

Abstract
Trifluridine/tipiracil (FTD/TPI) is a combination of FTD, an antineoplastic thymidine-based nucleoside analog, and TPI, which acts to enhance the bioavailability of FTD in vivo. It is used to treat patients with unresectable advanced or recurrent colorectal cancer that is refractory to standard therapies. We investigated the anticancer activity of FTD/TPI combined with anti-mouse programed cell death 1 (PD-1) monoclonal antibody (mAb) against CMT-93 cells, which are microsatellite stable (MSS)-type murine colorectal cancer cells. Tumor growth inhibition (TGI) after treatment with anti-mouse PD-1 mAb monotherapy (0.1 mg, i.p., days 1, 5, 9) and FTD/TPI monotherapy (150 mg/kg/day, p.o., days 1-14) were 86.7% and 52.7%, respectively, and that of the combination was 98.4%. The TGI of the combination therapy was significantly greater than that of each monotherapy (P<0.05). The combination therapy caused complete tumor regression in four out of five mice without body-weight reduction, but neither of the monotherapies resulted in complete tumor regression. Low dose FTD/TPI (75 and 100 mg/kg) combined with anti-mouse PD-1 mAb also showed significant antitumor activity against CMT-93 tumors. Flow cytometric analysis revealed that a higher CD8+ T cell ratio among total lymphocytes and a lower regulatory T cells (Tregs) ratio in CD4+ T cells in the combination group compared with that in the control group. These results suggested that the combination therapy induced a cytotoxic response from infiltrated cytotoxic CD8+ T cells and reduced immunosuppressive activity as indicated by decreased Tregs. In this study, the combination therapy was found to have synergistically greater antitumor activity against CMT-93 cells. These preclinical findings indicated that FTD/TPI and anti-mouse PD-1 mAb combination therapy may be a promising treatment option, even for MSS-type colorectal cancer.
Keywords: TAS-102, trifluridine, tipiracil, anti-mouse PD-1 antibody, MSS, MSI, colorectal cancer
Introduction
Trifluridine/tipiracil (FTD/TPI or TFTD), also known as TAS-102, is a combination drug used for the treatment of metastatic colorectal cancer [1]. It comprises a mixture of two distinct chemicals, FTD and TPI, at a molar ratio of 1:0.5. FTD, an analog of thymidine, exhibits two mechanisms of antitumor action. It inhibits thymidylate synthase [2] and is also incorporated into DNA, thereby inducing DNA dysfunction [3]. TPI enhances the bioavailability of FTD by inhibiting its enzymatic degradation by thymidine phosphorylase. TPI is therefore beneficial for producing a more durable and sustained response to FTD [4]. The primary cytotoxic mechanism of FTD/TPI in the preclinical colon cancer model or in patients with colorectal cancer is thought to be incorporation of FTD into DNA [5]. FTD/TPI significantly improved overall survival and has a favorable safety profile in patients with metastatic colorectal cancer refractory to standard chemotherapies in the phase III study (RECOURSE) [6]. Recent studies indicated that tumor cells have acquired several methods of escaping from host immunity in the tumor microenvironment [7]. One of the most important components of the mechanism is an immunosuppressive co-signal (immune checkpoint) mediated by programmed cell death 1 (PD-1)/PD-1 ligand 1 (PD-L1) in the tumor microenvironment. Several clinical trials of PD-1/PD-L1 signal-blockade agents have exhibited dramatic antitumor efficacy in patients with certain types of cancers. In a phase II clinical trial of pembrolizumab in 50 patients with colon cancer, RR was higher in the mismatch repair (MMR)-deficient group than in the MMR-proficient group, 40% versus 0% in the initial report [8]. Recently, pembrolizumab was approved for the treatment of adult and pediatric patients with unresectable or metastatic solid tumors that have been identified as microsatellite instability (MSI)-high or MMR-deficient. MMR deletion has become an important indicator of therapeutic efficacy of anti-PD-1 antibody. However, only 4% to 6% of metastatic colorectal cancers are associated with MSI-H [9,10]. There is room for treatment development for MSS, which account for the majority.
To enhance the antitumor effect, many clinical trials have sought to determine the PD-1 inhibitors combination with other antitumor therapy, such as chemotherapy, targeted therapy, radiotherapy, or other immunotherapy [11]. Most clinical studies in colorectal cancer patients were performed with MSI-type cancer [12]. Therefore, new developments in the treatment of MSS-type colorectal cancer were thought to be a crucial clinical issue related to PD-1 inhibitors.
