

ABSTRACT
The Nrf2 transcription factor is a master regulator of the cellular anti-stress response. A population of the transcription factor associates with the mitochondria through a complex with KEAP1 and the mitochondrial outer membrane histidine phosphatase, PGAM5. To determine the function of this mitochondrial complex, we knocked down each component and assessed mitochondrial morphology and distribution. We discovered that depletion of Nrf2 or PGAM5, but not KEAP1, inhibits mitochondrial retrograde trafficking induced by proteasome inhibition. Mechanistically, this disrupted motility results from aberrant degradation of Miro2, a mitochondrial GTPase that links mitochondria to microtubules. Rescue experiments demonstrate that this Miro2 degradation involves the KEAP1–cullin-3 E3 ubiquitin ligase and the proteasome. These data are consistent with a model in which an intact complex of PGAM5–KEAP1–Nrf2 preserves mitochondrial motility by suppressing dominant-negative KEAP1 activity. These data further provide a mechanistic explanation for how age-dependent declines in Nrf2 expression impact mitochondrial motility and induce functional deficits commonly linked to neurodegeneration.
KEY WORDS: Nrf2, Mitochondria, Miro2, Clustering, Proteasome, Ubiquitin
Summary: Mitochondria undergo retrograde trafficking in response to proteasome inhibition. Disruption of the mitochondrial PGAM5-KEAP1-Nrf2 complex leads to degradation of the mitochondrial GTPase Miro2 and inhibition of stress-induced mitochondrial trafficking.
INTRODUCTION
Nuclear factor erythroid 2 p45-related factor 2 (Nrf2) is a basic leucine zipper transcription factor, and its downstream gene products contribute to cellular antioxidant and xenobiotic defenses. During redox homeostasis, Nrf2 levels are suppressed by the KEAP1–cullin-3 E3 ubiquitin ligase (Cul3) and proteasomal degradation (Cullinan et al., 2004; Itoh et al., 2003; Kobayashi et al., 2004; Zhang, 2006; Zhang et al., 2004). Oxidative stress triggers the stabilization and activation of Nrf2 by releasing the transcription factor and the substrate adaptor Kelch-like erythroid cell-derived protein with CNC homology (ECH)-associated protein 1 (KEAP1) from the Cul3 scaffold (Itoh et al., 1997). Stabilized Nrf2 translocates into the nucleus and induces the transcription of its cognate target genes, the protein products of which neutralize the oxidative stress and restore redox homeostasis (Hayes and Dinkova-Kostova, 2014).
A mitochondrial population of Nrf2 has been described that forms a complex containing a KEAP1 dimer and the mitochondrial histidine phosphatase PGAM5 (Lo and Hannink, 2008). The DLG and ETGE motifs located in the N-terminal, Neh2 domain of Nrf2 mediate binding to KEAP1 and, likewise, PGAM5 contains an ESGE motif that binds to KEAP1 (Lo and Hannink, 2008; Tong et al., 2006). Although the existence of this complex has been described in overexpression studies (Lo and Hannink, 2008), the functional role of this complex has not been studied.
Mitochondria regulate a host of cellular functions ranging from ATP production and intracellular calcium buffering to redox homeostasis and apoptosis (Griffiths and Rutter, 2009; Wang and Youle, 2009). Paradoxically, this organelle is the principal generator of intracellular reactive oxygen species (ROS) (Cadenas and Davies, 2000). Proper regulation of mitochondrial function and distribution is, therefore, necessary to optimize ATP production and meet energetic needs while simultaneously minimizing excess ROS production. One mechanism by which cells achieve this equilibrium is by localizing mitochondria to areas of high metabolic demand. Spermatocytes concentrate mitochondria near the base of the flagellum and provide microtubule-based flagellar motors with energy for motility (Santel et al., 1998). Likewise, neurons deliver mitochondria along axons towards the nerve terminal and active zone for ATP-driven neurotransmission (Saxton and Hollenbeck, 2012; Schwarz, 2013; Sheng and Cai, 2012).
Mitochondria primarily traffic on microtubules in mammalian cells (Heggeness et al., 1978). The molecular mechanisms governing this mode of mitochondrial movement are not completely understood, although key proteins have been identified. Bidirectional movement of mitochondria along microtubules is mediated by kinesin and dynein motor proteins (Leopold et al., 1992; Varadi et al., 2004). Combinations of TRAK1 and TRAK2 (also known as Milton in Drosophila melanogaster) and the small mitochondrial Rho GTPases, Miro1 or Miro2, form adapter complexes that link mitochondria to the microtubule motor proteins. Miro1 and Miro2 are integral proteins localized to the mitochondrial outer membrane (MOM), and TRAK1/2 physically link the Miro proteins to kinesin-1 and the dynein–dynactin complex (Brickley et al., 2005; Glater et al., 2006; van Spronsen et al., 2013). Selectivity in the recruitment of TRAK1/2 and motor proteins by the two Miro proteins provides directionality and magnitude to mitochondrial trafficking (van Spronsen et al., 2013). In response to irreparable mitochondrial depolarization, the motility of the damaged organelles becomes restricted via proteasomal destruction of Miro1 by the coordinated actions of the PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin (Ub) ligase parkin (McWilliams and Muqit, 2017).
Miro1 and Miro2 share structural and functional similarities; both contain two internal GTPase domains flanked by two Ca2+-binding, EF-hand domains (MacAskill et al., 2009; Reis et al., 2009; Saotome et al., 2008; Wang et al., 2011b). The functional distinctions between these two proteins are still being uncovered, but overexpression and mutation studies have revealed differential phenotypic outcomes (Fransson et al., 2006). Overexpression of Miro1 in COS-7 cells results in hyperfused mitochondria, dependent on both the GTPase and EF-hand activities. By contrast, Miro2 overexpression induces the formation of juxtanuclear mitochondria. In a separate study, selective knockout of either Miro1 or Miro2 in mice demonstrated that Miro1 is the principal regulator of mitochondrial trafficking in axons and dendrites (López-Domenech et al., 2016). These data imply that the Miro proteins serve functionally distinct roles in regulating the trafficking, dynamics and distribution of mitochondria.
Mitochondrial distribution, motility and dynamics are regulated by, and responsive to, cellular redox status, energy demands, and the cell cycle (Liesa and Shirihai, 2013; Norton et al., 2014; Yamano and Youle, 2011). In these contexts, cytosolic and MOM proteins that mediate mitochondrial dynamics are subject to regulation by enzymes of the ubiquitin proteasome system (UPS). Notable examples include the cytosolic fission factor dynamin-related protein 1 (Drp1, also known as Dnm1) (Karbowski et al., 2007; Wang et al., 2011a), the fusion factors Mfn1 and Mfn2 (Glauser et al., 2011; Leboucher et al., 2012; Tanaka et al., 2010), and Miro1 (Liu et al., 2012; Wang et al., 2011b). Cytosolic proteins are polyubiquitylated and shuttled directly to the proteasome for degradation, while MOM proteins must first be extracted by the AAA-ATPase VCP/p97 (Fang et al., 2015; Hemion et al., 2014; Kimura et al., 2013; Taylor and Rutter, 2011).
There are numerous gaps in our understanding of the integration between the UPS, microtubules and the mitochondrial network. To date, it is still not clear exactly how cells select combinations of Miro and TRAK proteins to achieve directed mitochondrial movement and redistribution. Further, a role(s) for the mitochondrial PGAM5–KEAP1–Nrf2 complex in mitochondrial function, motility and dynamics remains to be established. Here, we have utilized mitochondrial retrograde trafficking induced by acute proteasome inhibition in human cells as a reliable assay of mitochondrial motility, and have discovered that redistribution of the mitochondrial network requires an intact PGAM5–KEAP1–Nrf2 mitochondrial complex. Disrupting this complex by depleting Nrf2 or PGAM5 blocks mitochondrial clustering owing to degradation of the essential mitochondrial trafficking factor Miro2. Mitochondrial clustering deficits in cells depleted of Nrf2 or PGAM5 are fully rescued by co-knockdown of either KEAP1, or its E3 ligase scaffolding partner, Cul3, implicating this pair in the aberrant destruction of Miro2. Collectively, these data identify a distinct function and regulatory mechanism for Miro2 and, moreover, provide a molecular explanation for how the age-associated reduction of Nrf2 contributes to the mitochondrial motility deficits commonly observed in neurodegenerative diseases of the elderly (Bereiter-Hahn, 2014; Esteras et al., 2016; Kubben et al., 2016).
RESULTS
Disruption of the mitochondrial PGAM5-KEAP1-Nrf2 complex mitigates retrograde mitochondrial trafficking
A PGAM5–KEAP1–Nrf2 complex is associated with the mitochondria via PGAM5 (Fig. 1A), a resident mitochondrial histidine phosphatase (Lo and Hannink, 2008; Panda et al., 2016). In an effort to identify a function for this complex and specifically for the mitochondria-associated population of Nrf2, we tested whether the transcription factor modulates redox homeostasis in either the cytosol or the mitochondrial matrix. We generated telomerase (hTERT)-immortalized, human retinal pigment epithelial (RPE-1) cell lines stably expressing cytosolic or mitochondria-localized, redox-sensitive GFP (roGFP and mito-roGFP, respectively). roGFP is an enhanced variant harboring two redox-sensing cysteines in the beta barrel of the GFP. The redox status of these cysteines regulates the excitation profile of the roGFP, allowing for free radical production and cellular redox status to be indirectly assessed by quantifying fluorescence intensity at the reducing and oxidizing excitation wavelengths (Hanson et al., 2004). We confirmed the localization of each sensor (Fig. 1B) and their sensitivities to the oxidant, hydrogen peroxide (Fig. 1C,D). Each cell line was then transfected with either control (siCON) or Nrf2-specific (siNrf2) siRNAs and, 3 days later, exposed to vehicle or the proteasome inhibitor, MG132, for 2 h. The rationale for the MG132 treatment is that proteasome inhibition rapidly stabilizes Nrf2 (Itoh et al., 2003; Zhang and Hannink, 2003; Zhang et al., 2004), and therefore provides the opportunity to assess whether increasing cellular Nrf2 levels impacts redox homeostasis. These studies demonstrated that Nrf2 depletion did not oxidize the cytosol or the mitochondrial matrix (Fig. 1E). Curiously, the matrix became slightly more reducing in siNrf2 cells treated with MG132 (Fig. 1E). These data show that Nrf2 is not required for the constitutive maintenance of redox homeostasis.
Fig. 1.
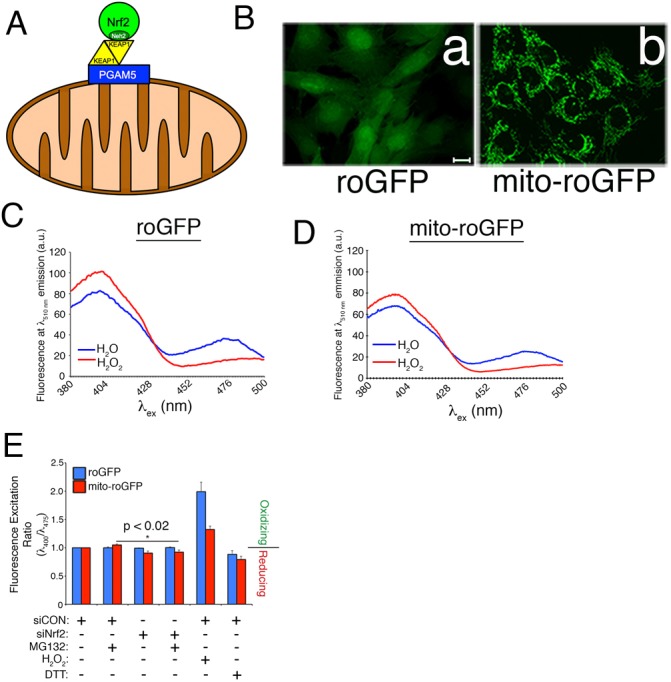
Nrf2 is not required for constitutive redox homeostasis. (A) Schematic of the mitochondrial PGAM5–KEAP1–Nrf2 complex, illustrating that a KEAP1 dimer bridges Nrf2 and PGAM5, and that the Neh2 domain of Nrf2 mediates binding to KEAP1. (B) 63× epifluorescence images confirming the expression and localization of the cytosolic roGFP and mitochondrial mito-roGFP reporter probes. Scale bar: 10 µm. (C) Representative excitation trace at 510 nm emission of RPE-1 cells stably expressing roGFP. Cells were treated with H2O (blue line) or 1 mM H2O2 (red line). (D) Same as in C but for mito-roGFP. (E) roGFP- or mito-roGFP-expressing RPE-1 cells (8000 cells/well) were transfected with siRNA, treated with DMSO or 10 µM MG132 for 2 h, or alternatively with 1 mM H2O2 or 1 mM dithiothreitol (DTT) for 30 min, and then analyzed at excitation wavelengths of 400 nm and 475 nm and emission wavelength of 510 nm. Values are normalized to siCON-transfected cells treated with vehicle (i.e. fold over control). Fluorescence excitation ratios >1.0 are oxidizing and those <1.0 are reducing. Data are mean±s.e.m. from five independent experiments. Statistical significance determined by two-way ANOVA with Tukey's post-hoc correction.
