

Abstract
Mechanical stress is one of the major aetiological factors underlying soft-tissue remodelling, especially for the mitral valve (MV). It has been hypothesized that altered MV tissue stress states lead to deviations from cellular homeostasis, resulting in subsequent cellular activation and extracellular matrix (ECM) remodelling. However, a quantitative link between alterations in the organ-level in vivo state and in vitro-based mechanobiology studies has yet to be made. We thus developed an integrated experimental–computational approach to elucidate MV tissue and interstitial cell responses to varying tissue strain levels. Comprehensive results at different length scales revealed that normal responses are observed only within a defined range of tissue deformations, whereas deformations outside of this range lead to hypo- and hyper-synthetic responses, evidenced by changes in α-smooth muscle actin, type I collagen, and other ECM and cell adhesion molecule regulation. We identified MV interstitial cell deformation as a key player in leaflet tissue homeostatic regulation and, as such, used it as the metric that makes the critical link between in vitro responses to simulated equivalent in vivo behaviour. Results indicated that cell responses have a delimited range of in vivo deformations that maintain a homeostatic response, suggesting that deviations from this range may lead to deleterious tissue remodelling and failure.
Keywords: valve interstitial cells, collagen, myofibroblast
1. Introduction
In soft-tissue biomechanics, the presence of normal tissue stress is considered to be closely related to the regulation of tissue homeostasis [1–5]. Several studies have shown that pathophysiological alterations in mechanical loading lead to stress changes and subsequent tissue adaptations that affect tissue structure and composition [6,7]. The fundamental question common to all soft collagenous tissues is the relationship between tissue remodelling and cell-level deformations. Tendons, for example, have demonstrated altered functional activity, tissue architecture, tenocyte shape, and collagen fibre alignment with increasing age and strain [8,9]. These changes were found to be regulated by extracellular matrix (ECM) components, such as decorin and biglycan, which are known to decrease with age [10,11]. In a similar way, mechanical stimuli were found to alter the homeostatic balance that exists between collagen biosynthesis and catabolism in the meniscus. In meniscal cells, static and dynamic compression regulated gene expression of key ECM components, such as types I and II collagen as well as decorin [12]. While tissue responses to different mechanical stimuli have been studied in isolation in vitro, complete elucidation of the underlying mechanisms of disease development and progression [13–22] requires the link to the in vivo organ-level functional state. Moreover, when tightly integrated with and informed by experimental studies, computational models can give unique insights into the function and physiology and provide a powerful platform for predictive simulations. Thus, we must first understand the underlying mechanisms of compensatory responses to altered organ-level loading by linking tissue-level stresses to cellular mechanobiological responses.
The above is especially true in heart valve leaflet tissues, where alterations in tissue stresses due to physiological conditions, disease, and surgical repair have long been suspected to play a major role in valvular remodelling. The average adult heart beats at approximately 3 billion times during the lifetime, with blood flow directed within its chambers by the four heart valves [23]. Heart valves function within a highly demanding intrinsically mechanical environment; the movement of their structures is coordinated by their complex geometry [24–27] and the underlying, intricate, and highly organized ECM. Of the four heart valves, the mitral valve (MV) is subjected to the greatest haemodynamic forces. Its leaflets consist of four histologically distinct layers: the atrialis and the ventricularis, both of which are thin elastin-rich layers, the spongiosa, which consists mainly of proteoglycans (PGs) and glycosaminoglycans (GAGs), and the fibrosa, the main load-bearing layer, composed of circumferentially oriented collagen fibres. The structure–function relationship between the MV interstitial cells (MVICs) and the surrounding ECM enables the valvular tissue to maintain a homeostatic state in different haemodynamic and biomechanical environments [1–5]. While the ECM provides the tissue with its structural integrity and influences cellular processes through matricellular, matricrine, and mechanical processes [28], it is the MVICs residing within the valve's four layers [29,30] that maintain the structural integrity of the leaflet tissue through protein synthesis and enzymatic degradation. VICs respond to mechanical stimuli by undergoing phenotypic activation: the transition to a myofibroblast-like phenotype leads to increased synthesis of PGs and GAGs [13,14,31], collagen [20,32], and α-smooth muscle actin (α-SMA) as well as increased proteolytic enzyme expression and activity [20,33] and a higher cellular elastic modulus [34]. This suggests that the in vivo mechanical environment plays a fundamental role in the mechanobiology and overall VIC responses to external mechanical stimuli caused by pathological factors or surgical repair. As such, the link between organ-, tissue-, and cellular-level deformations with VIC biosynthesis is essential in understanding valvular response to stress overload and the long-term outcome of surgical repair. Thus, heart valves provide a unique test bed for investigating the multi-scale response (figure 1) of soft tissue to mechanical stimuli and linking the experimental findings to the in vivo loading state.
Figure 1.

This study enables us to investigate the effect of mechanical stimuli on VICs at different length scales that encompass organ-level simulations, tissue-level deformations, cell-level deformation and extracellular matrix biosynthesis, and sub-cellular behaviour and gene expression. Included in the diagram are the following: (1) three-dimensional rendition of the MVIC micro-environment from serial focused ion beam scanning electron microscopy micrographs. (2) Transmission electron micrograph of a representative myofibroblast, characterized by the highly convoluted rough endoplasmic reticulum (rER). White arrows: rER, blue arrows: collagen fibrils, red arrows: amorphous elastin. EC, euchromatin; HC, heterochromatin; Mi, mitochondria. Scale bar, 1 µm. (Online version in colour.)
For the MV, ischaemic mitral regurgitation (IMR) occurs when the abnormal reversal of blood flow from the left ventricle to the left atrium is induced by a distorted or dilated left ventricle. The current clinical treatment is surgical repair by ring annuloplasty, which restores normal valve leaflet coaptation and motion [35,36]. The MV annulus, however, flattens and undergoes significant conformational changes after repair. This increase in stress results in the disruption of suture lines in both leaflets and annulus [37], inducing irreversible tissue damage and, ultimately, leading to repair failure [38]. Moreover, patients with IMR have a 30% recurrence rate at six months post-surgery, which increases to 60% after three to five years [39]. Mechanical stress is one of the main aetiological factors: alterations in mechanical loading caused by surgical repair lead to stress-induced changes in MVIC function that in turn affect both tissue structure and composition, ultimately leading to repair failure [6,7]. Biosynthetic analyses show evidence of cellular activation, highlighting the active adaptation of the MV to alterations in its local stress levels [16,40–43]. The remodelling capacity of mature MV leaflets is also observed in non-pathological conditions, such as pregnancy, whereby heart valves adapt to the volume overload and cardiac expansion associated with pregnancy [44,45] and exhibit a layer-specific compensatory response where collagen remodelling occurs mainly in the fibrosa layer [46]. These pregnancy-induced changes parallel those observed in pathological and surgically repaired heart valves. As such, understanding the underlying mechanobiology associated with the compensatory response of MVICs to non-physiological mechanical stimuli is necessary for the development of improved modelling tools and surgical repair, and paves the path for future work on degenerative valvular disease.
We thus hypothesized that tissue level-induced deformation is a major driver for MVIC mechanoregulation, and deformation levels outside of the normal physiological homeostatic range can lead to phenotypic activation and either hypo- or hyper-biosynthetic activity. To address this hypothesis, we used an integrated experimental–computational approach as follows. A bioreactor system (figure 2c) was used to deform valvular tissue specimens to physiological and non-physiological strain levels [47], and a macro–micro finite-element (FE) model of the MV [48] was used to translate these findings to the in vivo state. We report comprehensive results at different length scales (organ-, tissue-, cell-, sub-cell levels) and use MVIC nuclear deformation as a metric that relates in vitro experimental results to the in vivo functional state of the valve (figure 1). As such, we also report a range of MVIC deformations that capture both physiological behaviour as well as deviations from the homeostatic response caused by mechanical stimuli induced by surgical repair. This novel experimental–computational approach allowed us to elucidate the link between MVIC deformation level and biosynthetic response to stress under- or overload experienced after surgical repair.
Figure 2.

Mitral valve anterior leaflets (MVAL) samples were subjected to circumferential cyclic strain using a custom-designed load-sensing tissue strip bioreactor. (a) MVAL with the clear zone labelled. All samples for the bioreactor treatment are taken from the clear zone due to tissue uniformity in that region. A: sample from the clear zone used as a post-mortem control sample, B: sample from the clear zone used for the bioreactor treatment. Samples to be treated in the bioreactor are sutured with springs on the two radial sides and strained along the circumferential direction. R, radial; C, circumferential. (b) Viability index data for post-mortem and cultured samples (10, 20, and 30%) measured using the MTT assay. Data are mean ± SEM, n = 4, *p < 0.05, **p < 0.01 compared to post-mortem. (c) Left: side view of the bioreactor and the different components: linear actuator (black), load cell (white), specimen chamber, and specimen chamber lid with T-flask caps. Right: top view of the bioreactor. (Online version in colour.)
