

Abstract
Lipopolysaccharide‐responsive beige‐like anchor protein (LRBA) belongs to the enigmatic class of BEACH domain‐containing proteins, which have been attributed various cellular functions, typically involving intracellular protein and membrane transport processes. Here, we show that LRBA deficiency in mice leads to progressive sensorineural hearing loss. In LRBA knockout mice, inner and outer hair cell stereociliary bundles initially develop normally, but then partially degenerate during the second postnatal week. LRBA deficiency is associated with a reduced abundance of radixin and Nherf2, two adaptor proteins, which are important for the mechanical stability of the basal taper region of stereocilia. Our data suggest that due to the loss of structural integrity of the central parts of the hair bundle, the hair cell receptor potential is reduced, resulting in a loss of cochlear sensitivity and functional loss of the fraction of spiral ganglion neurons with low spontaneous firing rates. Clinical data obtained from two human patients with protein‐truncating nonsense or frameshift mutations suggest that LRBA deficiency may likewise cause syndromic sensorineural hearing impairment in humans, albeit less severe than in our mouse model.
Keywords: cochlear amplification, hair bundle degeneration, progressive hearing loss, radixin, stereociliar protein transport
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Development & Differentiation; Neuroscience
Introduction
Mechanosensory hair cells of the mammalian inner ear faithfully transduce sound waves into neural code with high temporal precision and frequency selectivity. This demanding task relies on the seamless interplay of two types of hair cells. While (i) outer hair cells (OHCs) amplify the sound‐induced oscillations of the basilar membrane and fine‐tune frequency resolution, (ii) inner hair cells (IHCs)—the true sensory cells of the inner ear—encode sound through graded release of glutamate from their ribbon‐type presynaptic active zones onto afferent spiral ganglion neurons (SGNs).
At their apical poles, both types of hair cells are equipped with highly organized arrays of stereocilia, a specialized type of actin‐rich microvilli that form the characteristic hair bundle and harbor the components of the mechanotransduction machinery. Upon physical hair bundle deflection, the coordinated opening of mechanically gated ion channels in the stereociliary tips mediates hair cell depolarization, which in turn leads to synchronized electromotile contraction of OHCs and presynaptic glutamate release from the 5–30 IHC active zones. Each IHC synapse is characterized by a synaptic ribbon, a highly specialized structure which enables the turnover of hundreds to thousands of vesicles per second with utmost temporal precision 1, 2.
At the apical pole, the development and maintenance of the stereociliar arrays critically depend on efficient targeting and interaction of the diverse protein and lipid components of the hair bundles (reviewed in Refs 3, 4, 5, 6). Perturbations of this molecular choreography underlie a number of severe hereditary hearing disorders, most commonly due to failure of either the mechanotransduction machinery itself, or corrupted stereociliar orientation, organization, or elongation 3, 4, 5, 7, 8. For example, the cause of human autosomal recessive hearing impairment DFNB24 9, 10, 11 is the genetic deletion of the ezrin–radixin–moesin (ERM) family protein, radixin (RDX)—a cross‐linker between plasma membranes and the stereociliar cytoskeleton. Radixin deficiency in mice leads to early postnatal hair bundle degeneration 12. Here, we describe a similar phenotype in another mouse mutant, the Lrba knockout (KO) mouse. LRBA is one of nine members of the BEACH protein family. These proteins are characterized by the presence of a BEACH domain (“Beige and Chediak‐Higashi”; ~260 amino acids; 13) and are typically large in size (~3,000 amino acids). Even though this protein family was discovered nearly 20 years ago, its primary molecular and cellular functions remain to be identified. The smallest common denominator at the current state of knowledge is a role in orchestrating the assembly of intricate arrays of biological macromolecules, typically in the context of membrane dynamics or membrane protein targeting. BEACH protein deficiency mutants in various species display complex phenotypes, and several of the nine identified human BEACH proteins, including LRBA, are involved in human diseases [reviewed in Ref. 14]. The physiological importance of LRBA has so far mostly been studied in stimulated immune cells 15, 16, but it is also expressed in many other tissues, where its function is not yet understood.
