

Abstract
Huntington's disease (HD) is a progressive neurodegenerative disorder characterized by motor, cognitive and psychiatric problems. Previous studies indicated that levels of brain gangliosides are lower than normal in HD models and that administration of exogenous ganglioside GM1 corrects motor dysfunction in the YAC128 mouse model of HD. In this study, we provide evidence that intraventricular administration of GM1 has profound disease‐modifying effects across HD mouse models with different genetic background. GM1 administration results in decreased levels of mutant huntingtin, the protein that causes HD, and in a wide array of beneficial effects that include changes in levels of DARPP32, ferritin, Iba1 and GFAP, modulation of dopamine and serotonin metabolism, and restoration of normal levels of glutamate, GABA, L‐Ser and D‐Ser. Treatment with GM1 slows down neurodegeneration, white matter atrophy and body weight loss in R6/2 mice. Motor functions are significantly improved in R6/2 mice and restored to normal in Q140 mice, including gait abnormalities that are often resistant to treatments. Psychiatric‐like and cognitive dysfunctions are also ameliorated by GM1 administration in Q140 and YAC128 mice. The widespread benefits of GM1 administration, at molecular, cellular and behavioural levels, indicate that this ganglioside has strong therapeutic and disease‐modifying potential in HD.
Keywords: behaviour, gangliosides, HD mouse models, huntingtin, neuroprotection
Subject Categories: Neuroscience, Pharmacology & Drug Discovery
Introduction
Huntington's disease (HD) is a dominantly inherited neurodegenerative disorder caused by the pathological expansion of a trinucleotide (CAG) repeat in the gene that codes for huntingtin (HTT) (Group THsDCR 1993). This mutation results in an abnormally long poly‐glutamine stretch at the N‐terminus of the mutant HTT (mHTT) protein, which in turn causes mHTT to misfold into toxic species and to aggregate. A plethora of molecular, cellular and network dysfunctions ensue, eventually leading to neuronal death (Imarisio et al, 2008). Cerebral cortex and corpus striatum are the most affected brain regions (Vonsattel et al, 2011), while more subtle pathological changes occur in other brain areas (Petersen & Bjorkqvist, 2006; Aziz et al, 2009; Vonsattel et al, 2011).
Motor dysfunction is the hallmark of HD and defines disease onset (Roos, 2010). However, cognitive and psychiatric problems often precede the appearance of motor symptoms and are frequently the most distressing for patients and their families (Paulsen et al, 2008; Roos, 2010).
To date, there is no cure or disease‐modifying therapy for HD. Clinical management of HD symptoms is possible to a certain extent with the use of tetrabenazine to reduce chorea, and with traditional antidepressant and anti‐psychotic drugs (Ross & Tabrizi, 2011). However, the use of these symptomatic treatments is often limited by their potential side effects (Rosenblatt, 2007; Killoran & Biglan, 2014), and the overall efficacy of antidepressants in HD patients is controversial (Moulton et al, 2014). Furthermore, none of these treatments target the underlying causes of dysfunction nor are able to slow down HD progression.
In previous studies, we showed that the synthesis of gangliosides—sialic acid‐containing glycosphingolipids—is affected in cellular and animal models of HD (Desplats et al, 2007; Denny et al, 2010; Maglione et al, 2010), resulting in lower levels of ganglioside GM1 and, to a lesser extent, other major brain gangliosides (Maglione et al, 2010).
Gangliosides perform a plethora of modulatory functions in cell signalling, cell–cell interactions and calcium homeostasis (Sonnino et al, 2007; Posse de Chaves & Sipione, 2010; Ledeen & Wu, 2015). Their importance in the central nervous system is underscored by the fact that knockout mouse models that lack complex gangliosides undergo neurodegeneration (Proia, 2003) and display motor impairment (Chiavegatto et al, 2000), depression‐like behaviour (Wang et al, 2014) and learning and memory deficits (Sha et al, 2014). This suggests that reduced GM1 levels in HD may contribute to disease pathogenesis and/or progression. In support of this hypothesis, we showed that administration of exogenous GM1 decreases HD cell susceptibility to apoptosis in vitro (Maglione et al, 2010) and corrects motor dysfunction in the YAC128 mouse model (Di Pardo et al, 2012). However, whether the treatment was able to ameliorate other important aspects of the disease—including non‐motor symptoms—and to attenuate the underlying molecular dysfunctions and neurodegeneration remained to be investigated.
In this study, we used three different—and for many aspects complementary—HD mouse models (R6/2, Q140 and YAC128 mice) (Mangiarini et al, 1996; Menalled et al, 2003; Slow et al, 2003) to show that chronic intraventricular infusion of GM1 has profound disease‐modifying effects that include a reduction in brain levels of toxic mHTT, restoration of normal brain concentrations of specific neurotransmitters, as well as decreased neurodegeneration and white matter atrophy, among others. These changes are accompanied by improvement or even restoration of motor function and by correction of behavioural abnormalities related to depression, anxiety and cognition.
Results
Intracerebroventricular infusion of GM1 slows down neurodegeneration, improves neuropathology and decreases body weight loss in R6/2 mice
To determine the effects of GM1 on HD brain neuropathology, we performed chronic intracerebral infusion of this ganglioside in R6/2 mice (Appendix Fig S1). Compared to other models used in this study, R6/2 mice present with an accelerated disease phenotype and develop widespread neurodegeneration from a young age (Mangiarini et al, 1996). Treatment with GM1 significantly attenuated striatal atrophy (effect of treatment: F 1,40 = 152.4, P < 0.0001—see also Appendix Table S1 for P‐values relative to pairwise comparisons; Fig 1A) and loss of striatal neurons (effect of treatment: F 1,35 = 21.0, P < 0.001) compared to vehicle (artificial cerebro‐spinal fluid, CSF)‐treated R6/2 mice (Fig 1B). Brain volume (between bregma 1.98 and −2.3; Fig 1C) and total brain weight (Fig 1D) were also significantly higher in R6/2 mice treated with GM1 compared to vehicle‐treated mice (effect of treatment: F 1,41 = 14.33, P < 0.001; effect on brain volume: P = 0.0092). While brain weight decreased progressively in R6/2 mice from 6 to 10 weeks of age (effect of age: F 3,33 = 10.86, P < 0.0001), administration of GM1 for 4 weeks significantly slowed down this process, maintaining brain weight at levels observed at 8 weeks of age without treatment (P = 0.99). Altogether, these data suggest that treatment with GM1 significantly slowed down neurodegeneration in R6/2 mice.
Figure 1. GM1 decreases neuropathology and weight loss in R6/2 mice.
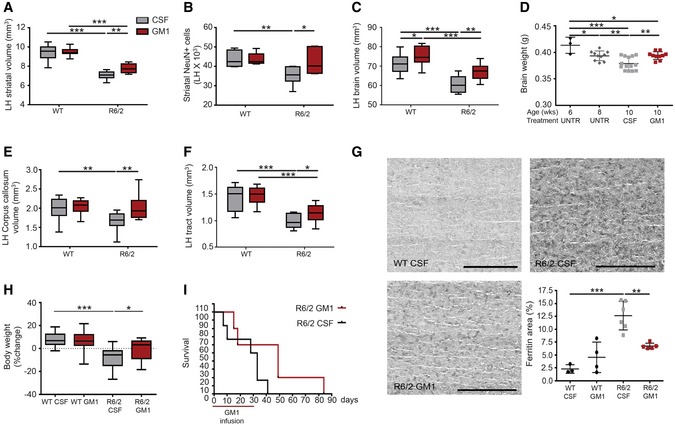
- Striatal volume in the brain left hemisphere (LH, contralateral to infusion site). N = 13 WT CSF, 11 WT GM1, 11 R6/2 CSF, 9 R6/2 GM1.
- Number of neurons (NeuN+ cells) in the LH striatum between bregma 0.02 mm and −2.3 mm. N = 8 WT CSF, 5 WT GM1, 7 R6/2 CSF, 6 R6/2 GM1.
- Volume of the brain (LH) from bregma 2.1 mm to −2.3 mm. N = 13 WT CSF, 11 WT GM1, 11 R6/2 CSF, 9 R6/2 GM1.
- Time course of brain weight loss in R6/2 mice. N = 3 6‐week R6/2, 10 8‐week R6/2, 14 10‐week R6/2 CSF and 10 10‐week R6/2 GM1.
- Corpus callosum volume (LH) between bregma 2.1 mm and 0.02 mm. N = 13 WT CSF, 11 WT GM1, 11 R6/2 CSF, 9 R6/2 GM1.
- Total white matter tract volume in the striatum, from 0.02 mm to bregma to −1.06 mm. N = 13 WT CSF, 11 WT GM1, 11 R6/2 CSF, 9 R6/2 GM1.
- Representative microscopy images of the striatum after immunostaining with anti‐ferritin antibodies. Scale bars are 0.62 mm in length. Quantification of the immunoreactive area is shown in the graph. Eight serial sections were analysed and averaged for each mouse. N = 3 WT CSF, 4 WT GM1, 6 R6/2 CSF, 5 R6/2 GM1.
- Percent change in body weight at day 21 of treatment compared to baseline (day 0). N = 23 WT CSF, 21 WT GM1, 14 R6/2 CSF, 11 R6/2 GM1.
- Survival curve for R6/2 mice treated with CSF (N = 6) or GM1 (N = 5). X‐axis shows days after the beginning of GM1 treatment. The horizontal red line indicates the duration of GM1 treatment.
GM1 treatment resulted also in decreased atrophy of the corpus callosum (Fig 1E) (interaction: F 1,42 = 4.26, P = 0.0451; GM1‐treated R6/2 vs. WT: P = 0.9553) and of cortico‐striatal white matter tracts (Fig 1F) (effect of genotype: F 1,43 = 54.01, P < 0.0001; R6/2 CSF vs. GM1: P = 0.0473) in R6/2 mice that received the ganglioside compared to CSF‐treated R6/2 mice. Changes in these white matter structures predate neuronal death and the onset of motor symptoms in HD patients (Paulsen et al, 2006; Jech et al, 2007; Tabrizi et al, 2011, 2012), and correlate with cognitive (Crawford et al, 2013; Novak et al, 2014; Matsui et al, 2015) and cortico‐striatal functional deficits (Sapp et al, 1999; Wolf et al, 2008; Douaud et al, 2009), respectively.
Dysregulation of iron metabolism and increased ferritin expression are pathological changes that occur in HD patients (Bartzokis et al, 2007b) and R6/2 mice (Simmons et al, 2007) and are likely linked to oxidative stress (Chen et al, 2013a) and dystrophic microglia (Simmons et al, 2007). Upon treatment with GM1, ferritin levels in the brain of R6/2 mice were significantly decreased (interaction: F 1,14 = 14.49, P = 0.0019) (Fig 1G).
Survival and body weight are frequently used as indices of treatment efficacy in R6/2 mice (Li et al, 2005). Body weight loss was significantly attenuated by administration of GM1 (interaction: F 1,65 = 6.43, P < 0.01; Fig 1H). This was not due to increased food intake, which was not significantly different among groups (Appendix Fig S2). Furthermore, GM1 treatment did not affect the weight of WT mice (P = 0.553), suggesting that weight increase in R6/2 mice is the result of overall improved health conditions. R6/2 mice receiving GM1 also showed a trend towards increased lifespan, although these results were not statistically significant (P = 0.10; Fig 1I).
GM1 attenuates pathological cellular and molecular changes in HD mice
Because of the increasingly recognized role of non‐neuronal cell populations in HD pathogenesis (Lobsiger & Cleveland, 2007; Ehrlich, 2012), we next measured the effects of GM1 on astrocytic and microglial markers that are affected by neuroinflammation and in HD (Singhrao et al, 1999; Laurine et al, 2003; Stack et al, 2006; Dalrymple et al, 2007; Giampa et al, 2010; Politis et al, 2011; Baune, 2015; Olejniczak et al, 2015). Immunohistochemistry (Fig 2A and B) and protein immunoblotting (Fig 2C) showed similar levels of glial fibrillary acid protein (GFAP) immunoreactivity in the striatum of WT and R6/2 mice, regardless of treatment. In cortical sections from both CSF‐ and GM1‐treated R6/2 mice, the area covered by GFAP immunoreactivity was slightly higher than in WT littermates (effect of genotype, F 1,22 = 4.32, P = 0.049; Fig 2B). In spite of this, GFAP protein expression measured by immunoblotting of cortical lysates was significantly decreased in CSF‐treated R6/2 mice compared to WT (effect of genotype: F 1,24 = 13, P = 0.0014; Fig 2C). This apparent discrepancy between immunohistochemistry and protein immunoblotting data (i.e. decreased protein expression of GFAP in spite of a larger area covered by GFAP+ astrocytes in cortical tissue) might reflect the coexistence of quantitative as well as qualitative changes in HD astroglia. Administration of GM1 restored GFAP protein expression to WT levels (P = 0.028; Fig 2C).
Figure 2. Effects of GM1 on astroglial and microglial markers.
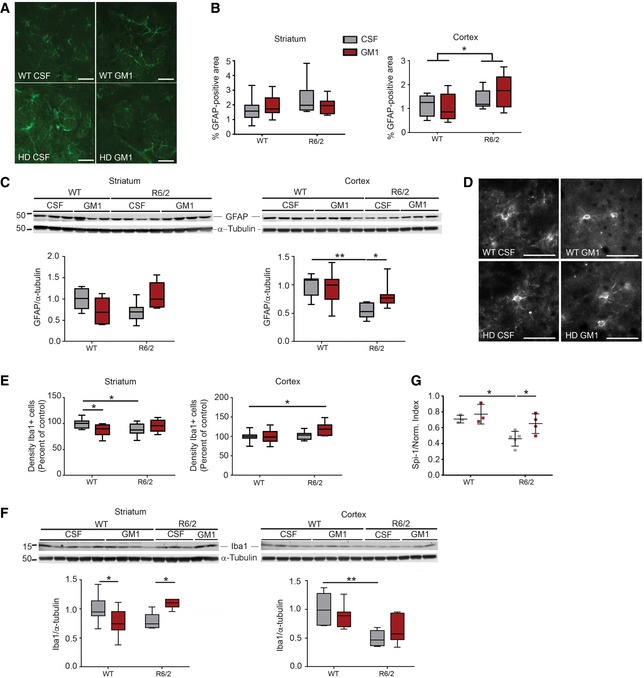
-
ARepresentative brain section staining with anti‐GFAP antibodies. Areas shown are in the corpus striatum. Scale bars are 50 μm in length.
-
BGraphs show the quantification of GFAP‐immunoreactive area in micrographs of coronal serial sections. For each mouse, eight serial sections were analysed and averaged. N = 11 WT CSF, 9 WT GM1, 10 R6/2 CSF, 8 R6/2 GM1.
-
CGFAP protein expression in tissue lysates. Representative immunoblots and densitometric analysis are shown. N = 7 WT CSF, 7 WT GM1, 7 R6/2 CSF, 7 R6/2 GM1. The immunoblot showing α‐tubulin in the cortex is the same as for the cortex in (F), since GFAP and Iba1 were run in the same gel.
-
D, ERepresentative micrographs (D) (from the striatum) and quantification (E) of Iba1+ cell density in the cortex and striatum. For each mouse, eight serial sections were analysed and averaged. Scale bars are 50 μm in length. N = 11 WT CSF, 10 WT GM1, 10 R6/2 CSF, 9 R6/2 GM1.