2. Material and methods
2.1. Bioreactor design and validation
The tissue strip bioreactor (figure 2c) used in this study is a modified version of the previously described tension bioreactor used for aortic valve (AV) stimulation [32]. Cyclic stretch is applied with a stepper motor linear actuator (Haydon Kerk Motion Solutions, Waterbury, CT). The actuator is connected to a metal rod that is coupled to a load cell (Futek Sensor and Electronics, Inc., Irvine, CA) and the tissue attachment system, which is housed within the environmentally sealed specimen chamber that is rigidly attached to the system base. The attachment system used, which consists of spring ends, along with the 1 : 0.75 specimen aspect ratio (AR) facilitated a uniform strain field. The springs have a total of five contact points along the 7.5 mm radial side with less than 2 mm between each pin/attachment puncture into the tissue. The large number of contact points, along with the small distance between each point ensures that the strain is distributed with reasonable uniformity. An in-house LabVIEW (National Instruments, Austin, TX) virtual instrument (VI) is used to control both the linear actuator and the load cell. The VI collects both load data and actuator displacement every 10 min. All components that are housed within the specimen chamber are sterilized with heat (i.e. autoclave) and ultraviolet exposure. The system base is cleaned with ethanol. We investigated tissue compliance during the 48-h circumferential cyclic stretch treatment by incorporating a load cell into the tissue strip bioreactor and tracking load readings with respect to time. Results indicate that there is increased tissue compliance over time (electronic supplementary material, figure S1). In line with previous reports on ovine MV properties after a radial cyclic stretch treatment [29], the degree of compliance is especially higher for the 30% strain group, whereby the load reading decreased from 3.8 to 1.8 lbf after the 48-h cyclic stretch treatment.
2.2. Tissue preparation and bioreactor treatment
Fresh porcine hearts were collected from young hogs (10 months, 250 lbs) from a local USDA-approved abattoir (Harvest House Farms, Johnson City, TX) within 30–45 min of slaughter. MV anterior leaflets (MVALs) were isolated on-site and submerged in ice-cold phosphate-buffered saline (PBS) (Thermo Fischer Scientific, Waltham, MA) for transport to the laboratory. Once in the laboratory, rectangular strips (12 × 7.5 mm, circumferential × radial) were dissected from the clear zone of the anterior leaflet, placed in sterile, ice-cold Hypothermosol FRS (Sigma-Aldrich, St Louis, MO) and later used for the different treatment groups (figure 2a). The remaining tissue from the clear zone (7 × 7.5 mm) was snap-frozen with liquid nitrogen, stored at −80°C and later used as the post-mortem sample for each valve. For each bioreactor treatment group, eight (n = 8) MVs were used. Placement of the tissue attachment springs was carried out aseptically in a sterile laminar flow hood. Spring dimensions, materials, and attachment technique have been described in detail previously [32]. The attachment system has a total of five contact points along the radial side. The large number of contact points, along with the small distance between each point, ensures that the strain is distributed uniformly. The eight culture wells of the specimen chamber were filled with 8 ml of serum-free DMEM (Life Technologies, Carlsbad, CA) supplemented with 1% (v/v) Pen/Strep antibiotics (Invitrogen, Carlsbad, CA), 1% (v/v) l-glutamine (BioWhittaker, Houston, TX), 10% (v/v) fetal bovine serum (Hyclone, Logan, UT) and 0.4% (v/v) Fungizone (Invitrogen, Carlsbad, CA). All eight specimens, with two springs sutured on either side, were placed in individual wells within the specimen chamber and their position secured with the pins attached to either the chamber or the pin holder. The specimen chamber was then attached to the system base, which is housed within the incubator maintained at 37°C and 5% CO2. Samples were then stretched for a total of 48 h. At the end of each treatment, samples were either fixed or snap-frozen for further analysis as described in the subsequent subsections.
2.3. Cell deformation and collagen fibre alignment
In separate experiments, a group of n = 9 valves was used for micro-environment and cell deformation analysis. Similarly, MVALs were isolated and stored in Hypothermosol. Rectangular strips were dissected from the clear zone and sutured with springs on both radial sides and spring-to-spring distance measured and inputted into the system. The individual wells of the specimen chamber were filled with PBS and the specimen placed in each well as described above. Each group of n = 9 samples was subjected to a single strain (10%, 20% or 30%). Once strained, PBS was removed from the wells and replaced with grade II EM grade glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA). Samples were fixed for a total of 4 h. A total of n = 3 samples were used for small angle light scattering (SALS) to quantify collagen fibre architecture (CFA). The rest of the samples were used to measure cellular deformation using transmission electron microscopy (TEM).
2.3.1. Small Angle Light Scattering
Samples used for SALS underwent a gradient of glycerolization as described previously [49]. Briefly, the fixed samples were dehydrated, cleared in a graded glycerol/water solution and stored in 100% glycerol. A 5 mW non-polarized continuous HeNe laser (JDS Uniphase Corporation, Milpitas, CA) was passed through the tissue samples and the normalized orientation index (NOI) was used to quantify tissue deformation. The NOIs that represent different regions of interest within the specimen were averaged and used to compute an average NOI that represents the CFA of the sample. An NOI value of 100% represents a highly oriented fibre network, whereas an NOI value of 0% represents a more randomly oriented network.
2.3.2. Electron Microscopy
Fixed samples were stained with osmium tetroxide for 4 h, dehydrated with alcohol and acetone, infiltrated in epoxy resin overnight, and cured for 48 h in a 60°C oven. The resulting resin blocks were trimmed with a razor blade into a trapezoid block face such that the y-axis represents the transmural/thickness axis and the x-axis represents the circumferential axis of the MVAL. Thin sections (70 nm) were cut using a Diatome Ultra Diamond Knife 35° (Diatome Diamond Knives, Hatfield, PA), picked up with Formvar-coated slot grids (Electron Microscopy Sciences, Hatfield, PA) and imaged with an FEI Tecnai TEM (FEI, Hillsboro, OR). Cytoplasmic and nuclear aspect ratios (NARs) were quantified with ImageJ (NIH, Bethesda, MD).
2.4. Cell viability
Cellular viability of cultured MVAL tissue was analysed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) viability assay (Sigma-Aldrich, St Louis, MO). The procedure has been described in detail previously [31]. In brief, a total of n = 4 control samples (obtained within 30–45 min of slaughter) and n = 12 cultured samples (removed from the bioreactor after a 48 h treatment at different strain levels, four samples for each strain level) were washed twice with PBS containing antibiotics, weighed, and placed in a 24-well plate containing 200 µl/well of MTT solution. The 24-well plate was covered with aluminium foil and incubated for 3 h at 37°C and 5% CO2 in the dark. After incubation, samples were carefully removed, blotted dry, transferred to a new 24-well plate with 1 ml per well of MTT extraction solution, covered with aluminium foil, and incubated for 1 h at 37°C and 5% CO2. At the end of the incubation, the plate was carefully mixed and the samples discarded. For each sample, 200 µl aliquots (n = 2 per sample) were transferred to a 96-well plate, and absorbance measured at 570 nm and background at 690 nm (VWR International, Radnor, PA). The viability index was calculated as such: viablity index = OD570nm – OD690nm/sample weight (milligrams), where OD570 nm and OD690 nm are the absorbance measurements at 570 nm and 690 nm, respectively.
2.5. Layer microdissection
To quantify layer-specific changes, a group of n = 3 samples were strained cyclically at two levels: 10% (physiological) and 30% (super-physiological). The atrialis, ventricularis and fibrosa were separated as follows: using dissecting forceps, the atrialis was gently pulled upwards and with ophthalmic scissors, the fibrous connections between the atrialis and the fibrosa were exposed and cut. In a similar way, on the opposite side, the ventricularis was separated from the fibrosa. To verify the effectiveness of the microdissection, 2 × 10 mm sections of the intact and separated MVAL layers were cut in the circumferential and radial direction, fixed in 10% buffered formalin, embedded in paraffin wax, stained with Movat's pentachrome, sectioned (5 µm), and examined with a light microscope.
2.6. Histology and immunohistochemistry
Tissue preparation for histology and immunohistochemistry has been described previously [20,32]. Briefly, MVAL samples (both control and treated) were fixed with 10% buffered formalin, embedded in paraffin, cut into 5 µm sections and mounted on glass slides. Slides were then stained with Movat's Pentachrome for layer dimensional analysis and haematoxylin and eosin (H&E) to assess cell density. Slides were imaged using a light microscope at 4× magnification (Leica, Wetzlar, Germany). Immunohistochemistry was performed to determine VIC phenotypic activation using the myofibroblast marker, α-SMA. In brief, slides were deparaffinized, hydrated and incubated in 10% goat serum and 0.03% hydrogen peroxide for 15 min each. The primary rabbit polyclonal α-SMA antibody (1 : 100, Abcam, Cambridge, MA) was applied for 40 min followed by the anti-rabbit peroxidase-labelled polymer for 30 min. Slides were then rinsed in PBS containing 0.01% Tween-20 after each incubation/staining step. All slides were counterstained with Mayer's haematoxylin (Dako, Carpenteria, CA), dehydrated, and mounted. We stained the samples for the endothelial marker, CD31 using the monoclonal mouse anti-human CD31 marker (1 : 25, Dako) to both verify complete endothelial cell (EC) denudation, but to also explore the role of ECs in possible endothelial mesenchymal transition (EMT). Immunohistochemical staining procedure for CD31 is the same as for α-SMA.