We previously reported that FTD/TPI in combination with irinotecan hydrochloride [13], oxaliplatin [14], bevacizumab, cetuximab, or panitumumab [15], and nintedanib [16] had superior in vivo activity against human colorectal cancer xenografts, including 5FU (5-fluorouracil)-resistant tumors, compared with any of these drugs alone. In this study, we aimed to evaluate the effects of FTD/TPI in combination with anti-PD-1 monoclonal antibody (mAb) in an MSS-type syngeneic mouse colorectal cancer model. We also investigated tumor infiltrating lymphocytes (TILs) and regulatory T cells (Tregs) after the same drug treatment used in the anticancer evaluations to examine the enhancement of antitumor efficacy by the combination therapy.
Materials and methods
Reagents
FTD and TPI were obtained from Taiho Pharmaceutical Co., Ltd. (Tokyo, Japan). Purified anti-mouse PD-1 mAb (clone RMP1-14) was purchased from BioXCell (West Lebanon, NH, USA). Hydroxypropyl methylcellulose (HPMC) was obtained from Shin-Etsu Chemical Co., Ltd (Tokyo, Japan).
Cancer cell lines
The murine male rectal CMT-93 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were cultured in Dulbecco’s modified Eagle’s cell culture media supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2 in air. The culture media and fetal bovine serum were obtained from Sigma-Aldrich Co., LLC (Tokyo, Japan).
MSI genotyping. The D1Mit15, D1Mit36, D3Mit22, D9Mit2, D10Mit2, D15Mit16, D16Mit10, and D19Mit33 repeats were amplified using fluorescent dye-labeled primer pairs flanking the (CA)n microsatellites, then the PCR fragments were analyzed using a 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) [17].
Next generation sequencing (NGS) and data processing
Exome capture from murine cell lines was carried out using the Agilent SureSelect Mouse All Exon kit. Libraries (Agilent Technologies International Japan, Ltd., Tokyo, Japan) were prepared using the SureSelect XT Target Enrichment System, sequenced on an Illumina HiSeq2500 (Illumina, Inc., San Diego, CA, USA). DNA-derived sequence reads were aligned to the mm9 genome.
Animals
Five-week-old male mice (C57BL/6J) were purchased from CLEA Japan (Tokyo, Japan) and housed under specific pathogen-free conditions, with food and water provided ad libitum. All animal studies were performed according to the protocol and guidelines of the Institutional Animal Care and Use Committee of Taiho Pharmaceutical Co. Ltd and with their approval.
Antitumor activity in vivo
CMT-93 tumor cells (approximately 5 × 106 cells/mouse) were transplanted subcutaneously into the dorsal region of each mouse. Approximately 9 days later, the animals were grouped so that the mean and standard deviation of the tumor volume (calculated using the equation below) would be as uniform as possible in all groups. Each group consisted of 6 mice on day 0.
FTD/TPI was prepared by mixing FTD and TPI at a molar ratio of 1:0.5 in 0.5% HPMC solution. The dose of FTD/TPI was expressed based on FTD content. FTD/TPI was administered orally from day 1 to 14 at the reported optimal effective dose (150 mg/kg/day) [5] or at a decreased dose (100 or 75 mg/kg/day). Anti-mouse PD-1 mAb was injected intraperitoneally at a dose of 0.1, 0.2, or 0.5 mg on days 1, 5, and 9 [18]. For the control group, 10 mL/kg of vehicle (0.5% HPMC solution) was administered orally from days 1 to 14 and 0.1 mL of saline was injected intraperitoneally on days 1, 5, and 9.
Tumor diameters were measured twice a week and the tumor volume was estimated as follows: 0.5 × length × width2. The tumor growth inhibition ratio (TGI, %) was calculated using the following formula: TGI (%) = [1 - (TV of the treated group)/(TV of the control group)] × 100. The antitumor effect of the drugs was evaluated 14 days after the final drug administration (day 28).
To evaluate toxicity, body weight change (BWC) was calculated using the following formula: BWC (%) = [(body weight on measured day) - (body weight on day 0)]/(body weight on day 0) × 100. Toxicity was defined as a BWC indicating weight loss of >20% or toxic death. The experimental endpoint was defined as the day on which the average tumor volume in the average body weight within each group reached more than 10%.
Flow cytometry analysis of tumor-infiltrating T cells
The separated tumor tissues were cut into small pieces and digested at 37°C for at least 20 min in RPMI 1640 supplemented with 0.05% collagenase Type IV (Worthington Biochemical Corporation, NJ, USA), 0.002% DNase I (Sigma-Aldrich Japan, Cat 10104159001 grade II), and 20% fetal calf serum (Sigma-Aldrich Japan). Dissociated cells were then filtered through 100, 70, and 40-μm mesh using 10 mL of RPMI 1640 supplemented with 20% fetal bovine serum (Sigma-Aldrich Japan), then washed twice with PBS for 10 min at 360 × g and 4°C.