-
FIba1 protein expression in tissue lysates. Representative immunoblots and densitometric analysis are shown. The immunoblot showing α‐tubulin in the cortex is the same as for the cortex in (C), since GFAP and Iba1 were run in the same gel. N = 7 WT CSF, 7 WT GM1, 6 R6/2 CSF, 7 R6/2 GM1.
-
GSpi‐1 gene expression in the striatum, analysed by qPCR and normalized over the geometric mean of three stably expressed reference genes. N = 3 WT CSF, 3 WT GM1, 5 R6/2 CSF, 4 R6/2 GM1.
Similar effects of GM1 were observed on the microglial marker Iba1. Although the number of Iba1+ cells was similar in the cortex of R6/2 and WT mice (P = 0.69) and only slightly decreased in R6/2 striatum (P = 0.03; Fig 2E), unexpectedly Iba1 protein expression was lower than normal in cortical tissue from R6/2 mice (effect of genotype: F 1,23 = 20.62, P = 0.001) (Fig 2F). A similar trend was also observed in striatal tissue from R6/2 mice, although here statistical significance was not reached (P = 0.10). GM1 treatment had genotype‐dependent effects, decreasing Iba1 protein levels in the striatum of WT animals, but increasing them to control levels in R6/2 mice (interaction: F 1,24 = 13.32, P = 0.001; Fig 2F). Like in the case of GFAP expression, our data suggest qualitative differences in the molecular signature of R6/2 microglia that were at least in part mitigated by administration of GM1. In support of this conclusion, expression of the gene encoding the transcription factor Spi‐1, a master regulator of microglia biogenesis and function (Heinz et al, 2010; Kierdorf et al, 2013), was lower in vehicle‐treated R6/2 mice than in WT animals, and restored to WT levels by GM1 (Fig 2G). Our data also suggest the absence of an overt neuroinflammatory microglia phenotype in our R6/2 mice and experimental conditions. In line with these data, brain levels of major inflammatory cytokines were similar (and low) across genotype and treatment groups (Appendix Fig S3).
Next, we determined the effects of GM1 on DARPP32, a key regulator of dopamine (DA) signalling and striatum output pathways (Greengard et al, 1999; Svenningsson et al, 2004). Downregulation—as well as decreased phosphorylation—of DARPP32 marks early dysfunction of HD medium spiny neurons (Bibb et al, 2000). For the analysis of DARPP32 levels (as for GFAP analysis), we only used male mice, to avoid potential confounding effects of the oestrous cycle in female animals (Bode et al, 2008; Hajos, 2008). Chronic treatment with GM1 for 42 days increased expression of DARPP32 and pDARPP32 to WT levels in heterozygous Q7/140 mice (for DARPP32: F 4,65 = 23.88, P < 0.0001; Q7/140 CSF vs. GM1 P < 0.001; Q7/7 vs. Q7/140 GM1, P = 0.71. For pDARPP32: F 4,65 = 23.2, P < 0.0001; Q7/140 CSF vs. GM1 P < 0.001; Q7/7 vs. Q7/140 GM1, P = 0.93) (Fig EV1). This suggests that GM1 has overall beneficial effects on HD medium spiny neurons signalling and functionality. These specific effects, however, did not extend to homozygous Q140/140 mice, for reasons that are currently unknown (see Discussion).
Figure EV1. GM1 increases DARPP32 and its phosphorylation in Q7/Q140 mice.
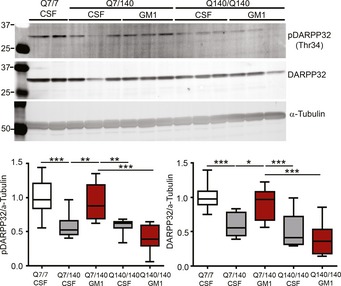
Representative immunoblots and densitometric analysis of DARPP32 expression and phospo‐Thr34 DARPP32 (p‐DARPP32) after treatment with GM1. N = 20 Q7/7 CSF, 13 Q7/140 CSF, 9 Q7/140 GM1, 14 Q140/140 CSF, 15 Q140/140 GM1. Box‐and‐whisker plots show median, maximum and minimum values. One‐way ANOVA with Tukey's post‐test. *P < 0.05, **P < 0.01, ***P < 0.001.
Mutant HTT levels are reduced by administration of GM1
Mutant HTT is arguably a primary therapeutic target in HD. GM1 treatment caused a reduction of mHTT protein levels in the striatum of both Q7/140 and Q140/140 mice (effect of treatment: F 1,11 = 8.798, P = 0.012; HD CSF vs. HD GM1 P = 0.023) after 42 days of treatment (Fig 3A; but not yet after 28 days—data not shown). Since Htt mRNA expression was not affected by GM1 (P = 0.45; Fig 3B), decreased mHTT levels were likely due to increased protein clearance. Wild‐type HTT was not significantly affected by the treatment (P = 0.95).
Figure 3. Mutant HTT protein levels are reduced by administration of GM1.
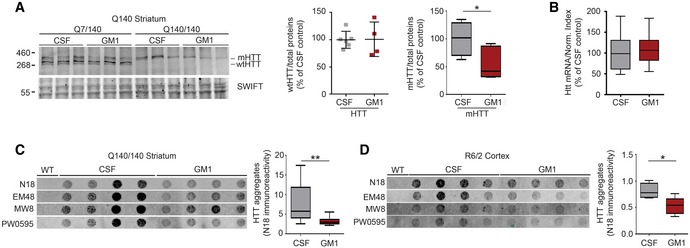
-
ARepresentative immunoblots and densitometric analysis of wtHTT and mHTT in striatal tissue lysates from Q140 mice after 42 days of treatment with artificial cerebro‐spinal fluid (CSF, vehicle) or GM1. Swift™ total protein staining was used as loading control and for data normalization. Homozygous and heterozygous Q140 mice were pooled for the analysis of mHTT as GM1 had similar effects in both genotypes. For the box‐and‐whisker plot: N = 9 Q140 CSF and 7 Q140 GM1.
-
BAnalysis of total HTT mRNA expression in CSF‐ and GM1‐treated Q140 mice. Data from heterozygous Q7/140 and homozygous Q140/140 mice were similar and were combined. N = 18 Q140 CSF, 17 Q140 GM1.
-
C, DFilter‐trap assay for mHTT insoluble aggregates in tissue lysates. Representative immunoblots and densitometric analysis are shown. N = 12 Q140/140 CSF, 11 Q140/140 GM1, five R6/2 CSF, six R6/2 GM1. SDS‐insoluble mHTT aggregates were detected with the indicated anti‐HTT antibodies. Only the densitometric analysis for N18 immunoreactivity is shown.
GM1 also decreased accumulation of SDS‐insoluble aggregates of mHTT in the striatum of Q140/140 mice (P = 0.005; Fig 3C). A similar effect was observed in the cortex (P = 0.001), although not in the striatum, of GM1‐treated R6/2 mice (Fig 3D). In heterozygous Q7/140 mice, a low amount of SDS‐insoluble aggregates and high inter‐animal variability likely prevented us from detecting any significant effect of GM1 treatment (data not shown).
Treatment with GM1 improves motor performance in R6/2 and Q140 mice
The beneficial action of GM1 on HD neuropathology in R6/2 and Q140 mice correlated with amelioration of motor dysfunction in both animal models (Figs 4 and 5), in line with our previous observations in YAC128 mice (Di Pardo et al, 2012).
Figure 4. GM1 improves motor behaviour in R6/2 and Q140 mice.
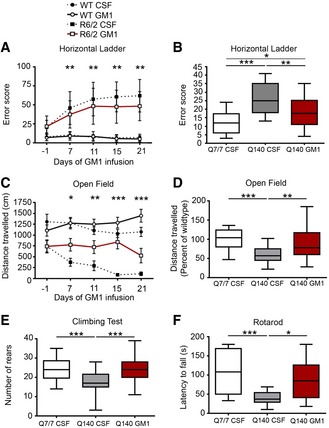
-
A, BHorizontal ladder test. Total error score from five consecutive passes is shown. N = 24 WT CSF, 21 WT GM1, 20 R6/2 CSF, 18 R6/2 GM1; N = 26 Q7/7 CSF, 23 Q140 CSF, 26 Q140 GM1.
-
C, DOpen field activity test. The distance travelled during 5‐min‐long sessions is reported. For Q140 mice, the distance travelled relative to Q7/7 is shown. N = 23 WT CSF, 21 WT GM1, 20 R6/2 CSF, 17 R6/2 GM1; N = 27 Q7/7 CSF, 30 Q140 CSF, 22 Q140 GM1.
-
EClimbing test. Number of rears performed in 5 min of placement in a wire mesh container. N = 25 Q7/7 CSF, 29 Q140 CSF, 27 Q140 GM1.
-
FFixed‐speed (12 RPM) rotarod test. Latency to fall is the average of three consecutive trials for each animal. N = 14 Q7/7 CSF, 15 Q140 CSF, 9 Q140 GM1, all females.
Figure 5. GM1 corrects gait abnormalities in Q140 mice.
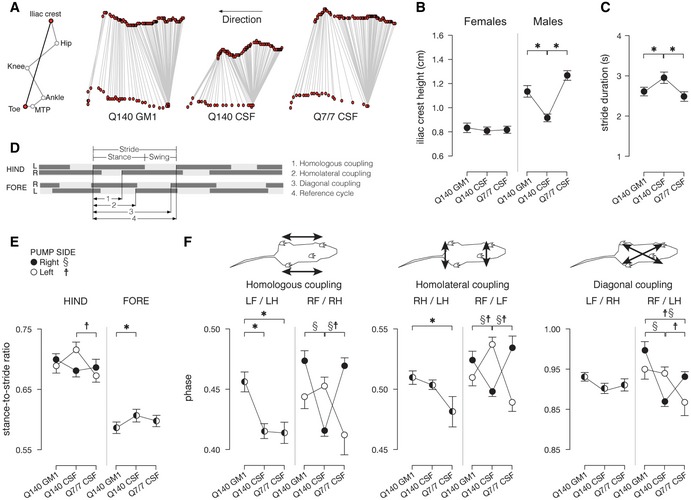
-
ARepresentative stick diagram decompositions (5 ms between sticks) of the left iliac crest and toe motion for a Q7/7 CSF mouse, a Q140 CSF mouse and a Q140 GM1 mouse during walking on a walkway.
-
BMean (± s.e.m.) values for iliac crest height. N = 9 Q7/7 CSF (five males, four females), 17 Q140 CSF (seven males, 10 females), 18 Q140 GM1 (eight males, 10 females).
-
CMean (± s.e.m.) values for stride duration. N = 9 Q7/7 CSF (five males, four females), 17 Q140 CSF (seven males, 10 females), 18 Q140 GM1 (eight males, 10 females).
-
D–FFootfall diagrams obtained from video analysis of locomotion were used to calculate: (E) stance‐to‐stride ratios and (F) coupling phase values (homologous LF/LH and RF/RH, homolateral RH/LH and RF/LF and diagonal LF/RH and RF/LH). Mean (± s.e.m.) values. N = 9 Q7/7 CSF (five males, four females), 17 Q140 CSF (seven males, 10 females), 18 Q140 GM1 (eight males, 10 females).
In the horizontal ladder test, a measure of skilled motor control (Metz & Whishaw, 2002), GM1 significantly improved performance of R6/2 mice at each time point from the beginning of treatment (P ≤ 0.01, day 7 95% CI: −19.4 to −0.1; day 11 95% CI: −19.9 to −1.6; day 15 95% CI: −21.1 to −2.5, day 21 95% CI: −23.9 to −2.7; Fig 4A). In the same test, we also observed a significant improvement in Q140 mice after treatment with GM1 past 28 days (effect of treatment: F 2,73 = 21.19, P ≤ 0.0001; Fig 4B). For this test, data from male Q7/140 and Q140/140 mice were combined as their performance was similar (Appendix Table S2). Female Q7/140 and Q140/140 were not significantly different from WT (P > 0.99). Compared to vehicle, GM1 administration also prevented a progressive decline of ambulatory and exploratory activity in R6/2 mice (Fig 4C; day 7 of treatment, 95% CI: 79.1–673.2, P ≤ 0.001; day 11, 95% CI: 68.3–828.2, P ≤ 0.001; day 15, 95% CI: 23.8–1017.0 P ≤ 0.01; day 21, 95% CI: 85.6–1171.6, P ≤ 0.01). Similar effects were observed in Q140 mice (treatment: F 2,76 = 15.58, P < 0.0001), where GM1 increased performance to wild‐type levels (P < 0.01 compared to vehicle‐treated Q140; P = 0.2 compared to Q7/7; Fig 4D), and restored normal rearing behaviour (P = 0.0005 compared to vehicle‐treated Q140; P > 0.99 compared to Q7/7; Fig 4E). For both tests, where we did not detect differences in performance between males and females and between Q7/140 and Q140/140 mice (Appendix Table S2), data from these groups were pooled and analysed together.
To determine the effects of GM1 on balance and motor coordination, we tested mice on the rotarod. Treatment with GM1 improved the performance of Q140 mice to Q7/7 levels (effect of treatment: F 2,36 = 8.992, P = 0.0007; Q140 GM1 vs. Q7/7: P > 0.9; Fig 4F). Interestingly, only female Q140 mice (both Q7/140 and Q140/140), but not males, displayed impaired behaviour in this test (effect of sex: F 1,56 = 6.7, P = 0.01); therefore, data shown in Fig 4F include females only. In a previous study (Hickey et al, 2012), male and female Q140 mice were shown to perform similarly in the rotarod test, at least up to 4.5 months of age. Since our mice were 7.5 months old at the time of testing on the rotarod, our data suggest that Q140 females might develop a progressive motor impairment on this test as they age.
Gait analysis provides an additional sensitive method to assess motor impairment and basal ganglia deficits (Amende et al, 2005; Vandeputte et al, 2010; Abada et al, 2013). Gait abnormalities are well documented in HD patients (Delval et al, 2008a,b; Kegelmeyer et al, 2014; Casaca‐Carreira et al, 2015) and are often resistant to treatments that improve other motor symptoms (Ferrara et al, 2012; Kegelmeyer et al, 2014). Q140 mice displayed several gait abnormalities compared to Q7/7 littermates (Fig 5). The height of the iliac crest during spontaneous walking was normal in female Q140 mice, but significantly decreased in vehicle‐treated Q140 male mice compared to Q7/7 male mice (P = 0.007) (interaction between group and sex: P = 0.048), indicating a decline in weight support. GM1 treatment restored the height of the iliac crest to wild‐type levels (P = 0.007 compared to CSF‐treated Q140; P = 0.39 compared to Q7/7; Fig 5A and B). The average stride duration, a measure of gait speed also affected in HD patients (Thaut et al, 1999), was significantly longer in both male and female Q140 mice compared to Q7/7 littermates (P = 0.007), and corrected to normal by GM1 (Q140 GM1 vs. Q140 CSF: P = 0.014; Q140 GM1 vs. Q7/7 CSF: P = 0.719; Fig 5C). The stance‐to‐stride ratio, where a longer stance phase duration is indicative of reduced weight support (American Physiological Society, 1996), was significantly higher in CSF‐treated Q140 mice compared to CSF‐treated Q7/7 (P = 0.002) but restored to normal in GM1‐treated Q140 mice (Fig 5E). The coupling between limbs, a measure of motor coordination and synchrony during walking, was established based on the gait diagram illustrated in Fig 5D (Leblond et al, 2003). Although some gait measurements were affected by the position (right or left) of the infusion pump implanted on the back of the animals (interaction between group and pump position: P = 0.009)—in which case data for right or left position of the pump were shown separately in Fig 5E and F—phase values in CSF‐treated Q140 mice were clearly different from those in CSF‐treated Q7/7 mice, and treatment with GM1 restored overall coupling to what was measured in Q7/7 mice (Fig 5E and F). Altogether, our data suggest a strong restorative effect of GM1 on motor function in HD animals, especially in Q140 mice.
