

Abstract
A wide variety of cell death mechanisms, such as ferroptosis, have been proposed in mammalian cells, and the classification of cell death attracts global attention because each type of cell death has the potential to play causative roles in specific diseases. However, the precise molecular mechanisms leading to cell death are poorly understood, particularly in ferroptosis. Here, we show that continuous severe cold stress induces ferroptosis and the ASK1‐p38 MAPK pathway in multiple cell lines. The activation of the ASK1‐p38 pathway is mediated by critical determinants of ferroptosis: MEK activity, iron ions, and lipid peroxide. The chemical compound erastin, a potent ferroptosis inducer, also activates the ASK1‐p38 axis downstream of lipid peroxide accumulation and leads to ASK1‐dependent cell death in a cell type‐specific manner. These lines of evidence provide mechanistic insight into ferroptosis, a type of regulated necrosis.
Keywords: ASK1, cold stress, ferroptosis, p38 MAPK
Subject Categories: Autophagy & Cell Death, Signal Transduction
Introduction
Cells are persistently exposed to the fluctuation of diverse environmental cues, including temperature, pH and redox status; hence, it is critical for cells to manage the perturbation in order to maintain their homeostasis. Extracellular temperature is one major parameter easily varied depending on the situation, even within homothermal animals. Ablation of thermoregulation of the core body temperature is a life‐threatening event accompanied with multiple defects such as hypotension 1, indicating that temperature homeostasis is vital for living organisms. Only a slight rise in temperature causes protein misfolding, cytoskeleton defects, and changes in membrane fluidity, which provide deleterious effects to the cell. To mitigate this detrimental situation, heat‐shock proteins (HSPs) reign at the center of the heat‐shock responses 2. Numerous studies have revealed the mechanisms of heat‐shock responses; however, only a few studies have investigated the cold stress responses in mammals 3. Most studies have focused on the responses to mild hypothermia (25–35°C); however, a more severe cold stress response appears to be an important issue in the context of organ transplantation, a significant therapeutic method to replace dysfunctional organs 4, 5, 6. Even though a wide variety of organ preservation methods have been extensively examined, such as the machine perfusion system, cold stress‐induced tissue damage is still a significant issue to be solved. Several lines of evidence have shown that the accumulation of reactive oxygen species (ROS) is one of the main damaging factors in cold‐storage tissues, which induces cell death 7. However, the signaling mechanism by which cell toxicities are induced under severe cold situations remains elusive.
Diverse signaling cascades are liable to convey the sensed physicochemical stress, such as thermal stress, to the effector molecules; the stress‐responsive mitogen‐activated protein kinase (MAPK) cascade is one of the major pathways in which the signals converge onto two MAPKs, p38 MAPK and c‐jun N‐terminal kinase (JNK). Several MAPK kinases (MAP2Ks) are responsible for activating MAPKs, and over a dozen of MAPK kinase kinases (MAP3Ks) exist as upstream kinases of MAP2Ks 8. Apoptosis signal‐regulating kinase 1 (ASK1) was first discovered in 1997 as a novel member of the MAP3K family 9. Many studies have revealed that ASK1 is activated by a wide range of extra/intracellular stresses such as oxidative stress, ER stress and UV irradiation 10. Overall, the unique mechanism of the oxidative stress responses has been investigated extensively in the past two decades. Briefly, in response to oxidative stress, an ASK1 inhibitory protein thioredoxin (Trx) is conformationally changed into the oxidized form and dissociates from ASK1, which leads to ASK1 activation via enhancement of ASK1 homo‐oligomerization 11, 12, 13. Hydrogen peroxide (H2O2), a type of ROS, is a potent ASK1 activator that induces cell death; a number of studies have revealed that ASK1 is involved in H2O2‐induced apoptosis and necrosis 14, 15, 16. Considering that generation of ROS seems to be enhanced under cold circumstances 7, 17, ASK1 might respond to cold stress. According to a previous study, heat shock activates ASK1 in an ROS‐independent manner in concert with the dissociation of glutathione S‐transferase mu 1‐1 (GSTM1‐1), an inhibitory protein of ASK1, which results in p38 MAPK activation 18. Conversely, while ASK1‐deficient mice exhibit vulnerability to cold shock 19, the role of ASK1 signaling in a low‐temperature environment has not been studied at the cellular level. Regarding the downstream kinases, one study has revealed that severe cold stress leads to p38 MAPK activation and subsequent IL‐8 expression in bronchial epithelial cells 20, suggesting the involvement of MAPK signaling in cold stress responses.
The cell possesses the capacity to maintain its homeostasis through the response to diverse environmental cues including ambient temperature; however, drastic changes in the extracellular environment cause severe damage to the cell, which results in cell death. Cell death has long been divided into two classes: apoptosis and necrosis; the former is a regulated cell death, and the latter is an accidental one. However, the emergence of necroptosis has caused a paradigm shift in the field of cell death research; necroptosis is a regulated cell death with necrotic features 21, 22. In addition to necroptosis, other forms of regulated necrosis including ferroptosis, pyroptosis and parthanatos have recently been discovered 23. However, the precise molecular mechanisms to induce cell death of these emerging types of regulated necrosis remain elusive, especially in ferroptosis.
Ferroptosis was first defined in 2012 as a novel type of regulated necrosis requiring iron 24 and has been implicated in the context of several pathophysiological conditions such as tumorigenesis and ischemia–reperfusion injury 25, 26. The specific feature of ferroptosis is the requirement of lipid peroxide accumulation for cell death, and iron and MEK activity are prerequisites for the generation of lipid peroxide. A potent and specific inhibitor of ferroptosis was also identified in the same study and named ferrostatin‐1 (Fer‐1). Fer‐1 does not chelate iron or inhibit MEK activity but possesses an ability to scavenge free radicals 24. Although growing evidence has revealed the mechanism for lipid peroxide accumulation 27, 28, 29, the downstream signaling pathway of lipid peroxide accumulation responsible for cell death remains largely unknown. Only one report has suggested that MAP kinases are involved in this process 30.
Here, we show that continuous severe cold stress induces ferroptosis through lipid peroxide accumulation, and ASK1‐p38 signaling plays a pivotal role in regulating the cell death.
Results
Sustained cold stress induces cell death through the ASK1‐p38 axis
To examine the responsiveness of the ASK1‐MAPK pathways under cold stress, we applied direct cold stress to A549 cells in a CO2 incubator and assessed the activation status of ASK1, JNK, and p38 by phospho‐specific antibodies. Although ASK1 is not activated in the 25°C condition, an apparent increase in ASK1 activity was observed within 2 h by cold stress on ice (Fig 1A). p38 was strongly activated within 30 min on ice and weakly activated at 25°C, while JNK was not activated in response to cold stress (Fig 1A). We measured the actual temperature of the medium on ice and revealed that placing cell culture plates on top of ice for 10 min was enough to cool the medium down to ~8 to 10°C (Fig 1B), which we define as a “cold stress” hereafter. The temporal transition of the activity of ASK1 and MAPK was monitored for 5 h. The results showed that ASK1 and p38 were activated in a time‐dependent manner, but JNK was not activated even after 5 h of cold stress (Fig 1C). We also evaluated the effect of siRNA‐mediated knockdown of ASK1 on p38 activation. Cold stress‐induced p38 activation was abolished by ASK1 knockdown (Fig 1D), which indicates that the ASK1‐p38 pathway is activated in response to continuous and severe cold stress. Likewise, cold stress also induces ASK1‐p38 pathway activation in HEK293A (Fig 1E). To further analyze the activation mechanism of ASK1, we focused on the critical regulator of ASK1, thioredoxin (Trx). Trx is a redox‐regulatory protein, which binds to the N‐terminal region of ASK1 and suppresses ASK1 activity through the inhibition of homo‐oligomerization 11, 13. Oxidative stress such as H2O2 converts Trx into the oxidized form, which results in the dissociation of Trx from ASK1. In a similar fashion, Trx dissociation from ASK1 was also observed in response to cold stress (Fig 1F). These results suggest that continuous and severe cold stress induces the activation of the ASK1‐p38 axis presumably through ROS signaling.
Figure 1. Sustained cold stress activates the ASK1‐p38 pathway.