We next examined whether Nrf2 depletion impacts the morphology of the mitochondrial network. Strikingly, Nrf2 knockdown resulted in mitochondria that appear thinner and less intensely labeled with anti-Tom20 (Fig. 2A, panel c and inset c′). This morphological phenotype resembles that observed in cells treated with nocodazole, an agent that depolymerizes the microtubule network and thus releases microtubule cargoes (De Brabander et al., 1977). Nocodazole restricts mitochondrial trafficking due to the dependence of mitochondrial motility on intact microtubules and microtubule-associated molecular motors (Movies 1 and 2). This morphological change of the mitochondria in siNrf2 cells prompted us to interrogate the role of the transcription factor in mitochondrial trafficking. We utilized the retrograde trafficking of mitochondria induced by proteasome inhibition as a readout because this assay requires only an acute treatment of cells (e.g. 2 h) and can be reliably scored based on the redistribution of the mitochondrial network into juxtanuclear clusters (Fig. 2A, panel b; Movies 3 and 4). Remarkably, 2 h of MG132 treatment induced robust mitochondrial clustering in control cells but this redistribution was mitigated by 40–50% in siNrf2 cells (Fig. 2A, panel d, Fig. 2B; Movies 5 and 6). Knockdown efficiency and the stabilization of Nrf2 by MG132 were both verified by anti-Nrf2 western blotting (Fig. 2C). Depletion of Nrf2 similarly reduced the mitochondrial clustering brought about by stressing cells with the electron transport chain uncoupler, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (Fig. S1A–C), indicating a general role for Nrf2 in mitochondrial motility beyond proteasome inhibition. These data also revealed that Nrf2 contributes to mitochondrial retrograde trafficking in the absence of an overt redox stress.
Fig. 2.
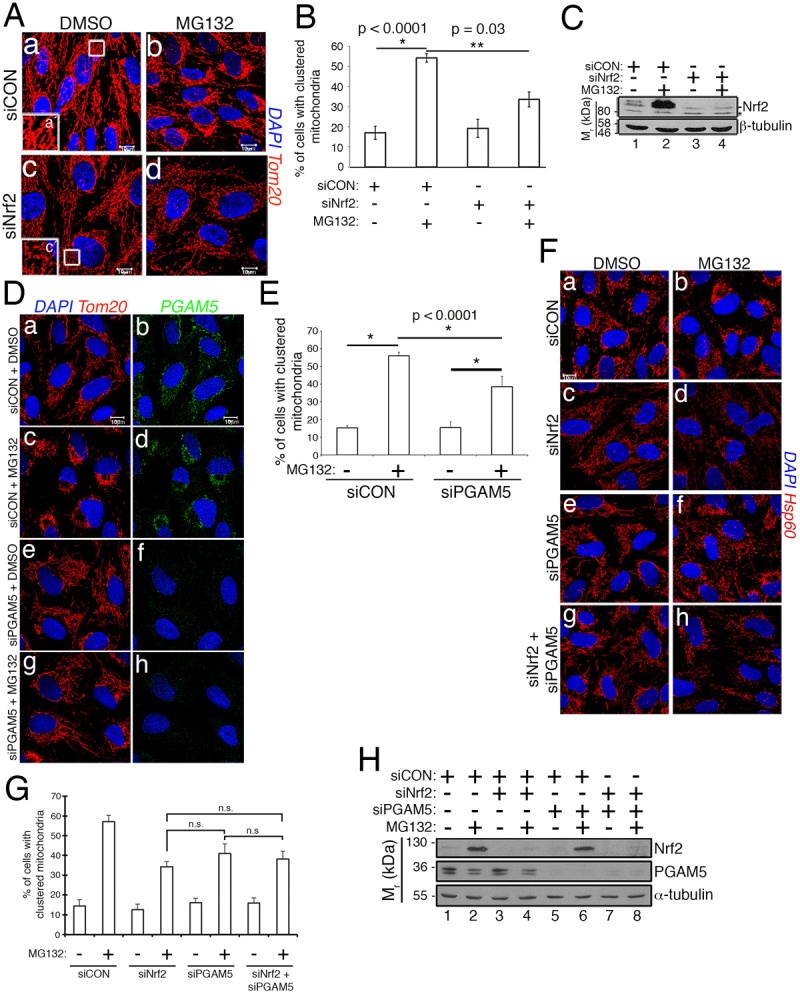
Nrf2 and PGAM5 are required for stress-induced mitochondrial retrograde trafficking. (A) RPE-1 cells transfected with siCON or siNrf2 were treated with DMSO or 10 µM MG132 for 2 h. Mitochondria are labeled with anti-Tom20 (red) and nuclei counterstained with DAPI (blue). Insets show higher magnification views of the boxed areas in panels a and c. (B) Quantification of mitochondrial clustering in siCON versus siNrf2 cells. Data are mean±s.d. from three independent experiments, in which >100 cells per condition were scored for each experiment. (C) Anti-Nrf2 and anti-β-tubulin loading control western blots to confirm the efficacy of Nrf2 knockdown and stabilization of Nrf2 by MG132. The asterisk denotes the nonspecific band, and the migration of molecular weight markers is indicated on the left. (D) RPE-1 cells transfected with control or siPGAM5 were treated with DMSO or 10 µM MG132 for 2 h. Mitochondria are labeled with anti-Tom20 (red) and nuclei with DAPI (blue). PGAM5 silencing demonstrated by loss of anti-PGAM5 immunoreactivity (green; panels f and h). (E) Data are mean±s.d. from four independent experiments, in which >100 cells per condition were scored for mitochondrial clustering per experiment. (F) Photomicrographs of siCON, siNrf2, siPGAM5 and siNrf2/siPGAM5 co-knockdown cells following exposure to DMSO or 10 µM MG132 for 2 h. Mitochondria and nuclei are labeled as in A. (G) Quantification of the mitochondrial clustering in F. Data are mean±s.d. from three independent experiments, in which >100 cells per condition were scored per experiment. (H) Western blots confirming knockdown of Nrf2 and PGAM5 in F. Scale bars: 10 µm. Statistical significance determined by two-way ANOVA with Sidak's or Tukey's post hoc correction.
This redox-independent connection of Nrf2 to mitochondrial trafficking implicated involvement of the mitochondrial population of the transcription factor. This population of Nrf2 is in a complex with a dimer of KEAP1 and is anchored at the MOM by PGAM5 (Fig. 1A). This complex was initially identified using overexpressed human proteins (Lo and Hannink, 2008), and an orthologous complex has been described in C. elegans (Paek et al., 2012). We confirmed the existence of the human complex using overexpressed proteins (Fig. S1D, lane 5). These data also demonstrated that a deletion mutant of Nrf2 lacking the ETGE domain, and therefore with reduced binding to KEAP1, fails to co-precipitate PGAM5 (Fig. S1D, lane 6). This further validates the bridging function of KEAP1 in the PGAM5–KEAP1–Nrf2 complex. To selectively target this mitochondria-associated complex, we depleted PGAM5 with siRNA. Knockdown of PGAM5 phenocopied Nrf2 knockdown by decreasing mitochondrial clustering ∼40% in response to proteasome inhibition (Fig. 2D,E). Co-knockdown of both Nrf2 and PGAM5 yielded a similar decrease in MG132-induced mitochondrial clustering as depleting either protein individually (Fig. 2F–H). These findings are consistent with both proteins acting in a common pathway and with an intact PGAM5–KEAP1–Nrf2 complex being required for mitochondrial retrograde trafficking.
Mitochondrial clustering depends on an intact microtubule network and the Miro2 GTPase
To further investigate the role of the PGAM5–KEAP1–Nrf2 complex in mitochondrial motility, we extensively characterized mitochondrial clustering in response to proteasome inhibition. We observed that clustering was induced within 30 min of treatment with MG132 and was complete by 2 h (Fig. S2A,B). This redistribution was induced using the reversible proteasome inhibitor, MG132, as well as the irreversible inhibitor, epoxomicin (Fig. 3A). Notably, the clustering phenotype was not an artifact of fixation as there was no visible difference in the appearance of the mitochondria before and after fixation (Fig. S2C). Masked scoring revealed a threefold increase in clustering induced by each inhibitor (Fig. 3B), and this redistribution was not caused by reduced cell area (Fig. S2D), although we observed cell shape changes irrespective of treatment (Movies 1–6). Live-cell microscopy of RPE-1 cells stably expressing a mitochondria-targeted GFP (mito-GFP) revealed that proteasome inhibition caused the normally reticular mitochondrial network surrounding the entire nucleus to redistribute into a juxtanuclear cluster on one side of the nucleus (compare Movies 3 and 4).
Fig. 3.

Miro2 is required for mitochondrial retrograde trafficking. (A) Representative photomicrographs of RPE-1 cells treated with DMSO or the indicated proteasome inhibitors (10 µM MG132 or 1 µM epoxomicin) for 2 h. Mitochondria are labeled with anti-Tom20 (red) and nuclei with DAPI (blue). (B) The percentage of cells with clustered mitochondria as a function of treatment. Data are mean±s.d. from three independent experiments utilizing >100 cells per condition per experiment. (C) Confocal, 3D reconstruction of MitoTracker-labeled mitochondria (red) and microtubule stalk (green) exclusively observed in proteasome inhibitor-treated cells. (D) Representative photomicrographs of cells treated with DMSO or proteasome inhibitor (10 µM MG132 or 1 µM epoximicin) ± 4 µg/ml nocodazole. Mitochondria and nuclei are labeled as in A. (E) The % of cells with clustered mitochondria as a function of the treatments described in D. Data are mean±s.d. from three independent experiments, in which >100 cells per condition were scored for each experiment. (F) RPE-1 cells transfected with siCON or siMiro1 were treated with DMSO or 10 µM MG132 for 2 h. Mitochondria are labeled with anti-Tom20 (red) and nuclei with DAPI (blue). (G) Quantification of mitochondrial clustering in siCON versus siMiro1 cells. Data are mean±s.d. from three independent experiments, in which >100 cells per condition were scored for each experiment. (H) Representative western blot demonstrating that siMiro1 siRNA knocks down Miro1, but not Miro2. (I) RPE-1 cells transfected with siCON or siMiro2 were treated and processed as in F. (J) Quantification of mitochondrial clustering in siCON versus siMiro2 cells. Data are mean±s.d. from four independent experiments, in which >100 cells per condition were scored per experiment. (K) Representative western blot demonstrating Miro2 knockdown. Scale bars: 10 µm. Statistical significance determined by one-way (B) or two-way (E,G,J) ANOVA with Sidak's or Tukey's post hoc correction.
As mitochondria in mammalian cells travel along microtubules, we hypothesized that the juxtanuclear clusters were surrounding centrosomes. Co-staining for mitochondria and the centrosomal marker, γ-tubulin, confirmed this notion (Fig. S2E). Furthermore, the ring-like formation of clustered mitochondria indicated that the organelle was wrapped around a structure. Co-staining with the mitochondrial dye MitoTracker and antibodies against the microtubule building block, β-tubulin, followed by reconstruction of confocal Z-stacks revealed that the clustered mitochondria formed a collar around a stalk of microtubules (Fig. 3C). Similar microtubule stalks piercing through mitochondrial ring-like structures were not observed in vehicle-treated cells. As predicted, depolymerization of the microtubule network with nocodazole abolished the redistribution and clustering of the mitochondria and led to an overall reduction in mitochondrial motility (Fig. 3D,E; Movies 1 and 2). Collectively, these data indicate that mitochondrial clustering is an early response to proteasome inhibition and requires an intact microtubule network.
Mitochondria are anchored to microtubules via attachments made by the GTPases Miro1 and Miro2 in the MOM. To interrogate the relative contributions of these proteins to MG132-induced mitochondrial clustering, we knocked down each with siRNA and exposed the cells to MG132. Curiously, Miro1 was completely dispensable for mitochondrial clustering (Fig. 3F–H), while Miro2 was essential (Fig. 3I–K). Knockdown efficiency was validated by western blotting (Fig. 3H,K). These data delineate nonredundant roles for these GTPases in mammalian retrograde trafficking (Tang, 2015).