Treatment with GM1 decreases anxiety‐ and depression‐related behaviour in HD mice
Although motor problems are the hallmark of HD and define disease onset, neuropsychiatric changes occur in nearly all HD patients and are frequently the most distressing for patients and their families (Paulsen et al, 2001a, 2008; Roos, 2010).
To assess the effect of GM1 treatment on non‐motor behaviour, we used YAC128 and Q140 mice, which differ from each other for genetic background and strain, and where cognitive and psychiatric‐like dysfunctions have been well characterized (Lione et al, 1999; Pouladi et al, 2009; Pla et al, 2014). R6/2 mice were only used in the spontaneous defecation test, as their profound motor impairment would have confounded results obtained with other tests of anxiety and depression.
In the elevated plus maze, vehicle‐treated YAC128 mice spent significantly more time in the closed arms compared to wild‐type (WT) littermates (Fig 6A; effect of genotype: F 1,57 = 5.7, P < 0.05). This behaviour was not due to differences in overall exploratory activity (Appendix Fig S4A), but rather to increased anxiety, since it could be corrected by acute administration of an anxiolytic drug (adinazolam, 2.5 mg/kg; Appendix Fig S4B; Owens et al, 1989). Treatment with GM1 increased the time YAC128 mice spent in light arms (interaction: F 1,57 = 4.31, P < 0.05; Fig 6A), suggesting that it exerted an anxiolytic effect.
Figure 6. GM1 improves anxiety‐like and depression‐like behaviour in HD mice.
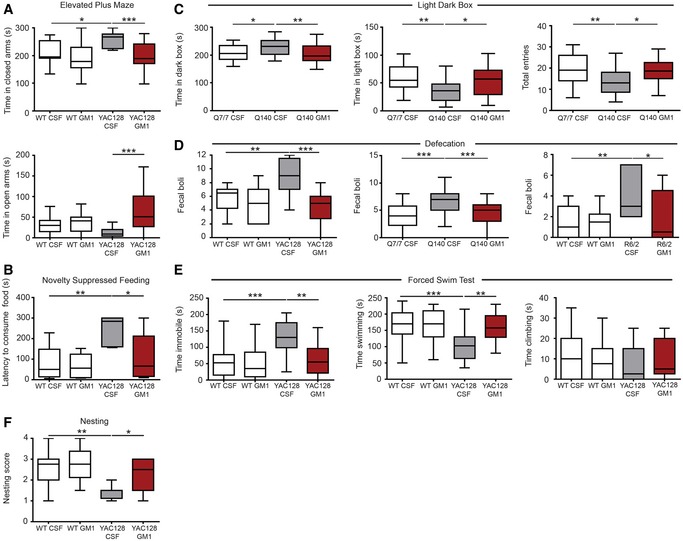
- Time spent by YAC128 mice and wild‐type (WT) littermates in the various compartments of an elevated plus maze during a 5‐min‐long session. N = 15 WT CSF, 14 WT GM1, 13 YAC128 CSF, 19 YAC128 GM1.
- Novelty‐suppressed feeding. Latency to consume sweetened condensed milk in a novel environment is shown. N = 10 WT CSF, 8 WT GM1, 6 YAC128 CSF, 8 YAC128 GM1.
- Time spent in each compartment of a light–dark box by Q7/7 and Q140. Total number of entries is also shown. N = 28 Q7/7 CSF, 26 Q140 CSF, 29 Q140 GM1.
- Faecal boli spontaneously excreted in 30‐min‐long (YAC128 and Q140 mice) or 5‐min‐long sessions (R6/2) in an open field arena. N = 12 WT CSF, 11 WT GM1, 9 YAC128 CSF, 10 YAC128 GM1; N = 24 Q7/7 CSF, 26 Q140 CSF, 29 Q140 GM1; N = 11 WT CSF, 14 WT GM1, 7 R6/2 CSF, 6 R6/2 GM1.
- Forced swim test. Time spent swimming, immobile or climbing is shown for 6‐month‐old YAC128 mice and WT littermates. N = 20 WT CSF, 19 WT GM1, 19 YAC128 CSF, 14 YAC128 GM1.
- Nest building was scored on a five‐point scale (0–4) based on the amount of the nestlet shredded and the height and shape of the nest. N = 12 WT CSF, 12 WT GM1, 8 YAC128 CSF, 10 YAC128 GM1.
In the novelty‐suppressed feeding test, which is based on a different anxiogenic/depressive paradigm than the elevated plus maze (Gross et al, 2000), vehicle‐treated YAC128 mice took more time than WT littermates to approach and consume food placed in a novel environment, while GM1‐treated YAC128 mice behaved as WT controls (genotype: F 1,28 = 13.6, P < 0.001; treatment: F 1,28 = 6.2, P < 0.05; interaction: F 1,28 = 4.3, P < 0.05; Fig 6B). The anxiolytic effects of GM1 in HD were further confirmed in Q140 mice using the light/dark box test (effect on time in open arm: F 2,79 = 7.47, P = 0.001; Fig 6C).
Treatment with GM1 also decreased open field defecation in YAC128 (F 1,38 = 6.980, P < 0.05), Q140 (F 2,76 = 12.59, P < 0.0001) and R6/2 mice (F 1,34 = 4.302, P < 0.05) (Fig 6D). Open field defecation is a sympathetic‐driven response to anxiogenic stress shared by rodents (Kinn et al, 2008; Tache & Brunnhuber, 2008) and humans (Rao et al, 1998). Altogether, our data demonstrate that GM1 normalizes anxiety‐related behaviours caused by the HD mutation across mouse models with different genetic backgrounds and disease severity.
Depression‐like behaviour has been reported in most HD models using the forced swim test (Pla et al, 2014). In line with previous reports (Pouladi et al, 2009), 6‐month‐old YAC128 mice spent more time immobile (genotype: F 1,67 = 13, P < 0.001) and less time swimming (genotype: F 1,67 = 9.35, P < 0.01) compared to WT littermates, a behaviour that indicates resignation and despair (Cryan et al, 2002; Petit‐Demouliere et al, 2005). Behavioural differences between WT and YAC128 mice were abolished by treatment with GM1 (interaction: F 1,67 = 7.3, P < 0.01 for time immobile; interaction: F 1,67 = 5.7, P < 0.05 for time swimming; Fig 6E). The therapeutic activity of GM1 was not due to its effects on motor function, since all mice performed equally well in control tests that measured swimming speed and endurance (Appendix Fig S5A and B). Moreover, acute treatment of YAC128 mice with 10 mg/kg imipramine, a tricyclic antidepressant, dramatically decreased the time YAC128 mice spent immobile and increased time spent swimming, confirming the depression‐like nature of YAC128 behaviour in the forced swim test (Appendix Fig S5C). Differently from imipramine, however, GM1 needed to be administered for longer than 10 days to improve depression‐like behaviour in YAC128 mice (Appendix Fig S6). Interestingly, GM1 also corrected the behaviour of older WT mice (9 months old; Fig EV2A), which spent more time immobile than younger (6‐month‐old) mice (41% increase, t‐test, P = 0.006). GM1 also improved the score of YAC128 mice in the nest‐building test (genotype: F 1,38 = 12.7, P < 0.01; treatment: F 1,38 = 5.3, P < 0.05; Fig 6F). This test primarily assesses instinctual species’ typical behaviour and general wellness in both male and female rodents (Deacon, 2012), but it can also reveal depression‐like behaviour (Nollet et al, 2013; Belzung, 2014) and nigrostriatal sensorimotor dysfunction (Sedelis et al, 2001; Fleming et al, 2004).
Figure EV2. GM1 decreases depression‐like behaviour in Q140 mice and in older WT mice.
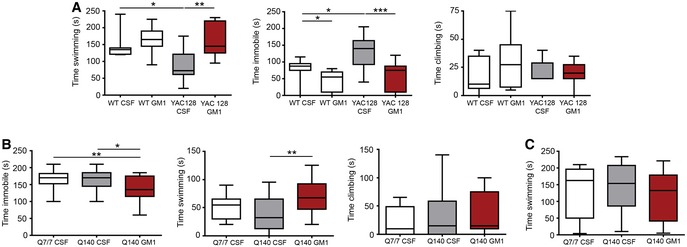
-
AThe behaviour of 9‐month‐old YAC128 mice and WT littermates was assessed in the forced swim test during treatment with cerebro‐spinal fluid (CSF, vehicle) or GM1. N = 12 WT CSF, 12 WT GM1, 8 YAC128 CSF, 9 YAC128 GM1.
-
B,CQ140 mice and Q7/7 littermates were tested in the forced swim test (B) as well as in the open pool test (C) to measure swimming endurance. N = 20 Q7/7 CSF, 20 Q140 CSF, 19 Q140 GM1.
As recently reported (Ciamei et al, 2015), the performance of Q140 mice in the forced swim test was similar to Q7/7 littermates, but GM1 still decreased time immobile (F 2,56 = 6.14, P < 0.01; Fig EV2B). Potential confounding effects related to differences in overall motor activity were excluded with control tests that measured swimming endurance (Fig EV2C).
GM1 improves cognitive performance in HD mice
Cognitive signs are common in HD and include decreased recognition memory and impaired ability to shift strategy and to plan at earlier disease stages (Paulsen et al, 2001b; Snowden et al, 2002; Stout et al, 2011), as well as deficits in procedural, working and long‐term memory with disease progression (Wilson et al, 1987; Knopman & Nissen, 1991; Lawrence et al, 2000).
In the Crawley's social approach test (Moy et al, 2004; Crawley, 2007), all mice showed similar sociability, but YAC128 mice spent more time with a familiar mouse rather than a new mouse (Fig 7A, preference for social novelty), a behaviour that is also observed in AD mouse models (Faizi et al, 2012; Cheng et al, 2014) and that is generally attributed to altered recognition memory and impaired social discrimination (Moy et al, 2004; Faizi et al, 2012). Treatment with GM1 restored normal preference for social novelty in YAC128 mice (interaction: F 1,53 = 7.275, P < 0.01).
Figure 7. GM1 improves the performance of HD mice in social and cognitive tests.
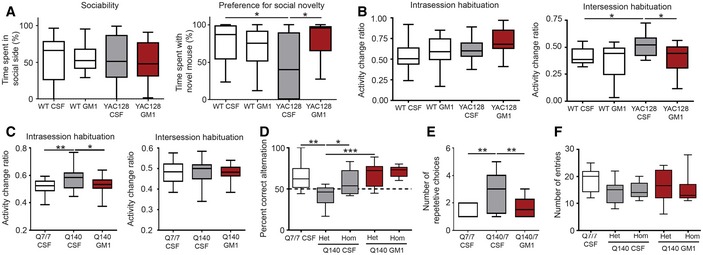
-
AThree‐chamber test. The percent of time mice spent in the chamber containing another mouse in the first part of the test (sociability) is reported. Preference for social novelty in the second part of the test is measured as the time mice spent with a second novel mouse. N = 18 WT CSF, 16 WT GM1, 16 YAC128 CSF, 17 YAC128 GM1.
-
B, CMice were placed in an open field apparatus on two consecutive days for 30 min each day. The change in activity between the first and last 5 min of testing on day 1 was used as a measure of intrasession habituation, while the change in total distance travelled between day 1 and day 2 reflected intersession habituation. N = 15 WT CSF, 14 WT GM1, 19 YAC128 CSF and 19 YAC128 GM1; N = 24 Q7/7 CSF, 24 Q140 CSF, 25 Q140 GM1.
-
D–FY‐maze. Mice were allowed to freely explore a Y‐maze apparatus for five minutes, during which all arm entries were recorded. Spatial working memory was assessed based on the percent correct alternation mice made (D). A correct alternation was defined as any sequence of three entries where no arm entry was repeated. N = 14 WT CSF, 14 Q7/140 CSF, 7 Q140/140 CSF, 14 Q7/140 GM1, 6 Q140/140 GM1. The number of repetitive arm entries (moving back and forth between two arms) is reported in (E) (N = 13 WT CSF, 12 Q7/140 CSF, 14 Q7/140 GM1), and the total number of arm entries is reported in (F).
Next, we assessed habituation. This is a complex behaviour that depends on non‐associative learning and long‐term memory (Leussis & Bolivar, 2006; Bolivar, 2009). Habituation can be measured as the change in exploratory activity that occurs over the time as mice explore a novel open field arena. None of the mice we tested showed intrasession habituation, a measure of non‐associative learning (Leussis & Bolivar, 2006; Bolivar, 2009) (indicated by an activity change ratio < 0.5; Fig 7B). Intersession habituation, which depends on memory of the previous session/s (Leussis & Bolivar, 2006; Bolivar, 2009), was evident on the second day of testing in both WT and GM1‐treated YAC128 mice (as measured by an activity change ratio below 0.5), but not in vehicle‐treated YAC128 animals (genotype: F 1,62 = 4.5, P < 0.05; treatment: F 1,62 = 6.5, P < 0.05; Fig 7B). Q140 and Q7/7 mice performed poorly in this test and did not show intrasession or intersession habituation (Fig 7C). However, vehicle‐treated Q140 mice displayed increased activity at the end of the first 30‐min trial (as measured by increased activity change ratio) when compared to other groups (F 1,70 = 5.2, P < 0.05). This unusual behaviour, likely due to increased anxiety (Bolivar, 2009), was not present in GM1‐treated Q140 mice (Fig 7C).
Next, we assessed the performance of Q7/7 and Q140 mice in the Y‐maze (Kokkinidis & Anisman, 1976; Paul et al, 2009; Fig 7D–F). Only data for male mice are shown, as all females scored similarly in this test, regardless of genotype and treatment. Because in this test we observed a difference in behaviour between heterozygous and homozygous Q140 mice, data for the two groups are shown separately. As expected, Q7/7 mice scored above chance level (correct alternation rate > 50%; chi‐square test, P < 0.05). The average alternation rate for vehicle‐treated heterozygous Q7/140 mice was significantly worse (F 4,50 = 8.1, P < 0.01; Fig 7D), and these mice made significantly more repetitive choices than Q7/7 mice prior to performing a correct alternation (F 2,32 = 6.373, P < 0.01; Fig 7E). This behaviour—which can also be observed in models of obsessive‐compulsive disorders (Yadin et al, 1991; Ulloa et al, 2004) or following impairment of serotonergic pathways (Geyer et al, 1976)—is indicative of perseveration, or cognitive inflexibility (Kokkinidis & Anisman, 1977; Kokkinidis, 1987), and reflects a common executive dysfunction in HD patients (Lawrence et al, 1996; Paulsen, 2011). Treatment with GM1 restored normal behaviour (Fig 7D and E). The total number of arm entries was similar across experimental groups, indicating similar exploratory and locomotor activity in this test (Fig 7F). Surprisingly, vehicle‐treated homozygous Q140/140 mice performed as well as Q7/7 in this test.