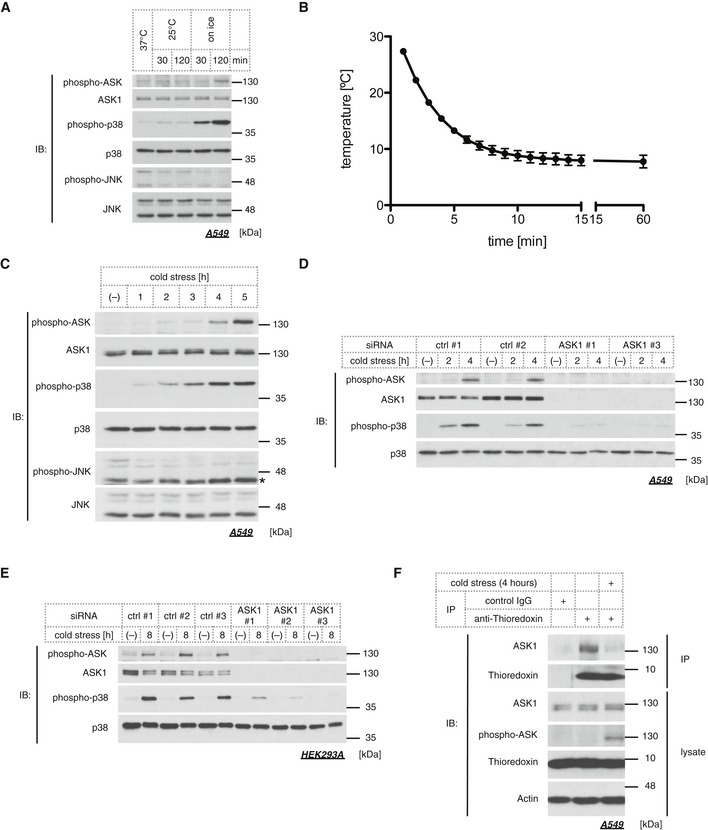
-
AImmunoblots of endogenous phospho‐ASK, phospho‐p38, and phospho‐JNK signals in the lysate from A549 cells treated with cold stress for the indicated time.
-
BThe temperature transition of culture medium placed on ice. Mean ± SEM, N = 3.
-
CImmunoblots of endogenous MAPK signals in A549 lysate treated with cold stress on ice for the indicated time. A star indicates non‐specific signal.
-
D, EImmunoblots of endogenous phospho‐ASK and phospho‐p38 signals in siRNA‐treated A549 (D) or in HEK293A cells (E). Cold stress on ice was applied for the indicated time.
-
FEndogenous thioredoxin was immunoprecipitated from A549 lysate with an anti‐thioredoxin antibody after 4 h of cold stress on ice, and the immune complex was examined by Western blot analysis.
Source data are available online for this figure.
Several lines of evidence have shown that prolonged cold stress eventually leads to cell death 17, 31, 32; hence, we confirmed whether cell death is observed in our system using A549 cells. As previously reported 32, cells treated with cold stress for 8 h underwent cell death as monitored by LDH release (Fig 2A). To investigate the requirement of the ASK1‐p38 axis in cold stress‐induced cell death, we utilized siRNA‐mediated knockdown and inhibitors. The knockdown of ASK1 clearly attenuated cold‐induced cell death (Fig 2B and C). Additionally, two p38 inhibitors (SB202190 and SB203580) also abolished cell death without affecting ASK1 activity, while their inactive analog (SB202474) did not suppress cell death (Figs 2D and EV1A). Conversely, a JNK inhibitor (SP600125) did not suppress cell death without affecting the phospho‐ASK and phospho‐p38 signals (Fig EV1B and C). The requirement of ASK1 for cold‐induced cell death was also observed in HEK293A cells (Fig 2E and F), and the requirement of p38 activity was also observed in HEK293A, HT‐1080, and HepG2 cells (Fig 2G–I). We observed that an inactive analog SB202474 exhibited only a marginal inhibitory effects on cold‐induced cell death in HEK293A cells with unknown mechanisms (Fig 2G). These data suggest that the ASK1‐p38 axis is required for cold stress‐induced cell death in multiple cell lines.
Figure 2. The ASK1‐p38 pathway is involved in cold stress‐induced cell death.
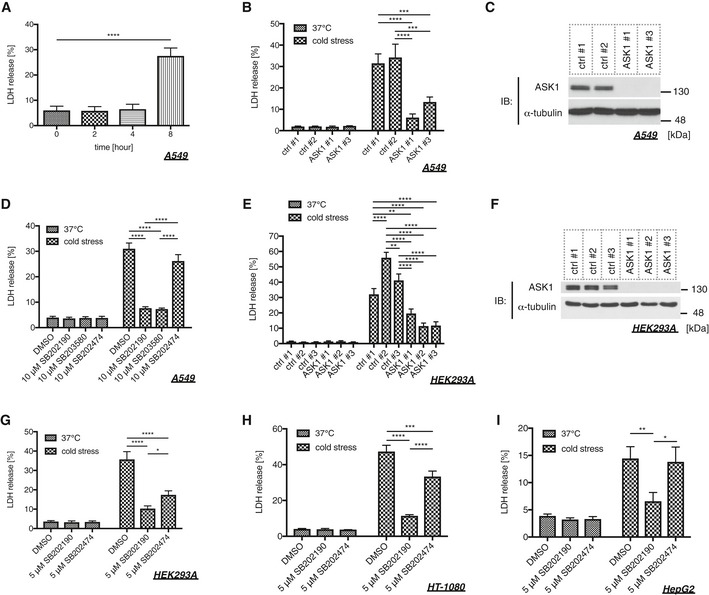
-
ACold stress was applied to A549 cells on ice for the indicated time, and cell death was determined by LDH assay. Mean ± SEM, N = 4. ****P < 0.0001 by one‐way ANOVA followed by Dunnett's multiple comparisons test. See also Appendix Table S1A.
-
B, C, E, FAn LDH assay was performed after 8 (B) or 12 (E) hours of cold stress on ice using siRNA‐treated A549 (B) or HEK293A (E) cells. Mean ± SEM, N = 4 (B), 5 (E). **P < 0.01, ***P < 0.001, ****P < 0.0001 by two‐way ANOVA followed by Tukey's multiple comparisons test. Immunoblots of endogenous ASK1 and α‐tubulin in A549 (C) or HEK293A (F) cells for confirming the knockdown efficiency of the samples used in (B) or (E), respectively. See also Appendix Table S1B and D.
-
D, G–ICells were pretreated with p38 inhibitors (10 μM SB203580, 10 μM SB202190) and their inactive analog (10 μM SB202474) 30 min before cold stress application. Cell death of A549 (D), HEK293A (G), HT‐1080 (H), and HepG2 (I) cells was determined by LDH assay after cold stress on ice for 8 (D, H) or 12 (G, I) hours. Mean ± SEM, N = 5 (D), 6 (G), 4 (H), 3 (I). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two‐way ANOVA followed by Tukey's multiple comparisons test. See also Appendix Table S1C, E, F, and G.
Source data are available online for this figure.
Figure EV1. Lipid peroxide‐ASK1‐p38 axis induces ferroptosis without mediating glutathione depletion.
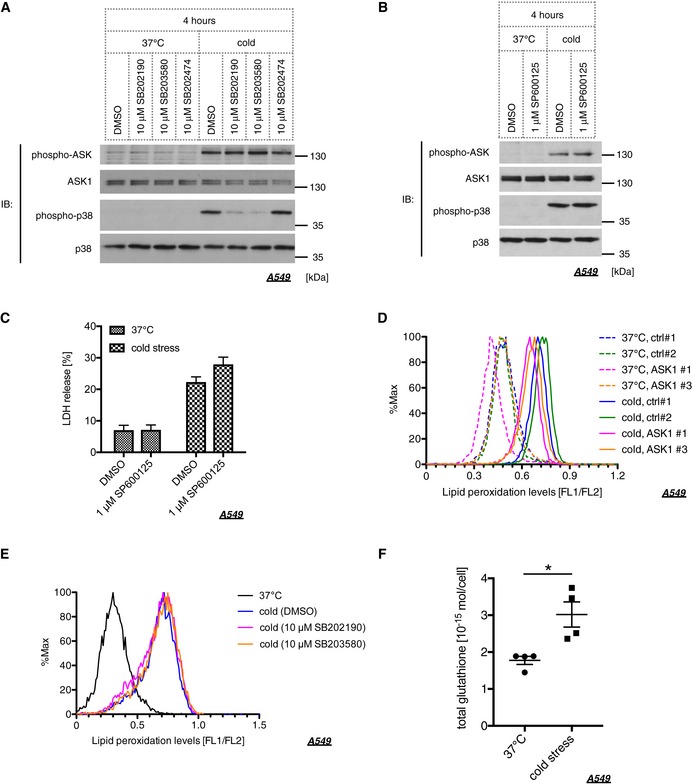
-
A, BCells were pretreated with p38 inhibitors (10 μM SB203580, 10 μM SB202190), their inactive analog (10 μM SB202474), and a JNK inhibitor (1 μM SP600125) 30 min before cold stress application. Endogenous phospho‐ASK and phospho‐p38 signals were assessed by Western blot analysis using A549 lysates treated with cold stress on ice for 4 h.