To define the percentage of tumor-infiltrating CD4+ T cells and Tregs, we obtained a mouse regulatory T cell staining kit (Cat. 130-094-164, Miltenyi Biotec, Bergisch-Gladbach, Germany) [19]. To immunostain CD8+ T cells, we obtained PE/Cy7 anti-mouse CD8 (BioLegend, San Diego, CA, USA). Briefly, 90 μL of the prepared cells (containing 1 × 106 cells) in BSA-EDTA buffer (0.5% bovine serum albumin, 2 mM EDTA in PBS, pH 7.2) was added to each tube (test or control). To stain surface molecules, 10 μL of CD4 antibody and CD25 antibody and 1 μL of PE/Cy7 anti-mouse CD8 antibody were added to each tube and the tubes were incubated for 30 min on ice. After washing twice using 1 mL BSA-EDTA buffer, the cells were resuspended and 1 mL of fixation buffer was added. The mixture was kept for 30 min on ice in the dark. The cells were then washed twice using BSA-EDTA buffer and 80 μL of permeabilization buffer was added. Ten microliters of anti-mouse Foxp3 antibody was added and the cells were incubated for 30 min on ice. Thereafter, they were washed with permeabilization buffer and resuspended in flow cytometry staining buffer. The samples were analyzed using a BD Accuri C6 flow cytometer (BD, San Jose, CAUSA). A total of 5 × 105 events was read for each sample and the results were analyzed using the BD Accuri C6 software (ver.1.0.264.21) software.
Statistical analysis
Differences in the mean TV between the treated and control groups on day 28 were assessed with a closed testing procedure using the Aspin-Welch two-sided t-test [20]. The combinatorial antitumor effect of FTD/TPI and anti-mouse PD-1 mAb was analyzed with a closed testing procedure using the Aspin-Welch two-tailed t-test. Statistical significance was assigned at P<0.05. P-values were calculated using EXSUS, ver. 8.1 (CAC Exicare Corp., Osaka, Japan). Differences in TILs in the drug treated group and the control group were assessed using the Student’s one-sided t-test.
Results
Genomic characterization of the CMT-93 cell line
First, we performed genotyping on the CMT-93 mouse tumor cell line using eight microsatellite markers (Supplementary Figure 1) [17]. All eight microsatellite markers were found to be stable in CMT-93 cells. We then performed whole-exome sequencing on CMT-93 cells to identify tumor-specific point mutations. Coding variants were measured relative to the reference mouse genome to identify 2108 and 1979 with non-synonymous and synonymous variants, respectively. DNA repair-related genes, such as Mlh1, Mlh3, Msh2, Msh6, and Pms, were found to be wild-type. Therefore, the genomic characterization of the CRC cell line CMT-93 demonstrated its validity as a model for MSS colorectal cancer.
Dose determination for anti-mouse PD-1 mAb alone in CMT-93 cells
We first performed a preliminary study to confirm the antitumor activity and toxicity of FTD/TPI monotherapy and anti-mouse PD-1-mAb monotherapy in CMT-93 cells (Figure 1A and 1B). We treated C57BL/6J mice bearing CMT-93 tumors with FTD/TPI and anti-mouse PD-1 mAb. On day 28, both FTD/TPI alone and anti-mouse PD-1 mAb alone exhibited significant antitumor activity (P<0.01). TGI on day 28 was 53.3% for FTD/TPI at 150 mg/kg and 86.4, 90.1, and 90.7% for anti-mouse PD-1 mAb at 0.1, 0.2, and 0.5 mg, respectively. There were no changes in body weight. From these results, the 0.1 mg dose of anti-mouse PD-1 on days 1, 5, and 9 was further used to evaluate combination therapy with FTD/TPI.
Figure 1.