GM1 treatment induces neurochemical changes in the brain of HD mice
Changes in levels of glutamate, γ‐aminobutyric acid (GABA; Pearson & Reynolds, 1994; Rosas et al, 2008; Estrada‐Sanchez & Rebec, 2013; Padowski et al, 2014) and monoamines (Yohrling et al, 2002; Dang et al, 2012; Du et al, 2013; Pla et al, 2014; Vinther‐Jensen et al, 2015) have been linked to psychiatric and cognitive dysfunctions in HD and can affect performance in the behavioural tests performed in this study (Detke et al, 1995; Cryan et al, 2005; Leussis & Bolivar, 2006). Therefore, we sought to determine whether the beneficial effects of GM1 on HD mouse behaviour were accompanied by neurochemical changes. Neurochemical analysis was performed in male mice only, to avoid potential confounding effects of oestrous cycle on the dopaminergic system (Thompson & Moss, 1997; Yu & Liao, 2000).
Levels of glutamate were lower in cortical tissue from YAC128 mice compared to WT (genotype: F 1,26 = 6.7, P < 0.05; Fig 8), likely reflecting early cortical pathology in mice and in HD patients (Rosas et al, 2002, 2008, 2011; Estrada‐Sanchez & Rebec, 2013). Our study also revealed a small but significant decrease in GABA levels in the cortex of YAC128 mice compared to WT, in line with earlier studies in HD patients (Pearson & Reynolds, 1994), and with more recent reports of reduced GABAergic inhibitory input from cortical interneurons in HD models (Gu et al, 2005; Spampanato et al, 2008; Cummings et al, 2009). Administration of GM1 restored control levels of cortical glutamate and GABA (F 1,27 = 5.2, P < 0.05), and it also increased levels of glycine (F 1,27 = 7.0, P < 0.05), D‐serine (F 1,27 = 10.2, P < 0.01) and L‐serine (F 1,27 = 6.8, P < 0.05; Fig 8), suggesting widespread disease‐modifying effect of GM1 in YAC128. In the striatum, amino acid levels were similar across groups (data not shown).
Figure 8. GM1 restores wild‐type levels of neuroactive amino acid levels in the cortex of YAC128 mice.
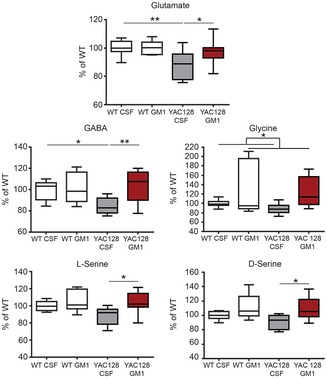
Amino acid levels were measured in the cortex of YAC128 mice and are expressed as percentage of WT control (CSF‐treated). N = 8 WT CSF, 7 WT GM1, 8 YAC128 CSF, and 8 YAC128 GM1, with the exception of Glu and GABA in the WT GM1 group (N = 6) and GABA in the YAC128 CSF group (N = 7).
Data information: Box‐and‐whisker plots show median, maximum and minimum values. Two‐way ANOVA with Bonferroni post‐test. *P < 0.05, **P < 0.01.
Psychiatric and cognitive problems—including cognitive inflexibility—have been linked to changes in monoaminergic systems in both HD and non‐HD populations (Castro et al, 1998; Yohrling et al, 2002; Du et al, 2013; Dang et al, 2012). As summarized in Table 1, treatment with GM1 resulted in region‐dependent changes in monoamines and/or their catabolites. In the striatum—where dopamine (DA) and metabolites resulting from DA turnover, 3,4‐dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), were decreased compared to WT—there was a significant effect of GM1 treatment on DA (increased by GM1) and DOPAC levels (decreased by GM1) in both WT and YAC128 mice. At the same time, GM1 decreased DOPAC/DA and DOPAC + HVA/DA ratios, suggesting an effect of this ganglioside on DA turnover. In the cortex, levels of DA were similar across groups (Table 1). DOPAC levels were significantly elevated in vehicle‐treated YAC128 mice compared to WT, and normalized by GM1 treatment. Therefore, GM1 treatment appeared to have an overall modulatory action on the dopaminergic system and dopamine turnover, with the direction of the effects likely being dependent on overall tissue dopaminergic tone.
Table 1.
Effect of GM1 treatment on biogenic amines
| WT | YAC128 | Two‐way ANOVA (Bonferroni post‐test) | |||
|---|---|---|---|---|---|
| CSF | GM1 | CSF | GM1 | ||
| Striatum | |||||
| DA | 100 ± 8.7 | 105.2 ± 6.6 | 88.2 ± 12.0v | 99.2 ± 11.3 |
Effect of genotype: F(1, 27) = 6.16, P < 0.05; Effect of treatment: F(1, 27) = 5.092, P < 0.05 v: HD CSF ≠ WT GM1 |
| DOPAC | 100 ± 13.7 | 81.5 ± 9.8§ | 78.0 ± 12.7** | 66.0 ± 13.9### |
Effect of genotype F(1, 27) = 16.8, P < 0.001; Effect of treatment F(1, 27) = 11.11, P < 0.01 **: HD CSF ≠ WT CSF; §: WT GM1 ≠ WT CSF; ###: HD GM1 ≠ WT CSF |
| HVA | 100 ± 11.7 | 97.9 ± 9.6 | 81.3 ± 6.7**,v | 84.0 ± 12.6# |
Effect of genotype F(1, 27) = 18.92, P < 0.001 **: HD CSF ≠ WT CSF; v: HD CSF ≠ WT GM1; #: HD GM1 ≠ WT CSF |
| DOPAC/DA | 100 ± 18 | 77.0 ± 12.4§ | 88.0 ± 13.6 | 66.3 ± 13^,### |
Effect of genotype F(1, 27) = 4.704, P < 0.05; Effect of treatment F(1, 27) = 18.22, P < 0.001 §: WT GM1 ≠ WT CSF; ^: HD GM1 ≠ HD CSF; ###: HD GM1 ≠ WT CSF |
| HVA/DA | 100 ± 7.8 | 93.2 ± 9.8 | 92.8 ± 7.8 | 85.3 ± 15# | #: HD GM1 ≠ WT CSF |
| 5HT | 100 ± 14.8 | 97.5 ± 11.9 | 92.9 ± 22.1 | 93.3 ± 11.0 | N.S. |
| 5HIAA | 100 ± 11.3 | 90.5 ± 11.3 | 85.1 ± 8.3* | 76.9 ± 9.3### | Effect of genotype F(1, 27) = 15.35, P < 0.001; Effect of treatment: F(1, 27) = 5.961, P < 0.05 *: HD CSF ≠ WT CSF; ###: HD GM1 ≠ WT CSF |
| 5HIAA/5HT | 100 ± 9.5 | 92.7 ± 10.2 | 95.8 ± 26.8 | 83.1 ± 17.2 | N.S. |
| NA | 100 ± 39.6 | 133.9 ± 64.4 | 137.3 ± 67.9 | 120.7 ± 42.1 | N.S. |
| Cortex | |||||
| DA | 100 ± 32.7 | 113.6 ± 19.0 | 106.4 ± 27.6 | 97.5 ± 14.1 | N.S. |
| DOPAC | 100 ± 26.7 | 123.1 ± 31.0 | 142.1 ± 31.7 ** | 92.1 ± 16.0^^ | Interaction F(1, 27) = 14.26, P < 0.001 **: HD CSF ≠ WT CSF; ^^: HD GM1 ≠ HD CSF |
| HVA | 100 ± 9.9 | 118.3 ± 17.9§ | 105.6 ± 17.6 | 97.2 ± 11.9∞ | Interaction: F(1, 27) = 6.403, P < 0.05 §: WT GM1 ≠ WT CSF; ∞: HD GM1 ≠ WT GM1 |
| DOPAC/DA | 100 ± 33.3 | 100.0 ± 19.4 | 133.3 ± 57.6 | 88.0 ± 5.9^ | ^: HD GMI ≠ HD CSF |
| HVA/DA | 100 ± 19.5 | 96.5 ± 16.6 | 95.7 ± 25.9 | 94.7 ± 15.1 | N.S. |
| 5HT | 100 ± 14.0 | 101.6 ± 12.8 | 84.5 ± 19.2 | 105.4 ± 7.5^ | Effect of treatment: F(1, 26) = 4.623, P < 0.01 ^: HD GM1 ≠ HD CSF |
| 5HIAA | 100 ± 9.7 | 102.7 ± 19.1 | 104.5 ± 17.0 | 83.6 ± 15.8^ | Interaction F(1, 27) = 4.369, P < 0.05 ^: HD GM1 vs. HD CSF |
| 5HIAA/5HT | 100 ± 23.0 | 98.4 ± 17.5 | 129.0 ± 52.2 | 80.4 ± 16.7^ | Effect of treatment: F(1, 27) = 4.911, P < 0.05; Interaction F(1, 27) = 4.324, P < 0.05 ^: HD GM1 ≠ HD CSF |
| NA | 100 ± 19.9 | 94.3 ± 12.9 | 95.3 ± 11.5 | 98.0 ± 14.8 | N.S. |
N.S. = Not Significant.
Biogenic amine levels were expressed as percentage of WT control (CSF). Values are means ± STDEV. Symbols used to indicate statistically significant differences between groups (P < 0.05) were repeated twice or three times to indicate different P‐values (P < 0.01 and P < 0.001, respectively). For all amines measured unless otherwise indicated: N = 8 WT CSF, 7 WT GM1, 8 HD CSF, 8 HD GM1. N = 7 for cortical 5‐HT measured in the HD GM1 group. N = 6 for striatal NA measured in the WT GM1 group.
Serotonin (5HT) and its metabolites were also affected by GM1 administration. GM1 increased cortical, but not striatal, 5HT levels in YAC128 mice, perhaps depending on local serotonergic tone. Concomitantly, GM1 decreased 5‐hydroxyindoleacetic acid (5HIAA), a product of serotonin catabolism, both in the striatum and in the cortex of YAC128 mice. Overall, these changes suggest slower or decreased 5HT turnover (San‐Martin‐Clark et al, 1993) as a result of GM1 administration and are in line with the antidepressant effects of GM1 in HD models. Levels of noradrenaline were normal in YAC128 mice and not affected by GM1 (Table 1).
Discussion
Our studies provide evidence of widespread therapeutic and disease‐modifying effects of GM1 across different HD mouse models, independently from mouse strain and genetic make‐up.
In R6/2 mice, GM1 administration decreased striatal atrophy and neuronal loss, hallmarks of HD that correlate with disease severity and progression in HD patients (Tabrizi et al, 2011, 2012). The number of striatal neurons in R6/2 mice that received GM1 was restored to WT levels, suggesting that the relatively early start of treatment (6 weeks of age), when neurodegeneration is still modest in this mouse model (Dodds et al, 2014), prevented neuronal loss during the period of treatment. Whether neurogenesis might have also contributed to the maintenance of striatal neuron number remains to be determined.
GM1 had trophic effects on corpus callosum and cortico‐striatal white matter tracts. Improved brain connectivity through these structures would predict cognitive improvement (Crawford et al, 2013; Novak et al, 2014; Matsui et al, 2015), which was in fact observed upon administration of GM1 in YAC128 and Q140 mice. GM1 is found in high concentrations in white matter in the mouse brain (Vajn et al, 2013), and gangliosides are required for the maintenance of myelinated axons (Yang et al, 1996; Sheikh et al, 1999; Pan et al, 2005). Whether in our study GM1 exerted a trophic/protective action on oligodendrocytes and myelin directly, or indirectly, through improved neuronal health, remains to be investigated.
One of the most striking changes observed in R6/2 mice upon administration of GM1 was the reduction of ferritin accumulation in the brain. Ferritin levels are an indirect measure of iron accumulation, which correlates, in HD patients, with CAG repeat length and with the degree of cortical and basal ganglia atrophy (Muller & Leavitt, 2014; Sanchez‐Castaneda et al, 2015). Furthermore, in R6/2 mice, high levels of ferritin and ferric iron accumulate within dystrophic microglia and elicit pro‐inflammatory microglia activation (Simmons et al, 2007). Thus, the ability of GM1 treatment to reduce ferritin levels in R6/2 mice might lead to decreased oxidative stress and damage, as well as to an improvement of microglia function.
The role of microglia in HD pathogenesis is still unclear. Reactive microglia are found in all grades of HD pathology, including pre‐symptomatic patients, in correlation with increased levels of pro‐inflammatory cytokines (Dalrymple et al, 2007; Bjorkqvist et al, 2008; Silvestroni et al, 2009; Politis et al, 2011). However, the relative contribution of microglial cell autonomous dysfunction (Bjorkqvist et al, 2008; Crotti et al, 2014) compared to microglial changes that develop secondary to the disease process in HD is still unclear. Conflicting findings have been reported for R6/2 mice, with some studies describing microglia activation (Stack et al, 2006; Giampa et al, 2010) and another reporting decreased microglia density (Laurine et al, 2003). In our study, and in spite of ferritin accumulation, we did not detect inflammatory markers nor an increase in the number of microglia cells in R6/2 mice; in cortical tissue, we actually observed decreased expression of Iba1, a microglia marker and calcium‐binding protein important for phagocytosis and microglia motility (Kanazawa et al, 2002). This observation is in line with, and could contribute to explain, reports of abnormal microglia motility in HD (Kwan et al, 2012). Furthermore, it suggests the presence of a previously unrecognized aberrant “molecular” signature (or activity state) of R6/2 microglia that develops independently from pro‐inflammatory changes and that we speculate could potentially affect protective housekeeping functions of microglia. This conclusion is also supported by our data showing decreased expression of Spi‐1—the gene that encodes the master regulator of microglia differentiation and function (Heinz et al, 2010; Kierdorf et al, 2013)—in R6/2 mice. The apparent discrepancy between our data and previous reports that showed increased expression of Spi‐1 in R6/2 and other HD models (Crotti et al, 2014) might reflect differences in the overall health status of the mouse colonies used in different laboratories, and/or differences in the amount of environmental enrichment provided, since both these factors are known to affect microglia phenotype (Bilbo et al, 2007; Hoogland et al, 2015; Rodríguez et al, 2015). Importantly, GM1 treatment restored Spi‐1 expression levels to normal and increased Iba‐1 levels in the striatum of R6/2 mice.
We also observed an effect of GM1 on the expression of GFAP, an intermediate filament protein with various functions in astrocytes (Bartzokis et al, 2007a) that has been proposed to have a role in the regulation of blood–brain barrier and myelin integrity as well as in the pathophysiology of depression (Middeldorp & Hol, 2011). We found that in spite of a slight increase in GFAP‐immunoreactive area in the cortex of CSF‐treated R6/2 mice, GFAP protein expression was lower than normal in cortical lysates from these mice. Similar findings were previously described in a rat model of HD (Cong et al, 2012). Although the overall implications of these findings remain to be elucidated, studies in a human astrocytoma cell line indicate that reduced GFAP expression affects astrocyte maturation and their ability to respond to neurons (Weinstein et al, 1991). The fact that GM1 treatment restored GFAP protein expression to wild‐type levels suggests a beneficial effect on astrocyte function. In light of the emerging roles of non‐neuronal cell populations in HD pathogenesis (Lobsiger & Cleveland, 2007; Ehrlich, 2012), we speculate that the effects exerted by GM1 on HD astrocytes and microglia may contribute to the disease‐modifying properties of this ganglioside in HD models.
Striatal dysfunction precedes striatal atrophy in HD patients and animal models, and is associated with the disruption of signalling pathways that converge on DARPP32 (Bibb et al, 2000; Svenningsson et al, 2004). Expression and phosphorylation (Thr34) of DARPP32 were restored to wild‐type levels after treatment with GM1 in heterozygous Q7/140 mice but not in homozygous Q140/140. The inability of GM1 to increase DARPP32 levels in the latter is intriguing and might point at the requirement for expression of some wild‐type HTT to mediate the effects of GM1 on DARPP32. Other beneficial effects of GM1 on molecular and behavioural HD phenotypes occurred in both Q7/140 and Q140/140 mice, suggesting that expression of wild‐type HTT was not required in all cases. Interestingly, we found that in Q140 mice, restoration of normal behaviour required a longer treatment with GM1 (28 days or longer) compared to R6/2 and YAC128 mice (Di Pardo et al, 2012), where significant benefits were already measurable after 14 days of treatment. Whether this was due to the different genetic background of these mice, or reflects the involvement of specific HTT functions (and therefore the requirement for wild‐type HTT expression) in mediating at least some of GM1 benefits, remains to be investigated.