-
CCells were pretreated with a JNK inhibitor (1 μM SP600125) 30 min before cold stress application. Cell death of A549 cells was determined by LDH assay after cold stress on ice for 8 h. Mean ± SEM, N = 5. Two‐way ANOVA followed by Bonferroni's multiple comparisons test was performed.
-
DLipid peroxidation levels in siRNA‐treated A549 cells were determined by Bodipy 581/591 after 4 h of cold stress on ice.
-
EA549 cells were pretreated with p38 inhibitors (10 μM SB203580, 10 μM SB202190) for 30 min and then subjected to cold stress on ice for 4 h. Lipid peroxidation levels were determined by Bodipy 581/591.
-
FCold stress on ice was applied to A549 cells for 4 h, and total glutathione was measured. Mean ± SEM, N = 4. *P < 0.05 by unpaired two‐tailed Student's t‐test with Welch correction.
Source data are available online for this figure.
Cold stress evokes ferroptosis through the ASK1‐p38 axis
Diverse classification of cell death is emerging and is a focus of recent research because each type of cell death has a potential to play a causative role in specific diseases. We therefore sought to reveal the type of cell death caused by sustained cold stress. First, we assessed the involvement of apoptotic pathways 33; however, the pan‐caspase inhibitor Z‐VAD‐FMK did not inhibit cold‐induced cell death (Fig 3A), and caspase‐3 activity was not enhanced under cold stress conditions (Fig 3B). We next assessed the involvement of RIP1, which is a crucial regulator of necroptosis, using RIP1 kinase inhibitors: necrostatin‐1 (Nec‐1) and Nec‐1 stable (Nec‐1s) 21, 34, 35. Neither inhibitor suppressed the cold‐induced cell death, whereas both exhibited potent inhibitory effects on canonical necroptosis in L929 cells induced by Smac‐mimetic and Z‐VAD‐FMK (Fig 3C and D) 36. MLKL is a key downstream effector of RIP1‐ and RIP3‐dependent necroptosis, and necrosulfonamide (NSA) was identified as a direct inhibitor of MLKL 37. Treatment with NSA did not suppress the cell death either, whereas it abolished canonical necroptosis in HT‐29 cells (Fig 3E and F). These data recapitulate the hypothesis that canonical apoptosis or necroptosis pathways are not the cause of cold stress‐induced cell death.
Figure 3. Cold stress induces neither caspase‐dependent apoptosis nor RIP1‐dependent necroptosis.
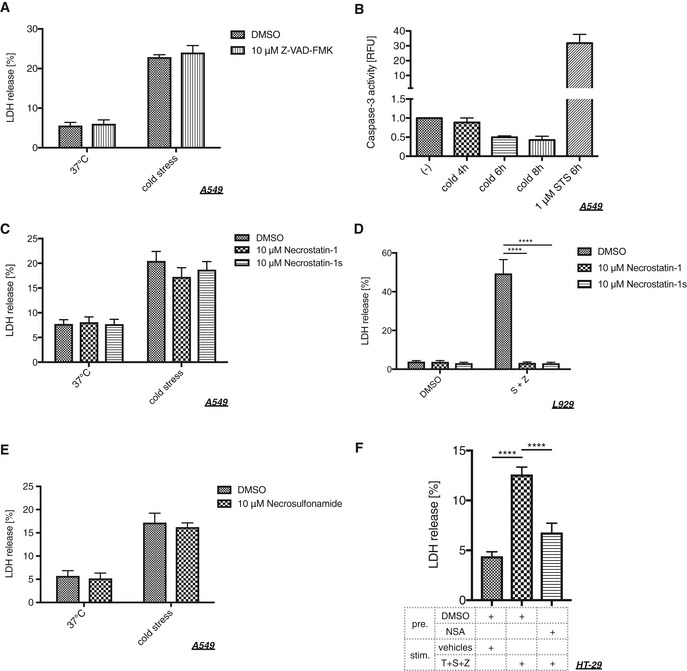
-
A, C, ECold stress was applied to A549 cells on ice for 8 h after pretreatment with a pan‐caspase inhibitor (10 μM Z‐VAD‐FMK), RIP1 inhibitors (10 μM necrostatin‐1, 10 μM necrostatin‐1s), or an MLKL inhibitor (10 μM necrosulfonamide) for 30 min. Cell death was assessed by LDH assay. Mean ± SEM, N = 3 (A), 4 (C), 5 (E). Two‐way ANOVA followed by Bonferroni's (A, E) or Tukey's (C) multiple comparisons test was performed. No significant difference was observed in all pairs.
-
BA caspase‐3 assay using A549 cells after cold stress treatment on ice for the indicated time. All samples were normalized by the protein amount and then normalized to the value of non‐treated cells in each experiment, and 1 μM staurosporine (STS) was used as a positive control. Mean ± SEM, N = 3.
-
DL929 cells were pretreated with RIP1 inhibitors (10 μM necrostatin‐1, 10 μM necrostatin‐1s) for 30 min. Then, necroptotic stimuli were applied using 50 nM Smac mimetics (S) and 10 μM Z‐VAD‐FMK (Z) for 8 h. Mean ± SEM, N = 3. ****P < 0.0001 by two‐way ANOVA followed by Dunnett's multiple comparisons test.
-
FHT‐29 cells were pretreated with an MLKL inhibitor (1 μM necrosulfonamide) for 30 min. Then, necroptotic stimuli were applied using 20 ng/μl TNF‐α (T), 100 nM Smac mimetics (S), and 20 μM Z‐VAD‐FMK (Z) for 24 h. Mean ± SEM, N = 7. ****P < 0.0001 by one‐way ANOVA followed by Tukey's multiple comparisons test.
In the context of organ preservation, several studies have investigated the mechanisms underlying cold stress‐induced cell death. The studies showing that lipid peroxide and MEK activity are involved in cell viability under cold circumstances 7, 17 led us to explore if cold stress evokes ferroptotic pathway. Three key determinants for ferroptosis have been identified in the previous report: lipid peroxide, iron ions, and MEK activity 24. Hence, we examined the involvement of these three factors using the lipid peroxide scavenger Fer‐1, the iron chelator deferoxamine (Dfx), and the MEK inhibitor U0126. Treatment with these three compounds clearly abolished cold‐induced cell death, while U0124, an inactive analog of U0126, failed to suppress cell death (Fig 4A and B). Even if cold stress was applied for longer time periods, such as 12 or 20 h, Fer‐1 completely inhibited cell death (Fig 4C). Activation of ASK1 and p38 was also suppressed by Fer‐1, Dfx, and U0126 but not by U0124, suggesting that the ASK1‐p38 axis is the downstream effector of the ferroptotic pathway (Fig 4D and E). To further examine whether lipid peroxide is accumulated within the cell under cold stress, we utilized Bodipy 581/591, which is commonly used to measure lipid peroxide levels 38, 39. As we expected, lipid peroxide was accumulated in a time‐dependent manner in response to cold stress (Fig 4F). Furthermore, the accumulation of lipid peroxide was completely abolished by Fer‐1, Dfx, and U0126 but not by U0124, in agreement with the previous report (Fig 4G and H) 24. However, knockdown of ASK1 or p38 inhibition did not affect lipid peroxide accumulation (Fig EV1D and E), which supports the model that the ASK1‐p38 axis is the downstream effector of lipid peroxide accumulation. Since glutathione depletion within the cell is the hallmark of ferroptotic pathway 26, we also measured the glutathione levels in A549 cells after 4 h of cold stress application. However, the total glutathione rather increased in response to cold stress (Fig EV1F), suggesting that cold stress‐induced cell death may differ in part from canonical ferroptosis.