2.7. Protein synthesis
Acid–pepsin soluble collagen, sulfated GAG (s-GAG), and elastin content were quantified using the Sircol, Blyscan, and Fastin quantitative dye-binding assay kits, respectively (Biocolor, Belfast, UK). Samples were weighed before and after overnight lyophilization. Samples were digested in 1% w/v pepsin in 0.5 M acetic acid (Sigma-Aldrich, St Louis, MO) for 24 h at 4°C for collagen quantification, 1% papain (Sigma-Aldrich, St Louis, MO) for 15 h at 60°C for s-GAG content, and in 0.25 M of oxalic acid at 100°C for 1 h (total of four extractions) for elastin assessment. Resulting extracts were then quantified using the manufacturers' protocols.
2.8. RT2 profiler PCR array and RT-qPCR
RNA was isolated from porcine MV tissue from the 0% (static), 10%, 20%, and 30% groups using the Qiagen fibrous tissue RNeasy kit (Qiagen, Valencia, CA). RNA was reverse transcribed using the Qiagen RT2 First Strand Kit and the Qiagen RT2 profiler PCR array was used to quantify the expression of 84 ECM and cell adhesion molecules (CAM) (electronic supplementary material, tables S2–S4). Data were analysed using the online Qiagen Data Analysis Center. RNA (2 µg) was reverse transcribed using the Applied Biosystems TaqMan Reverse Transcription reagents. Real-Time PCR (RT-qPCR) was performed in triplicate (n = 3 samples) using the Fast SYBR Green protocol on the ABI 7500 Fast thermocycler (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions and the following primers: porcine collagen type I (COL1A1): 5′GATCCTGCTGACGTGGCCAT′3, 5′ACTCGTGCAGCCGTCGTAGA′3; porcine α-SMA (ACTA2): 5′TGTGACAATGGTTCTGGGCTCTGT′3, 5′TTCGTCACCCACGTAGCTGTCTTT′3; porcine GAPDH: 5′GCAAAGTGGACATGGTCGCCATCA′3, 5′AGCTTCCCATTCTCAGCCTTGACT′3. Subsequent data analysis were performed using the SABiosciences qPCR analysis package. Fold change was calculated using the standard ddCt method. Software parameters/thresholds were as follows: Threshold 0.2, Auto Ct and Manual baseline. To maintain the most consistent and systematic approach with the abattoir-derived tissue, we used the 10% group, which represents the normal homeostatic range, for normalization [20,33].
2.9. Simulating in vivo mitral valve interstitial cell nuclear aspect ratio
MVAL functional deformations in both a normal (i.e. non-repaired) and surgically repaired (i.e. flat ring) valve were previously acquired using sonomicrometry array localization [50] and integrated into our established MVIC micro-environment model [48]. Briefly, one representative volume element (RVE) model (100 × 100 µm) was constructed for the fibrosa layer of the MVAL. MVICs were included as uniformly distributed ellipsoidal inclusions with cell dimensions, cell density, and cell orientations in accordance with previously acquired experimental data [48,51]. A computational FE mesh was generated using HyperMesh (Altair Engineering Inc.). The RVE mechanical behaviour was modelled by using the simplified structural constitutive model for the ECM regions [52] and the St Venant–Kirchhoff (SVK) material model for MVICs [48]. We note that our choice of the SVK model as opposed to the more commonly adopted neo-Hookean model was examined in detail in [48] (c.f. §4.3) by comparing predicted NARs of the MVICs in the fibrosa layer between the two models. Moreover, the SVK material model has a serious drawback when dealing with large compressive strains. As such, we adopted the modified SVK model in our implementation in ABAQUS UMAT. Nonlinear FE simulations were performed using ABAQUS v. 6.13 (SIMULIA, Dassault Systèmes, Providence, RI) by prescribing time-dependent tissue-level deformations acquired from [50] as equivalent boundary conditions on the edges of the RVE. Prediction of the deformation field was analysed to evaluate the overall in vivo MVIC deformations, denoted by a dimensionless indicator, the NAR, defined as the ratio of the MVIC circumferential length to its transmural length.
2.10. Statistical analysis
Colorimetric assay values (collagen, s-GAG, and elastin), thickness, ARs, normalized orientation indices and viability indices are reported as mean ± s.e. of the mean. For statistical comparison, colorimetric values were analysed with a repeated measures one-way ANOVA for strain level, followed by a post hoc pairwise comparison with a Tukey honest significant difference test for normally distributed data. Nuclear and cytoplasmic AR, viability, normalized orientation indices, and qPCR fold change were analysed with a one-way ANOVA. A linear regression, whereby all replicate Y values are considered, was performed on the qPCR data for both collagen and SMA. All statistical analyses were carried out in GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA).
3. Results
3.1. Collagen fibre structure–tissue strain relationship
The micromechanical interaction of MVICs with the surrounding collagen fibres is clearly fundamental to mechanical signalling and modulating subsequent cellular response. We first examined the ultrastructural characterization of the VIC micro-environment via electron microscopy, which highlighted the interconnections of collagen fibrils with the MVICs (figure 1). Several pathological factors, linked with abnormal mechanical stimuli, have been shown to affect valve tissue structure and composition [16,40,53]. As such, the first step in understanding heart valve disease is to link mechanical stimuli to CFA. We showed previously that AV leaflet CFA increases linearly with strain and successfully linked tissue-level deformation and collagen alignment to cell deformation [18,54]. To gain insights into how our applied strain configuration affects MV leaflet collagen alignment, we quantified changes in CFA with the applied strain level. Our previous computational work on the MV [47,48] enabled us to estimate the valve leaflet's in vivo stresses and tissue properties. This allowed us to delimitate a range of strains, from 0% to 30%, that can be used in our in vitro bioreactor studies, whereby 0% is the hypo-physiological strain level and 30% is the hyper-physiological strain level. According to our FE simulations [47] and other in vivo [50,55] and in vitro [56] measurements, 10% is the physiological circumferential strain level of the MVAL. CFA maps revealed predictably higher alignment at higher strain levels (figure 3a); interestingly, CFA alignment remained the same at lower strain levels (0%, 10%, and 20%), then increased sharply at 30% (p < 0.05). This result highlights the highly nonlinear changes that occur in the valve tissue micromechanical environment and the unique effects at hyper-physiological strain levels.
Figure 3.

Collagen fibre alignment and cellular deformation demonstrate nonlinear tissue micromechanics at higher strain levels. (a) Collagen fibre alignment maps at 0% (unstrained), 10% (physiological strain), 20%, and 30% (hyper-physiological strain). NOI, normalized orientation index. Warmer colours represent more highly aligned tissue. (b) Representative mitral valve interstitial cells from the fibrosa layer from samples stretched at different strain levels. T, transmural/thickness; C, circumferential. Scale bar: 5 µm. (c) NOI was averaged for all regions of the sample to find a representative NOI for the sample and averaged for all three samples. The NOI value at 0%, 10%, and 20% are significantly higher than at 30% (p < 0.05). Data are mean ± SEM, n = 3, *p < 0.05. (d) Nuclear and cytoplasmic aspect ratios (n = 3, 10 cells per each) at different strain levels. Data are mean ± SEM. Cytoplasmic AR is significantly higher than nuclear AR (p < 0.001) at hyper-physiological strain levels (30%). (Online version in colour.)
3.2. Cytoplasmic deformation uncouples from nuclear deformation at hyper-physiological strain levels
Moving to the cellular functional level, we examined the NAR, a dimensionless indicator defined as the ratio of the VIC nuclear circumferential dimension to its transmural dimension, as a metric that is used to quantify overall VIC deformation (electronic supplementary material, table S1) [18,48,57]. While there is no apparent cellular damage at higher strain levels, elongation of both the cytoplasm and nucleus along the circumferential direction was evident (figure 3b). Results show a linear trend and coupling of both nuclear and cytoplasmic ARs in the fibrosa at 0%, 10%, and 20% (figure 3d). However, a sharp increase occurred at the 30% strain level, whereby the cytoplasm deformed significantly more than the nucleus (p < 0.01), reaching an AR of 11.10, while the nuclear AR is only 7.23. A closer examination of the minor and major axes deformation allows us to examine the mechanism of NAR change and designate cellular deformation that results from increasing strain as either undergoing elongation or compression. Results show that, in the physiological range (10–20%), cells undergo both compression and elongation, but that at the hyper-physiological strain level (30%), the cells undergo very little or no elongation along the transmural direction and, thus, minimal compression (figure 4). These results indicate that cellular deformation is driven largely by circumferential elongation of the cytoplasm and is coordinated by collagen alignment, which increased substantially at the hyper-physiological strain level. These observations are in line with our previous findings on the aortic VIC deformation, which suggested that additional micromechanical and fibre compaction effects can occur at high pressure levels [54].
Figure 4.
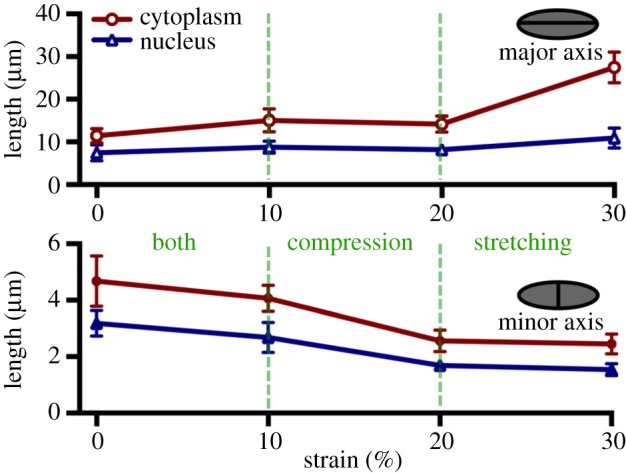
Length in micrometres of cytoplasm (red) and nucleus (blue) along the major (circumferential) and minor (transmural) axes. Deformation occurs mainly along the direction of the applied circumferential strain with minimal compression along the transmural direction: at higher strain levels, the deformation along the major axis accounts for the large aspect ratio. Data are mean ± SEM. (Online version in colour.)