A crucial finding in our studies was that GM1 treatment affects mHTT itself. We previously showed that GM1 administration modifies levels of HTT phosphorylation at Ser13 and Ser16 (Di Pardo et al, 2012), a post‐translation modification associated with a reduction in mHTT aggregation (Gu et al, 2009) and toxicity (Gu et al, 2009; Thompson et al, 2009; Atwal et al, 2011). In line with these reports, our study provides evidence that administration of GM1 decreases levels of soluble and aggregated (SDS‐insoluble) mHTT, without affecting wild‐type HTT levels. Since the expression of HTT mRNA was not altered by GM1, we speculate that the observed reduction of mHTT levels was likely due to increased protein clearance. The underlying mechanism remains to be investigated. GM1 might activate autophagy, as was shown in other disease models (Hwang et al, 2010; Garofalo et al, 2016; Dai et al, 2017), thus potentially facilitating autophagic degradation of mutant HTT. Future experiments will test this hypothesis.
The disease‐modifying effects of GM1 were mirrored by a profound improvement of motor and non‐motor dysfunctions in HD mice. In Q140 mice, motor skills were restored by GM1 to wild‐type levels, including gait abnormalities, which are often resistant to treatments that improve other motor symptoms (Ferrara et al, 2012; Kegelmeyer et al, 2014). In R6/2 mice, motor improvement was not as marked as in Q140 mice, but comparable to or better than that reported in another study where expression of mHTT had been targeted (decreased) in the same animal model (Cheng et al, 2015). Modest motor improvement in R6/2 mice is not surprising considering the much more widespread neuropathology and disease severity that characterize this model. Nevertheless, the behavioural effects of GM1 treatment in R6/2 mice were consistent with a disease‐modifying action of this ganglioside. While a rapidly progressing motor dysfunction characterized CSF‐treated R6/2 mice, those that received GM1 maintained stable performance throughout the study. In line with these findings, GM1 also prevented weight loss in R6/2 mice.
In addition to motor symptoms, GM1 corrected a large spectrum of emotionality‐related and cognitive phenotypes—along with underlying neurochemical defects—that are particularly difficult to treat in HD and that are often overlooked in pre‐clinical studies. Restorative effects of GM1 administration on anxiety‐like behaviour in HD mice were measured with a battery of tests that make use of anxiogenic paradigms that are based on distinct—although overlapping—neurobiological substrates (Cryan & Holmes, 2005; Ramos, 2008). GM1 administration was also able to decrease depression‐like behaviour in HD mice. Interestingly, GM1 decreased a depression‐like behaviour that developed in WT mice with ageing (Turner et al, 2012). These data suggest that GM1 targets pathways that are affected in depressive behaviour and that are shared between HD and ageing WT mice. GM1 also improved the performance of HD mice in the novelty‐suppressed feeding test and in nest building. Although not specific for depression, both these tests are affected by and can reveal depression‐like behaviours in rodents (Santarelli et al, 2003; David et al, 2009; Samuels & Hen, 2011; Nollet et al, 2013; Belzung, 2014). Therefore, the improved performance of HD mice in these tests after treatment with GM1 might be due to a decrease of both anxiety and depression.
In the forced swim test, GM1 treatment increased the time mice spent swimming, but not the time spent climbing. This is reminiscent of the effects of selective serotonin reuptake inhibitors (SSRI) and is in contrast to the effects of antidepressants that elevate noradrenaline levels (which increase time spent climbing; Detke et al, 1995; Cryan et al, 2005). In line with these observations, GM1 increased cortical levels of serotonin but not adrenaline in YAC128 mice (Table 1). Concomitantly, GM1 decreased 5HIAA levels, suggesting decreased serotonin turnover (San‐Martin‐Clark et al, 1993). In addition to its effects on serotonin turnover and levels, we speculate that treatment with GM1 could potentially improve overall serotonergic transmission, since this and other gangliosides were shown to bind serotonin and facilitate its interaction with serotonin receptors (Yandrasitz et al, 1980; Matinyan et al, 1989; Fantini & Barrantes, 2009), and to increase functional coupling of serotonin receptors with adenylate cyclase (Berry‐Kravis & Dawson, 1985). Altogether, these data suggest that GM1 has a modulatory effect on the serotonergic system that could at least in part explain its antidepressant effects in animal models.
Administration of GM1 improved cognitive behaviour in both YAC128 and Q140 mice, as shown by improved performance in Crawley's test, Y‐maze and open field habituation test. Administration of GM1 also improved intersession habituation in HD mice, a complex learning behaviour that is also compromised in AD models (Muller et al, 1994) and by drugs that affect memory (Platel & Porsolt, 1982; Hess et al, 1986). Serotonergic, cholinergic and glutamatergic activities play a major role in the habituation process (Leussis & Bolivar, 2006). Although we did not measure acetylcholine levels, in our study, GM1 increased serotonin, glutamate, D‐serine (an NMDA receptor co‐agonist) and its precursor L‐serine, a finding that contributes to explain the beneficial effects of GM1 on habituation in HD mice.
Psychiatric and cognitive problems, including cognitive inflexibility, have been linked to changes in monoaminergic systems in both HD and non‐HD populations (Yohrling et al, 2002; Dang et al, 2012; Du et al, 2013). The exact contribution of the DA system to HD pathology remains unclear (Chen et al, 2013b; Schwab et al, 2015). Although DA release is decreased in HD animal models (Hickey et al, 2002; Johnson et al, 2006; Callahan & Abercrombie, 2011), in HD patients, the DA system undergoes more complex dynamic and biphasic changes (Spokes, 1980; Kish et al, 1987; Garrett & Soares‐da‐Silva, 1992). In this study, we found decreased DA levels in the striatum of YAC128 mice compared to WT. We also observed region‐specific differences in the levels of DA metabolites between YAC128 and WT littermates, with DOPAC (and HVA) being decreased in the YAC128 striatum, but increased in the YAC128 cortex. These findings might reflect changes in MAOs expression and/or activity, as reported in other studies (Richards et al, 2011; Ooi et al, 2014; Laprairie et al, 2015), while the direction of these changes (whether up or down) might depend on regional differences in dopaminergic tone. Of note, polymorphisms in the genes that code for monoamine oxidase A (MAOA) and catechol‐O‐methyltransferase (COMT) have been recently shown to act as modifiers of cognitive and psychiatric symptoms in a Danish HD population (Vinther‐Jensen et al, 2015), thus strengthening the link between MAOA activity, dopaminergic tone and cognitive and psychiatric manifestations of HD.
GM1 treatment had variable effects on DA turnover, depending on brain region and mouse genotype. GM1 decreased DOPAC and DOPAC/DA ratio to WT levels in the cortex of YAC128 mice, but further decreased DOPAC and DOPAC/DA ratio below WT levels in the striatum. GM1 also affected DOPAC levels (and DOPAC/DA ratio) in WT animals. We speculate that changes induced by GM1 might reflect a modulatory and stabilizing—rather than inhibitory—action of GM1 on DA turnover. A modulatory role of GM1 was also suggested in previous studies, where administration of GM1 was shown to increase dopaminergic transmission in aged rats, but not in young animals (Goettl et al, 2003).
To the best of our knowledge, the widespread therapeutic and disease‐modifying effects of GM1 observed in our studies are only comparable to (and even exceed in certain cases) those exerted by antisense oligonucleotide therapies to decrease HTT production (Kordasiewicz et al, 2012). Such profound effects are likely the result of GM1 effects on HTT phosphorylation and overall levels, as discussed above. These crucial disease‐specific effects of GM1 are likely to be accompanied by additional general neuroprotective activities known to be exerted by this ganglioside (Mocchetti, 2005; Posse de Chaves & Sipione, 2010; Ledeen & Wu, 2015). The therapeutic effects of GM1 shown in this study warrant clinical investigations in HD patients. Past clinical trials for the treatment of other conditions have shown that GM1 administration in patients is relatively safe (Alter, 1998; Chinnock & Roberts, 2005; Schneider et al, 2010, 2013). In spite of poor permeability of the blood–brain barrier to GM1 (Ghidoni et al, 1986; Saulino & Schengrund, 1994), sustained benefits of GM1 were observed in a small randomized double‐blind placebo‐controlled trial in Parkinson's disease patients even when this ganglioside was administered by subcutaneous injections (Schneider et al, 2010, 2013, 2015). Potential drug delivery challenges in HD patients could be circumvented through intrathecal (or intraventricular) drug administration, as in recent clinical trials with antisense oligonucleotides, or with strategies and formulations to improve drug delivery to the brain. Co‐administration of GM1 with HTT‐lowering agents could also be envisioned, to increase therapeutic benefits and potentially extend “HTT holiday” periods.
Materials and Methods
Study design
Controlled animal experiments were performed in YAC128, Q140 and R6/2 mice.
Detailed information on mouse strains is provided in the Appendix—Materials and Methods. Throughout the paper, the abbreviation Q140 is used to refer to both Q7/140 heterozygous and Q140/140 homozygous mice, while wild‐type littermates are referred to as Q7/7. All mice were maintained in our animal facility at the University of Alberta on a 14‐/10‐h light–dark cycle in a temperature‐ and humidity‐controlled room. All procedures on animals were approved by the University of Alberta's Animal Care and Use Committee and were in accordance with the guidelines of the Canadian Council on Animal Care.
Chronic intraventricular administration of semi‐synthetic GM1 (provided by Seneb BioSciences Inc., Holliston, MA) or vehicle (artificial cerebro‐spinal fluid, CSF, Harvard Apparatus) was performed as previously published (Di Pardo et al, 2012), with minor modifications (detailed in the Appendix—Materials and Methods). Treatment duration was 28 days for YAC128 and R6/2 mice and 42 days for Q140 mice. Mouse age at the start of treatment was: 6 (cohort 1) or 8 weeks (cohort 2) for R6/2 mice, 6.4 ± 0.86 months for Q140, 6.4 ± 0.35 (cohort 1) and 9.2 ± 0.37 (cohort 2) months for YAC128 mice (see also Appendix Fig S1 for further experimental details). All mice were already symptomatic (as assessed by motor tests) at the beginning of treatment. R6/2 mice and their WT littermates were all males. Both male and female Q140 mice were used. In most tests, no significant sex‐specific differences in behaviour were observed; therefore, data from males and females were combined. The only exceptions were as follows: the rotarod test, where male mice showed no impairment compared to Q7/7 littermates and were therefore excluded from the analysis, and the Y‐maze and the nest‐building tests, where female Q140 mice show no deficit and were excluded from the analysis. Data from homozygous and heterozygous Q140 mice were combined, unless otherwise indicated, as these mice performed similarly in most tests (see also Appendix Table S1 for statistical analysis). All mice were housed individually throughout the period of treatment to avoid accidental displacement of the infusion kit and/or wound infection.
Histology and volumetric analysis
Mice were euthanized by cervical dislocation, and brains were immediately removed and flash‐frozen in isopentane (2‐methylbutane, Acros Organics) pre‐cooled at −80°C and kept on dry ice. Brains were then stored at −80°C until cryosectioning. Twenty micrometre‐thick serial coronal sections were obtained using a cryostat and thaw‐mounted onto charged slides (Superfrost Plus, Fisher). Slides were left to dry at RT overnight, prior to storage at −20°C. All sections were post‐fixed in formalin (10% buffered formalin phosphate, Fisher) for 5 min at RT prior to use. Volumetric analyses were performed from serial photomicrographs taken with an Olympus BX60 microscope coupled to a Cool Snap Image Pro colour camera, using the Cavalieri principle (Henery & Mayhew, 1989). Details are provided in the Appendix—Materials and Methods.
Immunohistochemistry and immunofluorescence staining
For immunohistochemistry, sections were dehydrated in serial alcohol dilutions, cleared with xylene and then rehydrated. Endogenous peroxidase activity was quenched in 1% hydrogen peroxide in 50% methanol for 10 min. Sections were then blocked in 0.3% Triton X‐100 (for NeuN staining) or 0.2% Triton X‐100 (for ferritin staining) in Universal Blocker (DakoCytomation) for 20 min (NeuN) or 1 h (ferritin) at RT, before incubating with the primary antibody overnight at 4°C. Incubation with biotinylated secondary antibodies was for 30 min at RT where the primary antibody was not conjugated to biotin. The signal was amplified by incubation with the avidin biotin complex (ABC) system for 30 min at RT and then detected with diaminobenzidine (DAB) (Sigma). Slides were mounted with Permount (Fisher Scientific. Waltham, MA). For immunofluorescence staining, brain sections were blocked with 0.2% Triton X‐100 (Fluka) in Universal Blocker (DakoCytomation) for 1 h at RT and then incubated in primary antibodies overnight in a humidity chamber at 4°C. After washing with phosphate‐buffered saline (PBS), sections were incubated with secondary antibodies for 30 min at RT, washed and mounted in ProLong Gold mounting media (Life Technologies).
Eriochrome staining
To identify white matter structures, sections were stained with Eriochrome Cyanine R as previously described (Kiernan, 1984) with minor modifications as detailed in the Appendix—Materials and Methods.
Tissue lysate preparation and immunoblotting
Brains were dissected immediately after mouse euthanasia and flash‐frozen in liquid N2. For immunoblotting, samples were immediately homogenized in ice‐cold lysis buffer (20 mM Tris, pH 7.4, 1% Igepal, 1 mM EDTA, 1 mM EGTA, 50 μM MG132, 1× Roche complete protease inhibitor cocktail and 1× Roche PhosStop phosphatase inhibitor cocktail), using a motor‐driven Potter‐Elvehjem homogenizer. Samples were sonicated twice for 10 s each using a Sonic Dismembrator Model 100 and then centrifuged at 20,000 g for 10 min at 4°C. Protein concentration in the supernatants was measured using the bicinchoninic acid (BCA) assay (Thermo). Thirty micrograms of proteins was separated by electrophoresis on a 4–20% SDS–polyacrylamide gel and transferred onto Immobilon‐FL polyvinylidene fluoride (PVDF) membrane (Millipore). For HTT immunoblotting, proteins were separated in 4–12% SDS–polyacrylamide gels and transferred overnight onto an Immobilon‐FL PVDF membrane in transfer buffer containing 0.01% sodium dodecyl sulphate (SDS) and 16% methanol. All membranes were blocked with 5% bovine serum albumin (BSA) in Tris‐buffered saline containing 0.1% Tween‐20 (TBS‐T) and incubated overnight at 4°C with primary antibodies, followed by the appropriate IRDye secondary antibody (1:40,000, LI‐COR Biotechnology) for 45 min at RT. Infrared signal was acquired and quantified using the Odyssey Imaging System. Swift™ total protein stain (G Biosciences) was performed according to manufacturer's instructions as the loading control.