Figure 4. Cold stress mediates ferroptosis through the ASK1‐p38 axis.
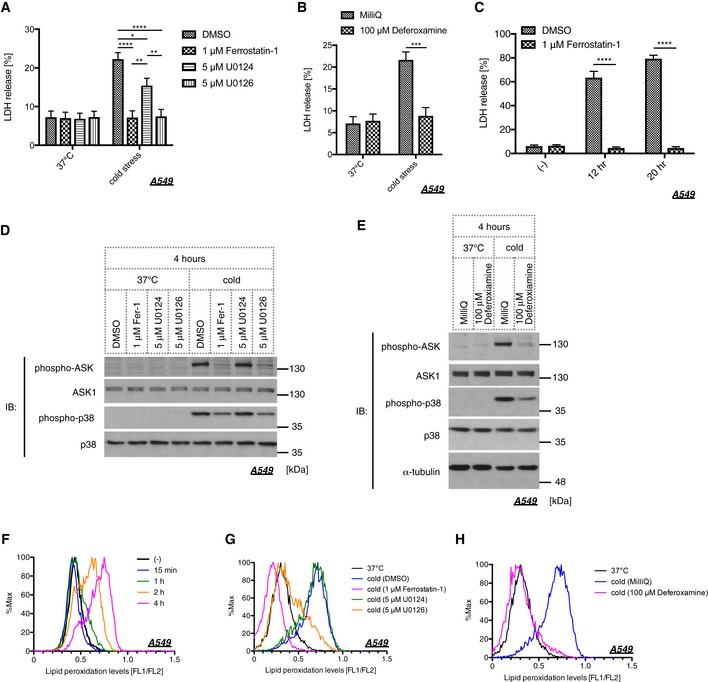
-
A, BCell death of A549 cells was examined by LDH assay after 8 h of cold stress on ice. Cells were pretreated with a lipid peroxide scavenger (1 μM ferrostatin‐1), MEK inhibitor (5 μM U0126), its inactive analog (5 μM U0124), or an iron chelator (100 μM deferoxamine). Mean ± SEM, N = 5. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two‐way ANOVA followed by Tukey's (A) or Bonferroni's (B) multiple comparisons test. See also Appendix Table S1H.
-
CA549 cells were pretreated with 1 μM ferrostatin‐1 for 30 min and then applied to cold stress on ice. Cell death was examined by LDH assay after the indicated time. Mean ± SEM, N = 4. ****P < 0.0001 by two‐way ANOVA followed by Bonferroni's multiple comparisons test.
-
D, EImmunoblots of endogenous phospho‐ASK and phospho‐p38 signals after 4 h of cold stress on ice. A549 cells were pretreated with the indicated compound for 30 min before cold stress was applied.
-
FLipid peroxidation levels in A549 cells were determined by Bodipy 581/591 after cold stress for the indicated time.
-
G, HCold stress on ice was applied to A549 cells after 30 min of pretreatment with the indicated compound. Lipid peroxidation was measured after 4 h of treatment.
Source data are available online for this figure.
Cold‐induced ferroptosis was observed not only in A549 cells but also in HEK293A, L929, HepG2, and HT‐1080 cells although the sensitivity to cold stress varies among the cell lines (Fig EV2A–E). These results suggest that ferroptosis is a common mechanism for cold‐induced cell death.
Figure EV2. Cold stress induces ferroptosis also in HEK293A, L929, HepG2, and HT‐1080 cells.
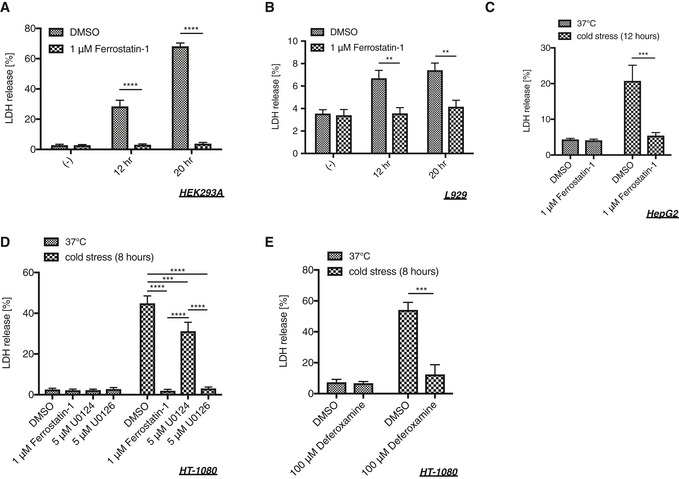
-
A–EHEK293A (A), L929 (B), HepG2 (C), or HT‐1080 (D, E) cells were pretreated with a lipid scavenger (1 μM ferrostatin‐1), MEK inhibitor (5 μM U0126), its inactive analog (5 μM U0124), or an iron chelator (100 μM deferoxamine) for 30 min followed by cold stress on ice for the indicated time. Cell death was measured by LDH assay. Mean ± SEM, N = 3 (A, E), 5 (B, D), 6 (C). **P < 0.01, ***P < 0.001, ****P < 0.0001 by two‐way ANOVA followed by Bonferroni's (A–C, E) or Tukey's (D) multiple comparisons test. See also Appendix Table S2A.
Taken together, these data indicate that cold stress evokes ferroptosis, and the ASK1‐p38 pathway is activated downstream of lipid peroxide, leading to the cell death.
The ASK1‐p38 axis is involved in erastin‐ and RSL3‐induced ferroptosis
To elucidate whether the ASK1‐p38 axis regulates ferroptosis in universal settings other than cold stress, we analyzed erastin‐induced cell death, one of the most established models of ferroptosis 24. Erastin inhibits cystine uptake by the Na+‐independent cystine/glutamate antiporter, system , which results in the depletion of glutathione, a major antioxidant within the cell, thereby facilitating toxic lipid peroxide accumulation 24. More than 16 h of treatment of A549 cells with erastin induced activation of ASK1 and p38 (Fig 5A), and Fer‐1 effectively prevented their activation (Fig 5B). Moreover, erastin‐induced p38 activation was suppressed in the absence of ASK1 as evidenced by the siRNA‐mediated knockdown experiment (Fig 5C). Thus, we concluded that the ASK1‐p38 axis is also activated in the erastin‐induced ferroptosis model as a downstream consequence of lipid peroxide accumulation.
Figure 5. The ASK1‐p38 pathway is activated through the course of erastin‐induced ferroptosis.
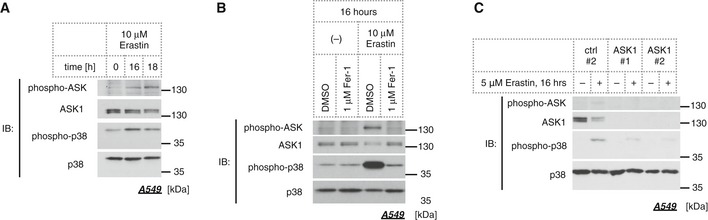
- Immunoblot of endogenous phospho‐ASK and phospho‐p38 signals in the lysate of A549 cells treated with 10 μM erastin for 16 or 18 h.
- Immunoblot of endogenous phospho‐ASK and phospho‐p38 signals in A549 cells after 16 h of treatment with 10 μM erastin in the presence or absence of 1 μM ferrostatin‐1. The pretreatment was for 30 min prior to erastin administration.
- The effect of siRNA‐mediated knockdown of ASK1 was assessed by Western blot analysis. A549 cells were treated with 5 μM erastin for 16 h.
Source data are available online for this figure.