Mitochondrial fusion and fission are dispensable for clustering
Mitochondria undergo fusion and fission, and these dynamics are closely linked to the status of the network (van der Bliek et al., 2013). We therefore tested whether disrupting the fusion and fission machinery impacts mitochondrial redistribution caused by proteasome inhibition. Because the morphological state of the mitochondrial network is determined by a regulated balance between fusion and fission, blockade of fission causes a fused phenotype, and vice-versa. siRNA knockdown of either the fission factor, Drp1, or the fusion factor, Mfn2, resulted in severely fused or fragmented mitochondria, respectively. However, neither knockdown attenuated mitochondrial clustering (Fig. 4A,B,D,E). The efficiency of the knockdowns was confirmed by western blotting (Fig. 4C,F).
Fig. 4.
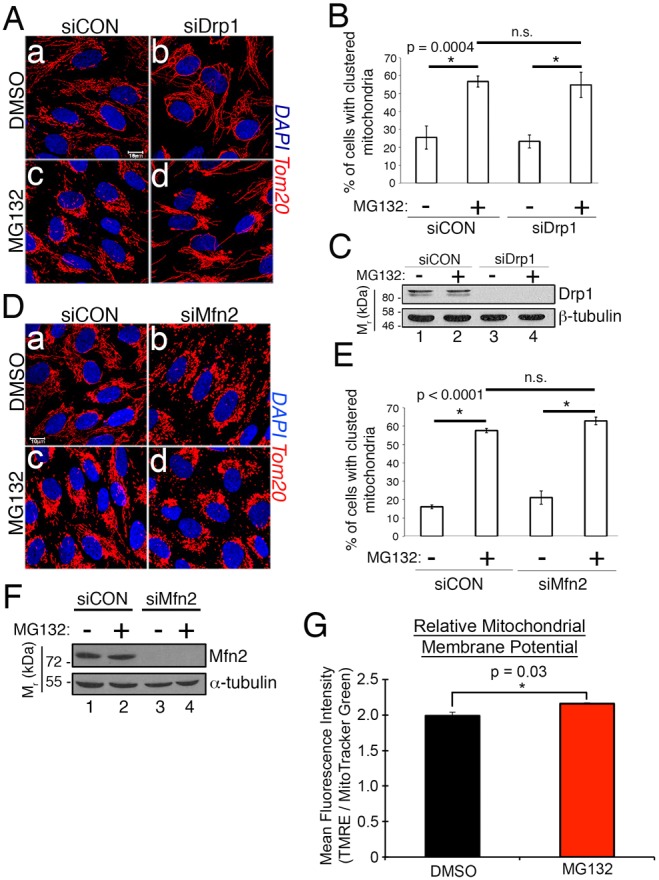
Intact fusion and fission machinery are not required for mitochondrial clustering. (A) Photomicrographs of siCON and Drp1-depleted (siDrp1) cells following 2 h of treatment with DMSO or 10 µM MG132. Mitochondria are labeled with anti-Tom20 (red) and nuclei with DAPI (blue). (B) The % of cells from A with clustered mitochondria as a function of treatment. Data are mean±s.d. from three independent experiments utilizing >100 cells per condition per experiment. (C) Anti-Drp1 western blot showing the efficacy of siDrp1 knockdown. β-tubulin blot shows comparable loading. The migration of molecular weight markers is indicated on the left. (D) Same as in A except knocking down Mfn2 (siMfn2). (E) Same as in B for siMfn2 experiment. (F) Western blot confirming Mfn2 knockdown. (G) Ratio of TMRE to MTG uptake as a function of proteasome inhibitor treatment (10 µM MG132 for 2 h). Data are mean±s.d. from three independent experiments, acquired by FACS. Scale bar: 10 µm. Statistical significance determined by one-way (G) or two-way (B,E) ANOVA with Tukey's or Sidak's post hoc correction.
Mitochondrial membrane potential is integral to mitochondrial dynamics and must be maintained for optimal oxidative phosphorylation. To determine whether acute treatment with MG132 impacts mitochondrial membrane potential, we established a fluorescence activated cell sorting (FACS) assay that simultaneously measures the uptake of tetramethylrhodamine, ethyl ester (TMRE) and MitoTracker Green (MTG). TMRE is selectively taken up and retained by mitochondria with an intact membrane potential, whereas MTG uptake is independent of the mitochondrial membrane potential to allow for normalization of mitochondrial mass. Comparison of the TMRE to MTG ratio in RPE-1 cells treated with DMSO or 10 µM MG132 for 2 h showed that proteasome inhibition induced a very modest increase in membrane potential (Fig. 4G).
Unconstrained mitochondrial KEAP1 suppresses mitochondrial clustering
Additional insights into the contribution of the PGAM5–KEAP1–Nrf2 complex to retrograde mitochondrial trafficking came from experiments targeting depletion of KEAP1. Based on our findings that knocking down either PGAM5 or Nrf2, or co-knockdown of both proteins, mitigated mitochondrial clustering (Fig. 2), we predicted that depleting KEAP1 would yield similar results. Surprisingly, KEAP1 depletion had no impact on mitochondrial clustering (Fig. 5A, panel d, Fig. 5B) but, as expected, stabilized Nrf2 (Fig. 5C, top blot, lane 3 versus lane 1).
Fig. 5.
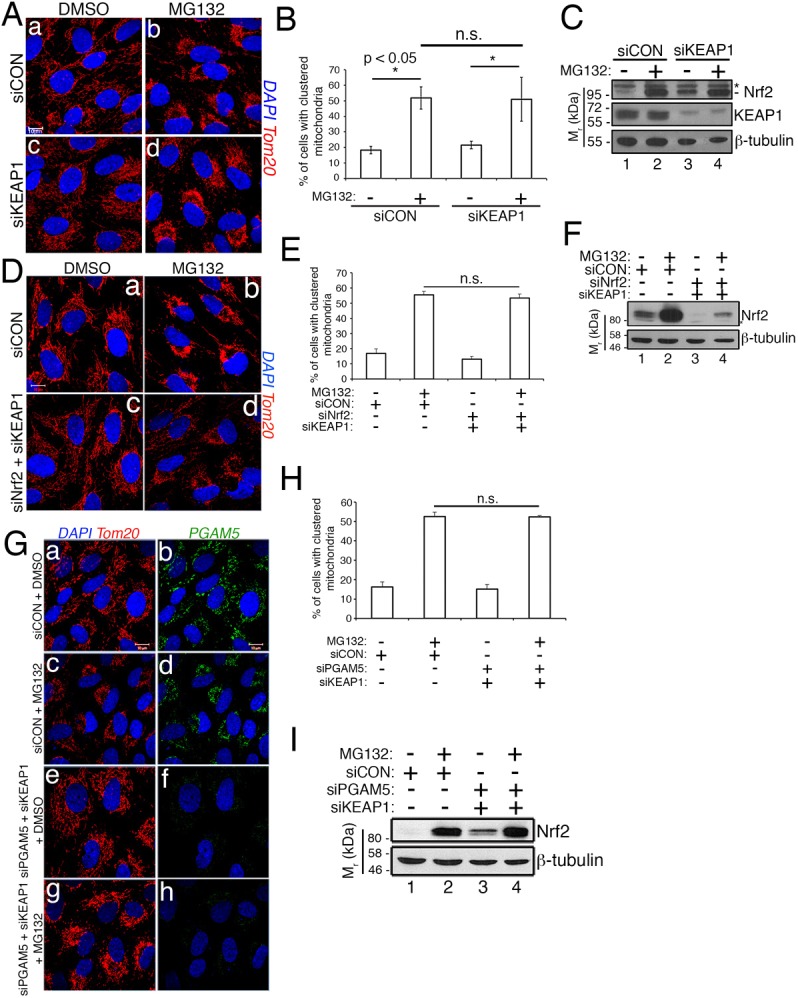
Unconstrained mitochondrial KEAP1 abrogates mitochondrial clustering. (A) Photomicrographs of siCON and siKEAP1 cells treated with DMSO or 10 µM MG132 for 2 h. Mitochondria are visualized with anti-Tom20 (red) and nuclei with DAPI (blue). (B) Data are mean±s.d. from five independent experiments, quantifying mitochondrial clustering in >100 cells per condition per experiment in A. (C) Representative western blot showing KEAP1 knockdown (lanes 3 and 4) and Nrf2 stabilization by siKEAP1 and MG132 (lanes 2 and 4). The asterisk indicates nonspecific band. (D) RPE-1 cells transfected with siCON or siNrf2/siKEAP1 combination were treated with DMSO or 10 µM MG132 for 2 h. Mitochondria are labeled with anti-Tom20 (red) and nuclei with DAPI (blue). (E) Quantification of mitochondrial clustering from D. Data are mean±s.d. from three independent experiments, in which >100 cells per condition were scored per experiment. (F) Anti-Nrf2 western blot demonstrating the efficacy of siNrf2 and siKEAP1 treatments. The anti-β-tubulin blot is shown as a loading control and migration of molecular weight markers is indicated on the left. (G) Photomicrographs of control and siPGAM5/siKEAP1 co-treated RPE-1 cells following exposure to DMSO or 10 µM MG132. Mitochondria and nuclei are labeled as in D. Endogenous PGAM5 (green) was detected with an anti-PGAM5 antibody. (H) Data are mean±s.d. from three independent experiments in G quantifying mitochondrial clustering and utilizing >100 cells per condition per experiment. (I) Anti-Nrf2 western blot demonstrating the impact of siPGAM5/siKEAP1 co-knockdown on Nrf2 levels. Scale bars: 10 µm. Statistical significance determined by two-way ANOVA with Tukey's post hoc correction.
At the mitochondria, PGAM5 and Nrf2 are mutually exclusively bound to monomers of a KEAP1 dimer (Fig. 1A) (Lo and Hannink, 2008). Thus, the requirement of Nrf2 and PGAM5, but not KEAP1, for mitochondrial clustering suggested that in cells depleted of Nrf2 or PGAM5, KEAP1 might be acting in a dominant-negative manner to suppress clustering. A direct prediction of this hypothesis is that co-knockdown of KEAP1 and either Nrf2 or PGAM5 should rescue clustering. This was the case, as co-knockdown of KEAP1 and Nrf2 (Fig. 5D–F), or KEAP1 and PGAM5 (Fig. 5G–I), restored mitochondrial clustering to control levels in response to MG132. These data support a model in which Nrf2 occupancy of mitochondrial KEAP1 restricts this population of KEAP1 from inappropriately suppressing a factor(s) required for mitochondrial clustering.
The nontranscriptional, Neh2 domain of Nrf2 is necessary and sufficient to support mitochondrial clustering
To further test the unconstrained KEAP1 hypothesis, we focused on the amino-terminal, Neh2 domain of Nrf2, which mediates binding to KEAP1 (Eggler et al., 2005; Itoh et al., 1999; McMahon et al., 2004; Tong et al., 2006). If Nrf2 occupancy of KEAP1 is required for mitochondrial clustering and Nrf2 transcriptional activity is dispensable, then the Nrf2 Neh2 domain should be necessary and sufficient to mediate retrograde trafficking. We first utilized mouse embryonic fibroblasts (MEFs) derived from wild-type and strain-matched Nrf2 knockout (Nrf2−/−) mice. We detected faint nuclear labeling of a Nrf2 immunoreactive protein in a subpopulation of Nrf2 knockout MEFs exposed to MG132 (Fig. 6A, panels h and l, white arrowheads). Because the Nrf2 knockout strain was generated by replacing part of exon 4 and all of exon 5 with a β-galactosidase (β-Gal) cassette, the protein expressed from the endogenous Nrf2 knockout allele consists of the first 125 residues of murine Nrf2 fused in-frame to β-Gal (Chan et al., 1996) (Fig. 6B). This fragment encompasses the Neh2 domain of Nrf2, which lacks transcriptional activity (Itoh et al., 1999), but is sufficient to target Nrf2 (or a heterologous protein to which the Neh2 is fused) for degradation via KEAP1–Cul3 and the proteasome (Itoh et al., 1999; McMahon et al., 2003). Chan and colleagues reported the instability of the chimeric Nrf2–LacZ transcript, and did not detect expression of the fusion or any β-Gal activity in Nrf2 knockout mice (Chan et al., 1996). We also did not detect the fusion protein in untreated Nrf2−/− MEFs (Fig. 6A, panel d) but visualized low levels of it by both immunofluorescence (Fig. 6A, panels h and l) and western blotting (Fig. S3A, lane 4) following 4 h or 6 h of MG132 treatment. This longer incubation time with MG312 was required for mitochondrial clustering in the MEFs. Stabilization of this fusion protein by MG132 is consistent with the Neh2 domain directing turnover of Nrf2 by KEAP1–Cul3. Stratification of mitochondrial clustering showed that cells expressing detectable levels of Neh2-β-Gal clustered their mitochondria similarly to control cells, whereas those without detectable levels of the fusion did not (Fig. 6C). Notably, we did not detect Nrf2 immunoreactivity in a small population of MG132-treated wild-type cells for reasons that are unclear; these same cells also failed to cluster their mitochondria (Fig. 6C, diagonally striped bars). Together, these data indicate that mitochondrial clustering in response to proteasome inhibition requires the amino-terminal 125 residues of mouse Nrf2.
