weeks JC, Antin JH. Predicting costs of stem-cell transplantation. J Clin Oncol. 2000;18(1):64–71. [DOI] [PubMed] [Google Scholar]
- 16.Svahn BM, Remberger M, Alvin O, Karlsson H, Ringden O. Increased costs after allogeneic haematopoietic SCT are associated with major complications and re-transplantation. Bone Marrow Transplant. 2012;47(5):706–715. [DOI] [PubMed] [Google Scholar]
- 17.Shenoy S, Eapen M, Wu J, et al. A multicenter phase II trial of unrelated donor reduced intensity bone marrow transplantation for children with severe sickle cell disease (SCURT): results of the blood and Marrow Transplant Clinical Trials Network (BMT CTN 0601) study. Blood. 2015;126(23):619–619. [Google Scholar]
- 18.Meier ER, Dioguardi JV, Kamani N. Current attitudes of parents and patients toward hematopoietic stem cell transplantation for sickle cell anemia. Pediatr Blood Cancer. 2015;62(7):1277–1284. [DOI] [PubMed] [Google Scholar]
- 19.Bhatia M, Sheth S. Hematopoietic stem cell transplantation in sickle cell disease: patient selection and special considerations. J Blood Med. 2015;6:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majhail NS, Mothukuri JM, Brunstein CG, Weisdorf DJ. Costs of hematopoietic cell transplantation: comparison of umbilical cord blood and matched related donor transplantation and the impact of post-transplant complications. Biol Blood Marrow Transplant. 2009;15(5):564–573. [DOI] [PubMed] [Google Scholar]
- 21.Horwitz ME, Chao NJ, Rizzieri DA, et al. Umbilical cord blood expansion with nicotinamide provides long-term multilineage engraftment. J Clin Invest. 2014;124(7):3121–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner JE, Brunstein CG, Boitano AE, et al. Phase I/II trial of StemRegenin-1 Expanded umbilical cord blood hematopoietic stem cells supports testing as a stand-alone graft. Cell Stem Cell. 2015;18(1):144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner JE, Jr, Eapen M, Carter S, et al. One-unit versus two-unit cord-blood transplantation for hematologic cancers. N Engl J Med. 2014;371(18):1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolanos-Meade J, Fuchs EJ, Luznik L, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285–4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll CP, Haywood C, Jr, Lanzkron S. Prediction of onset and course of high hospital utilization in sickle cell disease. J Hosp Med. 2011;6(5):248–255. [DOI] [PubMed] [Google Scholar]
- 26.Bhatia M, Kolva E, Cimini L, et al. Health-related quality of life after allogeneic hematopoietic stem cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2015;21(4):666–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly MJ, Pennarola BW, Rodday AM, Parsons SK, Journeys to Recovery Study H-CS. Health-related quality of life (HRQL) in children with sickle cell disease and thalassemia following hematopoietic stem cell transplant (HSCT). Pediatr Blood Cancer. 2012;59(4):725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ware RE, Davis BR, Schultz WH, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial Doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet. 2016;387(10019):661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang WC, Oyeku SO, Luo Z, et al. Hydroxyurea is associated with lower costs of care of young children with sickle cell anemia. Pediatrics. 2013;132(4):677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernaudin F, Robin M, Ferry C, et al. Related myeloablative stem cell transplantation (SCT) to cure sickle cell anemia (SCA): update of French results. Blood. 2010:116(21):3518. [Google Scholar]
- 31.Syrjala KL, Langer SL, Abrams JR, Storer BE, Martin PJ. Late effects of hematopoietic cell transplantation among 10-year adult survivors compared with case-matched controls. J Clin Oncol. 2005;23(27):6596–6606. [DOI] [PubMed] [Google Scholar]
- 32.Jaime-Pérez JC, Heredia-Salazar AC, Cantú-Rodríguez OG, et al. Cost structure and clinical outcome of a stem cell transplantation program in a developing country: the experience in northeast Mexico. Oncologist. 2015;20(4):386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma SK, Choudhary D, Gupta N, et al. Cost of hematopoietic stem cell transplantation in India. Mediterr J Hematol Infect Dis. 2014;6(1):e2014046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.