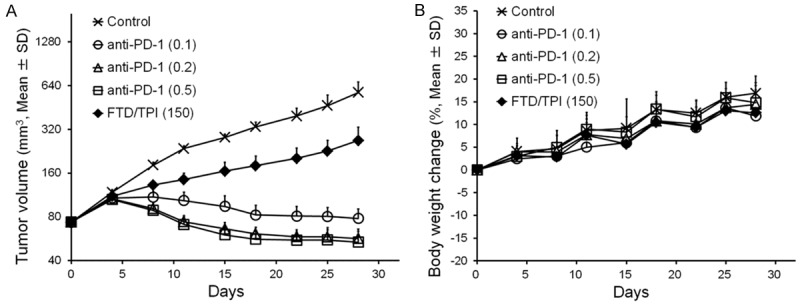
Effect of anti-mouse PD-1 mAb alone and FTD/TPI alone in mouse colorectal CMT-93 tumors. Change in tumor volume (A) and body weight in CMT-93 tumor-bearing mice (B). Mice were treated with vehicle (x), anti-mouse PD-1 mAb (0.1 mg, intraperitoneal administration on days 1, 5, and 9, ○), anti-mouse PD-1 mAb (0.2 mg, intraperitoneal administration on days 1, 5, and 9, Δ), anti-mouse PD-1 mAb (0.5 mg, intraperitoneal administration at days 1, 5, and 9, □), or FTD/TPI (150 mg/kg, orally daily from days 1 to 14, ♦). Values indicate means ± SD (n = 6). Tumor volume and body weight were measured twice weekly.
Antitumor efficacy of FTD/TPI and anti-mouse PD-1 mAb combination therapy in vivo
We next assessed TGI and changes in body weight after treatment with anti-mouse PD-1 mAb monotherapy, FTD/TPI monotherapy, and the combination in the CMT-93 cancer model (Figure 2A and 2B). TGI on day 28 was 87.0% for anti-mouse PD-1 mAb alone, 52.7% for FTD/TPI alone, and 98.4% for anti-mouse PD-1 mAb plus FTD/TPI. On day 28, suppression of tumor growth was significantly greater after the combination therapy compared to that with each monotherapy (P<0.01). No signs of toxicity were observed in the mice.
Figure 2.
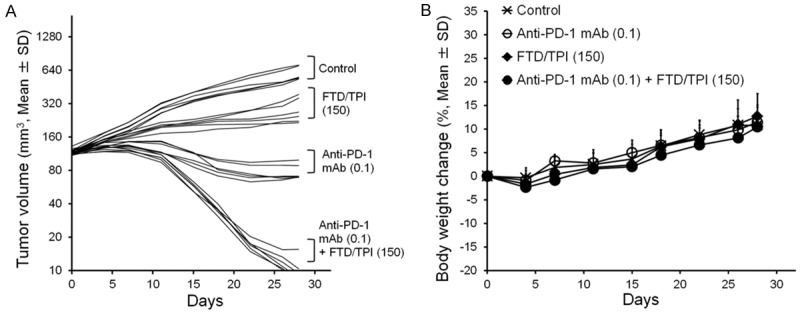
Effect of anti-mouse PD-1 mAb and FTD/TPI combination therapy in mouse colorectal CMT-93 tumors. Change in tumor volume (A) and body weight in CMT-93 tumor-bearing mice (B). Anti-mouse PD-1 mAb was administered intraperitoneally on days 1, 5, and 9. FTD/TPI was administered orally daily from days 1 to 14. Mice were treated with vehicle (x), anti-mouse PD-1 mAb (0.1 mg, ○), FTD/TPI (150 mg/kg, ♦), or anti-mouse PD-1 mAb (0.1 mg) plus FTD/TPI (150 mg/kg) (●). Values indicate means ± SD (n = 6). Tumor volume and body weight were measured twice weekly.
Furthermore, we evaluated efficacy and body weight changes after treatment with anti-mouse PD-1 mAb combined with low-dose FTD/TPI (75 and 100 mg/kg) or the optimal effective dose of FTD/TPI (150 mg/kg) in the CMT-93 cancer model (Figure 3A and 3B). FTD/TPI had significant and dose-dependent antitumor activity against CMT-93 tumors (Table 1). The synergistic antitumor effect of anti-mouse PD-1 mAb (0.1 mg) plus FTD/TPI (150 mg/kg) combination therapy was consistent with that of the previous study (Figure 2A). Furthermore, when the observation period in this combination treatment group was prolonged from day 28 to 39, macroscopic examination indicated that four out of five mice were cured. No weight loss was observed.
Figure 3.