Filter retardation assay
Filter retardation assay was performed as described in (Wanker et al, 1999), with minor modifications. Briefly, 30 μg of protein lysates were diluted in PBS, denatured and reduced by adding 2% SDS and 100 mM DTT to the lysis buffer, followed by heating at 98°C for 10 min. Samples were filtered through a cellulose acetate membrane (0.2 μm pore size, Sterlitech) in a Bio‐Dot microfiltration unit (Bio‐Rad). Wells were washed twice with 200 μl PBS. After drying for 30 min, membranes were washed twice with 2% SDS in PBS and then blocked with 5% BSA in TBS‐T followed by incubation with anti‐HTT antibodies (antibodies used and dilutions are detailed in the Supplementary Material and Methods), and appropriate IRDye secondary antibodies (LI‐COR Biotechnology) were used at 1:20,000 for 1 h at RT. Infrared signal was acquired and quantified using the Odyssey Imaging System.
Quantitative polymerase chain reaction (qPCR)
RNA extraction was performed from brain tissue preserved in RNAlater Solution (Ambion), using a RNA extraction kit (Qiagen) according to manufacturer's instruction. cDNA was produced using SuperScript II Reverse Transcriptase (Thermo) and amplified with Power Up SYBR Green PCR Master Mix (Invitrogen). Quantitative PCR was carried out in a StepOnePlus instrument using the following primers: Htt Fw: 5′‐GGGAAAGAGCTTGAGACACAG, Htt Rev: 5′‐AAGGATGAACATCTCCAGCAC; and Spi‐1 Fw: 5′‐ATGGAGAAGCTGATGGCTTG, Spi‐1 Rev: 5′‐AGGTCCAGCAGGAACTGGTA. Mouse Atp5B, Cyclophilin A and eIF4a2 were used as reference genes and their geometric mean was used to calculate a normalization index (Pfaffl et al, 2004). Target gene expression was calculated using the relative standard curve method.
Analysis of biogenic amines and amino acids
At the end of treatment, mice were euthanized by cervical dislocation. Cortical and striatal tissues from the left brain hemisphere (contralateral to the site of cannulation) were collected, flash‐frozen in liquid nitrogen and stored at −80°C prior to neurochemical analysis. Amino acids were analysed by HPLC according to (Grant et al, 2006) with minor modifications. Tissues were homogenized in 5 volumes of MeOH, let sit on ice for 10 min and then centrifuged at 10,000 g for 4 min. Supernatants were diluted up to 30‐ or 60‐fold in Milli‐Q system‐filtrated water. Aliquots of the diluted material were derivatized with o‐phthaldialdehyde (OPA, Sigma‐Aldrich) and N‐isobutyryl‐L‐cysteine (IBC, Novachem) for HPLC analysis. Fluorescence detector was set at an excitation wavelength of 344 nm and emission at 433 nm. Biogenic amines and their metabolites were analysed according to Parent et al (2001). Briefly, 1/10th the volume of ice‐cold 1 N HClO4 containing 500 μM ascorbic acid and EDTA (100 mg%) was added to tissue aliquots in 5 volumes of water. Samples were then vortexed and centrifuged at 10,000 g for 4 min. Supernatants were used for HPLC analysis. Calibration curves were constructed for each HPLC run. Electrochemical detection was performed with an applied potential of 0.65V.
Analysis of behaviour
Behavioural testing was conducted in the light phase of the cycle between 08:00 h and 18:00 h. All tests were performed by experimenters who were blind to genotype and treatment. In all behavioural training and testing sessions, mice were allowed to acclimate to the testing room for 1 h. Additional details on specific tests and kinematic analysis are provided in the Appendix—Material and Methods.
Statistical analysis
All statistical analyses for behaviour tests in R6/2 mice were performed using a linear mixed effect regression model with 95% confidence intervals calculated at each time point. Analysis of normality for all data sets was performed using the D'Agostino–Pearson omnibus test. One‐way ANOVA followed by Tukey's multiple comparison test was used to compare brain weight at different ages in R6/2 mice and for the analysis of pDARPP32 and DARPP32 in Q140 mice. One‐way ANOVA followed by Bonferroni post‐tests was used in behavioural experiments involving Q140 mice. Two‐tailed t‐test was used when two groups were compared to each other. Two‐way ANOVA followed by Bonferroni or Holm–Sidak post‐test was used for comparisons including two genotypes and two treatments. The chi‐square test was used to determine whether arm alternation by mice in the Y‐maze was different than expected by chance. The non‐parametric Mann–Whitney test was used for analysis of HTT aggregates, where a normal distribution of data could not be demonstrated. For kinematics data, separate three‐factor ANOVAs (GROUP [3] × SEX [2] × PUMP [2]) were used to determine any effects of the sex (SEX), the side at which the pump hang (PUMP) and the treatment group (GROUP) on the different kinematic measures. All comparisons were performed using a statistical significance level of 0.05, and all P‐values were corrected for multiple comparisons.
Author contributions
MA and DG designed and performed experiments, analysed data and wrote the manuscript; JF, LCM and ADP performed experiments and analysed data; PK and SWL performed experiments; AH, BJK, GBB, KF and KGT provided methods and supervised experiments and data analysis; SS designed research and experiments, supervised experiments and data analysis and wrote the manuscript.
Conflict of interest
SS holds a non‐provisional US patent for the use of GM1 in treating HD. SS and the other authors declare no other financial interests or potential conflict of interest.
The paper explained.
Problem
Huntington's disease (HD) is a neurodegenerative disorder characterized by motor, psychiatric and cognitive problems. There is no cure for HD and treatments that slow down disease progression while attenuating all disease symptoms are urgently needed.
Results
Our studies show that a molecule called ganglioside GM1 is able to ameliorate all aspects of HD, from motor symptoms to cognitive problems and anxiety and depression, when it is administered chronically into the brain of animal models of the disease. The effects of GM1 extend beyond the treatment of HD symptoms. GM1 administration results in decreased levels of the disease‐causing protein, mutant huntingtin (mHTT), which is a primary target in HD therapy. This is accompanied by decreased neurodegeneration along with several other beneficial effects, including normalization of levels of key neurotransmitters in the brain and restoration of markers of striatal neuron, astrocyte and microglia functions.
Impact
The profound and widespread therapeutic effects of GM1 reported in this paper demonstrate that GM1 is a novel HD disease‐modifying treatment in mice that warrants clinical investigations in HD patients.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 2
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (CIHR, MOP 111219 and PDD 115217), from Alberta Innovates Health Solutions/Pfizer and from the Huntington Society of Canada to SS. MA was supported by a CIHR studentship. SS was supported by a Canada Research Chair (Tier 2).
EMBO Mol Med (2017) 9: 1537–1557
References
- Abada YS, Nguyen HP, Schreiber R, Ellenbroek B (2013) Assessment of motor function, sensory motor gating and recognition memory in a novel BACHD transgenic rat model for huntington disease. PLoS One 8: e68584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter M (1998) GM1 ganglioside for acute ischemic stroke. Trial design issues. Ann N Y Acad Sci 845: 391–401 [DOI] [PubMed] [Google Scholar]
- Amende I, Kale A, McCue S, Glazier S, Morgan JP, Hampton TG (2005) Gait dynamics in mouse models of Parkinson's disease and Huntington's disease. J Neuroeng Rehabil 2: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Physiological Society (1996) Proprioceptive feedback and movement regulation In Handbook of physiology. Exercise: regulation and integration of multiple systems, Rowell LB, Shepherd JT. (eds), pp 89–127. New York: American Physiological Society; [Google Scholar]
- Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, Truant R (2011) Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat Chem Biol 7: 453–460 [DOI] [PubMed] [Google Scholar]
- Aziz NA, Pijl H, Frolich M, van der Graaf AW, Roelfsema F, Roos RA (2009) Increased hypothalamic‐pituitary‐adrenal axis activity in Huntington's disease. J Clin Endocrinol Metab 94: 1223–1228 [DOI] [PubMed] [Google Scholar]
- Bartzokis G, Lu PH, Nuechterlein KH, Gitlin M, Doi C, Edwards N, Lieu C, Altshuler LL, Mintz J (2007a) Differential effects of typical and atypical antipsychotics on brain myelination in schizophrenia. Schizophr Res 93: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G, Lu PH, Tishler TA, Fong SM, Oluwadara B, Finn JP, Huang D, Bordelon Y, Mintz J, Perlman S (2007b) Myelin breakdown and iron changes in Huntington's disease: pathogenesis and treatment implications. Neurochem Res 32: 1655–1664 [DOI] [PubMed] [Google Scholar]
- Baune BT (2015) Inflammation and neurodegenerative disorders: is there still hope for therapeutic intervention? Curr Opin Psychiatry 28: 148–154 [DOI] [PubMed] [Google Scholar]
- Belzung C (2014) Innovative drugs to treat depression: did animal models fail to be predictive or did clinical trials fail to detect effects? Neuropsychopharmacology 39: 1041–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry‐Kravis E, Dawson G (1985) Possible role of gangliosides in regulating an adenylate cyclase‐linked 5‐hydroxytryptamine (5‐HT1) receptor. J Neurochem 45: 1739–1747 [DOI] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P (2000) Severe deficiencies in dopamine signaling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci USA 97: 6809–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Newsum NJ, Sprunger DB, Watkins LR, Rudy JW, Maier SF (2007) Differential effects of neonatal handling on early life infection‐induced alterations in cognition in adulthood. Brain Behav Immun 21: 332–342 [DOI] [PubMed] [Google Scholar]
- Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D et al (2008) A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med 205: 1869–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode FJ, Stephan M, Suhling H, Pabst R, Straub RH, Raber KA, Bonin M, Nguyen HP, Riess O, Bauer A et al (2008) Sex differences in a transgenic rat model of Huntington's disease: decreased 17beta‐estradiol levels correlate with reduced numbers of DARPP32 + neurons in males. Hum Mol Genet 17: 2595–2609 [DOI] [PubMed] [Google Scholar]
- Bolivar VJ (2009) Intrasession and intersession habituation in mice: from inbred strain variability to linkage analysis. Neurobiol Learn Mem 92: 206–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan JW, Abercrombie ED (2011) In vivo Dopamine Efflux is Decreased in Striatum of both Fragment (R6/2) and Full‐Length (YAC128) Transgenic Mouse Models of Huntington's Disease. Front Syst Neurosci 5: 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaca‐Carreira J, Temel Y, van Zelst M, Jahanshahi A (2015) Coexistence of Gait Disturbances and Chorea in Experimental Huntington's Disease. Behav Neurol 2015: 970204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro ME, Pascual J, Romon T, Berciano J, Figols J, Pazos A (1998) 5‐HT1B receptor binding in degenerative movement disorders. Brain Res 790: 323–328 [DOI] [PubMed] [Google Scholar]
- Chen J, Marks E, Lai B, Zhang Z, Duce JA, Lam LQ, Volitakis I, Bush AI, Hersch S, Fox JH (2013a) Iron accumulates in Huntington's disease neurons: protection by deferoxamine. PLoS One 8: e77023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Wang EA, Cepeda C, Levine MS (2013b) Dopamine imbalance in Huntington's disease: a mechanism for the lack of behavioral flexibility. Front Neurosci 7: 114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D, Low JK, Logge W, Garner B, Karl T (2014) Novel behavioural characteristics of female APPSwe/PS1DeltaE9 double transgenic mice. Behav Brain Res 260: 111–118 [DOI] [PubMed] [Google Scholar]
- Cheng HM, Chern Y, Chen IH, Liu CR, Li SH, Chun SJ, Rigo F, Bennett CF, Deng N, Feng Y et al (2015) Effects on murine behavior and lifespan of selectively decreasing expression of mutant huntingtin allele by supt4 h knockdown. PLoS Genet 11: e1005043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavegatto S, Sun J, Nelson RJ, Schnaar RL (2000) A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Exp Neurol 166: 227–234 [DOI] [PubMed] [Google Scholar]
- Chinnock P, Roberts I (2005) Gangliosides for acute spinal cord injury. Cochrane Database Syst Rev CD004444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciamei A, Detloff PJ, Morton AJ (2015) Progression of behavioural despair in R6/2 and Hdh knock‐in mouse models recapitulates depression in Huntington's disease. Behav Brain Res 291: 140–146 [DOI] [PubMed] [Google Scholar]
- Cong WN, Cai H, Wang R, Daimon CM, Maudsley S, Raber K, Canneva F, von Horsten S, Martin B (2012) Altered hypothalamic protein expression in a rat model of Huntington's disease. PLoS One 7: e47240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford HE, Hobbs NZ, Keogh R, Langbehn DR, Frost C, Johnson H, Landwehrmeyer B, Reilmann R, Craufurd D, Stout JC et al (2013) Corpus callosal atrophy in premanifest and early Huntington's disease. J Huntingtons Dis 2: 517–526 [DOI] [PubMed] [Google Scholar]
- Crawley JN (2007) Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol 17: 448–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti A, Benner C, Kerman BE, Gosselin D, Lagier‐Tourenne C, Zuccato C, Cattaneo E, Gage FH, Cleveland DW, Glass CK (2014) Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage‐determining factors. Nat Neurosci 17: 513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Markou A, Lucki I (2002) Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci 23: 238–245 [DOI] [PubMed] [Google Scholar]
- Cryan JF, Holmes A (2005) The ascent of mouse: advances in modelling human depression and anxiety. Nat Rev Drug Discovery 4: 775–790 [DOI] [PubMed] [Google Scholar]
- Cryan JF, Valentino RJ, Lucki I (2005) Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Biobehav Rev 29: 547–569 [DOI] [PubMed] [Google Scholar]
- Cummings DM, Andre VM, Uzgil BO, Gee SM, Fisher YE, Cepeda C, Levine MS (2009) Alterations in cortical excitation and inhibition in genetic mouse models of Huntington's disease. J Neurosci 29: 10371–10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai R, Zhang S, Duan W, Wei R, Chen H, Cai W, Yang L, Wang Q (2017) Enhanced Autophagy Contributes to Protective Effects of GM1 Ganglioside Against Abeta1‐42‐Induced Neurotoxicity and Cognitive Deficits. Neurochem Res 42: 2417–2426 [DOI] [PubMed] [Google Scholar]
- Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Bjorkqvist M, Petersen A, Jackson GS, Isaacs JD, Kristiansen M, Bates GP et al (2007) Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res 6: 2833–2840 [DOI] [PubMed] [Google Scholar]
- Dang LC, Donde A, Madison C, O'Neil JP, Jagust WJ (2012) Striatal dopamine influences the default mode network to affect shifting between object features. J Cogn Neurosci 24: 1960–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP et al (2009) Neurogenesis‐dependent and ‐independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62: 479–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon R (2012) Assessing burrowing, nest construction, and hoarding in mice. J Vis Exp 59: e2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delval A, Krystkowiak P, Delliaux M, Blatt JL, Derambure P, Destee A, Defebvre L (2008a) Effect of external cueing on gait in Huntington's disease. Mov Disord 23: 1446–1452 [DOI] [PubMed] [Google Scholar]
- Delval A, Krystkowiak P, Delliaux M, Dujardin K, Blatt JL, Destee A, Derambure P, Defebvre L (2008b) Role of attentional resources on gait performance in Huntington's disease. Mov Disord 23: 684–689 [DOI] [PubMed] [Google Scholar]