We next examined the cell viability of erastin‐treated cells using water‐soluble tetrazolium salt. As previously reported, erastin dramatically reduced cell viability in A549 cells in a dose‐dependent manner (Fig 6A), which is regarded as the consequence of ferroptosis 40. We then confirmed that the decrease in cell viability was clearly rescued by treatment with Fer‐1, U0126, or Dfx but not U0124 (Fig 6A and B), suggesting that the dramatic decrease in the cell viability evoked by erastin is caused by ferroptosis. A recent study has suggested that p38 MAPK might be involved in this process in HL‐60 cells 30; hence, we analyzed the p38 requirement for erastin‐induced ferroptosis in our model. The p38 inhibitor SB202190 significantly suppressed erastin‐dependent ferroptosis, but its inactive analog SB202474 failed (Fig 6C). We further analyzed the importance of ASK1 by siRNA‐mediated knockdown and revealed that erastin‐induced ferroptosis was partially but significantly suppressed in the absence of ASK1, which was confirmed using three different siRNAs (Fig 6D and E). To confirm the requirement of ASK1 for erastin‐induced cell death, we also performed an LDH assay. Similar to the results found using the Cell Counting Kit‐8 assay in Fig 6D, ASK1 knockdown significantly attenuated LDH release (Fig 6E and F). RSL3 is another major ferroptosis inducer, which inhibits the activity of glutathione peroxidase 4 (GPX4) 26, 41. We then analyzed the ASK1‐dependency of RSL3‐induced ferroptosis in A549. The knockdown of ASK1 partially attenuated RSL3‐mediated ferroptosis in a similar fashion with erastin‐induced model (Fig 6E and G). To further analyze whether ASK1‐p38 pathway is the general mediator of ferroptosis, we also examined using HEK293A and HT‐1080 cells. p38 inhibition partially suppressed both erastin‐ and RSL3‐induced ferroptotic cell death in HEK293A and HT‐1080 cells, where Fer‐1, U0126, or Dfx clearly abolished the cell death (Fig EV3A–F). However, although ASK1‐p38 pathway is activated in response to RSL3 in HT‐1080 cells (Fig EV4A), siRNA‐mediated knockdown of ASK1 did not exhibit any effect for erastin‐ and RSL3‐induced cell death in HEK293A and HT‐1080 cells unlike in A549 cells (Fig EV4B–G). Moreover, the ASK1 knockdown did not suppress GPX4 deficiency‐dependent ferroptosis in HT‐1080 cells (Fig EV5A and B). These lines of evidence indicate that the ASK1‐p38 pathway is a signal mediator of ferroptosis in a cell type‐specific manner (Fig 6H).
Figure 6. The ASK1‐p38 pathway is involved in erastin‐induced ferroptosis.
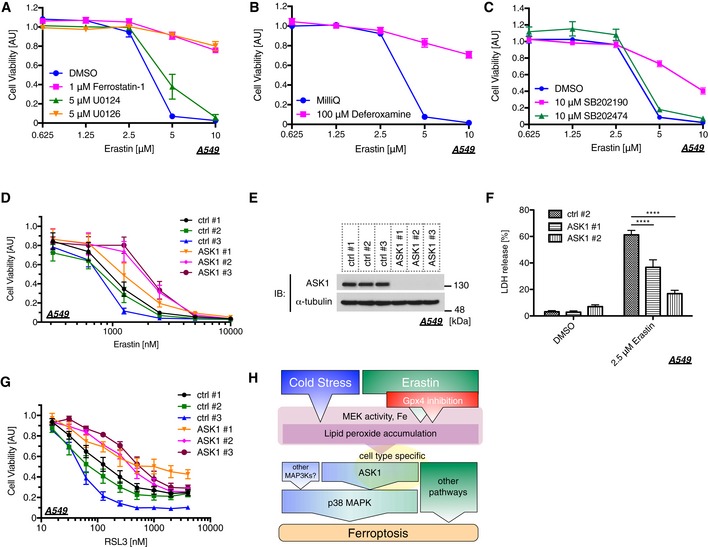
-
A–CA549 cells were treated with a lipid scavenger (1 μM ferrostatin‐1), a MEK inhibitor (5 μM U0126), its inactive analog (5 μM U0124), an iron chelator (100 μM deferoxamine), p38 inhibitor (10 μM SB202190), or its inactive analog (10 μM SB202474), followed by erastin treatment for 24 h at the indicated concentration. A Cell Counting Kit‐8 was utilized to measure cell viability. Mean ± SEM, N = 5. Two‐way ANOVA followed by Tukey's multiple comparisons test. See also Appendix Table S1I–K.
-
D–GA549 cells were transfected with siRNAs followed by erastin (D, F) or RSL3 (G) treatment. Cells were reseeded a day prior to erastin or RSL3 treatment in order to match the number of cells among each sample. Cells were treated with erastin or RSL3 at the indicated concentration for 24 h. Cell viability was measured using the Cell Counting Kit‐8 (D, G), and cell death was determined by LDH assay (F). Mean ± SEM, N = 4 (D), 6 (F, G). Two‐way ANOVA followed by Tukey's (D, G) or by Dunnett's (F) multiple comparisons test. ****P < 0.0001. See also Appendix Table S1L and M. (E) Immunoblots of endogenous ASK1 and α‐tubulin in A549 cells for confirming the knockdown efficiency of the samples used in (D, F, and G).
-
HSchematic diagram of the ferroptosis pathway through the ASK1‐p38 axis.
Source data are available online for this figure.
Figure EV3. p38 MAPK is partially involved in erastin‐ and RSL3‐induced ferroptotic cell death.
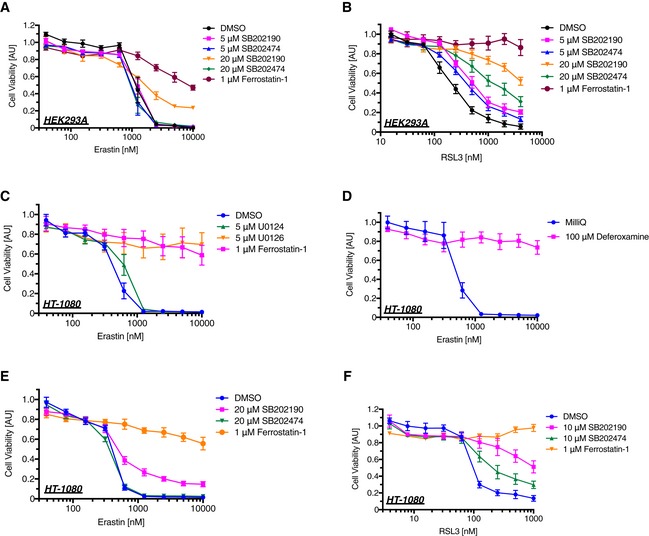
-
A–FHEK293A (A, B) or HT‐1080 (C–F) cells were treated with a lipid scavenger (1 μM ferrostatin‐1), a MEK inhibitor (5 μM U0126), its inactive analog (5 μM U0124), an iron chelator (100 μM deferoxamine), p38 inhibitor (SB202190), or its inactive analog (SB202474), immediately followed by erastin or RSL3 treatment for 24 h at the indicated concentration. A Cell Counting Kit‐8 was utilized to measure cell viability. Mean ± SEM, N = 4 (A–D), 5 (E), 3 (F). Two‐way ANOVA followed by Tukey's multiple comparisons test. See also Appendix Table S2B–G.
Figure EV4. ASK1 does not affect ferroptotic cell death in HEK293A and HT‐1080 cells.
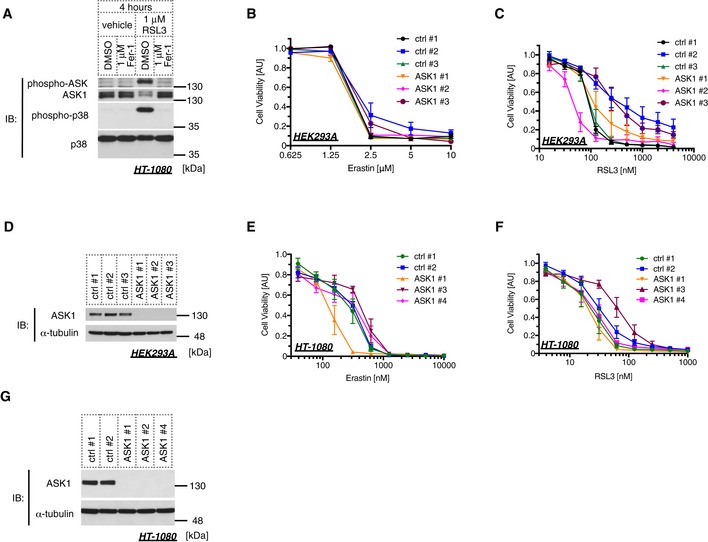
-
AImmunoblot of endogenous phospho‐ASK and phospho‐p38 signals in HT‐1080 cells after 4 h of treatment with 1 μM RSL3 in the presence or absence of 1 μM ferrostatin‐1. RSL3 administration was performed immediately after ferrostatin‐1 treatment.