Fig. 6.
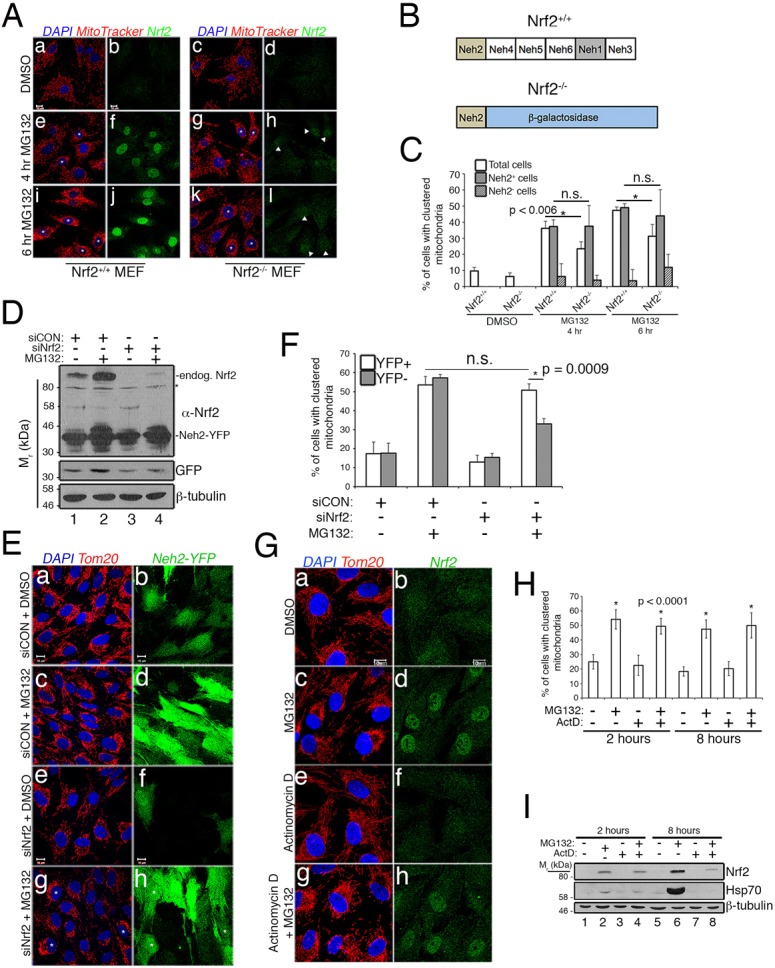
The nontranscriptional, Neh2 domain of Nrf2 restores mitochondrial clustering. (A) MEFs from wild-type and strain-matched Nrf2−/− mice treated with DMSO or 5 μM MG132 for 4 h or 6 h. Mitochondria are labeled with MitoTracker (red), Nrf2 with an anti-Nrf2 antibody (green) and nuclei with DAPI (blue). Arrowheads indicate Nrf2−/− MEFs showing anti-Nrf2 immunoreactivity; asterisks indicate examples of cells scored as having clustered mitochondria. (B) Diagrams of wild-type protein (Nrf2+/+) and the Neh2-β-galactosidase fusion produced in Nrf2−/− mice. (C) Data are mean±s.d. from four independent experiments showing mitochondrial clustering in MEFs, with >50 cells analyzed per condition per experiment. Data are stratified for all cells (white bars), cells with Nrf2-immunoreactivity (gray bars) and cells without Nrf2-immunoreactivity (diagonally striped bars). (D) Anti-Nrf2, anti-GFP and anti-β-tubulin western blots of RPE-1 cells stably expressing siRNA-resistant Neh2-YFP. Cells were transfected with siCON or siNrf2 siRNA and treated with DMSO or 10 µM MG132 for 2 h. Endogenous (endog.) Nrf2 and Neh2-YFP are marked. The asterisk indicates nonspecific band detected by anti-Nrf2 antibody, and the migration of molecular weight markers is indicated on the left. (E) Neh2-YFP-expressing RPE-1 cells treated as indicated; mitochondria are labeled with anti-Tom20 (red) and nuclei with DAPI (blue). Examples of Neh2-YFP-expressing cells scored as having clustered mitochondria are indicated with asterisks. (F) Mitochondrial clustering quantified from E and stratified based on YFP-positive cells (white bars) and YFP-negative cells (gray bars). Data are mean±s.d. from three independent experiments, in which >100 cells per condition were analyzed for each experiment. (G) Photomicrographs of RPE-1 cells treated with vehicle, 10 µM MG132, 1 µM Actinomycin D or MG132+Actinomycin D. Mitochondria are labeled with anti-Tom20 (red), endogenous Nrf2 with anti-Nrf2 (green) and nuclei with DAPI (blue). (H) Quantification of % of cells with clustered mitochondria in G. Data are mean±s.d. from three independent experiments utilizing >100 cells per condition per experiment. (I) Anti-Nrf2, anti-Hsp70 and anti-β-tubulin western blots confirming the efficacy of Actinomycin D (ActD) in blocking Hsp70 induction by MG132. Scale bars: 10 µm. Statistical significance determined by three-way ANOVA with Tukey's or Sidak's post hoc correction (C,H) or two-way ANOVA with Tukey's post hoc correction (F).
We independently validated the necessity of the Neh2 domain by generating a stable RPE-1 cell line expressing the human Neh2 domain (amino acids 1–86) fused to yellow fluorescent protein (Neh2-YFP). The construct was designed such that the resulting mRNA encoding Neh2–YFP is siRNA-resistant. We then knocked down endogenous Nrf2 in these cells (Fig. 6D, lanes 3 and 4) to determine if the Neh2 domain could rescue MG132-induced mitochondrial clustering. Stratification of the results based on the presence or absence of YFP epifluorescence showed that cells depleted of full-length, endogenous Nrf2, but expressing detectable levels of Neh2–YFP, were typically rescued for clustering (Fig. 6E, panels g and h, and Fig. 6F).
Because the Neh2 domain of Nrf2 lacks the Cap ‘n’ Collar bZIP regions and thus any DNA binding or transcriptional activity (Itoh et al., 1999), our data indicate a nontranscriptional role for Nrf2 in preserving mitochondrial trafficking. Consistent with this, de novo transcription was not required for mitochondrial clustering following 2 h or 8 h of proteasome inhibition in RPE-1 cells (Fig. 6G,H). Transcriptional blockade with Actinomycin D was confirmed by showing that, in contrast to control cells (Fig. 6I, lane 6), Hsp70 induction was ablated in cells co-treated with Actinomycin D and MG132 (Fig. 6I, lane 8). These studies confirm a nontranscriptional, KEAP1 occupancy role for Nrf2 in regulating mitochondrial retrograde trafficking.
Aberrant KEAP1 activity at mitochondria is mediated by the availability of Nrf2, PGAM5 and p62/SQSTM1
Our model predicts that, in the absence of Nrf2 and/or PGAM5, KEAP1 mediates the degradation of an essential mitochondrial trafficking factor. To identify this factor, we initially focused on p62/sequestosome1 (hereafter referred to as p62), a central mediator of cargo recruitment into autophagosomes (Kirkin et al., 2009), which competes with Nrf2 for binding to KEAP1 (Bjørkøy et al., 2006; Komatsu et al., 2010) and has been linked to clustering in response to depolarization of the mitochondria (Narendra et al., 2010). Knockdown of p62, however, did not mitigate mitochondrial clustering in response to proteasome inhibition (Fig. 7A, panel f, Fig. 7B). Moreover, Nrf2 depletion did not reduce p62 levels (Fig. 7C, lanes 3 and 4), as would be expected if KEAP1 targeted p62 for degradation in the absence of Nrf2. These experiments did, however, reveal that co-knockdown of Nrf2 and p62 resulted in a complete attenuation of mitochondrial clustering induced by MG132, as compared to the ∼40–50% reduction observed with siNrf2 alone (Fig. 7A,B). In fact, cells co-depleted of Nrf2 and p62 and treated with MG132 had the same level of clustered mitochondria as untreated cells (∼10%). This finding suggests that, in the absence of Nrf2, mitochondrial KEAP1 becomes occupied with p62 (Komatsu et al., 2010), and this partially protects an essential mitochondrial clustering factor from being targeted by unconstrained KEAP1 activity. Removal of this protection by depleting Nrf2 and p62 results in fully unrestricted KEAP1 and a complete suppression of mitochondrial clustering. Additional support for this model came from the finding that knockdown of KEAP1 in cells co-depleted of Nrf2 and p62 rescued clustering to near wild-type levels (Fig. 7D,E). The efficiency of the combinatorial knockdown was confirmed by western blotting (Fig. 7F).
Fig. 7.
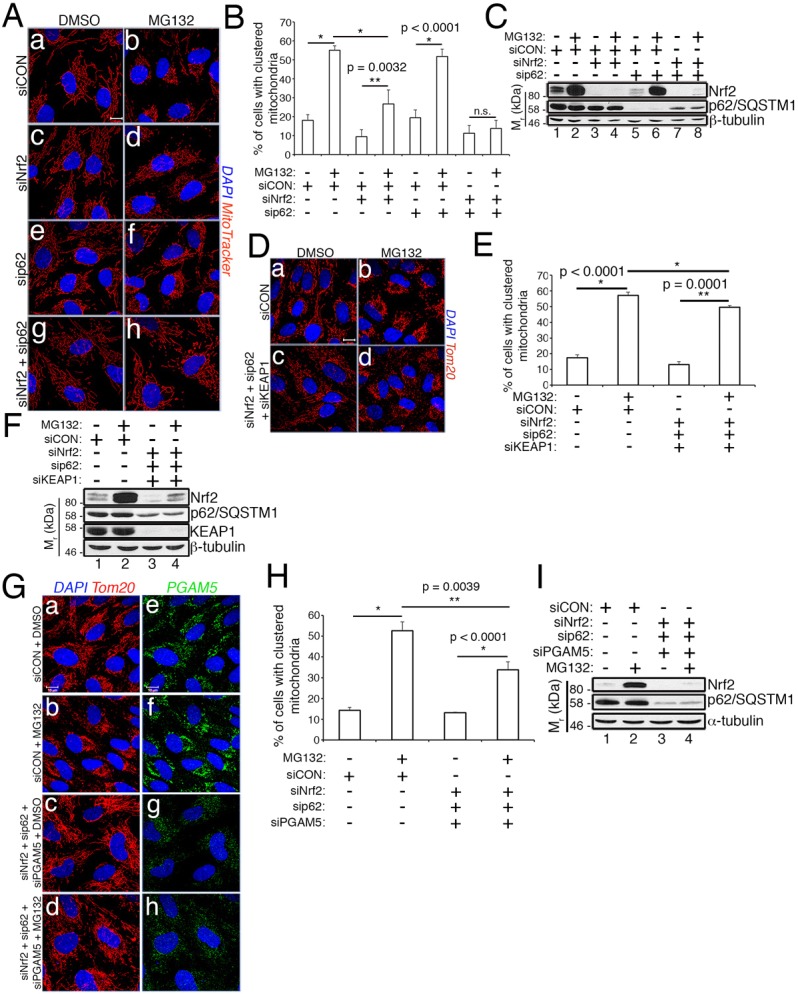
Nrf2 and p62 cooperatively suppress aberrant KEAP1 activity. (A) RPE-1 cells transfected with siCON, siNrf2, sip62 or siNrf2/sip62 combination were treated with DMSO or 10 µM MG132 for 2 h. Mitochondria are labeled with MitoTracker (red), and nuclei with DAPI (blue). (B) Quantification of clustering in A. Data are mean±s.d. from three independent experiments utilizing >100 cells per condition per experiment. (C) Anti-Nrf2, anti-p62 and anti-β-tubulin western blots to demonstrate the efficacy of knockdowns in A and B. (D) RPE-1 cells transfected with siCON or siNrf2/sip62/siKEAP1 combination were treated and processed as in A. (E) Quantification of clustering from D, performed as described in B. (F) Anti-Nrf2, anti-p62, anti-KEAP1 and anti-β-tubulin western blots to demonstrate the efficacy of knockdowns in D and E. (G) RPE-1 cells transfected with siCON or siNrf2/sip62/siPGAM5 combination were treated with DMSO or 10 µM MG132 for 2 h. Mitochondria labeled with anti-Tom20 (red), endogenous PGAM5 with anti-PGAM5 (green) and nuclei with DAPI (blue). (H) Quantification of clustering in G. Data are mean±s.d. from three independent experiments utilizing >100 cells per condition per experiment. (I) Representative western blot from G demonstrating knockdown of Nrf2 and p62. Scale bars: 10 µm. Statistical significance was determined by two-way ANOVA with Tukey's post hoc correction.