- Denny CA, Desplats PA, Thomas EA, Seyfried TN (2010) Cerebellar lipid differences between R6/1 transgenic mice and humans with Huntington's disease. J Neurochem 115: 748–758 [DOI] [PubMed] [Google Scholar]
- Desplats PA, Denny CA, Kass KE, Gilmartin T, Head SR, Sutcliffe JG, Seyfried TN, Thomas EA (2007) Glycolipid and ganglioside metabolism imbalances in Huntington's disease. Neurobiol Dis 27: 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detke MJ, Rickels M, Lucki I (1995) Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. Psychopharmacology 121: 66–72 [DOI] [PubMed] [Google Scholar]
- Di Pardo A, Maglione V, Alpaugh M, Horkey M, Atwal RS, Sassone J, Ciammola A, Steffan JS, Fouad K, Truant R et al (2012) Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc Natl Acad Sci USA 109: 3528–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds L, Chen J, Berggren K, Fox J (2014) Characterization of Striatal Neuronal Loss and Atrophy in the R6/2 Mouse Model of Huntington's Disease. PLOS Currents Huntington Disease https://doi.org/10.1371/currents.hd.48727b68b39b82d5fe350f753984bcf9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douaud G, Behrens TE, Poupon C, Cointepas Y, Jbabdi S, Gaura V, Golestani N, Krystkowiak P, Verny C, Damier P et al (2009) In vivo evidence for the selective subcortical degeneration in Huntington's disease. NeuroImage 46: 958–966 [DOI] [PubMed] [Google Scholar]
- Du X, Pang TY, Hannan AJ (2013) A Tale of Two Maladies? Pathogenesis of Depression with and without the Huntington's Disease Gene Mutation. Front Neurol 4: 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich ME (2012) Huntington's disease and the striatal medium spiny neuron: cell‐autonomous and non‐cell‐autonomous mechanisms of disease. Neurotherapeutics 9: 270–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada‐Sanchez AM, Rebec GV (2013) Role of cerebral cortex in the neuropathology of Huntington's disease. Front Neural Circuits 7: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faizi M, Bader PL, Saw N, Nguyen TV, Beraki S, Wyss‐Coray T, Longo FM, Shamloo M (2012) Thy1‐hAPP(Lond/Swe+) mouse model of Alzheimer's disease displays broad behavioral deficits in sensorimotor, cognitive and social function. Brain Behav 2: 142–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini J, Barrantes FJ (2009) Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim Biophys Acta 1788: 2345–2361 [DOI] [PubMed] [Google Scholar]
- Ferrara JM, Mostile G, Hunter C, Adam OR, Jankovic J (2012) Effect of tetrabenazine on motor function in patients with huntington disease. Neurol Ther 1: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF (2004) Early and progressive sensorimotor anomalies in mice overexpressing wild‐type human alpha‐synuclein. J Neurosci 24: 9434–9440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo T, Matarrese P, Manganelli V, Marconi M, Tinari A, Gambardella L, Faggioni A, Misasi R, Sorice M, Malorni W (2016) Evidence for the involvement of lipid rafts localized at the ER‐mitochondria associated membranes in autophagosome formation. Autophagy 12: 917–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett MC, Soares‐da‐Silva P (1992) Increased cerebrospinal fluid dopamine and 3,4‐dihydroxyphenylacetic acid levels in Huntington's disease: evidence for an overactive dopaminergic brain transmission. J Neurochem 58: 101–106 [DOI] [PubMed] [Google Scholar]
- Geyer MA, Puerto A, Menkes DB, Segal DS, Mandell AJ (1976) Behavioral studies following lesions of the mesolimbic and mesostriatal serotonergic pathways. Brain Res 106: 257–269 [DOI] [PubMed] [Google Scholar]
- Ghidoni R, Trinchera M, Venerando B, Fiorilli A, Sonnino S, Tettamanti G (1986) Incorporation and metabolism of exogenous GM1 ganglioside in rat liver. Biochem J 237: 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR (2010) Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS One 5: e13417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goettl VM, Zhang H, Burrows AC, Wemlinger TA, Neff NH, Hadjiconstantinou M (2003) GM1 enhances dopaminergic markers in the brain of aged rats. Exp Neurol 183: 665–672 [DOI] [PubMed] [Google Scholar]
- Grant SL, Shulman Y, Tibbo P, Hampson DR, Baker GB (2006) Determination of d‐serine and related neuroactive amino acids in human plasma by high‐performance liquid chromatography with fluorimetric detection. J Chromatogr B Analyt Technol Biomed Life Sci 844: 278–282 [DOI] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC (1999) Beyond the dopamine receptor: the DARPP‐32/protein phosphatase‐1 cascade. Neuron 23: 435–447 [DOI] [PubMed] [Google Scholar]
- Gross C, Santarelli L, Brunner D, Zhuang X, Hen R (2000) Altered fear circuits in 5‐HT(1A) receptor KO mice. Biol Psychiat 48: 1157–1163 [DOI] [PubMed] [Google Scholar]
- Group THsDCR (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983 [DOI] [PubMed] [Google Scholar]
- Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I et al (2005) Pathological cell‐cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 46: 433–444 [DOI] [PubMed] [Google Scholar]
- Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R, Yang XW (2009) Serines 13 and 16 are critical determinants of full‐length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64: 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos F (2008) Changes in glial fibrillary acidic protein (GFAP) immunoreactivity reflect neuronal states. Neurochem Res 33: 1643–1650 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henery CC, Mayhew TM (1989) The cerebrum and cerebellum of the fixed human brain: efficient and unbiased estimates of volumes and cortical surface areas. J Anat 167: 167–180 [PMC free article] [PubMed] [Google Scholar]
- Hess EJ, Albers LJ, Le H, Creese I (1986) Effects of chronic SCH23390 treatment on the biochemical and behavioral properties of D1 and D2 dopamine receptors: potentiated behavioral responses to a D2 dopamine agonist after selective D1 dopamine receptor upregulation. J Pharmacol Exp Ther 238: 846–854 [PubMed] [Google Scholar]
- Hickey MA, Reynolds GP, Morton AJ (2002) The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington's disease. J Neurochem 81: 46–59 [DOI] [PubMed] [Google Scholar]
- Hickey MA, Zhu C, Medvedeva V, Lerner RP, Patassini S, Franich NR, Maiti P, Frautschy SA, Zeitlin S, Levine MS et al (2012) Improvement of neuropathology and transcriptional deficits in CAG 140 knock‐in mice supports a beneficial effect of dietary curcumin in Huntington's disease. Mol Neurodegener 7: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland ICM, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D (2015) Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 12: 114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Lee S, Lee JT, Kwon TK, Kim DR, Kim H, Park HC, Suk K (2010) Gangliosides induce autophagic cell death in astrocytes. Br J Pharmacol 159: 586–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood BR et al (2008) Huntington's disease: from pathology and genetics to potential therapies. Biochem J 412: 191–209 [DOI] [PubMed] [Google Scholar]
- Jech R, Klempir J, Vymazal J, Zidovska J, Klempirova O, Ruzicka E, Roth J (2007) Variation of selective gray and white matter atrophy in Huntington's disease. Mov Disord 22: 1783–1789 [DOI] [PubMed] [Google Scholar]
- Johnson MA, Rajan V, Miller CE, Wightman RM (2006) Dopamine release is severely compromised in the R6/2 mouse model of Huntington's disease. J Neurochem 97: 737–746 [DOI] [PubMed] [Google Scholar]
- Kanazawa H, Ohsawa K, Sasaki Y, Kohsaka S, Imai Y (2002) Macrophage/microglia‐specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C‐gamma ‐dependent pathway. J Biol Chem 277: 20026–20032 [DOI] [PubMed] [Google Scholar]
- Kegelmeyer DA, Kloos AD, Fritz NE, Fiumedora MM, White SE, Kostyk SK (2014) Impact of tetrabenazine on gait and functional mobility in individuals with Huntington's disease. J Neurol Sci 347: 219–223 [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C et al (2013) Microglia emerge from erythromyeloid precursors via Pu.1‐ and Irf8‐dependent pathways. Nat Neurosci 16: 273–280 [DOI] [PubMed] [Google Scholar]
- Kiernan JA (1984) Chromoxane cyanine R. II. Staining of animal tissues by the dye and its iron complexes. J Microsc 134: 25–39 [DOI] [PubMed] [Google Scholar]
- Killoran A, Biglan KM (2014) Current therapeutic options for Huntington's disease: good clinical practice versus evidence‐based approaches? Mov Disord 29: 1404–1413 [DOI] [PubMed] [Google Scholar]
- Kinn AM, Gronli J, Fiske E, Kuipers S, Ursin R, Murison R, Portas CM (2008) A double exposure to social defeat induces sub‐chronic effects on sleep and open field behaviour in rats. Physiol Behav 95: 553–561 [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O (1987) Elevated serotonin and reduced dopamine in subregionally divided Huntington's disease striatum. Ann Neurol 22: 386–389 [DOI] [PubMed] [Google Scholar]
- Knopman D, Nissen MJ (1991) Procedural learning is impaired in Huntington's disease: evidence from the serial reaction time task. Neuropsychologia 29: 245–254 [DOI] [PubMed] [Google Scholar]
- Kokkinidis L, Anisman H (1976) Interaction between cholinergic and catecholaminergic agents in a spontaneous alternation task. Psychopharmacology 48: 261–270 [DOI] [PubMed] [Google Scholar]
- Kokkinidis L, Anisman H (1977) Perseveration and rotational behavior elicited by d‐amphetamine in a Y‐maze exploratory task: differential effects of intraperitoneal and unilateral intraventricular administration. Psychopharmacology 52: 123–128 [DOI] [PubMed] [Google Scholar]
- Kokkinidis L (1987) Amphetamine‐elicited perseverative and rotational behavior: evaluation of directional preference. Pharmacol Biochem Behav 26: 527–532 [DOI] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS et al (2012) Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron 74: 1031–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R, Miller A, Weiss A, Giorgini F, Cheah C et al (2012) Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest 122: 4737–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Precious SV, Denovan‐Wright EM (2015) Components of the endocannabinoid and dopamine systems are dysregulated in Huntington's disease: analysis of publicly available microarray datasets. Pharmacol Res Perspect 3: e00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurine E, Gregoire C, Fandrich M, Engemann S, Marchal S, Thion L, Mohr M, Monsarrat B, Michel B, Dobson CM et al (2003) Lithostathine quadruple‐helical filaments form proteinase K‐resistant deposits in Creutzfeldt‐Jakob disease. J Biol Chem 278: 51770–51778 [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Sahakian BJ, Hodges JR, Rosser AE, Lange KW, Robbins TW (1996) Executive and mnemonic functions in early Huntington's disease. Brain 119(Pt. 5): 1633–1645. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Watkins LH, Sahakian BJ, Hodges JR, Robbins TW (2000) Visual object and visuospatial cognition in Huntington's disease: implications for information processing in corticostriatal circuits. Brain 123(Pt. 7): 1349–1364. [DOI] [PubMed] [Google Scholar]
- Leblond H, L'Esperance M, Orsal D, Rossignol S (2003) Treadmill locomotion in the intact and spinal mouse. J Neurosci 23: 11411–11419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeen RW, Wu G (2015) The multi‐tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem Sci 40: 407–418 [DOI] [PubMed] [Google Scholar]
- Leussis MP, Bolivar VJ (2006) Habituation in rodents: a review of behavior, neurobiology, and genetics. Neurosci Biobehav Rev 30: 1045–1064 [DOI] [PubMed] [Google Scholar]
- Li JY, Popovic N, Brundin P (2005) The use of the R6 transgenic mouse models of Huntington's disease in attempts to develop novel therapeutic strategies. NeuroRx: the journal of the American Society for Experimental. Neurotherapeutics 2: 447–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lione LA, Carter RJ, Hunt MJ, Bates GP, Morton AJ, Dunnett SB (1999) Selective discrimination learning impairments in mice expressing the human Huntington's disease mutation. J Neurosci 19: 10428–10437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsiger CS, Cleveland DW (2007) Glial cells as intrinsic components of non‐cell‐autonomous neurodegenerative disease. Nat Neurosci 10: 1355–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglione V, Marchi P, Di Pardo A, Lingrell S, Horkey M, Tidmarsh E, Sipione S (2010) Impaired ganglioside metabolism in Huntington's disease and neuroprotective role of GM1. J Neurosci 30: 4072–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW et al (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493–506 [DOI] [PubMed] [Google Scholar]
- Matinyan NS, Melikyan GB, Arakelyan VB, Kocharov SL, Prokazova NV, Avakian TM (1989) Interaction of ganglioside‐containing planar bilayers with serotonin and inorganic cations. Biochem Biophys Acta 984: 313–318 [DOI] [PubMed] [Google Scholar]
- Matsui JT, Vaidya JG, Wassermann D, Kim RE, Magnotta VA, Johnson HJ, Paulsen JS, Investigators P‐H, Coordinators of the Huntington Study G (2015) Prefrontal cortex white matter tracts in prodromal Huntington disease. Hum Brain Mapp 36: 3717–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF (2003) Time course of early motor and neuropathological anomalies in a knock‐in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol 465: 11–26 [DOI] [PubMed] [Google Scholar]
- Metz GA, Whishaw IQ (2002) Cortical and subcortical lesions impair skilled walking in the ladder rung walking test: a new task to evaluate fore‐ and hindlimb stepping, placing, and co‐ordination. J Neurosci Methods 115: 169–179 [DOI] [PubMed] [Google Scholar]
- Middeldorp J, Hol EM (2011) GFAP in health and disease. Prog Neurobiol 93: 421–443 [DOI] [PubMed] [Google Scholar]
- Mocchetti I (2005) Exogenous gangliosides, neuronal plasticity and repair, and the neurotrophins. Cell Mol Life Sci 62: 2283–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton CD, Hopkins CW, Bevan‐Jones WR (2014) Systematic review of pharmacological treatments for depressive symptoms in Huntington's disease. Mov Disord 29: 1556–1561 [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN (2004) Sociability and preference for social novelty in five inbred strains: an approach to assess autistic‐like behavior in mice. Genes Brain Behav 3: 287–302 [DOI] [PubMed] [Google Scholar]
- Muller U, Cristina N, Li ZW, Wolfer DP, Lipp HP, Rulicke T, Brandner S, Aguzzi A, Weissmann C (1994) Behavioral and anatomical deficits in mice homozygous for a modified beta‐amyloid precursor protein gene. Cell 79: 755–765 [DOI] [PubMed] [Google Scholar]
- Muller M, Leavitt BR (2014) Iron dysregulation in Huntington's disease. J Neurochem 130: 328–350 [DOI] [PubMed] [Google Scholar]
- Nollet M, Le Guisquet AM, Belzung C (2013) Models of depression: unpredictable chronic mild stress in mice. Curr Protoc Pharmacol Chapter 5: Unit 5.65 [DOI] [PubMed] [Google Scholar]
- Novak MJ, Seunarine KK, Gibbard CR, Hobbs NZ, Scahill RI, Clark CA, Tabrizi SJ (2014) White matter integrity in premanifest and early Huntington's disease is related to caudate loss and disease progression. Cortex 52: 98–112 [DOI] [PubMed] [Google Scholar]
- Olejniczak M, Urbanek MO, Krzyzosiak WJ (2015) The Role of the Immune System in Triplet Repeat Expansion Diseases. Mediators Inflamm 2015: 873860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi J, Hayden MR, Pouladi MA (2014) Inhibition of Excessive Monoamine Oxidase A/B Activity Protects Against Stress‐induced Neuronal Death in Huntington Disease. Mol Neurobiol 52: 1850–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens MJ, Bissette G, Nemeroff CB (1989) Acute effects of alprazolam and adinazolam on the concentrations of corticotropin‐releasing factor in the rat brain. Synapse 4: 196–202 [DOI] [PubMed] [Google Scholar]