-
B, C, E, FHEK293A (B, C) or HT‐1080 (E, F) cells were transfected with siRNAs followed by erastin (B, E) or RSL3 (C, F) treatment. Cells were reseeded a day prior to erastin or RSL3 treatment in order to match the number of cells among each sample. Cells were treated with erastin or RSL3 at the indicated concentration for 24 h. Cell viability was measured using the Cell Counting Kit‐8. Mean ± SEM, N = 5 (B, F), 3 (C, E). Two‐way ANOVA followed by Tukey's multiple comparisons test. See also Appendix Table S2H–K.
-
D, GImmunoblots of endogenous ASK1 and α‐tubulin in HEK293A (D) or HT‐1080 (G) cells for confirming the knockdown efficiency of the samples used in (B and C), and (E and F), respectively.
Source data are available online for this figure.
Figure EV5. ASK1 is not involved in GPX4 deficiency‐dependent ferroptosis.
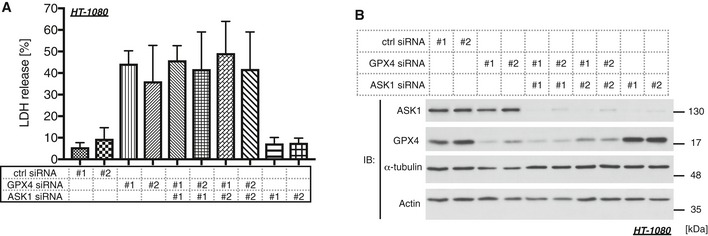
- Cell death of HT‐1080 cells was examined by LDH assay 2 days after siRNA transfection. Mean ± SEM, N = 4. One‐way ANOVA followed by Tukey's multiple comparisons test. See also Appendix Table S2L.
- Immunoblots of endogenous ASK1, GPX4, α‐tubulin, and actin in HT‐1080 cells for confirming the knockdown efficiency of the samples that was obtained 1 day after siRNA transfection.
Source data are available online for this figure.
Discussion
Homeothermal animals maintain their body temperature at ~37°C even under cold circumstances through shivering or non‐shivering thermogenesis. Most cells themselves, however, have difficulty producing heat in order to maintain their intracellular temperature; hence, a dramatic drop in ambient temperature easily causes cell damage. Many studies have focused on the cold ischemia–warm reperfusion model because it is worth analyzing as it relates to organ transplantation. In addition, several studies have attempted to discover the mechanism of cold stress‐induced cell death; however, whether apoptosis or necrosis occurs under cold circumstances remains controversial 17, 31, 42, 43. Here, we clearly show that cold stress induces ferroptosis in multiple cell lines (Figs 4 and EV2), and canonical apoptotic or necroptotic pathways are not major players in this process (Fig 3). There seems to be a discrepancy with the studies insisting that apoptosis is induced in response to cold 17, 43; however, previous reports only showed DNA fragmentation or changes in cell morphology as the evidence for apoptosis, which is apparently insufficient to demonstrate the requirement of apoptotic pathways. Although we cannot exclude the possibility that the type of cell death is dependent on cell type, cold stress‐dependent ferroptosis was commonly observed in at least five different cell types (Figs 4 and EV2).
Cells are rarely exposed to a severe cold environment for a long time period in physiological settings; however, sustained cold stress is regarded as one of the major problems in the context of organ transplantation 4. Many efforts have been made to improve the cold preservation solution or the apparatus used for continuous perfusion; however, the time period that a donor organ remains viable is still limited 44. Our results advocate that a ferroptosis inhibitor, such as Fer‐1, U0126, or Dfx, or p38 inhibitors are potential drugs for the prevention of cold‐induced injury, which might improve the method of organ preservation.
Since the discovery of ferroptosis in 2012, numerous studies have elucidated the mechanism by which lipid peroxide accumulates within the cell. Nevertheless, the exact effectors mediating cell death have remained largely unknown. In the present report, we revealed that the ASK1‐p38 pathway is one of the regulators of ferroptosis downstream of lipid peroxide. The fact that Trx clearly dissociates from ASK1 in response to cold stress suggests that the ASK1 complex may directly sense oxidative stress caused by lipid peroxidation (Fig 1F). Recent study revealed that NADPH depletion is one common biomarker of ferroptosis downstream of lipid peroxide accumulation; hence, NADPH depletion may lead to the activation of ASK1‐p38, or vice versa 45, 46. To understand the precise mechanism by which cold stress activates ASK1, the subcellular localization of the activation site of ASK1 would be an important issue to be elucidated. Furthermore, the downstream signaling of p38 also remains elusive. Taking one study with others, there is a possibility that p38 induces the nuclear translocation of AIF that was reported as one of the ferroptosis activators 47, 48, 49. Further analyses are required to elucidate the precise downstream mechanisms to induce ferroptosis including non‐cell‐autonomous mechanisms 25, 50.
A recent study suggested that the endoplasmic reticulum (ER) is an important site of lipid peroxidation 29. ASK1 adjacent to the ER might be activated in response to lipid peroxide. Considering that ASK1 activation is largely dependent on homo‐oligomerization‐dependent autophosphorylation 51, local activation of ASK1 may be propagated throughout the cytosol. There is also a possibility that lipid peroxide initiates further reactions that lead to redox imbalance at the other intracellular compartment where ASK1 is activated. From another point of view, many studies have suggested that cold stress evokes cell swelling dependent on the disruption of ionic integrity 42. Because ASK1 is activated by hypoosmotic stress that induces cell swelling 52, ASK1 may respond to cell expansion or ion imbalance under cold circumstances, provided that cold‐induced cell swelling requires lipid peroxide.
Growing evidence indicates that ferroptosis governs several pathophysiological settings including ischemia–reperfusion injury (IRI) and neurodegeneration 23. Based on our present data, the ASK1‐p38 axis potentially contributes to these pathologies. Indeed, several studies have already revealed that ASK1 deficiency ameliorates the pathogenesis of IRI in cardiomyocytes and the kidney 53, 54. These phenotypes may presumably be based on the suppression of ferroptotic cell death in ASK1‐deficient mice. Furthermore, our present dataset provides the insight that deletion of ASK1 or p38 inhibition is a promising target for the treatment of other ferroptosis‐related diseases such as cancer or neurological disorders.
Materials and Methods
Antibodies and reagents
A polyclonal antibody to phospho‐ASK1 (Thr838) was established as previously described 51. Phospho‐specific antibodies to p38 (Thr180/Tyr182) (#9211) and JNK (Thr183/Tyr185) (#9251) were purchased from Cell Signaling Technology. Antibodies to p38 (#9228) were also purchased from Cell Signaling Technology. The anti‐ASK1 antibody (ab45178) and the anti‐GPX4 antibody (ab125066) were purchased from Abcam, and the anti‐α‐tubulin antibody (MCA77G) was purchased from Bio‐Rad. The anti‐thioredoxin antibody (sc‐20146) and anti‐JNK1 antibody (sc‐571) were purchased from Santa Cruz Biotechnology. Normal rabbit IgG (sc‐2027) purchased from Santa Cruz Biotechnology was used for the immunoprecipitation assay.
SB202190 (559388), SB203580 (559389), SB202474 (559387), U0124 (662006), and necrosulfonamide (480073) were purchased from Calbiochem. SP600125 (194‐14823) and U0126 (211‐01051) were purchased from Wako. Ferrostatin‐1 (SML0583), necrostatin‐1 (N9037), and Z‐VAD‐FMK (V116) were purchased from Sigma. Deferoxamine (14595) was purchased from Cayman Chemical, necrostatin‐1s (2263‐1) from BioVision, Smac‐mimetic (LCL‐161) from Active Biochem, RSL3 (S8155) from Selleckchem, and Staurosporine (569396) from Millipore. Erastin was purchased from either Sigma (E7781) or Calbiochem (329600).