3.3. Phenotypic activation occurs at higher strain levels and begins in the outer layers
VICs consist of a heterogeneous population of cells that have a diverse and dynamic phenotype. In normal and healthy valves, VICs are smooth muscle cell- and fibroblast-like; they become activated and switch to the myofibroblast phenotype in development and disease [58]. This phenotypic activation is characterized by an upregulation of α-SMA [59]. When activated, MVICs become myofibroblast-like and begin to actively remodel the ECM, highlighting the fact that VIC phenotypic state is linked to the remodelling demands of the tissue [60]. To ensure that cell viability was maintained in the 48 h cyclic stretch experiments, we used the MTT viability assay and calculated the resulting viability index. Results show that the viability index increased in the 30% group (figure 2b). As the MTT assay is a measure of mitochondrial activity, the increase observed at the hyper-physiological strain level is an indication of phenotypic activation. As such, we sought to evaluate the effect of mechanical stimuli on phenotypic activation (figure 5a). Analysis of immunohistochemical stains at different strain levels (figure 5c–e) showed an upregulation of α-SMA expression at higher strain levels (electronic supplementary material, figure S2). These results correlate with the gene expression data (figure 6a), which showed a linear trend (R2 = 0.6794, p = 0.001), suggesting that VIC phenotypic activation is linked to the magnitude of the mechanical stimuli imposed on the tissue and, thus, the cell. A closer examination reveals that cellular activation begins in the atrialis and spongiosa layers at the lower strain levels (figure 5c,d). Given that EMT can occur in hyper-physiological strain scenarios [42,43,61], we sought to explore the role played by ECs in this experimental set-up by staining post-mortem control samples for CD31 (figure 5b; electronic supplementary material, figure S3). The immunohistochemical stain highlights the absence of ECs and demonstrates that this cell population does not interfere with VIC activation through EMT.
Figure 5.

Representative micrographs of α-SMA expression in post-mortem MV samples (a) and after a 48-h cyclic stretch treatment at 10% (c), 20% (d) and 30% (e). The higher levels of α-SMA expression in VICs within the fibrosa layer suggest cellular activation and phenotypic transition from a fibroblast-like phenotype to a myofibroblast-like phenotype at higher strain levels. (b) Representative micrographs of CD31 expression of post-mortem control sample demonstrate successful removal of endothelial cells from the outer layers of the valve samples prior to the bioreactor treatment. Scale bar, 250 µm. (Online version in colour.)
Figure 6.

Cyclic strain leads to an increase in α-SMA and type I collagen gene expression. qPCR reveals a linear increase with strain in both (a) α-SMA (R2 = 0.6794, p = 0.001) and (b) type I collagen gene expression (R2 = 0.5674, p = 0.0191). Data are mean ± SEM, n = 3 samples. All data are referenced to the average of the 10% group. (Online version in colour.)
3.4. Extracellular matrix biosynthesis at hyper- and hypo-physiological strain levels
VICs are critical to the remodelling demands of the valve and tissue homeostasis. The interaction of MVICs with the surrounding ECM, particularly the collagen fibrils, is fundamental in modulating cellular response. The ECM can transmit external mechanical stimuli, such as stretch, shear stress, and pressure, to the cell via integrins and other membrane-bound protein receptors. As in all soft tissues, the ECM of heart valves is a dynamic environment driven by the VICs, which act to maintain tissue homeostasis by continuously degrading and replacing it. Various studies have investigated VIC response to mechanical stimuli. Results follow a trend: phenotypic activation, increased synthesis of PGs, GAGs [13,14,31], and collagen [20,32], as well as increased proteolytic expression and activity [20,33]. Such results emphasize that the in vivo mechanical environment plays a fundamental role in the mechanobiology and overall VIC response to external mechanical stimuli caused by disease or surgical repair. Using colorimetric assays to assess ECM composition (figure 7a–c), we also detected a significant increase in acid–pepsin soluble collagen content (figure 7a) at the 30% strain level (p < 0.01). The results also indicated a decrease in soluble content in the static group (p < 0.05). This observation was made also at the gene level, whereby type I collagen gene expression exhibited a linear trend with increasing circumferential strain (R2 = 0.5674, p = 0.0191) (figure 6b). Sulfated GAG content also increased at the hyper-physiological strain level (p < 0.05) (figure 7b), whereas elastin content experienced a decrease (p < 0.05) (figure 7c). Previous work on the AV showed strain-level dependence on type I collagen gene expression, whereby a twofold increase in collagen gene expression was observed at hyper-physiological circumferential strain levels [33]. To gain insights into gene expression of collagen as well as other ECM and CAM, we used the RT2 Profiler PCR array for 84 ECM and CAM genes (figures 8 and 9). Results demonstrate an overall downregulation of some of these genes after a static incubation, mainly collagens, transmembrane and basement membrane molecules (electronic supplementary material, tables S2–S4). There is an upregulation in some ECM proteases in the static group, mainly matrix metalloproteinase (MMP)-8 and MMP-15 (electronic supplementary material, tables S2–S4). On the other hand, a general upregulation of ECM and CAM genes is observed at higher strain levels. This includes a twofold increase in type I collagen as well as an upregulation of ECM proteases such as MMP-2, -9, -14, and -15, and ECM protease inhibitors, such as tissue inhibitor of metalloproteinase (TIMP)-2 (electronic supplementary material, tables S2–S4). Collectively, these results demonstrate that a significant amount of alterations occur in ECM remodelling and synthesis by VICs when stimulated outside of their normal, physiological, homeostatic range. These alterations may translate to actual ECM changes over time.
Figure 7.

Extracellular matrix composition. All bar graphs, mean ± SEM, n = 8, *p < 0.05, **p < 0.01, white: post-mortem, black: post-stimulation samples (after 48 h). ECM content (s-GAG, collagen, and elastin) quantified with colorimetric assays is presented in microgram per milligram dry weight. (a) s-GAG content increases after the 30% cyclic strain treatment. (b) Sircol colorimetric assay demonstrates that static incubation at 0% strain resulted in a significant decrease (p < 0.05) in acid–pepsin soluble collagen, whereas a significant increase is observed at the 30% hyper-physiological strain level. (c) A significant decrease in elastin content is observed at the hyper-physiological strain level. (Online version in colour.)
Figure 8.

RT2 Profiler PCR array for 84 ECM and cell adhesion molecule genes indicates an overall downregulation of some of these genes after static (0% strain) incubation and an upregulation of gene expression at higher strain levels (20% and 30%). (a) Scatter plots compare the normalized expression of every gene on the RT2 Profiler PCR array between two groups: (i–iii) static versus 10%, 20% versus 10%, and 30% versus 10%. The central solid line indicates unchanged gene expression and the dotted lines indicate the fold regulation threshold (=2). Genes that are above and below the dotted line meet the fold regulation threshold and are significantly upregulated or downregulated, respectively. Fold change was calculated by using the 10% group as the calibrator. Specific genes and corresponding fold changes are included in the electronic supplementary material. (b) Total number of genes (N) that are either below (downregulated), within, or above (upregulated) the fold regulation threshold at different strain levels. (c) Heat map of gene expression magnitude for all four groups (0%, 10%, 20%, and 30%) with key genes highlighted: EMILIN1, TIMP1, MMP1, COL1A1, MMP9, and ITGB1. (Online version in colour.)
Figure 9.

Layer-specific analysis. (a) Layer separation technique: atrial and ventricular sides are shown. The atrialis is separated from the fibrosa as shown in the image. (b) Layer-specific analysis: Movat-stained sections of MVAL samples after layer separation. From left to right: intact anterior leaflet sample with all four layers, leaflet with the atrialis removed, leaflet with both ventricularis and atrialis removed. A, atrialis; S, spongiosa; F, fibrosa; V, ventricularis. Scale bar: 100 µm. (c) Acid–pepsin soluble collagen content (microgram per milligram wet weight) in the separated atrialis, fibrosa, and ventricularis quantified for the post-mortem, 10% and 30% samples. Data are mean ± SEM, n = 3 samples, *p < 0.05. A significant soluble collagen content increase is observed at the hyper-physiological strain level (30%) in both the atrialis and fibrosa layers. (d) Collagen content relative to the fibrosa for the post-mortem, 10%, and 30% groups. (e) Measured NAR for both non-pregnant (NP) and pregnant (P) bovine specimens (post-mortem), highlighting an initial increase in NAR in early pregnancy (EP) followed by a gradual recovery to the pre-pregnancy value. These data underscore the relevance of our experimental work by linking it to the physiological phenomenon of pregnancy. Adapted with permission from [46]. (f) Previous layer-specific findings of growth and remodelling during pregnancy from [44,45]. (Online version in colour.)