- Padowski JM, Weaver KE, Richards TL, Laurino MY, Samii A, Aylward EH, Conley KE (2014) Neurochemical correlates of caudate atrophy in Huntington's disease. Mov Disord 29: 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Fromholt SE, Hess EJ, Crawford TO, Griffin JW, Sheikh KA, Schnaar RL (2005) Myelin‐associated glycoprotein and complementary axonal ligands, gangliosides, mediate axon stability in the CNS and PNS: neuropathology and behavioral deficits in single‐ and double‐null mice. Exp Neurol 195: 208–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent M, Bush D, Rauw G, Master S, Vaccarino F, Baker G (2001) Analysis of amino acids and catecholamines, 5‐hydroxytryptamine and their metabolites in brain areas in the rat using in vivo microdialysis. Methods 23: 11–20 [DOI] [PubMed] [Google Scholar]
- Paul CM, Magda G, Abel S (2009) Spatial memory: theoretical basis and comparative review on experimental methods in rodents. Behav Brain Res 203: 151–164 [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Ready RE, Hamilton JM, Mega MS, Cummings JL (2001a) Neuropsychiatric aspects of Huntington's disease. J Neurol Neurosurg Psychiatry 71: 310–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Zhao H, Stout JC, Brinkman RR, Guttman M, Ross CA, Como P, Manning C, Hayden MR, Shoulson I (2001b) Clinical markers of early disease in persons near onset of Huntington's disease. Neurology 57: 658–662 [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Magnotta VA, Mikos AE, Paulson HL, Penziner E, Andreasen NC, Nopoulos PC (2006) Brain structure in preclinical Huntington's disease. Biol Psychiatry 59: 57–63 [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ et al (2008) Detection of Huntington's disease decades before diagnosis: the Predict‐HD study. J Neurol Neurosurg Psychiatry 79: 874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS (2011) Cognitive impairment in Huntington disease: diagnosis and treatment. Curr Neurol Neurosci Rep 11: 474–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson SJ, Reynolds GP (1994) Neocortical neurotransmitter markers in Huntington's disease. J Neural Transm Gen Sect 98: 197–207 [DOI] [PubMed] [Google Scholar]
- Petersen A, Bjorkqvist M (2006) Hypothalamic‐endocrine aspects in Huntington's disease. Eur J Neurosci 24: 961–967 [DOI] [PubMed] [Google Scholar]
- Petit‐Demouliere B, Chenu F, Bourin M (2005) Forced swimming test in mice: a review of antidepressant activity. Psychopharmacology 177: 245–255 [DOI] [PubMed] [Google Scholar]
- Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel‐based tool using pair‐wise correlations. Biotechnol Lett 26: 509–515 [DOI] [PubMed] [Google Scholar]
- Pla P, Orvoen S, Saudou F, David DJ, Humbert S (2014) Mood disorders in Huntington's disease: from behavior to cellular and molecular mechanisms. Front Behav Neurosci 8: 135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platel A, Porsolt RD (1982) Habituation of exploratory activity in mice: a screening test for memory enhancing drugs. Psychopharmacology 78: 346–352 [DOI] [PubMed] [Google Scholar]
- Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ, Tabrizi SJ, Barker RA, Piccini P (2011) Microglial activation in regions related to cognitive function predicts disease onset in Huntington's disease: a multimodal imaging study. Hum Brain Mapp 32: 258–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posse de Chaves E, Sipione S (2010) Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett 584: 1748–1759 [DOI] [PubMed] [Google Scholar]
- Pouladi MA, Graham RK, Karasinska JM, Xie Y, Santos RD, Petersen A, Hayden MR (2009) Prevention of depressive behaviour in the YAC128 mouse model of Huntington disease by mutation at residue 586 of huntingtin. Brain 132: 919–932 [DOI] [PubMed] [Google Scholar]
- Proia RL (2003) Glycosphingolipid functions: insights from engineered mouse models. Philos Trans R Soc Lond B Biol Sci 358: 879–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A (2008) Animal models of anxiety: do I need multiple tests? Trends Pharmacol Sci 29: 493–498 [DOI] [PubMed] [Google Scholar]
- Rao SS, Hatfield RA, Suls JM, Chamberlain MJ (1998) Psychological and physical stress induce differential effects on human colonic motility. Am J Gastroenterol 93: 985–990 [DOI] [PubMed] [Google Scholar]
- Richards G, Messer J, Waldvogel HJ, Gibbons HM, Dragunow M, Faull RL, Saura J (2011) Up‐regulation of the isoenzymes MAO‐A and MAO‐B in the human basal ganglia and pons in Huntington's disease revealed by quantitative enzyme radioautography. Brain Res 1370: 204–214 [DOI] [PubMed] [Google Scholar]
- Rodríguez JJ, Noristani HN, Verkhratsky A (2015) Microglial response to Alzheimer's disease is differentially modulated by voluntary wheel running and enriched environments. Brain Struct Funct 220: 941–953 [DOI] [PubMed] [Google Scholar]
- Roos RA (2010) Huntington's disease: a clinical review. Orphanet J Rare Dis 5: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, van der Kouwe A, Jenkins BG, Dale AM, Fischl B (2002) Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology 58: 695–701 [DOI] [PubMed] [Google Scholar]
- Rosas HD, Salat DH, Lee SY, Zaleta AK, Pappu V, Fischl B, Greve D, Hevelone N, Hersch SM (2008) Cerebral cortex and the clinical expression of Huntington's disease: complexity and heterogeneity. Brain 131: 1057–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Reuter M, Doros G, Lee SY, Triggs T, Malarick K, Fischl B, Salat DH, Hersch SM (2011) A tale of two factors: what determines the rate of progression in Huntington's disease? A longitudinal MRI study. Mov Disord 26: 1691–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt A (2007) Neuropsychiatry of Huntington's disease. Dialogues Clin Neurosci 9: 191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ (2011) Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10: 83–98 [DOI] [PubMed] [Google Scholar]
- Samuels BA, Hen R (2011) Novelty‐suppressed feeding in the mouse In Mood and anxiety related phenotypes in mice: characterization using behavioral tests, Gould TD. (ed) pp 107–122. New York: Humana Press; [Google Scholar]
- Sanchez‐Castaneda C, Squitieri F, Di Paola M, Dayan M, Petrollini M, Sabatini U (2015) The role of iron in gray matter degeneration in Huntington's disease: a magnetic resonance imaging study. Hum Brain Mapp 36: 50–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San‐Martin‐Clark O, Leza JC, Lizasoain I, Lorenzo P (1993) Changes induced by sodium cromoglycate on brain serotonin turnover in morphine dependent and abstinent mice. Psychopharmacology 111: 233–238 [DOI] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O et al (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301: 805–809 [DOI] [PubMed] [Google Scholar]
- Sapp E, Penney J, Young A, Aronin N, Vonsattel JP, DiFiglia M (1999) Axonal transport of N‐terminal huntingtin suggests early pathology of corticostriatal projections in Huntington disease. J Neuropathol Exp Neurol 58: 165–173 [DOI] [PubMed] [Google Scholar]
- Saulino MF, Schengrund CL (1994) Differential accumulation of gangliosides by the brains of MPTP‐lesioned mice. J Neurosci Res 37: 384–391 [DOI] [PubMed] [Google Scholar]
- Schneider JS, Sendek S, Daskalakis C, Cambi F (2010) GM1 ganglioside in Parkinson's disease: results of a five year open study. J Neurol Sci 292: 45–51 [DOI] [PubMed] [Google Scholar]
- Schneider JS, Gollomp SM, Sendek S, Colcher A, Cambi F, Du W (2013) A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson's disease patients. J Neurol Sci 324: 140–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JS, Cambi F, Gollomp SM, Kuwabara H, Brasic JR, Leiby B, Sendek S, Wong DF (2015) GM1 ganglioside in Parkinson's disease: pilot study of effects on dopamine transporter binding. J Neurol Sci 356: 118–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab LC, Garas SN, Drouin‐Ouellet J, Mason SL, Stott SR, Barker RA (2015) Dopamine and Huntington's disease. Expert Rev Neurother 15: 445–458 [DOI] [PubMed] [Google Scholar]
- Sedelis M, Schwarting RK, Huston JP (2001) Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behav Brain Res 125: 109–125 [DOI] [PubMed] [Google Scholar]
- Sha S, Zhou L, Yin J, Takamiya K, Furukawa K, Furukawa K, Sokabe M, Chen L (2014) Deficits in cognitive function and hippocampal plasticity in GM2/GD2 synthase knockout mice. Hippocampus 24: 369–382 [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, Griffin JW, Schnaar RL (1999) Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci USA 96: 7532–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestroni A, Faull RL, Strand AD, Moller T (2009) Distinct neuroinflammatory profile in post‐mortem human Huntington's disease. NeuroReport 20: 1098–1103 [DOI] [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G (2007) Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. Glia 55: 1074–1084 [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Morgan BP, Gasque P (1999) Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp Neurol 159: 362–376 [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ et al (2003) Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 12: 1555–1567 [DOI] [PubMed] [Google Scholar]
- Snowden JS, Craufurd D, Thompson J, Neary D (2002) Psychomotor, executive, and memory function in preclinical Huntington's disease. J Clin Exp Neuropsychol 24: 133–145 [DOI] [PubMed] [Google Scholar]
- Sonnino S, Mauri L, Chigorno V, Prinetti A (2007) Gangliosides as components of lipid membrane domains. Glycobiology 17: 1R–13R [DOI] [PubMed] [Google Scholar]
- Spampanato J, Gu X, Yang XW, Mody I (2008) Progressive synaptic pathology of motor cortical neurons in a BAC transgenic mouse model of Huntington's disease. Neuroscience 157: 606–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spokes EG (1980) Neurochemical alterations in Huntington's chorea: a study of post‐mortem brain tissue. Brain 103: 179–210 [DOI] [PubMed] [Google Scholar]
- Stack EC, Smith KM, Ryu H, Cormier K, Chen M, Hagerty SW, Del Signore SJ, Cudkowicz ME, Friedlander RM, Ferrante RJ (2006) Combination therapy using minocycline and coenzyme Q10 in R6/2 transgenic Huntington's disease mice. Biochim Biophys Acta 1762: 373–380 [DOI] [PubMed] [Google Scholar]
- Stout JC, Paulsen JS, Queller S, Solomon AC, Whitlock KB, Campbell JC, Carlozzi N, Duff K, Beglinger LJ, Langbehn DR et al (2011) Neurocognitive signs in prodromal Huntington disease. Neuropsychology 25: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P (2004) DARPP‐32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol 44: 269–296 [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, Landwehrmeyer GB, Fox NC, Johnson H, Hicks SL et al (2011) Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: the 12‐month longitudinal analysis. Lancet Neurol 10: 31–42 [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Reilmann R, Roos RA, Durr A, Leavitt B, Owen G, Jones R, Johnson H, Craufurd D, Hicks SL et al (2012) Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK‐HD study: analysis of 24 month observational data. Lancet Neurol 11: 42–53 [DOI] [PubMed] [Google Scholar]
- Tache Y, Brunnhuber S (2008) From Hans Selye's discovery of biological stress to the identification of corticotropin‐releasing factor signaling pathways: implication in stress‐related functional bowel diseases. Ann N Y Acad Sci 1148: 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaut MH, Miltner R, Lange HW, Hurt CP, Hoemberg V (1999) Velocity modulation and rhythmic synchronization of gait in Huntington's disease. Mov Disord 14: 808–819 [DOI] [PubMed] [Google Scholar]
- Thompson TL, Moss RL (1997) Modulation of mesolimbic dopaminergic activity over the rat estrous cycle. Neurosci Lett 229: 145–148 [DOI] [PubMed] [Google Scholar]
- Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez‐Vincente M, Arrasate M, O'Rourke JG, Khashwji H et al (2009) IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol 187: 1083–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RC, Seminerio MJ, Naser ZJ, Ford JN, Martin SJ, Matsumoto RR, Rosen CL, Huber JD (2012) Effects of aging on behavioral assessment performance: implications for clinically relevant models of neurological disease. J Neurosurg 117: 629–637 [DOI] [PubMed] [Google Scholar]
- Ulloa RE, Nicolini H, Fernandez‐Guasti A (2004) Sex differences on spontaneous alternation in prepubertal rats: implications for an animal model of obsessive‐compulsive disorder. Prog Neuropsychopharmacol Biol Psychiatry 28: 687–692 [DOI] [PubMed] [Google Scholar]
- Vajn K, Viljetic B, Degmecic IV, Schnaar RL, Heffer M (2013) Differential distribution of major brain gangliosides in the adult mouse central nervous system. PLoS One 8: e75720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeputte C, Taymans JM, Casteels C, Coun F, Ni Y, Van Laere K, Baekelandt V (2010) Automated quantitative gait analysis in animal models of movement disorders. BMC Neurosci 11: 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinther‐Jensen T, Nielsen TT, Budtz‐Jorgensen E, Larsen IU, Hansen MM, Hasholt L, Hjermind LE, Nielsen JE, Norremolle A (2015) Psychiatric and cognitive symptoms in Huntington's disease are modified by polymorphisms in catecholamine regulating enzyme genes. Clin Genet 89: 320–327 [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Keller C, Cortes Ramirez EP (2011) Huntington's disease – neuropathology. Handb Clin Neurol 100: 83–100 [DOI] [PubMed] [Google Scholar]
- Wang J, Cheng A, Wakade C, Yu RK (2014) Ganglioside GD3 is required for neurogenesis and long‐term maintenance of neural stem cells in the postnatal mouse brain. J Neurosci 34: 13790–13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H (1999) Membrane filter assay for detection of amyloid‐like polyglutamine‐containing protein aggregates. Methods Enzymol 309: 375–386 [DOI] [PubMed] [Google Scholar]
- Weinstein DE, Shelanski ML, Liem RK (1991) Suppression by antisense mRNA demonstrates a requirement for the glial fibrillary acidic protein in the formation of stable astrocytic processes in response to neurons. J Cell Biol 112: 1205–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Como PG, Garron DC, Klawans HL, Barr A, Klawans D (1987) Memory failure in Huntington's disease. J Clin Exp Neuropsychol 9: 147–154 [DOI] [PubMed] [Google Scholar]
- Wolf RC, Sambataro F, Vasic N, Schonfeldt‐Lecuona C, Ecker D, Landwehrmeyer B (2008) Aberrant connectivity of lateral prefrontal networks in presymptomatic Huntington's disease. Exp Neurol 213: 137–144 [DOI] [PubMed] [Google Scholar]
- Yadin E, Friedman E, Bridger WH (1991) Spontaneous alternation behavior: an animal model for obsessive‐compulsive disorder? Pharmacol Biochem Behav 40: 311–315 [DOI] [PubMed] [Google Scholar]
- Yandrasitz JR, Cohn RM, Masley B, DelRowe D (1980) Evaluation of the binding of serotonin by isolated CNS acidic lipids. Neurochem Res 5: 465–477 [DOI] [PubMed] [Google Scholar]
- Yang LJ, Zeller CB, Shaper NL, Kiso M, Hasegawa A, Shapiro RE, Schnaar RL (1996) Gangliosides are neuronal ligands for myelin‐associated glycoprotein. Proc Natl Acad Sci USA 93: 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohrling IG, Jiang GC, DeJohn MM, Robertson DJ, Vrana KE, Cha JH (2002) Inhibition of tryptophan hydroxylase activity and decreased 5‐HT1A receptor binding in a mouse model of Huntington's disease. J Neurochem 82: 1416–1423 [DOI] [PubMed] [Google Scholar]
- Yu L, Liao PC (2000) Sexual differences and estrous cycle in methamphetamine‐induced dopamine and serotonin depletions in the striatum of mice. J Neural Transm 107: 419–427 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 2