Cell culture and immunoblotting
A549, HEK293A, HT‐29 cells were cultured in DMEM–high glucose (D5796, Sigma) containing 10% FBS, and HT‐1080 cells were cultured in DMEM–high glucose (D5796, Sigma) containing 10% FBS and MEM Non‐Essential Amino Acids Solution (11140050, Thermo Fisher Scientific), and L929 and HepG2 cells were cultured in DMEM–low glucose (D6046, Sigma) containing 10% FBS in a 5% CO2 atmosphere at 37°C.
Lipofectamine RNAiMAX (Life Technologies) was utilized for transfection of siRNA. siRNAs were purchased from Invitrogen, and the sequences were as follows: human ASK1 #1, 5′‐GCCAACACUACAGUCAGGAAUUAAU‐3′; human ASK1 #2, 5′‐UGAAGCUAAGUAGUCUUCUUGGUAA‐3′; human ASK1 #3, 5′‐CCUGUGCUAACGACUUGCUUGUUGA‐3′; human ASK1 #4, 5′‐AUAUCUGAAGCAACUUGUCCUUCGC‐3′; human GPX4 #1, 5′‐UACUUGUCCAGGUUAACCAUGUGCC‐3′; human GPX4 #2, 5′‐UUUACUUCGGUCUUGCCUCACUGGG‐3′. Stealth RNAi siRNA Negative Control Med GC #1, #2, and #3 (Invitrogen) were used as controls. We avoided the use of the siRNAs that clearly affect cell proliferation.
Cells were lysed with IP lysis buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 4 mM EDTA pH 8.0, 1% w/v sodium deoxycholate, 1% v/v Triton X‐100, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 8 mM NaF, 1 mM Na3VO4, 12 mM β‐glycerophosphate, 1.2 mM Na2MoO4, 5 μM cantharidin, 2 mM imidazole), and immunoprecipitation was performed using nProtein A Sepharose 4 Fast Flow (GE Healthcare, 17‐5280‐04). Cell extracts and immunoprecipitates were resolved by SDS–PAGE and electroblotted onto polyvinylidene difluoride membranes (Pall or Millipore). The membranes were blocked with 2% skim milk (Yukijirushi) in TBS‐T (50 mM Tris–HCl, 150 mM NaCl, and 0.05% Tween 20, pH 8.0) and then probed with appropriate antibodies. Detection was performed using an ECL system.
Cold stress application
Cell culture plates were placed on top of metal covered with ice in a 5% CO2 atmosphere at ~26°C. The ice was replaced when it was melted. Cells were placed in a 5% CO2 atmosphere at 25°C if indicated. The temperature of the medium was measured using a microprobe thermometer (BAT‐12, Physitemp).
Cell death‐related assays
Cell death rate was measured using the LDH‐Cytotoxic Test (Wako) basically following the manufacturer's instruction. Briefly, culture medium was collected and centrifuged for 5 min at 400 × g to separate into the supernatant (medium sample) and a pellet. Adherent cells were lysed with PBS containing 0.1% Triton X‐100 (Sigma), and then the pellet was also lysed with the same solution, which was centrifuged for 5 min at 17,700 × g to separate it into the supernatant (lysate sample) and a pellet. Medium and lysate samples were individually mixed with reagents on microplates, and the absorbance was measured at 570 nm using Varioskan Flash (Thermo Fisher Scientific) or Multiskan BICHROMATIC (LabSystems) after a 10‐min incubation at room temperature.
Cell viability was measured using a Cell Counting Kit‐8 (CK04, Donjindo) following the manufacturer's instruction.
A caspase‐3 assay was performed using Caspase‐3 Substrate VII (264151, Calbiochem). Cells were lysed with RIPA buffer (150 mM NaCl, 50 mM Tris–HCl pH 8.0, 1% NP‐40, 0.5% DOC, 0.1% SDS), and the cell lysate was incubated with PBS containing reaction buffer (1068‐80, BioVision), dithiothreitol (D0632, Sigma) and Caspase‐3 Substrate VII for 2 h at 37°C. The luminescent signal was measured by Varioskan Flash (Thermo Fisher Scientific), and the signals were standardized by the protein amount. The protein concentration was determined using a DC Protein Assay (5000116JA, Bio‐Rad) following the manufacturer's instruction.
Lipid peroxide measurement
Here, 10 μM BODIPY 581/591 C11 (D3861, Thermo Fisher Scientific) was added to the culture media and incubated for an hour in a 5% CO2 atmosphere at 37°C. Cells were washed with PBS twice and then replaced with fresh culture media right before inhibitor treatment or cold stress application. In the erastin treatment experiments, 10 μM BODIPY 581/591 C11 was added to the culture media an hour before the analysis. After stimulation, cells were washed with PBS twice and trypsinized, followed by suspension in PBS. The cell suspension was filtered through a cell strainer (0.04 mm, Falcon) and then subjected to flow cytometer analysis (FACSCalibur, BD Biosciences) measuring 10,000 cells for each sample. The raw data were extracted by FlowPy (http://flowpy.wikidot.com) and calculated using Microsoft Excel, followed by depicting figures using GraphPad Prism. To calculate the FL1/FL2 ratio, we subtracted the median fluorescence values of unstained cells before dividing the values of each sample.
Determination of glutathione
Total glutathione was quantified using GSSG/GSH Quantification Kit (G257, Dojindo) following the manufacturer's instruction. The values were standardized by the cell number.
Statistical analysis
All experiments were independently repeated at least three times. All results are given as the mean ± SEM and N in each figure legend represent biological replicates. Unpaired two‐tailed Student's t‐test, one‐way ANOVA followed by Dunnett's or Tukey's multiple comparisons test, or two‐way ANOVA followed by Dunnett's, Bonferroni's or Tukey's multiple comparisons tests were used. The results of the statistical analyses are represented in Appendix Tables S1 and S2; stars are also indicated in some figures. Statistical analyses were performed using GraphPad Prism 7.
Author contributions
KH and HIs designed and performed the experiments. KH, CS, and ST performed the experiments for the revised version. IN and HIc supervised this study. KH and HIc wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank all of the members of our laboratory for meaningful discussion. This work was supported by a Grant‐in‐Aid for Scientific Research (KAKENHI) from Japan Society for the Promotion of Science (JSPS) Grant Numbers JP25221302, JP26111007, JP16K18872, JP15H04643, JP26114009, and Kowa Life Science Foundation.