3.5. Collagen remodelling is predominant in the atrialis and fibrosa layers
Under equibiaxial membrane tension, VICs in the fibrosa layer undergo significantly larger deformations than those in the ventricularis and orthogonally oriented atrialis layers of the MV [48]. A more recent study has shown that the volume overload present in pregnancy leads to increased remodelling activity and, as a result, significant MV dimensional changes. This includes an increase in total collagen content, mainly observed in the fibrosa layer [44,45]. To further elucidate the layer-specific compensatory response of the MV to deviations from homeostatic mechanical stimuli, we developed a layer separation protocol (figure 9a), verified it with histology (figure 9b), and quantified the acid–pepsin soluble collagen of separated atrialis, fibrosa, and ventricularis after a 48-h cyclic stretch treatment at a physiological strain level (10%) and a hyper-physiological strain level (30%). A significantly larger amount of soluble collagen was measured in the atrialis and fibrosa at the 30% strain level (p < 0.05) without any significant changes in the ventricularis (figure 8c,d). These results suggest that most of the collagen remodelling occurs in the atrialis and fibrosa layers of the MV, which agree well with the layer-specific remodelling that occurs in pregnancy (figure 9e,f) [44–46].
3.6. Linking in vitro results to the organ-level in vivo state
Though numerous studies have quantified the biosynthetic changes that are triggered by abnormal mechanical stimuli [18,20,31–33,61,62], little information exists on how these readily obtained in vitro data can be related to the in vivo state of the valve. To frame our in vitro results in a physiological context, we integrated previously reported [50] in vivo MVAL circumferential and radial deformations for both a normal and surgically repaired MV (figure 10a,b; electronic supplementary material, figures S4 and S5) into our downscale MVIC micro-environment model [48]. Briefly, a FE downscale model of the MV was used to simulate MVIC fibrosa NAR during ventricular systole (figure 10c,d). Circumferential deformation of the MVAL decreased after a flat-ring surgical repair, whereas the radial deformation remained relatively the same (figure 10b) [50]. The novelty of this study, however, lies in our ability to simulate in vivo cell deformation throughout the cardiac cycle and link it back to the in vitro benchtop results. Simulation results indicated that the MVIC NAR at peak end-systole in a normal, non-repaired MV is 4.92. This value decreased to 3.28 after a flat-ring surgical repair (figure 10d), suggesting that cellular deformations, to a large degree, parallel the tissue-level reduction in strain observed along the circumferential direction (figure 10b). These simulations further highlight how quickly the NAR changes with tissue strain: increasing from 2.5 to 4.92 (physiological) and 3.28 (repair) in less than 0.1 s—reinforcing the important role played by the valve's dynamic environment on cellular response. To provide additional insight into our hypothesis that VIC deformation is a major driver for mechanobiological responses, we used the simulated MVIC NAR values along with the experimental results to formulate a bracketed array of in vitro cell deformation ranges that capture MVIC homeostasis and, more importantly, deviations from homeostasis (figure 11). At the protein level, the homeostatic range is observed to be between 10% and 20% strain, whereby the NAR is at 3.47 ± 0.46 and 5.05 ± 0.43, respectively. As such, simulations combined with experimental results allowed us to use NAR as a metric to bracket the following levels: (i) hypo-physiological: NAR < 3.30, (ii) physiological: 3.30 < NAR < 5.0 and (iii) hyper-physiological: NAR > 5.0, whereby the hypo- and hyper-physiological levels represent deviations from the physiological VIC homeostatic range. Mechanical stimuli that lead to NAR in these ranges induce alterations in cell phenotypic and biosynthetic responses to re-establish homeostasis. Though the bracketing is performed with rigid NAR boundaries, the bracketed regions are flexible as indicated in the figure (figure 11).
Figure 10.

Using a multi-scale approach to quantify the effect of surgical repair on the MVAL and subsequent MVIC deformation. (a) Schematic of physiological (i.e. normal/non-repaired) MV with corresponding diagram of the position of the five sonocrystals and an MV during flat-ring surgical repair. Heart valve schematics acquired from [63]. (b) Ovine in vivo tissue-level circumferential (solid lines) and radial strains (dotted lines) during ventricular systole for physiological (green) and surgically repaired valve (blue). In vivo kinematic data were acquired through sonocrystal array micrometry [50]. Flat-ring surgical repair leads to a decrease in the maximum circumferential strain, whereas the radial strain remains relatively constant. (c) Schematic diagrams of the MVIC micro-environment model with tissue-level deformations prescribed as boundary conditions whereby cells are included in the tissue model as ellipsoidal inclusions. Nonlinear FE simulations were performed by prescribing the in vivo tissue-level deformations shown in (b) as boundary conditions on the edges of the RVE and predicted deformation fields were analysed to quantify MVIC NAR. Adapted with permission from [48]. (d) Simulated in vivo fibrosa MVIC deformation in a physiological (non-repaired) MV and a surgically repaired MV, plotted in green and blue, respectively. Right: RVE (80 × 80 × 80 µm) of the fibrosa region of the MVAL (10 × 10 mm) with MVICs represented as ellipsoidal inclusions in the loaded and unloaded configurations for a normal (non-repaired/physiological) valve. In the normal valve, fibrosa MVICs reach a maximum NAR of 4.92, whereas those in the flat-ring repair reach an NAR of 3.28, suggesting that VICs are under-loaded after a flat-ring surgical repair. R, radial; C, circumferential; T, transmural. (Online version in colour.)
Figure 11.

MVIC deformation is a major driver for cellular mechanoregulation. This plot shows the fibrosa NAR quantified in the experimental work at each strain level (shown in figure 3) and bracketed regions based on in vivo simulations from figure 9. A maximum fibrosa NAR that is less than 3.28 is bracketed as hypo-physiological, an NAR between 3.28 and 4.92 is bracketed as physiological and an NAR above 4.92 is hyper-physiological. This suggests that there is a narrow physiological range of MVIC NAR. (Online version in colour.)
4. Discussion
Prior work has focused on elucidating the role of mechanical stimuli on cells in their in vivo states using a wide variety of in vitro and ex vivo techniques [18,20,31–33,62]. Though these studies identified the strain-sensitive behaviour of VICs, they lacked scale-up organ-level considerations, which are critical when relating data to in vivo loading conditions. Moreover, these studies have focused on the AV and subsequent effects on aortic VICs [20,32,61]. While both the AV and MV are in the more mechanically demanding left side of the heart, their respective structures vary greatly. The local haemodynamic environment of the MV is very different from that of the AV. In fact, due to these differences and the osteoblast-like VIC phenotype and matrix mineralization, AVs are more susceptible to calcification. MV pathologies, on the other hand, lead to increased deposition of collagen and PGs, resulting in biomechanical weakening of the valvular tissue. As such, our work addresses two important needs in the field of heart valve mechanobiology: first, understanding the effect of mechanical deformation on MVICs, and second, investigating this same effect across different length scales that incorporate organ-, tissue-, and cell-level data unique to the MV.
Because of the complexities involved with measuring these quantities, we chose to take a more simulations-based approach. Here, we used an integrated experimental–computational approach, whereby a load-sensing tissue strip bioreactor was used to stimulate valve tissue at different strain levels and a macro–micro FE model was used to shed light on the multi-scale nature of soft tissue responses to mechanical stimuli and gain insights into VIC behaviour in the normal and post-surgical states. Our findings on collagen fibre alignment show that the collagen/VIC coupling and micromechanical interactions are major drivers for MVIC deformation and subsequent phenotypic activation. Our results indicate that cellular compression occurs in the physiological range, whereas cellular elongation drives mechanobiological response at higher strain levels, suggesting that compression and stretching could lead to different mechanotransduction pathways. The observation that there is minimal transmural compression at hyper-physiological strain levels is an indication that MVIC deformation is coordinated and largely driven by the micromechanical interactions with the collagen fibres, which are elongated at higher strain levels. As such, the sensitivity of MVIC NAR to larger strain levels is largely due to the pronounced mechanical coupling to the nonlinear MV mechanical properties. It is important to note that in the case of valvular tissue, VIC/collagen coupling, particularly through integrins, plays a large role in cell deformation. Integrins, which are transmembrane signal receptors that physically link the cell surface to the surrounding ECM, sort and process these signals, and play a key role in transducing them to the cell interior, leading to a network of intracellular signalling pathways that result in specific cell responses [64]. In the MV, the α2β1 integrin, which shows higher affinity for type I collagen, is the main collagen-binding integrin [65,66]. Work by other groups has shown that VIC/collagen coupling through α2β1 drives VIC deformation when the tissue is strained [65]. This, along with detailed 3D reconstructions of the tissue micro-environment and VIC micro-environment (figure 1) [64,67], suggests that a local mechanism drives cell deformation and encourages us to make the link between cell deformation and increased collagen alignment.