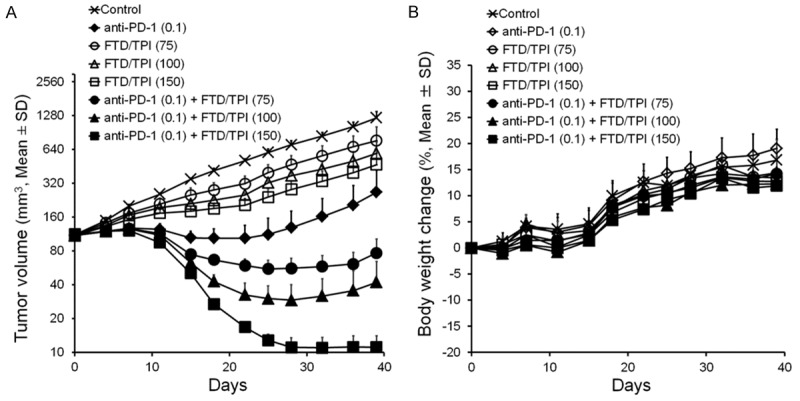
Effect of anti-mouse PD-1 mAb and low-dose FTD/TPI combination therapy in mouse colorectal CMT-93 tumors. Change in tumor volume (A) and body weight in CMT-93 tumor-bearing mice (B). Anti-mouse PD-1 mAb was administered intraperitoneally on days 1, 5, and 9. FTD/TPI was administered orally daily from days 1 to 14. Mice were treated with vehicle (x), anti-mouse PD-1 mAb (0.1 mg, ♦), FTD/TPI (75 mg/kg, ○), FTD/TPI (100 mg/kg, Δ), FTD/TPI (150 mg/kg, □), anti-mouse PD-1 mAb (0.1 mg) plus FTD/TPI (75 mg/kg) (●), anti-mouse PD-1 mAb (0.1 mg) plus FTD/TPI (100 mg/kg) (▲), or anti-mouse PD-1 mAb (0.1 mg) plus FTD/TPI (150 mg/kg) (■). Values indicate means ± SD (n = 6). Tumor volume and body weight were measured twice weekly.
Table 1.
Antitumor activity in mice implanted with mouse colorectal tumor CMT-93 after treatment with FTD/TPI and anti-mouse PD-1 mAb
| Group | Dose (mg/body or mg/kg)3 | TV1 (mean ± SD) | TGI2 (%) |
|---|---|---|---|
| Control | - | 705.2±63.0 | - |
| Anti-PD-1 mAb | 0.1 | 129.2±51.9** | 81.7 |
| FTD/TPI | 75 | 469.3±92.6** | 33.5 |
| FTD/TPI | 100 | 374.8±43.8** | 46.9 |
| FTD/TPI | 150 | 282.8±53.8** | 59.9 |
| Anti-PD-1 mAb+FTD/TPI | 0.1+75 | 56.0±12.7**,##,$ | 92.1 |
| Anti-PD-1 mAb+FTD/TPI | 0.1+100 | 29.2±10.8**,##,$$ | 95.9 |
| Anti-PD-1 mAb+FTD/TPI | 0.1+150 | 11.1±2.4**,##,$$ | 98.4 |
Tumor volume on day 28.
Tumor growth inhibition ratio on day 28.
Anti-PD-1 = mg/body, FTD/TPI = mg/kg.
p<0.01 with Aspin-Welch’s t-test as compared with the Control group.
p<0.01 with Aspin-Welch’s t-test as compared with the Anti-mouse PD-1 mAb group.
p<0.05, p<0.01 with Aspin-Welch’s t-test as compared with the FTD/TPI group.
p<0.05, p<0.01 with Aspin-Welch’s t-test as compared with the FTD/TPI group.
We also evaluated the antitumor activity of the anti-mouse PD-1 mAb combined with TPI, which prevents degradation of FTD as a thymidine phosphorylase inhibitor (Supplementary Figure 2). TPI alone had no antitumor activity and there was no enhancement of the antitumor activity of anti-mouse PD-1 mAb when combined with TPI in CMT-93 cells in vivo.
Change in tumor infiltrating lymphocytes (TILs) population after combination treatment with FTD/TPI and anti-mouse PD-1 mAb
We investigated, using flow cytometry, whether the FTD/TPI and anti-mouse PD-1 mAb combination therapy influenced the TIL population. Twenty-four tumor-bearing mice (four groups, each containing six mice) were used (Figure 4A-D). Using single cell suspensions prepared from the tumor tissues of the animals, three-color staining and flow cytometry analysis were performed. The CD8+ T cell ratio among total lymphocytes in the FTD/TPI and anti-mouse PD-1 mAb combination-treated group was significantly higher than that of the untreated control group. Although CD4+ T cells in the lymphocytes of the combination group tended to increase compared to that in the control group, Tregs in the CD4+ T cells of the combination group were significantly reduced compared to those of the control group (P<0.01).
Figure 4.