Here, we used a Lrba‐KO mouse line to comprehensively study the function of LRBA. Lrba‐KO mice proved to be viable and fertile with no obvious abnormalities, but a phenotyping screen at the German Mouse Clinic (GMC) detected reduced startle reactivity and prepulse inhibition, suggestive of hearing impairment. In order to clarify which step(s) of the auditory signal cascade is (are) perturbed by the genetic deletion of Lrba, we embarked on a detailed interdisciplinary analysis of auditory function in Lrba‐KO mice. We found that LRBA is highly expressed in wild‐type mouse cochlear IHCs and OHCs, where it clusters at the base of the kinocilium during development. Genetic ablation of Lrba left OHC electromotility and IHC synaptic transmission intact, but resulted in premature degeneration of the central parts of the OHC hair bundles, which is likely related to reduced abundance of the stereociliar ERM protein RDX and the RDX‐interacting protein Nherf2. Consequently, cochlear receptor potentials are reduced, entailing deficient cochlear amplification and impaired sound encoding. Finally, two human patients with LRBA deficiency were found to exhibit hearing impairment, an observation consistent with the described KO mouse phenotype, albeit less severe.
Results
LRBA expression in the murine cochlea and progressive hearing loss in Lrba‐KO mice
Lrba‐KO mice were generated by constitutive deletion of coding exon 3, thereby introducing a frameshift mutation predicted to lead to degradation of the truncated protein. Western blot analysis confirmed the complete loss of LRBA protein expression in all tested tissues (kidney, brain, olfactory epithelium, retina 17). Lrba‐KO mice were born at Mendelian ratios and showed no obvious abnormalities; however, phenotypic screening at the German Mouse Clinic revealed reduced auditory startle responses (Appendix Fig S1A), a finding suggestive of hearing impairment.
Immunofluorescence (IF) microscopy in cochleae of p15 wild‐type mice revealed prominent LRBA expression in IHCs and OHCs, which is in agreement with entries in the SHIELD database reporting RNA‐Seq‐based expression analysis 18, 19. Weaker immunostaining signals were observed in several types of supporting cells, the lining of the stria vascularis and Reissner's membrane, but not in SGNs (Fig 1A and B). Specific LRBA expression in IHC, OHC, and supporting cells was confirmed by positive LacZ staining of organs of Corti from gene‐trap mice expressing β‐galactosidase reporter activity under the control of the Lrba gene promoter (Appendix Fig S1B), and by the complete absence of staining in Lrba‐KO mice (Fig 1B′, Appendix Fig S1C and D).
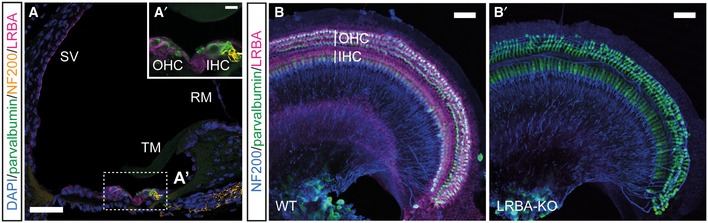
Figure 1. LRBA expression in the organ of Corti.
-
A, A′Representative immunofluorescence images from a WT p15 organ of Corti cryosection stained for LRBA (magenta), the hair cell marker parvalbumin (PV; green), and the spiral ganglion neuronal fiber marker neurofilament 200 (NF200; orange); nuclei are counterstained with DAPI. LRBA immunoreactivity could be detected in hair cells, multiple types of supporting cells, the marginal cells of the stria vascularis (SV), and Reissner's membrane (RM). (A′) Inset shows a higher magnification view of the sensory epithelium, but omitting the DAPI staining for clarity. Scale bars: 50 μm, in inset: 10 μm. IHC, inner hair cells; OHC, outer hair cells; TM, tectorial membrane.
-
B, B′Representative confocal maximum projections from acutely dissected (B) WT and (B′) Lrba‐KO p15 apical turn whole‐mount organs of Corti immunostained for LRBA (magenta), NF200 (blue), and PV (green). Scale bars: 100 μm.
Next, we assessed hearing of Lrba‐KO mice by recording auditory brainstem responses (ABR). ABR thresholds to tone bursts and click stimulation were moderately elevated in Lrba‐KO mice directly after hearing onset and progressively deteriorated with advancing age (Fig 2A). All ABR waves, reporting synchronous spiking in response to sound onset in SGNs (wave I) and neurons of the auditory brainstem (waves II–V), were markedly reduced in amplitude (Fig 2B and C). Moreover, ABR latencies were increased, as expected for the loss in sensitivity (Appendix Fig S2A). Moreover, electrocochleography recordings confirmed that the elevated thresholds and reduced amplitudes of the SGN compound action potential (CAP) amplitude (Appendix Fig S2B) were associated with an amplitude reduction in the cochlear microphonic and summating potentials (Fig 2D–F). In addition, distortion product otoacoustic emissions (DPOAE) were greatly reduced in Lrba mutants across all frequencies (Fig 2G). These data indicate that the loss in sound sensitivity is due to impaired peripheral auditory function and involves a primary or secondary defect of cochlear amplification, the mechanism of which remained to be determined.