Our demonstration of increased cellular deformation with increasing strain levels is compatible with the findings of others [18,48]. Previous work, however, has mainly focused on quantifying the NAR of valvular cells. The present study, for the first time, reported both cytoplasmic and nuclear deformations and reported evidence for their decoupling at hyper-physiological strain levels, providing insights into VIC mechanotransduction drivers that are in the cell membrane and thus, warranting future work on the transmembrane proteins that drive these mechanotransduction pathways, such as α2β1 [65]. The strong correlation between strain magnitude and α-SMA expression reinforces the concept that mechanical stimuli that are outside of the normal physiological range induce VIC phenotypic activation and subsequent transition into a biosynthetically active myofibroblast-like phenotype [20,32,33,61]. Though our group has quantified cell deformation and correlated it to collagen architecture in the past [18], this is the first time that such a correlation is also made to both the biosynthetic response and phenotypic activation. Although cell deformation correlates well with cellular activation and the biosynthetic response, it is important to note that it is not the only way for VICs to reach this activated, myofibroblast-like phenotype. Other pathologies, such as tachycardia-induced cardiomyopathy [40,68,69], can lead to similar remodelling without necessarily exhibiting significant changes in cell deformation. Another example includes myxomatous MV disease [17,58,70,71], which is defined by the accumulation of mucopolysaccharides responsible for the thickening of valve tissue. In myxomatous leaflets, the expression of PGs and catabolic enzymes is increased and VICs are mostly in an activated myofibroblast phenotype [43]. Though cell deformation has not been measured in these cases, it is known that cellular activation occurs through other mechanisms that are caused by the disease rather than mechanotransduction. In our current experimental–computational framework, cell deformation, more specifically the NAR, is used as a metric only because it allows us to link the benchtop experimental results and the in vivo state of the valve.
Several groups have previously demonstrated that increased mechanical stimuli in an in vitro setting lead to an upregulation of collagen [20,32] and sulfated-GAG [22,31] biosynthesis by aortic and mitral VICs, similar to the observations we made in this study. From a physiological perspective, such an increase in collagen mass fraction was observed in MVs of pregnant subjects [44–46], with a significant increase occurring mainly in the fibrosa layer of the valve. Our in vitro data provides key evidence of this layer-specific remodelling that is notably more pronounced in the fibrosa layer of the MV. This increase in collagen biosynthesis was associated with a decrease in elastin content [44]. Our work suggests that hyper-physiological strain levels can in fact lead to a decrease in elastin content, pointing to increased proteolytic activity by MMPs, TIMPs, and cathepsins [33]. These results also underscore the possible under-stimulation of valvular tissue in some pathologies and surgical repair scenarios and the subsequent downregulation of key ECM and CAM genes.
Though we could discern and quantify both gene- and protein-level changes after 48 h of mechanical stimulation, the duration of this study is far from the physiological timescale of valvular disease progression and post-surgical repair response. Our approach focuses on using a tissue strip system to induce a wide range of MVIC deformations that occur in different conditions in vivo that span the estimated physiological range. This system is best described as ‘strip biaxial’ rather than ‘uniaxial’, whereby λ1 varies while λ2 is constrained on the attachment edges, and λ3 decreases due to tissue incompressibility. Thus, it is closer to a pure-shear test. This said, the focus of this work is not on reproducing in vivo tissue-level deformation modes, which we have shown in the past to be multiaxial [55], but rather on using cellular deformation as a metric to make the link between our benchtop results and the in vivo state of the valve. Thus, the current experimental–computational approach remains a good first step to elucidating the role of mechanical stimuli induced by disease and surgical repair on VIC response while maintaining tissue viability and investigating the effect of key strain levels on VIC mechanobiological response. Finally, we note that our group has shown in multiple studies that valvular tissues, including the MVAL, are functionally elastic [72–74]. As such, MV leaflet in vivo behaviour is effectively rate-independent. In more recent work, we extended our previously developed VIC computational continuum mechanics model [75] to include stress fibre geometries, force–length relations, explicit α-smooth muscle actin (α-SMA) and F-actin expression levels, and strain rate. We integrated this model with micro-indentation measurements and showed that aortic VICs also exhibit rate-independence in the normal state [76]. Based on our previous findings, we do not expect rate effects to influence the deformation of the tissue nor the MVIC population.
The present results, taken together, identify VIC deformation as a key player in cell-based remodelling and valve tissue homeostatic regulation. This is in line with our more recent findings on MVICs in pregnancy, which demonstrate that cellular deformation, quantified using the NAR, plays an important role in the long-term remodelling of the MV (figure 9e) [46]. As such, we set out to use cellular deformation as a metric that links in vitro mechanobiological response to the in vivo state of the valve after surgical repair. We speculate the link to human conditions, particularly IMR, on the basis that a large animal model of IMR developed in sheep resembles the multiple presentations of the human disease [77] and has been used extensively in the surgical community to not only investigate IMR effects on MVIC mechanobiology [40,41,78,79], but to also make the clinical link by studying the effect of annuloplasty ring selection [80]. This novel experimental–computational approach, made possible by in vivo kinematic data [50] and our established macro–micro FE model of the MV [48], enables us to map in vitro cell deformation data to organ-level stimuli. The most important, however, is our ability to place this data in the proper physiological context. With this novel integrated approach, we can bracket cell deformation ranges that lie outside the physiological VIC homeostatic range and, ultimately, get an ‘insider's look’ into in vivo VIC response after surgical repair. Though we chose the MV/MVIC system as a test bed, our experimental–computational framework can be applied on other soft-tissue systems to provide further insights on the adaptation of cellular regulation and the long-term remodelling of soft tissues in both healthy and pathological conditions.
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
Acknowledgements
The authors would like also to acknowledge John Lesicko, Ethan Kwan, Michelle Lu and Karen Tsai.
Data accessibility
Additional data are available in the electronic supplementary material.
Authors' contributions
M.S.S., G.F., R.C.G. and J.H.G. designed the research. S.A., C.-H.L., K.H.D., W.A. and C.T.H. performed the research. G.F. contributed new reagents or analytic tools. S.A. and C.-H.L. analysed data. S.A. and M.S.S. wrote the paper.
Competing interests
The authors declare no conflict of interest.
Funding
This work was supported by the National Institutes of Health (NIH), grant nos R01HL119297, R01HL131872 and R01HL122805. C.-H.L. was in part supported by start-up funds from the School of Aerospace and Mechanical Engineering (AME) at the University of Oklahoma and the American Heart Association Scientist Development Grant Award (16SDG27760143).
References
- 1.Chuong CJ, Fung YC. 1986. On residual stress in arteries. J. Biomech. Eng. 108, 189–192. ( 10.1115/1.3138600) [DOI] [PubMed] [Google Scholar]
- 2.Fung YC. 1991. What are the residual stresses doing in our blood vessels? Ann. Biomed. Eng. 19, 237–249. ( 10.1007/BF02584301) [DOI] [PubMed] [Google Scholar]
- 3.Lanir Y. 2009. Mechanisms of residual stress in soft tissues. J. Biomech. Eng. 131, 044506 ( 10.1115/1.3049863) [DOI] [PubMed] [Google Scholar]
- 4.Ogden R. 2003. Nonlinear elasticity, anisotropy, material stability, and residual stresses in soft tissue. In Biomechanics of soft tissue in cardiovascular system (ed. Ogden RW.), pp. 65–108. New York, NY: Springer. [Google Scholar]
- 5.Taber LA, Humphrey JD. 2001. Stress-modulated growth, residual stress, and vascular heterogeneity. J. Biomech. Eng. 123, 528–535. ( 10.1115/1.1412451) [DOI] [PubMed] [Google Scholar]
- 6.Fung YC, Liu SQ. 1991. Changes of zero-stress state of rat pulmonary arteries in hypoxic hypertension. J. Appl. Physiol. 70, 2455–2470. ( 10.1063/1.349398) [DOI] [PubMed] [Google Scholar]