Further evidence that the mitochondrial population of KEAP1 aberrantly mediates the degradation of a clustering factor came from the observation that knockdown of PGAM5 (the mitochondrial anchor for the PGAM5–KEAP1–Nrf2 complex) partially rescued mitochondrial clustering in cells co-depleted of Nrf2 and p62 (Fig. 7G–I). We interpret this to indicate that in the absence of its primary binding partners, Nrf2 and p62, mitochondrial KEAP1 functions in a dominant-negative fashion to mediate the degradation of a factor(s) required for mitochondrial trafficking, but disrupting the mitochondrial localization of KEAP1 by depleting PGAM5 partially overcomes this aberrant activity by releasing KEAP1 from the mitochondria.
Unconstrained mitochondrial KEAP1 mediates the aberrant turnover of Miro2
Miro1 and Miro2 link mitochondria to the motor proteins of the microtubule network and have been implicated in stress-induced mitochondrial clustering (Tang, 2015), so we focused on them as substrates for KEAP1. We favored Miro2 because its overexpression induces mitochondrial clustering (Fransson et al., 2006) and, more importantly, knockdown of Miro2, but not Miro1, inhibits MG132-induced clustering (Fig. 3). Strikingly, Miro2 protein levels were dramatically reduced in cells lacking Nrf2 and p62 (Fig. 8A, lanes 3–6), and restored by co-knockdown of KEAP1 (Fig. 8A, lanes 7 and 8). Miro levels were likewise suppressed in Nrf2−/− MEFs (Fig. S3A) and in brain lysates from Nrf2−/− mice (Fig. 8B).
Fig. 8.
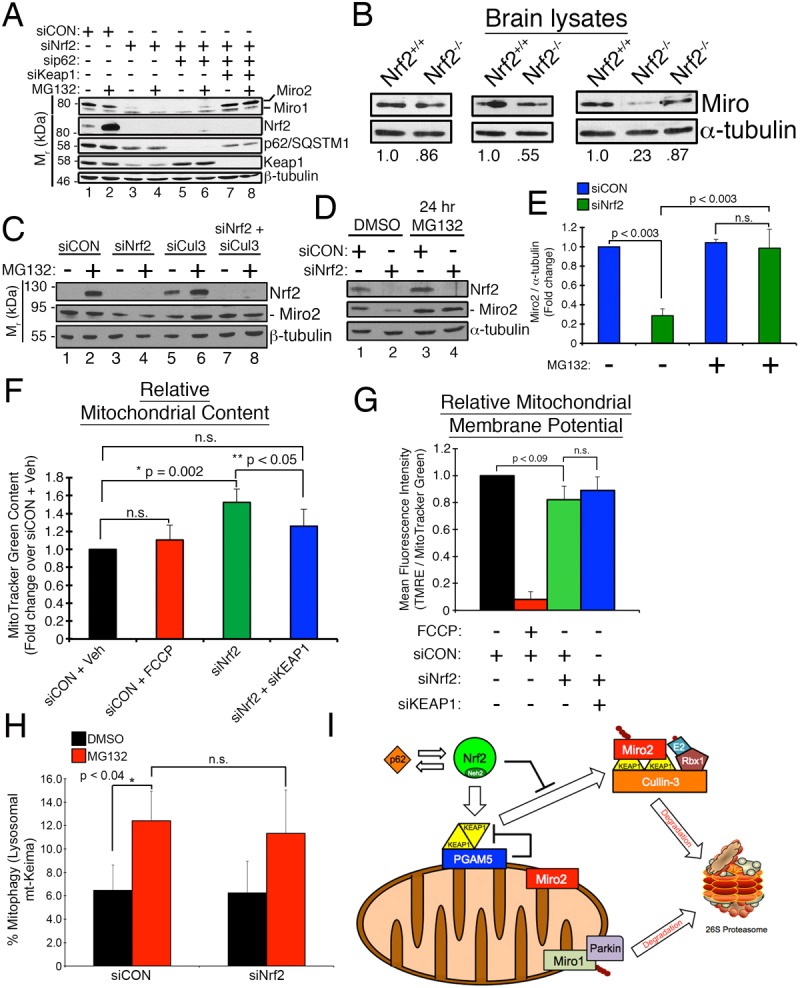
Unconstrained KEAP1 promotes loss of Miro2. (A) RPE-1 cells were transfected with the indicated siRNAs and treated with DMSO or 10 µM MG132 for 2 h. Western blots show knockdown of targeted proteins and levels of Miro1 and Miro2. (B) Anti-Miro western blots of lysates from Nrf2 knockout (Nrf2−/−) and age-matched, wild-type (Nrf2+/+) brains. Numbers indicate the fraction of Miro2 present compared to wild-type control for each age tested (wild type set at 1.0). Left and center panels from 11- to 12-month-old mice and right panel from 22- to 23-month-old mice. (C) Cells were transfected with siRNA and treated as in A. (D) RPE-1 cells were transfected with the indicated siRNAs for 12 h followed by exposure to DMSO or 1 µM MG132 for 24 h prior to western blotting with the indicated antibodies. (E) Densitometric quantification from D. Data are mean±s.e.m. from four independent experiments. (F) Graph of mean fluorescence intensity of MitoTracker Green (MTG) uptake in cells transfected with the indicated siRNAs and treated with ethanol or 5 µM FCCP prior to FACS analysis. MTG uptake is a proxy for mitochondrial content in cells and the acute FCCP treatment was used to dissipate the membrane potential to demonstrate that MTG uptake is independent of mitochondrial membrane potential. Data are mean±s.e.m. from five independent experiments. (G) Mean fluorescence intensity calculated by dividing the TMRE signal by the MTG signal with the siCON control set at a value of 1.0. Cells were treated as in F. Data are mean±s.e.m. from five independent FACS experiments utilizing >20,000 cell counts per condition per experiment. (H) Graph of FACS data pooled from three independent experiments demonstrating that Nrf2 depletion does not alter basal mitophagic flux. RPE-1 cells stably expressing mt-mKeima and YFP-parkin were treated with the indicated siRNAs and exposed to DMSO (black bars) or 10 µM MG132 (red bars) for 4 h. Excitation profiles of mt-mKeima at 440 nm (neutral pH) and at 586 nm (acidic pH) in siCON- and siNrf2-depleted RPE-1 cells were quantified by FACS analysis and used to define mitochondria as being mitochondrial (neutral) or lysosomal (acidic). (I) Schematic showing the roles of Nrf2 and p62 in suppressing aberrant degradation of Miro2 by the KEAP1-Cul3 E3 Ub ligase. Parkin mediates the degradation of Miro1 to halt mitochondrial motility following irreparable loss of mitochondrial membrane potential (Birsa et al., 2014; Kazlauskaite et al., 2014; Klosowiak et al., 2016; Wang et al., 2011b). Statistical significance was determined by two-way ANOVA with Tukey's post hoc correction (E,H) and by one-way ANOVA (F,G).
A further prediction of our model was that knockdown of a different component of the E3 Ub ligase complex that KEAP1 is a part of should likewise rescue Miro2 levels. Indeed, co-knockdown of the Cul3 scaffold in siNrf2 cells rescued Miro2 levels (Fig. 8C, lanes 3 and 4 versus lanes 7 and 8). Moreover, Miro2 loss in Nrf2-depleted cells was completely reversed following 24 h of 1 µM MG132 treatment (Fig. 8D, lane 2 versus 4, and Fig. 8E), but not with the lysosomal hydrolase inhibitor chloroquine (data not shown). Importantly, this rescue of Miro2 by extended treatment with a low amount of MG132 was not simply a result of stabilizing residual transcription factor as indicated by the lack of Nrf2 build up (Fig. 8D, top blot, lane 4).
These data led us to test whether the aberrant KEAP1–Cul3 activity generated in siNrf2 cells was also mediating the degradation of other mitochondrial proteins and, in doing so, promoting mitophagy, analogous to the manner in which activated parkin decorates irreparably depolarized mitochondria with polyubiquitin chains to initiate mitophagy (Narendra et al., 2008; Ordureau et al., 2014; Sarraf et al., 2013). We analyzed the expression levels of a panel of mitochondrial proteins derived from siNrf2 cell lysates and found that depletion of Nrf2 did not suppress wholesale mitochondrial protein expression, as would be predicted if Nrf2 loss promoted mitophagy. In fact, the levels of most proteins analyzed remained unchanged (Fig. S3B,C). These data are consistent with aberrant KEAP1–Cul3 ligase activity not promoting wholesale turnover of resident mitochondrial proteins but rather selectively mediating the degradation of some MOM proteins, most notably Miro2 and Tom20 (Fig. S3B,C).
This interpretation was corroborated independently by a FACS assay showing that loss of Nrf2 caused a 1.5-fold increase in mitochondrial mass, as measured by MTG uptake (Fig. 8F; Fig. S3D). Albeit, these organelles are slightly depolarized, as indicated by a small reduction in the ratio of TMRE to MTG uptake (Fig. 8G, black versus green bar; Fig. S3D). Interestingly, co-knockdown of KEAP1 attenuated the increase in MTG uptake without affecting membrane potential (Fig. 8F,G, green versus blue bar). Notably, the membrane potential sensitivity of TMRE and membrane potential independence of MTG were validated in a separate experiment, showing that FCCP, a compound that dissipates the mitochondrial membrane potential, restricts TMRE, but not MTG, uptake (Fig. 8F,G; Fig. S3E). Additional direct evidence that Nrf2 depletion does not induce mitophagy came from RPE-1 cells stably expressing mt-mKeima and YFP–parkin. Mt-mKeima is a coral-derived, acid-stable, red fluorescent protein with a mitochondria-targeting sequence that exhibits pH-dependent excitation properties and is resistant to lysosomal proteases (Katayama et al., 2011). It excites at 440 nm in neutral pH and at 586 nm in an acidic environment. These two populations of mitochondria can be quantified by FACS, with the 440 nm population considered to be cytosolic/mitochondrial and the 586 nm population to be lysosomal (Katayama et al., 2011; Sun et al., 2015). Co-expression of YFP-parkin was necessary because of the minimal mitophagic flux observed in cells not overexpressing parkin (G.B.O., K.S.P., W.L.B. et al., unpublished data; Katayama et al., 2011). The sensitivity of this assay was established by demonstrating the dramatic increase in lysosomal mt-mKeima signal induced by treating cells with a combination of FCCP and Oligomycin A (Fig. S3F). In both vehicle- and MG132-treated cells, depletion of Nrf2 did not increase the lysosomal population of mt-mKeima relative to siCON control cells, confirming that loss of the transcription factor does not promote constitutive mitophagy (Fig. 8H). Collectively, these findings support a model in which loss of Nrf2 from the mitochondrial PGAM5–KEAP1–Nrf2 complex results in aberrant KEAP1–Cul3 ligase activity, proteasomal destruction of Miro2, and subsequent loss of mitochondrial motility in response to proteasome inhibition (Fig. 8I).