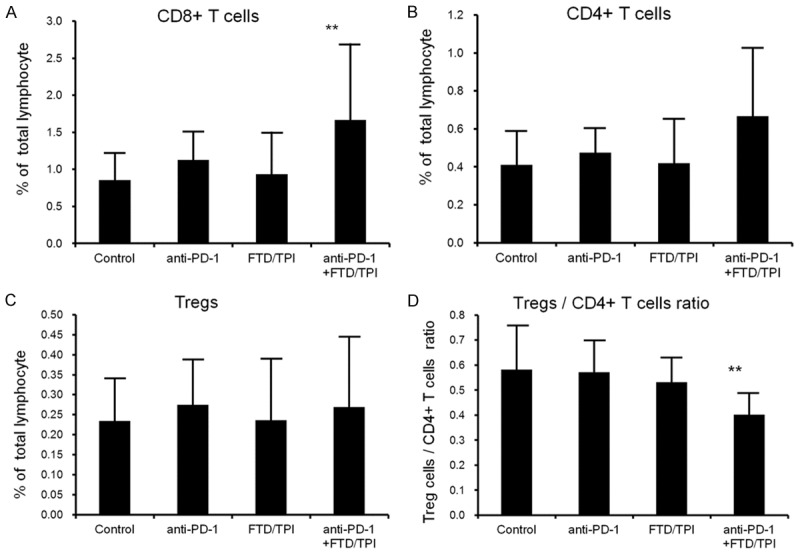
The proportion of each T cell subpopulation in the CMT-93 cells was determined by flow cytometry. FTD/TPI was administered orally from days 1 to 14 at the reported effective dose (150 mg/kg/day). Anti-mouse PD-1 mAb was injected intraperitoneally at a dose of 0.1 mg on days 1, 5, and 9. Combination therapy with FTD (150 mg/kg/day) and anti-mouse PD-1 mAb (0.1 mg/body) was administered on the same schedule as that of each monotherapy. CMT-93 tumors were collected 24 h after the final administration of the drugs (day 15). Values indicate means ± SD (n = 6). **P<0.01 by the Student’s one-sided t-test compared to the untreated control group.
Discussion
In the present study, we investigated the anticancer activity of FTD/TPI combined with anti-mouse PD-1 mAb in CMT-93 cancer cells. These cells are MSS-type colorectal cancer cells resulting from MSI typing and NGS sequencing of mismatch repair genes. The in vivo results indicated that the antitumor effect of FTD/TPI and anti-mouse PD-1 mAb combination therapy was synergistically greater than that of monotherapy with either FTD/TPI or anti-mouse PD-1 mAb. Furthermore, flow cytometric analysis suggested that the combination of both drugs enhanced antitumor activity by the infiltration of cytotoxic CD8+ T cells and reduction of immunosuppressive activity by reducing the Tregs in CMT-93 cells.
Interestingly, the greater anti-tumor activity of the combination therapy occurred without any increased toxicity in C57BL/6J mice bearing CMT-93 tumors. These results suggested that the combination of FTD/TPI and anti-mouse PD-1 mAb might be effective for MSS-type colorectal patients. Furthermore, in the present study, anti-mouse PD-1 mAb combined with low-dose FTD/TPI (half the optimal effective dose) exhibited potent antitumor activity (TGI >90%). TPI alone or combined with anti-mouse PD-1 mAb was not effective (Supplementary Figure 2), indicating this synergistic effect was primarily derived from FTD. FTD is more resistant to DNA glycosylase than 5FU [21] and its incorporation into DNA induces instability of the DNA [22]. The persistence of FTD in the DNA of tumor cells treated with FTD/TPI may underlie the anticancer activity in several preclinical cancer models or prolong survival in cancer patients [6,23]. Therefore, even at a low dose, the long-term presence of FTD in DNA might have initiated the anti-tumor activity.
Combination treatment with FTD/TPI and anti-mouse PD-1 mAb for 2 weeks resulted in a higher CD8+ T cell ratio among total lymphocytes and a lower Tregs ratio in CD4+ T cells compared with the control group. However, under the same conditions, FTD/TPI alone or anti-mouse PD-1 mAb alone did not significantly change the number of CD8+ T cells or Tregs compared with the control group. We previously demonstrated that FTD incorporation into DNA induces single-strand breaks followed by double strand breaks during the G2/M-phase of the cell cycle [24]. Recently, it was reported that FTD incorporation into DNA might induce double strand breaks followed by DNA repair, mainly through the homologous recombination pathway, after analysis of genomic DNA extracted from 233 colorectal cancer patients [25]. Although the precise mechanism for FTD/TPI-induced immunogenic activity in CMT-93 cells remains to be elucidated, it is possible that the increased antitumor efficacy of FTD/TPI is due to the DNA-instability and DNA-dysfunction caused by FTD. The efficacy of anti-mouse PD-1 blockade was reported to be correlated with neo-antigen count and mutations in the DNA-repair pathway, all of which are linked to the mutation level [26,27].