- 7.Liu SQ, Fung YC. 1989. Relationship between hypertension, hypertrophy, and opening angle of zero-stress state of arteries following aortic constriction. J. Biomech. Eng. 111, 325–335. ( 10.1115/1.3168386) [DOI] [PubMed] [Google Scholar]
- 8.Dunkman AA., et al. 2014. The injury response of aged tendons in the absence of biglycan and decorin. Matrix. Biol. 35, 232–238. ( 10.1016/j.matbio.2013.1010.1008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunkman AA, et al. 2013. Decorin expression is important for age-related changes in tendon structure and mechanical properties. Matrix. Biol. 32, 3–13. ( 10.1016/j.matbio.2012.11.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson PS, Derwin KA, Iozzo RV, Soslowsky LJ, Lin TW, Reynolds PR. 2004. Strain-rate sensitive mechanical properties of tendon fascicles from mice with genetically engineered alterations in collagen and decorin. J. Biomech. Eng. 126, 252–257. ( 10.1115/1.1695570) [DOI] [PubMed] [Google Scholar]
- 11.Robinson PS, Huang T-F, Kazam E, Iozzo RV, Soslowsky LJ, Birk DE. 2005. Influence of decorin and biglycan on mechanical properties of multiple tendons in knockout mice. J. Biomech. Eng. 127, 181–185. ( 10.1115/1.1835363) [DOI] [PubMed] [Google Scholar]
- 12.Upton ML, Chen J, Guilak F, Setton LA. 2003. Differential effects of static and dynamic compression on meniscal cell gene expression. J. Orthop. Res. 21, 963–969. ( 10.1016/S0736-0266(03)00063-9) [DOI] [PubMed] [Google Scholar]
- 13.Gupta V, Tseng H, Lawrence BD, Grande-Allen KJ. 2009. Effect of cyclic mechanical strain on glycosaminoglycan and proteoglycan synthesis by heart valve cells. Acta Biomater. 5, 531–540. ( 10.1016/j.actbio.2008.10.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta V, Grande-Allen KJ. 2006. Effects of static and cyclic loading in regulating extracellular matrix synthesis by cardiovascular cells. Cardiovasc. Res. 72, 375–383. ( 10.1016/j.cardiores.2006.08.017) [DOI] [PubMed] [Google Scholar]
- 15.Quick DW, Kunzelman KS, Kneebone JM, Cochran RP. 1997. Collagen synthesis is upregulated in mitral valves subjected to altered stress. ASAIO J. 43, 181–186. [PubMed] [Google Scholar]
- 16.Stephens EH, Nguyen TC, Itoh A, Ingels NB Jr, Miller DC, Grande-Allen KJ. 2008. The effects of mitral regurgitation alone are sufficient for leaflet remodeling. Circulation 118, S243–S249. ( 10.1161/circulationaha.107.757526) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grande-Allen KJ, Griffin BP, Ratliff NB, Cosgrove DM, Vesely I. 2003. Glycosaminoglycan profiles of myxomatous mitral leaflets and chordae parallel the severity of mechanical alterations. J. Am. Coll. Cardiol. 42, 271–277. ( 10.1016/S0735-1097(03)00626-0) [DOI] [PubMed] [Google Scholar]
- 18.Sacks MS, Merryman WD, Schmidt DE. 2009. On the biomechanics of heart valve function. J. Biomech. 42, 1804–1824. ( 10.1016/j.jbiomech.2009.05.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ku CH, Johnson PH, Batten P, Sarathchandra P, Chambers RC, Taylor PM, Yacoub MH, Chester AH. 2006. Collagen synthesis by mesenchymal stem cells and aortic valve interstitial cells in response to mechanical stretch. Cardiovasc. Res. 71, 548–556. ( 10.1016/j.cardiores.2006.03.022) [DOI] [PubMed] [Google Scholar]
- 20.Balachandran K, Konduri S, Sucosky P, Jo H, Yoganathan A. 2006. An ex vivo study of the biological properties of porcine aortic valves in response to circumferential cyclic stretch. Ann. Biomed. Eng. 34, 1655–1665. ( 10.1007/s10439-006-9167-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H-YS, Liao J, Sacks MS. 2007. In-situ deformation of the aortic valve interstitial cell nucleus under diastolic loading. J. Biomech. Eng. 129, 880–889. [DOI] [PubMed] [Google Scholar]
- 22.Gupta V, Werdenberg JA, Blevins TL, Grande-Allen KJ. 2007. Synthesis of glycosaminoglycans in differently loaded regions of collagen gels seeded with valvular interstitial cells. Tissue Eng. 13, 41–49. ( 10.1089/ten.2006.0091) [DOI] [PubMed] [Google Scholar]
- 23.Tao G, Kotick JF, Lincoln J, Lincoln J. 2012 Heart valve development, maintenance, and disease: the role of endothelial cells Curr. Top. Dev. Biol. 100, 203–232. [DOI] [PubMed] [Google Scholar]
- 24.Drach A, Khalighi AH, Sacks MS. 2017. A comprehensive pipeline for multi-resolution modeling of the mitral valve: validation, computational efficiency, and predictive capability. Inter. J. Num. Met. Biomed. Eng. e2921, 1–30. ( 10.1002/cnm.2921) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khalighi AH, Drach A, Bloodworth CHT, Pierce EL, Yoganathan AP, Gorman RC, Gorman JH 3rd, Sacks MS. 2017. Mitral valve chordae tendineae: topological and geometrical characterization. Ann. Biomed. Eng. 45, 378–393. ( 10.1007/s10439-016-1775-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khalighi AH, Drach A, ter Huurne FM, Lee CH, Bloodworth C, Pierce EL, Jensen MO, Yoganathan AP, Sacks MS. 2015. A comprehensive framework for the characterization of the complete mitral valve geometry for the development of a population-averaged model. In Functional Imaging and Modeling of the Heart: 8th International Conference, FIMH 2015, Maastricht, The Netherlands, June 25–27, 2015 (eds H van Assen, P Bovendeerd, T Delhaas), pp. 164–171. Proceedings, Springer International Publishing, Cham, 2015. [Google Scholar]
- 27.Bloodworth CH, Pierce EL, Easley TF, Drach A, Khalighi AH, Toma M, Jensen MO, Sacks MS, Yoganathan AP. 2017. Ex Vivo Methods for Informing Computational Models of the Mitral Valve. Annals of biomedical engineering. 45, 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen JH, Simmons CA. 2011. Cell–matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ. Res. 108, 1510–1524. ( 10.1161/circresaha.110.234237) [DOI] [PubMed] [Google Scholar]
- 29.David MW, Shadow HHY, Schoen FJ, Sacks MS. 2006. The effects of cellular contraction on aortic valve leaflet flexural stiffness. J. Biomech. 39, 88–96. ( 10.1016/j.jbiomech.2004.11.008) [DOI] [PubMed] [Google Scholar]
- 30.Bairati A, DeBiasi S. 1981. Presence of a smooth muscle system in aortic valve leaflets. Anat. Embryol. 161, 329–340. ( 10.1007/BF00301830) [DOI] [PubMed] [Google Scholar]
- 31.Lacerda CM, Kisiday J, Johnson B, Orton EC. 2012. Local serotonin mediates cyclic strain-induced phenotype transformation, matrix degradation, and glycosaminoglycan synthesis in cultured sheep mitral valves. Am. J. Physiol. 302, H1983–H1990. ( 10.1152/ajpheart.00987.2011) [DOI] [PubMed] [Google Scholar]
- 32.Merryman WD, Lukoff HD, Long RA, Engelmayr GC Jr, Hopkins RA, Sacks MS. 2007. Synergistic effects of cyclic tension and transforming growth factor-beta1 on the aortic valve myofibroblast. Cardio. Pathol. 16, 268–276. ( 10.1016/j.carpath.2007.03.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balachandran K, Sucosky P, Jo H, Yoganathan AP. 2009. Elevated cyclic stretch alters matrix remodeling in aortic valve cusps: implications for degenerative aortic valve disease. Am. J. Physiol 296, H756–H764. ( 10.1152/ajpheart.00900.2008) [DOI] [PubMed] [Google Scholar]
- 34.Wyss K, Yip CY, Mirzaei Z, Jin X, Chen J-H, Simmons CA. 2012. The elastic properties of valve interstitial cells undergoing pathological differentiation. J. Biomech. 45, 882–887. ( 10.1016/j.jbiomech.2011.11.030) [DOI] [PubMed] [Google Scholar]
- 35.Carpentier A. 1983. Cardiac valve surgery—the ‘French correction’. J. Thorac. Cardiovasc. Surg. 86, 323–337. [PubMed] [Google Scholar]
- 36.Carpentier A, et al. 1980. Reconstructive surgery of mitral valve incompetence: ten-year appraisal. J. Thorac. Cardiovasc. Surg. 79, 338–348. [PubMed] [Google Scholar]
- 37.Gillinov AM, Blackstone EH, White J, Howard M, Ahkrass R, Marullo A, Cosgrove DM. 2001. Durability of combined aortic and mitral valve repair. Ann. Thorac. Surg. 72, 20–27. ( 10.1016/S0003-4975(01)02677-7) [DOI] [PubMed] [Google Scholar]
- 38.Go AS, et al. 2014. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 129, e28–e292. ( 10.1161/01.cir.0000441139.02102.80) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGee EC, et al. 2004. Recurrent mitral regurgitation after annuloplasty for functional ischemic mitral regurgitation. J. Thorac. Cardiovasc. Surg. 128, 916–924. ( 10.1016/j.jtcvs.2004.07.037) [DOI] [PubMed] [Google Scholar]
- 40.Stephens EH, Timek TA, Daughters GT, Kuo JJ, Patton AM, Baggett LS, Ingels NB, Miller DC, Grande-Allen KJ. 2009. Significant changes in mitral valve leaflet matrix composition and turnover with tachycardia-induced cardiomyopathy. Circulation 120, S112–S119. ( 10.1161/circulationaha.108.844159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dal-Bianco JP, et al. 2009. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation 120, 334–342. ( 10.1161/circulationaha.108.846782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dal-Bianco JP, et al. 2016. Myocardial infarction alters adaptation of the tethered mitral valve. J. Am. Coll. Cardiol. 67, 275–287. ( 10.1016/j.jacc.2015.10.092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine RA, et al. 2015. Mitral valve disease-morphology and mechanisms. Nat. Rev. Cardiol. 12, 689–710. ( 10.1038/nrcardio.2015.161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pierlot CM, Lee JM, Amini R, Sacks MS, Wells SM. 2014. Pregnancy-induced remodeling of collagen architecture and content in the mitral valve. Ann. Biomed. Eng. 42, 2058–2071. ( 10.1007/s10439-014-1077-6) [DOI] [PubMed] [Google Scholar]