DISCUSSION
Mitochondria in mammalian cells primarily traffic along microtubules using kinesin and dynein motor proteins (Leopold et al., 1992; Varadi et al., 2004). This microtubule-based movement requires adaptor complexes consisting of TRAK1/2 and Miro1/2 that link the mitochondria to the microtubules (Schwarz, 2013). Net mitochondrial retrograde movement towards the centrosome has been observed in response to hypoxia (Al-Mehdi et al., 2012), oxidative stress (Hallmann et al., 2004), exposure to tumor necrosis factor (De Vos et al., 1998), Hepatitis B virus X protein (Kim et al., 2007), mitophagy (Narendra et al., 2010) and extended proteasome inhibition (Bauer and Richter-Landsberg, 2006; Zaarur et al., 2014). The functional significance of these stress-induced trafficking events is not completely understood, but a study of hypoxia provided evidence that the perinuclear accumulation of mitochondria can lead to the deposition of ROS into the nucleus. These ROS modify and activate the vascular endothelial growth factor promoter to ultimately relieve the hypoxia (Al-Mehdi et al., 2012). Additionally, mitochondria-derived ROS deposited into the nucleus induce DNA damage to drive a mitotic exit in postnatal cardiomyocytes (Puente et al., 2014). Herein, we have characterized the rapid mitochondrial clustering induced by acute proteasome inhibition. Characterization of this stress response revealed that mitochondrial clustering precedes many other early responses to proteotoxic stress. Clustering was readily observed 30 min after adding proteasome inhibitor and completed within 2 h, whereas other responses to proteasome blockade, including aggresome formation, autophagy induction, chaperone expression and E-zone formation, were only modestly, if at all, detectable at 2 h (data not shown). Further, mitochondrial clustering is dependent on the mitochondrial GTPase, Miro 2, but not on its paralogue, Miro 1 (Fig. 3), or on the fusion-fission status of the mitochondrial network (Fig. 4).
The major discovery of this manuscript is that the retrograde, microtubule-dependent transport of mitochondria requires an intact mitochondrial complex containing the transcription factor Nrf2, the substrate adaptor KEAP1, and the mitochondrial phosphatase, PGAM5. Disruption of this complex results in neomorphic KEAP1–Cul3 activity and the subsequent degradation of Miro2, an essential factor linking mitochondria to microtubule motor proteins (Fig. 8A). A number of approaches were taken to identify the mechanism by which unconstrained KEAP1 leads to Miro2 degradation. Collectively, the data implicate the KEAP1-Cul3 ligase in the turnover of Miro2 as co-knockdown of either Cul3 or KEAP1 in siNrf2 cells completely restored Miro2 to control levels (Fig. 8A,C). This interpretation is further supported by data showing that Miro2 levels are restored in Nrf2-depleted cells treated with proteasome inhibitor for 24 h (Fig. 8D,E), whereas inhibiting autophagy failed to rescue Miro2 expression (data not shown). Experiments to co-precipitate endogenous KEAP1 and Miro2 were unsuccessful; this may be attributable to either dissociation of the complex under conditions necessary to extract Miro2 from the MOM and/or the involvement of a second E3 Ub ligase, with parkin being a primary candidate (Bingol et al., 2014; Klosowiak et al., 2016; Ordureau et al., 2015, 2014; Sarraf et al., 2013). However, a different E3 ligase may well be involved as parkin-mediated Miro1 degradation is triggered by robust mitochondrial membrane depolarization (e.g. Birsa et al., 2014; Kazlauskaite et al., 2014; Klosowiak et al., 2016; Wang et al., 2011b), and only very modest changes in membrane potential were detectable in our experiments (Fig. 8G; Fig. S3D).
An additional advance from this study is the discovery that Miro2, but not Miro1, is required for mitochondrial retrograde trafficking in response to proteasome inhibition. Previous work indicated that although Miro1 and Miro2 share >60% homology and the same functional domains (two GTPase domains, two calcium-binding EF hands, and a transmembrane domain at the C-terminus) (Reis et al., 2009), the two GTPases serve nonoverlapping functions (Fransson et al., 2006; Nguyen et al., 2014). This notion derives from studies showing that overexpression of Miro1 in COS-7 cells caused both the aggregation of mitochondria and mitochondrial hyperfusion, whereas Miro2 overexpression resulted in only perinuclear clustering of the mitochondria (Fransson et al., 2006). In a separate study, genetic ablation of Miro1 in mice caused early postnatal death, indicating that Miro2 cannot functionally compensate for Miro1 (Nguyen et al., 2014). Work in Arabidopsis thaliana also demonstrated an unequal redundancy of the Miro proteins during female gametogenesis (Sormo et al., 2011). Our work delineates a clear functional distinction by demonstrating that Miro1 is dispensable for stress-induced mitochondrial retrograde trafficking, whereas Miro2 is essential for this motility (Fig. 3). When coupled with the discovery that disruption of the Nrf2–PGAM5–KEAP1 complex destabilizes Miro2, this distinction indicates that the nonredundancy of these two proteins could render cells vulnerable to changes in Miro2 protein expression or to the stability of the Nrf2–PGAM5–KEAP1 complex that arise by, for example, through pathological mechanisms or aging (Wang et al., 2014).
The model presented in this manuscript (Fig. 8I) describes a novel function for a complex containing the mitochondrial phosphatase PGAM5. A recent study described the development of a syndrome similar to Parkinson's disease in mice genetically ablated for PGAM5 and this phenotype was attributed, at least in part, to the loss of the PGAM5-dependent stabilization of PINK1 following mitochondrial depolarization (Lu et al., 2014). The authors highlight that this phenotype was unexpectedly more severe than that reported for PINK1 knockout animals (Gispert et al., 2009; Kitada et al., 2007), and speculated that this difference stems from microbiome differences, environmental enrichment, and/or genetic strain variability. Our findings imply that the increased severity of symptoms induced by PGAM5 loss may be attributable to disrupted mitochondrial motility (Fig. 2D–G), an essential feature of neuronal function and survival (Schwarz, 2013).
The finding that unconstrained KEAP1 leads to loss of Miro2 could have pathophysiological implications as multiple KEAP1 substrates and binding partners are decreased in specific disease states (Du et al., 2009a,b; Goven et al., 2008; Pauty et al., 2014; Sarlette et al., 2008). Therefore, not only do cells have deficits in the functions that these proteins carry out, but loss of these proteins could promote dominant-negative KEAP1 activity and a resulting disruption of mitochondrial trafficking. For example, the generation of neomorphic KEAP1 may underlie the perturbations in mitochondrial morphology and homeostasis that commonly accompany aging diseases, such as age-related macular degeneration, in which the primary KEAP1 substrate, Nrf2, declines (Feher et al., 2006; Wang et al., 2014). Consistent with this, lysates derived from Nrf2 knockout mouse brains consistently show decreased Miro expression compared to age-matched controls (Fig. 8B). In addition, the identification of a novel role for Nrf2 at the mitochondria could help clarify paradoxical observations made using the proteasome inhibitor Bortezomib. High basal levels of Nrf2 were associated with mantle cell lymphoma resistance to Bortezomib, whereas robust activation of the transcription factor and its cognate target genes was observed only in the tumor cells of patients that responded well to the proteasome inhibitor (Weniger et al., 2011). These findings imply that a stringent titration of Nrf2 levels, activity and intracellular distribution (Kubben et al., 2016; Plafker and Plafker, 2015), governs the cellular response to proteasome disruption. By identifying a novel molecular link between proteasome dysfunction and mitochondrial motility deficits, our data offer fresh insights into the intended (and possibly unintended) clinical consequences of proteasome inhibitors (Manasanch and Orlowski, 2017) and into the pathological impact(s) that proteasomal deficits contribute to age-related neurodegeneration (Zheng et al., 2016).
MATERIALS AND METHODS
Cell culture and transfections
Human retinal pigment epithelial cells transformed with telomerase (RPE-1) (CRL-4000, ATCC) were cultured in Dulbecco's modified Eagle medium (DMEM) containing 1 g/l glucose supplemented with penicillin (100 U/ml), streptomycin (100 U/ml), 1X nonessential amino acid cocktail (Life Technologies) and 10% fetal bovine serum (FBS, Life Technologies). MEFs and human embryonic kidney (HEK) 293T cells were cultured in DMEM containing 4.5 g/l glucose supplemented with penicillin (100 U/ml), streptomycin (100 µg/ml), and 10% FBS. For siRNA-transfections, 20,000–35,000 RPE-1 cells/ml were seeded in 12-well dishes overnight. Cells received 10 nM siRNA diluted in serum-free DMEM and combined with 0.3% Interferin transfection reagent (PolyPlus). For the PGAM5 and Miro2 knockdowns, 20 nM siRNA was required. Control siCON siRNA was added to samples such that all cells within a given experiment received equal amounts of siRNA. Cells were harvested 2–3 days post-transfection. All cell lines were regularly checked for contamination.
Chemicals, antibodies and siRNA oligomers
See Table S1 for a full list of antibodies, chemicals and siRNA oligomers, sources and dilutions/concentrations. Miro1 and Miro2 were simultaneously detected with an antibody that recognizes both enzymes.
Immunofluorescence
Cells seeded on 18 mm glass coverslips were treated with vehicle or inhibitors, fixed in 3.7% formaldehyde and then permeabilized in 0.2% Triton X-100/PBS on ice for 10 min. To visualize β-tubulin or γ-tubulin, cells were fixed with ice-cold methanol for 2 min. Primary antibodies were incubated in 3% bovine serum albumin (BSA) in PBS overnight at 4°C. Following PBS washes, cells were incubated for 1 h in species-appropriate Alexa Fluor 488-, Alexa Fluor 546- or Alexa Fluor 647-conjugated secondary antibodies (diluted 1:1000) and 0.1 µg/ml DAPI (Sigma-Aldrich) or 2 µg/ml Hoechst 33342 (Molecular Probes) in 3% BSA/PBS. Mitochondria were visualized either by anti-Tom20 immunofluorescence or by incubating cells in 200 nM MitoTracker Red CMXRos (Molecular Probes) in serum-free DMEM for 30 min at 37°C prior to fixation.
Flow cytometry-based mitophagy assay
RPE-1 cells stably expressing both mt-mKeima and YFP–parkin were transfected with siRNAs as described above. Three days post-siRNA transfection, cells were incubated with vehicle or 10 µM MG132. After 4 h at 37°C, cells were trypsinized, resuspended in 10% fetal calf serum and 0.5 mM EDTA in PBS, passed through 100 µm mesh, and subjected to flow cytometry on a Becton Dickinson LSR II. YFP-Parkin was detected with 488 nm excitation and a 530/30 nm emission filter. Mt-mKeima was detected with a 488 nm excitation laser and a 695/40 nm emission filter for the neutral or ‘mitochondrial’ localization, and a 561 nm laser with 670/30 nm filter for the acidic or ‘lysosomal’ localization. The resulting data were plotted in FlowJo as acidic against neutral, and two gates defined the cells exhibiting predominately lysosomal or mitochondrial mt-mKeima localization.
Flow cytometry measurements of mitochondrial membrane potential
To test the impact of MG132 on mitochondrial membrane potential, RPE-1 cells were incubated with 5 nM TMRE and 200 nM MitoTracker Green FM (MTG), for 30 min before excess dye was washed away. DMSO or 10 µM MG132 was then added for 2 h at 37°C prior to processing for FACS analysis. To determine whether depletion of Nrf2 impacted mitochondrial membrane potential, RPE-1 cells were transfected with siRNAs and 3 days later processed for TMRE and MTG uptake. As a positive control to establish the sensitivity of the FACS assay for monitoring mitochondrial membrane potential, 5 µM FCCP was added to dissipate the mitochondrial electrochemical gradient. For all FACS studies analyzing membrane potential, cells were trypsinized, resuspended in Phenol Red-free DMEM, filtered through 100 µm mesh and subjected to flow cytometry on a Becton Dickinson LSR II. TMRE was visualized by excitation with a 561 nm laser and a 582/12 nm emission filter, and MTG was visualized by excitation with a 488 nm laser and a 530/30 nm emission filter. Data were analyzed using FlowJo software. The mean TMRE and MTG fluorescence intensities were graphed as a ratio to show membrane potential (TMRE) per mitochondrial content (MTG).
Fluorimetry
RPE-1 cells stably expressing either roGFP or mito-roGFP were grown to confluence and treated with 1 mM hydrogen peroxide (H2O2) for 30 min or 10 µM MG132 for 2 h. After treatment, cells were washed with PBS, trypsinized, and resuspended at a density of 2×106 cells/ml in PBS containing 1 mM H2O2 or 10 µM MG132. The cell suspension was loaded into a Versa Fluor (Bio-Rad) cuvette with a stir bar and read in a Shimadzu RF-5301PC Spectrofluorophotometer. Excitation wavelengths spanning 380–500 nM were scanned at ‘slow’ speed with a slit width of 1.5 nm while emission wavelength was set at 510 nm with a slit width of 3 nm. For siRNA transfection experiments, roGFP- or mito-roGFP-expressing cells were seeded at a density of 8000 cells per well of a black 96-well plate and transfected the next day. After treatment, cells were analyzed in a BioTek Synergy H1 Microplate Reader using excitation wavelengths of 400 nm and 475 nm and an emission wavelength of 510 nm.