In this study, the growth of mouse MSS-type colorectal cancer CMT-93 cells treated with anti-mouse PD-1 mAb monotherapy was significantly suppressed compared to that of untreated control cells (P<0.01). The degree of TGI caused by anti-mouse PD-1 mAb (clone RMP1-14) monotherapy in CMT-93 cells was similar to that in colon cancer MC38 cells [18]. The publicly available genomic data from the MC38 cells submitted by Efemova M. et al. (GSE93018, SRP095725) revealed that MC38 cells had a mutation in the DNA mismatch repair genes, Msh3 and Msh6, and in Pold1. This result indicated that the MC38 cells were MSI-type tumor cells, unlike CMT-93 cells. However, the rate for non-synonymous mutations in CMT-93 and MC38 cells was 41 and 87 per 1 Mb, respectively. Therefore, both CMT-93 cells and MC38 cells had a higher mutation rate than the average human spontaneous mutation rate (more than 12 mutations per 1 Mb) [28]. Conversely, CT-26, which is another murine colorectal tumor cell line that was reported to be less sensitive to immune checkpoint blockade, had no mismatch repair genes [29,30], indicating that CT-26 cells were an MSS-type. Moreover, we confirmed that the CMT-93 mouse colorectal cancer model used in the present study yielded highly reproducible results. Therefore, this model along with the MC38 and CT-26 models can be used to predict the potential clinical benefits of anti-mouse PD-1 mAb combination therapy. However, the effects of the combination therapy were evaluated using a single cell line in MSS-type mouse colorectal cancer. Further studies are needed to investigate the other MSS-type mouse colorectal cancer cell lines.
Our preclinical findings indicated that combination therapy with FTD/TPI and PD-1/PD-L1 signal-blockade agents was a promising treatment option for a range of colorectal cancers. Although further research is needed to fully elucidate this promising combination in colorectal cancer therapy, the concept of combining FTD/TPI and anti-PD-1 mAb drugs seems to present a potential strategy in the field of immunotherapeutic drugs and antimetabolites combination therapeutics.
Supporting Information
References
- 1.Heidelberger C, Griesbach L, Cruz O, Schnitzer RJ, Grunberg E. Fluorinated pyrimidines. VI. Effects of 5-fluorouridine and 5-fluoro-2’-deoxyuridine on transplanted tumors. Proc Soc Exp Biol Med. 1958;97:470–475. doi: 10.3181/00379727-97-23777. [DOI] [PubMed] [Google Scholar]
- 2.Reyes P, Heidelberger C. Fluorinated pyrimidines. XXVI. Mammalian thymidylate synthetase: its mechanism of action and inhibition by fluorinated nucleotides. Mol Pharmacol. 1965;1:14–30. [PubMed] [Google Scholar]
- 3.Fujiwara Y, Oki T, Heidelberger C. Fluorinated pyrimidines. XXXVII. Effects of 5-trifluoromethyl-2’-deoxyuridine on the synthesis of deoxyribonucleic acid of mammalian cells in culture. Mol Pharmacol. 1970;6:273–280. [PubMed] [Google Scholar]
- 4.Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, Yamada Y, Asao T. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2’-deoxyribonucleosides. Biochem Pharmacol. 2000;59:1227–1236. doi: 10.1016/s0006-2952(00)00253-7. [DOI] [PubMed] [Google Scholar]
- 5.Emura T, Suzuki N, Yamaguchi M, Ohshimo H, Fukushima M. A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FUresistant human cancer cells through a mechanism involving FTD incorporation in DNA. Int J Oncol. 2004;25:571–578. [PubMed] [Google Scholar]
- 6.Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, Yamazaki K, Shimada Y, Tabernero J, Komatsu Y, Sobrero A, Boucher E, Peeters M, Tran B, Lenz HJ, Zaniboni A, Hochster H, Cleary JM, Prenen H, Benedetti F, Mizuguchi H, Makris L, Ito M, Ohtsu A. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372:1909–1919. doi: 10.1056/NEJMoa1414325. [DOI] [PubMed] [Google Scholar]
- 7.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 8.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lochhead P, Kuchiba A, Imamura Y, Liao X, Yamauchi M, Nishihara R, Qian ZR, Morikawa T, Shen J, Meyerhardt JA, Fuchs CS, Ogino S. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst. 2013;105:1151–1156. doi: 10.1093/jnci/djt173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujiyoshi K, Yamamoto G, Takenoya T, Takahashi A, Arai Y, Yamada M, Kakuta M, Yamaguchi K, Akagi Y, Nishimura Y, Sakamoto H, Akagi K. Metastatic pattern of stage IV colorectal cancer with high-frequency microsatellite instability as a prognostic factor. Anticancer Res. 2017;37:239–247. doi: 10.21873/anticanres.11313. [DOI] [PubMed] [Google Scholar]