- 45.Wells SM, Pierlot CM, Moeller AD. 2012. Physiological remodeling of the mitral valve during pregnancy. Am. J. Physiol 303, H878–H892. ( 10.1152/ajpheart.00845.2011) [DOI] [PubMed] [Google Scholar]
- 46.Rego BV, Wells SM, Lee CH, Sacks MS. 2016. Mitral valve leaflet remodelling during pregnancy: insights into cell-mediated recovery of tissue homeostasis. J. R. Soc. Interface 13, 20160709 ( 10.1098/rsif.2016.0709) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee CH, Amini R, Gorman RC, Gorman JH III, Sacks MS. 2014. An inverse modeling approach for stress estimation in mitral valve anterior leaflet valvuloplasty for in-vivo valvular biomaterial assessment. J. Biomech. 47, 2055–2063. ( 10.1016/j.jbiomech.2013.10.058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee CH, Carruthers CA, Ayoub S, Gorman RC, Gorman JH III, Sacks MS. 2015. Quantification and simulation of layer-specific mitral valve interstitial cells deformation under physiological loading. J. Theor. Biol. 373, 26–39. ( 10.1016/j.jtbi.2015.03.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sacks MS, Smith DB, Hiester ED. 1997. A small angle light scattering device for planar connective tissue microstructural analysis. Ann. Biomed. Eng. 25, 678–689. ( 10.1007/BF02684845) [DOI] [PubMed] [Google Scholar]
- 50.Amini R, Eckert CE, Koomalsingh K, McGarvey J, Minakawa M, Gorman JH, Gorman RC, Sacks MS. 2012. On the in vivo deformation of the mitral valve anterior leaflet: effects of annular geometry and referential configuration. Ann. Biomed. Eng. 40, 1455–1467. ( 10.1007/s10439-012-0524-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carruthers CA, Alfieri CM, Joyce EM, Watkins SC, Yutzey KE, Sacks MS. 2012. Gene expression and collagen fiber micromechanical interactions of the semilunar heart valve interstitial cell. Cell. Mol. Bioeng. 5, 254–265. ( 10.1007/s12195-012-0230-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fan R, Sacks MS. 2014. Simulation of planar soft tissues using a structural constitutive model: finite element implementation and validation. J. Biomech. 47, 2043–2054. ( 10.1016/j.jbiomech.2014.03.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stephens EH, et al. 2011. Differential proteoglycan and hyaluronan distribution in calcified aortic valves. Cardiovasc. Pathol. 20, 334–342. ( 10.1016/j.carpath.2010.10.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang HY, Liao J, Sacks MS. 2007. In-situ deformation of the aortic valve interstitial cell nucleus under diastolic loading. J. Biomech. Eng. 129, 880–889. ( 10.1115/1.2801670) [DOI] [PubMed] [Google Scholar]
- 55.Sacks MS, Enomoto Y, Graybill JR, Merryman WD, Zeeshan A, Yoganathan AP, Levy RJ, Gorman RC, Gorman JH 3rd. 2006. In-vivo dynamic deformation of the mitral valve anterior leaflet. Ann. Thorac. Surg. 82, 1369–1377. ( 10.1016/j.athoracsur.2006.03.117) [DOI] [PubMed] [Google Scholar]
- 56.Sacks MS, He Z, Baijens L, Wanant S, Shah P, Sugimoto H, Yoganathan AP. 2002. Surface strains in the anterior leaflet of the functioning mitral valve. Ann. Biomed. Eng. 30, 1281–1290. ( 10.1114/1.1529194) [DOI] [PubMed] [Google Scholar]
- 57.Carruthers CA, Good B, D'Amore A, Liao JA, Watkins SC, Sacks MS. 2012. Alterations in the microstructure of the anterior mitral valve leaflet under physiological stress. In ASME 2012 Summer Bioengineering Conference. American Society of Mechanical Engineers, pp. 227–228. Fajardo, Puerto Rico. [Google Scholar]
- 58.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. 2001. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 104, 2525–2532. ( 10.1161/hc4601.099489) [DOI] [PubMed] [Google Scholar]
- 59.Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. 2003. The cardiac valve interstitial cell. Inter. J. Biochem. Cell Biol. 35, 113–118. ( 10.1016/S1357-2725(02)00100-0) [DOI] [PubMed] [Google Scholar]
- 60.Rabkin E, Hoerstrup SP, Aikawa M, Mayer JE Jr, Schoen FJ. 2002. Evolution of cell phenotype and extracellular matrix in tissue-engineered heart valves during in-vitro maturation and in-vivo remodeling. J. Heart Valve Dis. 11, 308–314; discussion 314. [PubMed] [Google Scholar]
- 61.Balachandran K, Alford PW, Wylie-Sears J, Goss JA, Grosberg A, Bischoff J, Aikawa E, Levine RA, Parker KK. 2011. Cyclic strain induces dual-mode endothelial–mesenchymal transformation of the cardiac valve. Proc. Natl Acad. Sci. USA 108, 19 943–19 948. ( 10.1073/pnas.1106954108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thayer P, Balachandran K, Rathan S, Yap CH, Arjunon S, Jo H, Yoganathan AP. 2011. The effects of combined cyclic stretch and pressure on the aortic valve interstitial cell phenotype. Ann. Biomed. Eng. 39, 1654–1667. ( 10.1007/s10439-011-0273-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaiser LR, Kron IL, Spray TL. 2014. Mastery of cardiothoracic surgery, 3rd edn, xxii, 1210 p Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins. [Google Scholar]
- 64.Ayoub S, Ferrari G, Gorman RC, Gorman JH, Schoen FJ, Sacks MS. 2016. Heart valve biomechanics and underlying mechanobiology. Comprehen. Physiol. 6, 1743–1780. ( 10.1002/cphy.c150048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stephens E, Durst C, Swanson J, Grande-Allen KJ, Ingels N Jr, Miller DC. 2010. Functional coupling of valvular interstitial cells and collagen via α2β1 integrins in the mitral leaflet. Cel. Mol. Bioeng. 3, 428–437. ( 10.1007/s12195-010-0139-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barczyk M, Carracedo S, Gullberg D. 2010. Integrins. Cell Tissue Res. 339, 269–280. ( 10.1007/s00441-009-0834-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rego BV, Sacks MS. 2017. A functionally graded material model for the transmural stress distribution of the aortic valve leaflet J. Biomech. 54, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Timek TA, Dagum P, Lai DT, Liang D, Daughters GT, Tibayan F, Ingels NB Jr, Miller DC. 2003. Tachycardia-induced cardiomyopathy in the ovine heart: mitral annular dynamic three-dimensional geometry. J. Thorac. Cardiovasc. Surg. 125, 315–324. ( 10.1067/mtc.2003.80) [DOI] [PubMed] [Google Scholar]
- 69.Timek TA, Lai DT, Dagum P, Liang D, Daughters GT, Ingels NB Jr, Miller DC. 2006. Mitral leaflet remodeling in dilated cardiomyopathy. Circulation 114, I518–I523. ( 10.1161/CIRCULATIONAHA.105.000554) [DOI] [PubMed] [Google Scholar]
- 70.Cole WG, Chan D, Hickey AJ, Wilcken DE. 1984. Collagen composition of normal and myxomatous human mitral heart valves. Biochem. J. 219, 451–460. ( 10.1042/bj2190451) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. 2004. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J. Heart Valve Dis. 13, 841–847. [PubMed] [Google Scholar]
- 72.Grashow JS, Sacks MS, Liao J, Yoganathan AP. 2006. Planar biaxial creep and stress relaxation of the mitral valve anterior leaflet. Ann. Biomed. Eng. 34, 1509–1518. ( 10.1007/s10439-006-9183-8) [DOI] [PubMed] [Google Scholar]
- 73.Grashow JS, Yoganathan AP, Sacks MS. 2006. Biaxial stress–stretch behavior of the mitral valve anterior leaflet at physiologic strain rates. Ann. Biomed. Eng. 34, 315–325. ( 10.1007/s10439-005-9027-y) [DOI] [PubMed] [Google Scholar]
- 74.Liao J, Yang L, Grashow J, Sacks MS. 2007. The relation between collagen fibril kinematics and mechanical properties in the mitral valve anterior leaflet. J. Biomech. Eng. 129, 78–87. ( 10.1115/1.2401186) [DOI] [PubMed] [Google Scholar]
- 75.Sakamoto Y, Buchanan RM, Sacks MS. 2016. On intrinsic stress fiber contractile forces in semilunar heart valve interstitial cells using a continuum mixture model. J. Mech. Behav. Biomed. Mater. 54, 244–258. ( 10.1016/j.jmbbm.2015.09.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakamoto Y, Buchanan RM, Sanchez-Adams J, Guilak F, Sacks MS. 2017. On the functional role of valve interstitial cell stress fibers: a continuum modeling approach. J Biomech Eng 139, 021007 ( 10.1115/1.4035557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Llaneras MR, Nance ML, Streicher JT, Lima JA, Savino JS, Bogen DK, Deac RF, Ratcliffe MB, Edmunds LH Jr. 1994. Large animal model of ischemic mitral regurgitation. Ann. Thorac. Surg. 57, 432–439. ( 10.1016/0003-4975(94)91012-X) [DOI] [PubMed] [Google Scholar]
- 78.Bischoff J, et al. 2016. CD45 expression in mitral valve endothelial cells after myocardial infarction. Circ. Res. 119, 1215–1225. ( 10.1161/circresaha.116.309598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rego BV, Ayoub S, Khalighi AH, Drach A, Gorman JH III, Gorman RC, Sacks MS. Alterations in mechanical properties and in vivo geometry of the mitral valve following myocardial infarction. In Proc. of the 2017 Summer Biomechanics, Bioengineering and Biotransport Conf., 21–24 June, Tucson, AZ. [Google Scholar]
- 80.Gorman JH 3rd, Gorman RC, Jackson BM, Enomoto Y, St John-Sutton MG, Edmunds LH Jr. 2003. Annuloplasty ring selection for chronic ischemic mitral regurgitation: lessons from the ovine model. Ann. Thorac. Surg. 76, 1556–1563. ( 10.1016/S0003-4975(03)00891-9) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional data are available in the electronic supplementary material.