Isolation of MEFs and generation of brain lysates
Animal studies were performed in adherence with the guidelines of The Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee (IACUC). Pregnant female C57BL/6J mice (aged 3–6 months) were killed 13.5 days post coitus and embryos removed by manual dissection from amniotic sacs, washed with ethanol, and the limbs, tail, red organs and head above the eyes were removed. The remaining material was minced with a sterile razor blade in 4.5 g/l glucose-containing DMEM supplemented with penicillin (100 U/ml) and streptomycin (100 µg/ml), trypsinized for 10 min at 37°C, and nonsoluble material was removed by centrifugation. The remaining cells were cultured in 4.5 g/l glucose-containing DMEM with penicillin, streptomycin and 10% FCS. Brain lysates were generated from Nrf2+/+ and Nrf2−/− mice on a C57BL/6J background. Males and females aged 11–12 months or 22–23 months were used.
Microscopy and image analysis
Immunofluorescence samples were viewed on an LSM710 confocal microscope (Carl Zeiss). Micrographs were captured using 63× or 100× oil immersion objectives, and images adjusted and enhanced using Adobe Photoshop CS6. 3D reconstructions were made by compiling Z-stacks that covered all detectable fluorescence signals above and below the plane of the sample. Cell area analysis was performed on micrographs captured with a 20× phase objective using freeform ROIs to capture cell borders in OpenLab software. For all mitochondria clustering graphs, cells were scored by an observer masked to the identity of the samples. Live cell microscopy was performed with a 60× oil immersion objective on an LSM710 confocal microscope. Cells were treated with drugs for 30 min prior to imaging. Each movie represents 90 min of imaging time with a picture in series being taken every 60 s.
Statistical analysis
Statistical significance was assessed in GraphPad Prism 7.0 software using one-, two- or three-way ANOVA with post hoc correction as appropriate. Significant differences, corresponding to corrected P-values <0.05, are indicated in the figures by the use of asterisks. n.s. denotes a lack of statistical significance (P>0.05) between two means within a data set. Vertical error bars indicate either one standard deviation (s.d.) or one standard error of the mean (s.e.m.).
Western blotting
Cells were washed in PBS and solubilized in 2× Laemmli solubilizing buffer [100 mM Tris (pH 6.8), 2% SDS, 0.008% Bromophenol Blue, 2% 2-mercaptoethanol, 26.3% glycerol and 0.001% Pyronin Y]. Lysates were boiled for 5 min prior to loading on sodium dodecyl sulfate (SDS) polyacrylamide gels. Proteins were transferred to nitrocellulose membranes and the membranes were blocked for 1 h in 5% milk/TBST. Primary antibodies were diluted in 5% milk/TBST and incubated with the blot overnight at 4°C. Horseradish peroxidase (HRP)-conjugated secondary antibodies were diluted in 5% milk/TBST. Blots were processed with enhanced chemiluminescence and densitometric quantifications were performed using ImageJ software.
Creation of stable cell lines
Lentiviruses expressing roGFP, mito-roGFP, Neh2–YFP, YFP–parkin or mt-mKeima were generated in HEK293T cells by the polyethylenimine transfection method, as described (Kim et al., 2012). At 48–72 h post-transfection, supernatant was harvested, filtered through a 0.45 µm membrane and concentrated using polyethylene glycol (Marino et al., 2003). RPE-1 cells were transduced in the presence of 8 µg/ml polybrene (Sigma-Aldrich). Stable cell lines displayed equivalent growth curves to the parental cell line and underwent mitochondrial clustering in response to acute proteasome inhibition that was comparable to parental cells.
Supplementary Material
Acknowledgements
We thank Dr Allan Weissman (National Cancer Institute) for his generous gift of the anti-Mfn2 antibody, Dr Constantin Georgescu for consultation regarding the statistical analyses, and members of the Plafker laboratory for helpful discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: G.B.O., R.J., S.M.P.; Methodology: G.B.O., K.S.P., W.L.B., S.M.P.; Validation: K.S.P.; Formal analysis: G.B.O., K.S.P.; Investigation: G.B.O., S.M.P.; Resources: W.L.B., R.J., J.Y.C., S.M.P.; Data curation: G.B.O., K.S.P., S.M.P.; Writing - original draft: G.B.O., S.M.P.; Writing - review & editing: G.B.O., K.S.P., S.M.P.; Supervision: S.M.P.; Project administration: S.M.P.; Funding acquisition: S.M.P.
Funding
This work was supported by National Institutes of Health [R01GM092900 to S.M.P.] and the Oklahoma Center for the Advancement of Science and Technology [HR16-068 to S.M.P.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.203216.supplemental
References
- Al-Mehdi A. B., Pastukh V. M., Swiger B. M., Reed D. J., Patel M. R., Bardwell G. C., Pastukh V. V., Alexeyev M. F. and Gillespie M. N. (2012). Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 5, ra47 10.1126/scisignal.2002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer N. G. and Richter-Landsberg C. (2006). The dynamic instability of microtubules is required for aggresome formation in oligodendroglial cells after proteolytic stress. J. Mol. Neurosci. 29, 153-168. 10.1385/JMN:29:2:153 [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn J. (2014). Mitochondrial dynamics in aging and disease. Mitochondrion Aging Dis. 127, 93-131. 10.1016/B978-0-12-394625-6.00004-0 [DOI] [PubMed] [Google Scholar]
- Bingol B., Tea J. S., Phu L., Reichelt M., Bakalarski C. E., Song Q., Foreman O., Kirkpatrick D. S. and Sheng M. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370-375. 10.1038/nature13418 [DOI] [PubMed] [Google Scholar]
- Birsa N., Norkett R., Wauer T., Mevissen T. E. T., Wu H.-C., Foltynie T., Bhatia K., Hirst W. D., Komander D., Plun-Favreau H. et al. (2014). Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J. Biol. Chem. 289, 14569-14582. 10.1074/jbc.M114.563031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T. and Johansen T. (2006). p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2, 138-139. 10.4161/auto.2.2.2405 [DOI] [PubMed] [Google Scholar]
- Brickley K., Smith M. J., Beck M. and Stephenson F. A. (2005). GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J. Biol. Chem. 280, 14723-14732. 10.1074/jbc.M409095200 [DOI] [PubMed] [Google Scholar]
- Cadenas E. and Davies K. J. A. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222-230. 10.1016/S0891-5849(00)00317-8 [DOI] [PubMed] [Google Scholar]
- Chan K., Lu R., Chang J. C. and Kan Y. W. (1996). NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 93, 13943-13948. 10.1073/pnas.93.24.13943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan S. B., Gordan J. D., Jin J., Harper J. W. and Diehl J. A. (2004). The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 24, 8477-8486. 10.1128/MCB.24.19.8477-8486.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Brabander M., De May J., Joniau M. and Geuens G. (1977). Ultrastructural immunocytochemical distribution of tubulin in cultured cells treated with microtubule inhibitors. Cell Biol. Int. Rep. 1, 177-183. 10.1016/0309-1651(77)90038-8 [DOI] [PubMed] [Google Scholar]
- De Vos K., Goossens V., Boone E., Vercammen D., Vancompernolle K., Vandenabeele P., Haegeman G., Fiers W. and Grooten J. (1998). The 55-kDa tumor necrosis factor receptor induces clustering of mitochondria through its membrane-proximal region. J. Biol. Chem. 273, 9673-9680. 10.1074/jbc.273.16.9673 [DOI] [PubMed] [Google Scholar]
- Du Y., Wooten M. C., Gearing M. and Wooten M. W. (2009a). Age-associated oxidative damage to the p62 promoter: implications for Alzheimer disease. Free Radic. Biol. Med. 46, 492-501. 10.1016/j.freeradbiomed.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Wooten M. C. and Wooten M. W. (2009b). Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol. Dis. 35, 302-310. 10.1016/j.nbd.2009.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggler A. L., Liu G., Pezzuto J. M., van Breemen R. B. and Mesecar A. D. (2005). Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 102, 10070-10075. 10.1073/pnas.0502402102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteras N., Dinkova-Kostova A. T. and Abramov A. Y. (2016). Nrf2 activation in the treatment of neurodegenerative diseases: a focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 397, 383-400. 10.1515/hsz-2015-0295 [DOI] [PubMed] [Google Scholar]
- Fang L., Hemion C., Pinho Ferreira Bento A. C., Bippes C. C., Flammer J. and Neutzner A. (2015). Mitochondrial function in neuronal cells depends on p97/VCP/Cdc48-mediated quality control. Front. Cell Neurosci. 9, 16 10.3389/fncel.2015.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher J., Kovacs I., Artico M., Cavallotti C., Papale A. and Balacco Gabrieli C. (2006). Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 27, 983-993. 10.1016/j.neurobiolaging.2005.05.012 [DOI] [PubMed] [Google Scholar]
- Fransson S., Ruusala A. and Aspenström P. (2006). The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 344, 500-510. 10.1016/j.bbrc.2006.03.163 [DOI] [PubMed] [Google Scholar]
- Gispert S., Ricciardi F., Kurz A., Azizov M., Hoepken H.-H., Becker D., Voos W., Leuner K., Müller W. E., Kudin A. P. et al. (2009). Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 4, e5777 10.1371/journal.pone.0005777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater E. E., Megeath L. J., Stowers R. S. and Schwarz T. L. (2006). Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 173, 545-557. 10.1083/jcb.200601067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauser L., Sonnay S., Stafa K. and Moore D. J. (2011). Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 118, 636-645. 10.1111/j.1471-4159.2011.07318.x [DOI] [PubMed] [Google Scholar]
- Goven D., Boutten A., Lecon-Malas V., Marchal-Somme J., Amara N., Crestani B., Fournier M., Leseche G., Soler P., Boczkowski J. et al. (2008). Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax 63, 916-924. 10.1136/thx.2007.091181 [DOI] [PubMed] [Google Scholar]
- Griffiths E. J. and Rutter G. A. (2009). Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta-Bioenergetics 1787, 1324-1333. 10.1016/j.bbabio.2009.01.019 [DOI] [PubMed] [Google Scholar]
- Hallmann A., Milczarek R., Lipinski M., Kossowska E., Spodnik J. H., Wozniak M., Wakabayashi T. and Klimek J. (2004). Fast perinuclear clustering of mitochondria in oxidatively stressed human choriocarcinoma cells. Folia. Morphol. (Warsz) 63, 407-412. [PubMed] [Google Scholar]
- Hanson G. T., Aggeler R., Oglesbee D., Cannon M., Capaldi R. A., Tsien R. Y. and Remington S. J. (2004). Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 279, 13044-13053. 10.1074/jbc.M312846200 [DOI] [PubMed] [Google Scholar]
- Hayes J. D. and Dinkova-Kostova A. T. (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199-218. 10.1016/j.tibs.2014.02.002 [DOI] [PubMed] [Google Scholar]
- Heggeness M. H., Simon M. and Singer S. J. (1978). Association of mitochondria with microtubules in cultured cells. Proc. Natl. Acad. Sci. USA 75, 3863-3866. 10.1073/pnas.75.8.3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemion C., Flammer J. and Neutzner A. (2014). Quality control of oxidatively damaged mitochondrial proteins is mediated by p97 and the proteasome. Free Radic. Biol. Med. 75, 121-128. 10.1016/j.freeradbiomed.2014.07.016 [DOI] [PubMed] [Google Scholar]
- Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I. et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313-322. 10.1006/bbrc.1997.6943 [DOI] [PubMed] [Google Scholar]
- Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J. D. and Yamamoto M. (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76-86. 10.1101/gad.13.1.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K., Wakabayashi N., Katoh Y., Ishii T., O'Connor T. and Yamamoto M. (2003). Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379-391. 10.1046/j.1365-2443.2003.00640.x [DOI] [PubMed] [Google Scholar]
- Karbowski M., Neutzner A. and Youle R. J. (2007). The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 178, 71-84. 10.1083/jcb.200611064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H., Kogure T., Mizushima N., Yoshimori T. and Miyawaki A. (2011). A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 18, 1042-1052. 10.1016/j.chembiol.2011.05.013 [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A., Kondapalli C., Gourlay R., Campbell D. G., Ritorto M. S., Hofmann K., Alessi D. R., Knebel A., Trost M. and Muqit M. M. K. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127-139. 10.1042/BJ20140334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Kim H.-Y., Lee S., Kim S. W., Sohn S., Kim K. and Cho H. (2007). Hepatitis B virus X protein induces perinuclear mitochondrial clustering in microtubule- and dynein-dependent manners. J. Virol. 81, 1714-1726. 10.1128/JVI.01863-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.-D., Shin S., Berry W. L., Oh S. and Janknecht R. (2012). The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. J. Cell. Biochem. 113, 1368-1376. 10.1002/jcb.24009 [DOI] [PubMed] [Google Scholar]
- Kimura Y., Fukushi J., Hori S., Matsuda N., Okatsu K., Kakiyama Y., Kawawaki J., Kakizuka A. and Tanaka K. (2013). Different dynamic movements of wild-type and pathogenic VCPs and their cofactors to damaged mitochondria in a Parkin-mediated mitochondrial quality control system. Genes Cells 18, 1131-1143. 10.1111/gtc.12103 [DOI] [PubMed] [Google Scholar]