- 11.Chinai JM, Janakiram M, Chen F, Chen W, Kaplan M, Zang X. New immunotherapies targeting the PD-1 pathway. Trends Pharmacol Sci. 2015;36:587–595. doi: 10.1016/j.tips.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boland PM, Ma WW. Immunotherapy for colorectal cancer. Cancers (Basel) 2017;9 doi: 10.3390/cancers9050050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nukatsuka M, Nakagawa F, Saito H, Sakata M, Uchida J, Takechi T. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, with irinotecan hydrochloride on human colorectal and gastric cancer xenografts. Anticancer Res. 2015;35:1437–1445. [PubMed] [Google Scholar]
- 14.Nukatsuka M, Nakagawa F, Takechi T. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, with oxaliplatin on human colorectal and gastric cancer xenografts. Anticancer Res. 2015;35:4605–4615. [PubMed] [Google Scholar]
- 15.Tsukihara H, Nakagawa F, Sakamoto K, Ishida K, Tanaka N, Okabe H, Uchida J, Matsuo K, Takechi T. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, together with bevacizumab, cetuximab, or panitumumab on human colorectal cancer xenografts. Oncol Rep. 2015;33:2135–2142. doi: 10.3892/or.2015.3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki N, Nakagawa F, Matsuoka K, Takechi T. Effect of a novel oral chemotherapeutic agent containing a combination of trifluridine, tipiracil and the novel triple angiokinase inhibitor nintedanib, on human colorectal cancer xenografts. Oncol Rep. 2016;36:3123–3130. doi: 10.3892/or.2016.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dietrich W, Katz H, Lincoln SE, Shin HS, Friedman J, Dracopoli NC, Lander ES. A genetic map of the mouse suitable for typing intraspecific crosses. Genetics. 1992;131:423–447. doi: 10.1093/genetics/131.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. 2013;19:5626–5635. doi: 10.1158/1078-0432.CCR-13-0545. [DOI] [PubMed] [Google Scholar]
- 19.Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, Maeda M, Onodera M, Uchiyama T, Fujii S, Sakaguchi S. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:1643–1656. doi: 10.1093/intimm/dxh165. [DOI] [PubMed] [Google Scholar]
- 20.Bauer P, Rohmel J, Maurer W, Hothorn L. Testing strategies in multi-dose experiments including active control. Stat Med. 1998;17:2133–2146. doi: 10.1002/(sici)1097-0258(19980930)17:18<2133::aid-sim901>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki N, Emura T, Fukushima M. Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int J Oncol. 2011;39:263–270. doi: 10.3892/ijo.2011.1003. [DOI] [PubMed] [Google Scholar]
- 22.Markley JC, Chirakul P, Sologub D, Sigurdsson ST. Incorporation of 2’-deoxy-5-(trifluoromethyl) uridine and 5-cyano-2’-deoxyuridine into DNA. Bioorg Med Chem Lett. 2001;11:2453–2455. doi: 10.1016/s0960-894x(01)00461-9. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka N, Sakamoto K, Okabe H, Fujioka A, Yamamura K, Nakagawa F, Nagase H, Yokogawa T, Oguchi K, Ishida K, Osada A, Kazuno H, Yamada Y, Matsuo K. Repeated oral dosing of TAS-102 confers high trifluridine incorporation into DNA and sustained antitumor activity in mouse models. Oncol Rep. 2014;32:2319–2326. doi: 10.3892/or.2014.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki N, Nakagawa F, Nukatsuka M, Fukushima M. Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp Ther Med. 2011;2:393–397. doi: 10.3892/etm.2011.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suenaga M, Schirripa M, Cao S, Zhang W, Yang D, Murgioni S, Rossini D, Marmorino F, Mennitto A, Ning Y, Okazaki S, Berger MD, Miyamoto Y, Gopez R Jr, Barzi A, Yamaguchi T, Loupakis F, Lenz HJ. Genetic variants of DNA repair-related genes predict efficacy of TAS-102 in patients with refractory metastatic colorectal cancer. Ann Oncol. 2017;28:1015–1022. doi: 10.1093/annonc/mdx035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 28.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA Jr, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111:11774–11779. doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, Diken M, Kreiter S, Tureci O, Sahin U. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15:190. doi: 10.1186/1471-2164-15-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.