EMBO Reports (2017) 18: 2067–2078
References
- 1. Cheshire WP Jr (2016) Thermoregulatory disorders and illness related to heat and cold stress. Auton Neurosci 196: 91–104 [DOI] [PubMed] [Google Scholar]
- 2. Richter K, Haslbeck M, Buchner J (2010) The heat shock response: life on the verge of death. Mol Cell 40: 253–266 [DOI] [PubMed] [Google Scholar]
- 3. Al‐Fageeh MB, Smales CM (2006) Control and regulation of the cellular responses to cold shock: the responses in yeast and mammalian systems. Biochem J 397: 247–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guibert EE, Petrenko AY, Balaban CL, Somov AY, Rodriguez JV, Fuller BJ (2011) Organ preservation: current concepts and new strategies for the next decade. Transfus Med Hemother 38: 125–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi S, Xue F (2016) Current antioxidant treatments in organ transplantation. Oxid Med Cell Longev 2016: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishikawa J, Oshima M, Iwasaki F, Suzuki R, Park J, Nakao K, Matsuzawa‐Adachi Y, Mizutsuki T, Kobayashi A, Abe Y et al (2015) Hypothermic temperature effects on organ survival and restoration. Sci Rep 5: 9563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karhumaki P, Tiitinen SL, Turpeinen H, Parkkinen J (2007) Inhibition of ERK1/2 activation by phenolic antioxidants protects kidney tubular cells during cold storage. Transplantation 83: 948–953 [DOI] [PubMed] [Google Scholar]
- 8. Hotamisligil GS, Davis RJ (2016) Cell signaling and stress responses. Cold Spring Harb Perspect Biol 8: a006072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275: 90–94 [DOI] [PubMed] [Google Scholar]
- 10. Sakauchi C, Wakatsuki H, Ichijo H, Hattori K (2017) Pleiotropic properties of ASK1. Biochim Biophys Acta 1861: 3030–3038 [DOI] [PubMed] [Google Scholar]
- 11. Fujino G, Noguchi T, Matsuzawa A, Yamauchi S, Saitoh M, Takeda K, Ichijo H (2007) Thioredoxin and TRAF family proteins regulate reactive oxygen species‐dependent activation of ASK1 through reciprocal modulation of the N‐terminal homophilic interaction of ASK1. Mol Cell Biol 27: 8152–8163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell 2: 389–395 [DOI] [PubMed] [Google Scholar]
- 13. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal‐regulating kinase (ASK) 1. EMBO J 17: 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagai H, Noguchi T, Homma K, Katagiri K, Takeda K, Matsuzawa A, Ichijo H (2009) Ubiquitin‐like sequence in ASK1 plays critical roles in the recognition and stabilization by USP9X and oxidative stress‐induced cell death. Mol Cell 36: 805–818 [DOI] [PubMed] [Google Scholar]
- 15. Noguchi T, Ishii K, Fukutomi H, Naguro I, Matsuzawa A, Takeda K, Ichijo H (2008) Requirement of reactive oxygen species‐dependent activation of ASK1‐p38 MAPK pathway for extracellular ATP‐induced apoptosis in macrophage. J Biol Chem 283: 7657–7665 [DOI] [PubMed] [Google Scholar]
- 16. Watanabe T, Sekine S, Naguro I, Sekine Y, Ichijo H (2015) Apoptosis signal‐regulating Kinase 1 (ASK1)‐p38 pathway‐dependent cytoplasmic translocation of the orphan nuclear receptor NR4A2 is required for oxidative stress‐induced necrosis. J Biol Chem 290: 10791–10803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rauen U, Polzar B, Stephan H, Mannherz HG, Groot HD (1999) Cold‐induced apoptosis in cultured hepatocytes and liver endothelial cells: mediation by reactive oxygen species. FASEB J 13: 155–168 [DOI] [PubMed] [Google Scholar]
- 18. Dorion S, Lambert H, Landry J (2002) Activation of the p38 signaling pathway by heat shock involves the dissociation of glutathione S‐transferase Mu from Ask1. J Biol Chem 277: 30792–30797 [DOI] [PubMed] [Google Scholar]
- 19. Hattori K, Naguro I, Okabe K, Funatsu T, Furutani S, Takeda K, Ichijo H (2016) ASK1 signalling regulates brown and beige adipocyte function. Nat Commun 7: 11158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gon Y, Hashimoto S, Matsumoto K, Nakayama T, Takeshita I, Horie T (1998) Cooling and rewarming‐induced IL‐8 expression in human bronchial epithelial cells through p38 MAP kinase‐dependent pathway. Biochem Biophys Res Commun 249: 156–160 [DOI] [PubMed] [Google Scholar]
- 21. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G et al (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4: 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang D‐W, Shao J, Lin J, Zhang N, Lu B‐J, Lin S‐C, Dong M‐Q, Han J (2009) RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science 325: 332–336 [DOI] [PubMed] [Google Scholar]
- 23. Conrad M, Angeli JPF, Vandenabeele P, Stockwell BR (2016) Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 15: 348–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS et al (2012) Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell 149: 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, Zen FD, Prokai A, Zuchtriegel G, Krombach F, Welz P‐S et al (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA 111: 16836–16841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB et al (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156: 317–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti‐Furga G, Stockwell BR (2015) Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 10: 1604–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A et al (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13: 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB et al (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13: 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D (2015) The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2: e1054549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morishita S, Nishi Y, Sato EF, Yamaoka K, Manabe M, Inoue M (1997) Cold‐stress induces thymocyte apoptosis in the rat. Pathophysiology 4: 213–219 [Google Scholar]
- 32. Rauen U, Petrat F, Li T, Groot HD (2000) Hypothermia injury/cold‐induced apoptosis—evidence of an increase in chelatable iron causing oxidative injury in spite of low O2−/H2O2 formation. FASEB J 14: 1953–1964 [DOI] [PubMed] [Google Scholar]
- 33. Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119 [DOI] [PubMed] [Google Scholar]
- 35. Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB , Goossens V, Roelandt R, Van Hauwermeiren F, Libert C et al (2012) Necrostatin‐1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 3: e437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐α. Cell 137: 1100–1111 [DOI] [PubMed] [Google Scholar]
- 37. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X et al (2012) Mixed lineage kinase domain‐like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227 [DOI] [PubMed] [Google Scholar]
- 38. Drummen GPC, van Liebergen LCM, Op den Kamp JAF, Post JA (2002) C11‐BODIPY581/591, an oxidation‐sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic Biol Med 33: 473–490 [DOI] [PubMed] [Google Scholar]
- 39. Pap EHW, Drummen GPC, Winter VJ, Kooij TWA, Rijken P, Wirtz KWA, Op den Kamp JAF, Hage WJ, Post JA (1999) Ratio‐fluorescence microscopy of lipid oxidation in living cells using C11‐BODIPY581/591. FEBS Lett 453: 278–282 [DOI] [PubMed] [Google Scholar]
- 40. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS et al (2014) Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3: e02523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron‐dependent, nonapoptotic cell death in oncogenic‐RAS‐harboring cancer cells. Chem Biol 15: 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boutilier RG (2001) Mechanisms of cell survival in hypoxia and hypothermia. J Exp Biol 204: 3171–3181 [DOI] [PubMed] [Google Scholar]
- 43. Gregory CD, Milner AE (1994) Regulation of cell survival in burkitt lymphoma: implications from studies of apoptosis following cold‐shock treatment. Int J Cancer 57: 419–426 [DOI] [PubMed] [Google Scholar]
- 44. Cameron AM, Cornejo JFB (2015) Organ preservation review: history of organ preservation. Curr Opin Organ Transplant 20: 146–151 [DOI] [PubMed] [Google Scholar]
- 45. Tonnus W, Linkermann A (2016) ‘Death is my Heir’—Ferroptosis connects cancer pharmacogenomics and ischemia‐reperfusion injury. Cell Chem Biol 23: 202–203 [DOI] [PubMed] [Google Scholar]
- 46. Shimada K, Hayano M, Pagano NC, Stockwell BR (2016) Cell‐line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol 23: 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang Y‐H, Lee S‐J (2008) The role of p38 MAPK and JNK in Arsenic trioxide‐induced mitochondrial cell death in human cervical cancer cells. J Cell Physiol 217: 23–33 [DOI] [PubMed] [Google Scholar]
- 48. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W et al (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15‐lipoxygenase dependent‐ and AIF‐mediated cell death. Cell Metab 8: 237–248 [DOI] [PubMed] [Google Scholar]
- 49. Kim MJ, Woo JS, Kwon CH, Kim JH, Kim YK, Kim KH (2012) Luteolin induces apoptotic cell death through AIF nuclear translocation mediated by activation of ERK and p38 in human breast cancer cell lines. Cell Biol Int 36: 339–344 [DOI] [PubMed] [Google Scholar]
- 50. Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X et al (2016) Ultrasmall nanoparticles induce ferroptosis in nutrient‐deprived cancer cells and suppress tumour growth. Nat Nanotechnol 11: 977–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tobiume K, Saitoh M, Ichijo H (2002) Activation of apoptosis signal‐regulating kinase 1 by the stress‐induced activating phosphorylation of pre‐formed oligomer. J Cell Physiol 191: 95–104 [DOI] [PubMed] [Google Scholar]
- 52. Naguro I, Umeda T, Kobayashi Y, Maruyama J, Hattori K, Shimizu Y, Kataoka K, Kim‐Mitsuyama S, Uchida S, Vandewalle A et al (2012) ASK3 responds to osmotic stress and regulates blood pressure by suppressing WNK1‐SPAK/OSR1 signaling in the kidney. Nat Commun 3: 1285 [DOI] [PubMed] [Google Scholar]
- 53. Terada Y, Inoshita S, Kuwana H, Kobayashi T, Okado T, Ichijo H, Sasaki S (2007) Important role of apoptosis signal‐regulating kinase 1 in ischemic acute kidney injury. Biochem Biophys Res Commun 364: 1043–1049 [DOI] [PubMed] [Google Scholar]
- 54. Watanabe T, Otsu K, Takeda T, Yamaguchi O, Hikoso S, Kashiwase K, Higuchi Y, Taniike M, Nakai A, Matsumura Y et al (2005) Apoptosis signal‐regulating kinase 1 is involved not only in apoptosis but also in non‐apoptotic cardiomyocyte death. Biochem Biophys Res Commun 333: 562–567 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6