- Kirkin V., McEwan D. G., Novak I. and Dikic I. (2009). A role for ubiquitin in selective autophagy. Mol. Cell 34, 259-269. 10.1016/j.molcel.2009.04.026 [DOI] [PubMed] [Google Scholar]
- Kitada T., Pisani A., Porter D. R., Yamaguchi H., Tscherter A., Martella G., Bonsi P., Zhang C., Pothos E. N. and Shen J. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 104, 11441-11446. 10.1073/pnas.0702717104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosowiak J. L., Park S., Smith K. P., French M. E., Focia P. J., Freymann D. M. and Rice S. E. (2016). Structural insights into Parkin substrate lysine targeting from minimal Miro substrates. Sci. Rep. 6, 33019 10.1038/srep33019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A., Kang M.-I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K. and Yamamoto M. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130-7139. 10.1128/MCB.24.16.7130-7139.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y. S., Ueno I., Sakamoto A., Tong K. I. et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213-223. 10.1038/ncb2021 [DOI] [PubMed] [Google Scholar]
- Kubben N., Zhang W. Q., Wang L. X., Voss T. C., Yang J. P., Qu J., Liu G.-H. and Misteli T. (2016). Repression of the antioxidant NRF2 pathway in premature aging. Cell 165, 1361-1374. 10.1016/j.cell.2016.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboucher G. P., Tsai Y. C., Yang M., Shaw K. C., Zhou M., Veenstra T. D., Glickman M. H. and Weissman A. M. (2012). Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 47, 547-557. 10.1016/j.molcel.2012.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold P. L., McDowall A. W., Pfister K. K., Bloom G. S. and Brady S. T. (1992). Association of kinesin with characterized membrane-bounded organelles. Cell Motil. Cytoskeleton 23, 19-33. 10.1002/cm.970230104 [DOI] [PubMed] [Google Scholar]
- Liesa M. and Shirihai O. S. (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491-506. 10.1016/j.cmet.2013.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Sawada T., Lee S., Yu W., Silverio G., Alapatt P., Millan I., Shen A., Saxton W., Kanao T. et al. (2012). Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 8, e1002537 10.1371/journal.pgen.1002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo S.-C. and Hannink M. (2008). PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 314, 1789-1803. 10.1016/j.yexcr.2008.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Doménech G., Higgs N. F., Vaccaro V., Roš H., Arancibia-Cárcamo I. L., MacAskill A. F. and Kittler J. T. (2016). Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep. 17, 317-327. 10.1016/j.celrep.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Karuppagounder S. S., Springer D. A., Allen M. D., Zheng L., Chao B., Zhang Y., Dawson V. L., Dawson T. M. and Lenardo M. (2014). Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson's-like movement disorder. Nat. Commun. 5, 4930 10.1038/ncomms5930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill A. F., Brickley K., Stephenson F. A. and Kittler J. T. (2009). GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol. Cell. Neurosci. 40, 301-312. 10.1016/j.mcn.2008.10.016 [DOI] [PubMed] [Google Scholar]
- Manasanch E. E. and Orlowski R. Z. (2017). Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 14, 417-433. 10.1038/nrclinonc.2016.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M. P., Luce M. J. and Reiser J. (2003). Small- to large-scale production of lentivirus vectors. Methods Mol. Biol. 229, 43-55. 10.1385/1-59259-393-3:43 [DOI] [PubMed] [Google Scholar]
- McMahon M., Itoh K., Yamamoto M. and Hayes J. D. (2003). Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 278, 21592-21600. 10.1074/jbc.M300931200 [DOI] [PubMed] [Google Scholar]
- McMahon M., Thomas N., Itoh K., Yamamoto M. and Hayes J. D. (2004). Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 279, 31556-31567. 10.1074/jbc.M403061200 [DOI] [PubMed] [Google Scholar]
- McWilliams T. G. and Muqit M. M. K. (2017). PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 45, 83-91. 10.1016/j.ceb.2017.03.013 [DOI] [PubMed] [Google Scholar]
- Narendra D., Tanaka A., Suen D.-F. and Youle R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795-803. 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D., Kane L. A., Hauser D. N., Fearnley I. M. and Youle R. J. (2010). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090-1106. 10.4161/auto.6.8.13426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. T., Oh S. S., Weaver D., Lewandowska A., Maxfield D., Schuler M.-H., Smith N. K., Macfarlane J., Saunders G., Palmer C. A. et al. (2014). Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc. Natl. Acad. Sci. USA 111, E3631-E3640. 10.1073/pnas.1402449111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton M., Ng A. C.-H., Baird S., Dumoulin A., Shutt T., Mah N., Andrade-Navarro M. A., McBride H. M. and Screaton R. A. (2014). ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci. Signal. 7, ra10 10.1126/scisignal.2004374 [DOI] [PubMed] [Google Scholar]
- Ordureau A., Sarraf S. A., Duda D. M., Heo J.-M., Jedrychowski M. P., Sviderskiy V. O., Olszewski J. L., Koerber J. T., Xie T., Beausoleil S. A. et al. (2014). Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56, 360-375. 10.1016/j.molcel.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A., Heo J.-M., Duda D. M., Paulo J. A., Olszewski J. L., Yanishevski D., Rinehart J., Schulman B. A. and Harper J. W. (2015). Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 112, 6637-6642. 10.1073/pnas.1506593112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek J., Lo J. Y., Narasimhan S. D., Nguyen T. N., Glover-Cutter K., Robida-Stubbs S., Suzuki T., Yamamoto M., Blackwell T. K. and Curran S. P. (2012). Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 16, 526-537. 10.1016/j.cmet.2012.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S., Srivastava S., Li Z., Vaeth M., Fuhs S. R., Hunter T. and Skolnik E. Y. (2016). Identification of PGAM5 as a Mammalian protein histidine phosphatase that plays a central role to negatively regulate CD4(+) T cells. Mol. Cell 63, 457-469. 10.1016/j.molcel.2016.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauty J., Rodrigue A., Couturier A., Buisson R. and Masson J.-Y. (2014). Exploring the roles of PALB2 at the crossroads of DNA repair and cancer. Biochem. J. 461, 539 10.1042/BJ4610539 [DOI] [PubMed] [Google Scholar]
- Plafker K. S. and Plafker S. M. (2015). The ubiquitin-conjugating enzyme UBE2E3 and its import receptor importin-11 regulate the localization and activity of the antioxidant transcription factor NRF2. Mol. Biol. Cell 26, 327-338. 10.1091/mbc.E14-06-1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente B. N., Kimura W., Muralidhar S. A., Moon J., Amatruda J. F., Phelps K. L., Grinsfelder D., Rothermel B. A., Chen R., Garcia J. A. et al. (2014). The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565-579. 10.1016/j.cell.2014.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis K., Fransson A. and Aspenström P. (2009). The Miro GTPases: at the heart of the mitochondrial transport machinery. FEBS Lett. 583, 1391-1398. 10.1016/j.febslet.2009.04.015 [DOI] [PubMed] [Google Scholar]
- Santel A., Blumer N., Kampfer M. and Renkawitz-Pohl R. (1998). Flagellar mitochondrial association of the male-specific Don Juan protein in Drosophila spermatozoa. J. Cell Sci. 111, 3299-3309. [DOI] [PubMed] [Google Scholar]
- Saotome M., Safiulina D., Szabadkai G., Das S., Fransson A., Aspenstrom P., Rizzuto R. and Hajnoczky G. (2008). Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. USA 105, 20728-20733. 10.1073/pnas.0808953105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarlette A., Krampfl K., Grothe C., Neuhoff N., Dengler R. and Petri S. (2008). Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 67, 1055-1062. 10.1097/NEN.0b013e31818b4906 [DOI] [PubMed] [Google Scholar]
- Sarraf S. A., Raman M., Guarani-Pereira V., Sowa M. E., Huttlin E. L., Gygi S. P. and Harper J. W. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372-376. 10.1038/nature12043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton W. M. and Hollenbeck P. J. (2012). The axonal transport of mitochondria. J. Cell Sci. 125, 2095-2104. 10.1242/jcs.053850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz T. L. (2013). Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 5, a011304 10.1101/cshperspect.a011304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z. H. and Cai Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77-93. 10.1038/nrn3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sormo C. G., Brembu T., Winge P. and Bones A. M. (2011). Arabidopsis thaliana MIRO1 and MIRO2 GTPases are unequally redundant in pollen tube growth and fusion of polar nuclei during female gametogenesis. PLoS ONE 6, e18530 10.1371/journal.pone.0018530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N., Yun J., Liu J., Malide D., Liu C., Rovira I. I., Holmström K. M., Fergusson M. M., Yoo Y. H., Combs C. A. et al. (2015). Measuring in vivo mitophagy. Mol. Cell 60, 685-696. 10.1016/j.molcel.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D.-F., Karbowski M. and Youle R. J. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367-1380. 10.1083/jcb.201007013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B. L. (2015). MIRO GTPases in mitochondrial transport, homeostasis and pathology. Cells 5, 1 10.3390/cells5010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor E. B. and Rutter J. (2011). Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 39, 1509-1513. 10.1042/BST0391509 [DOI] [PubMed] [Google Scholar]
- Tong K. I., Katoh Y., Kusunoki H., Itoh K., Tanaka T. and Yamamoto M. (2006). Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell. Biol. 26, 2887-2900. 10.1128/MCB.26.8.2887-2900.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek A. M., Shen Q. and Kawajiri S. (2013). Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 5 10.1101/cshperspect.a011072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spronsen M., Mikhaylova M., Lipka J., Schlager M. A., van den Heuvel D. J., Kuijpers M., Wulf P. S., Keijzer N., Demmers J., Kapitein L. C. et al. (2013). TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485-502. 10.1016/j.neuron.2012.11.027 [DOI] [PubMed] [Google Scholar]
- Varadi A., Johnson-Cadwell L. I., Cirulli V., Yoon Y., Allan V. J. and Rutter G. A. (2004). Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J. Cell Sci. 117, 4389-4400. 10.1242/jcs.01299 [DOI] [PubMed] [Google Scholar]
- Wang C. X. and Youle R. J. (2009). The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95-118. 10.1146/annurev-genet-102108-134850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Song P., Du L., Tian W., Yue W., Liu M., Li D., Wang B., Zhu Y., Cao C. et al. (2011a). Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 286, 11649-11658. 10.1074/jbc.M110.144238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Winter D., Ashrafi G., Schlehe J., Wong Y. L., Selkoe D., Rice S., Steen J., LaVoie M. J. and Schwarz T. L. (2011b). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893-906. 10.1016/j.cell.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Kondo N., Cano M., Ebrahimi K., Yoshida T., Barnett B. P., Biswal S. and Handa J. T. (2014). Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic. Biol. Med. 70, 155-166. 10.1016/j.freeradbiomed.2014.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weniger M. A., Rizzatti E. G., Perez-Galan P., Liu D., Wang Q., Munson P. J., Raghavachari N., White T., Tweito M. M., Dunleavy K. et al. (2011). Treatment-induced oxidative stress and cellular antioxidant capacity determine response to bortezomib in mantle cell lymphoma. Clin. Cancer Res. 17, 5101-5112. 10.1158/1078-0432.CCR-10-3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K. and Youle R. J. (2011). Coupling mitochondrial and cell division. Nat. Cell Biol. 13, 1026-1027. 10.1038/ncb2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaarur N., Meriin A. B., Bejarano E., Xu X., Gabai V. L., Cuervo A. M. and Sherman M. Y. (2014). Proteasome failure promotes positioning of lysosomes around the aggresome via local block of microtubule-dependent transport. Mol. Cell. Biol. 34, 1336-1348. 10.1128/MCB.00103-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. D. (2006). Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 38, 769-789. 10.1080/03602530600971974 [DOI] [PubMed] [Google Scholar]
- Zhang D. D. and Hannink M. (2003). Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137-8151. 10.1128/MCB.23.22.8137-8151.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. D., Lo S.-C., Cross J. V., Templeton D. J. and Hannink M. (2004). Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24, 10941-10953. 10.1128/MCB.24.24.10941-10953.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q., Huang T., Zhang L., Zhou Y., Luo H., Xu H. and Wang X. (2016). Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front. Aging. Neurosci. 8, 303 10.3389/fnagi.2016.00303 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.