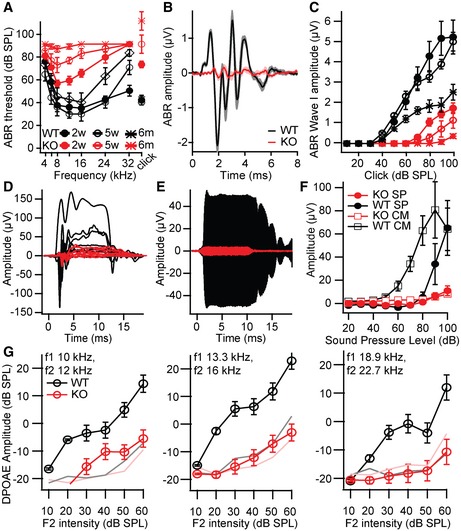
Figure 2. Progressive hearing loss in Lrba mutants.
-
AAuditory brainstem response (ABR) recordings from 2‐ to 3‐week‐old (p14–19; filled circles, n = 6 each), 4‐ to 5‐week‐old (open circles, n = 8 KO/9 WT), and 6‐month‐old (open diamonds, n = 5 KO/4 WT) Lrba‐KO mice (red) and WT littermates (black). Thresholds exceeding the maximum loudspeaker output (80 dB for tone bursts, 120 dB for clicks) were assigned values of 90/130 dB for calculation of the means ± s.e.m.
-
BGrand averages ± s.e.m. (shaded) of ABR to 80 dB click stimuli show a strong reduction in ABR waves I–V in Lrba‐KO mice (red, n = 9) compared to WT (black, n = 8, age 5 weeks).
-
CABR wave I has dramatically decreased amplitudes (mean ± s.e.m., same animals and markers as in A) with a shallower growth function in the mutants, progressively deteriorating with age.
-
D–FRepresentative traces of (D) summating potentials and (E) cochlear microphonic potentials to 90‐dB 16‐kHz tone bursts. (F) Quantification of the amplitude of cochlear microphonic potentials (open squares) and summating potentials (closed circles) for 16‐kHz tone bursts at varying intensities (mean ± s.e.m., n = 9 each, age 4–5 weeks). SP, summating potentials; CM, cochlear microphonic potentials.
-
GReduced amplitudes of distortion product otoacoustic emissions (DPOAE) in Lrba‐KO animals (red, n = 7) compared to WT littermates (black, n = 8) are suggestive of an amplifier defect (mean ± s.e.m., age 4–5 weeks). Pink and gray lines indicate the average noise floor.
Next, we set out to narrow down the site of lesion and further characterized the sound encoding deficit in the absence of LRBA by recording spontaneous and sound‐evoked action potential firing of postsynaptic SGNs in vivo. The distribution of spontaneous SGN firing rates was clearly biased towards high SR fibers in Lrba‐KO mice (Fig 3A). For each fiber, we next characterized the frequency tuning by determining sound thresholds for varying tone burst frequencies. While fibers from wild‐type animals had normal, sharp tuning curves with low thresholds, Lrba‐KO fibers showed broadened tuning curves with poorly defined characteristic frequencies and highly elevated thresholds (Fig 3B; median threshold; WT: 51 dB; n = 45; Lrba‐KO: 92 dB; n = 94; P < 0.0001, Mann–Whitney U‐test). As expected for a defect in cochlear amplification, the increase in spike rates with stimulus level was significantly steeper in Lrba‐KO than in wild‐type littermates (Fig 3C and C′). Moreover, the dynamic range of Lrba‐KO SGNs tended to be reduced (Fig 3C″). Nonetheless, the evoked spike rates and time courses of adaptation in response to tone bursts at high intensities—at which spike rates were saturated—were identical between Lrba‐KO and wild‐type littermate controls (Fig 3D). Also, IHC presynaptic vesicle depletion and recovery, as assessed in a paired pulse paradigm, were normal in Lrba‐KO mice (Fig 3E).
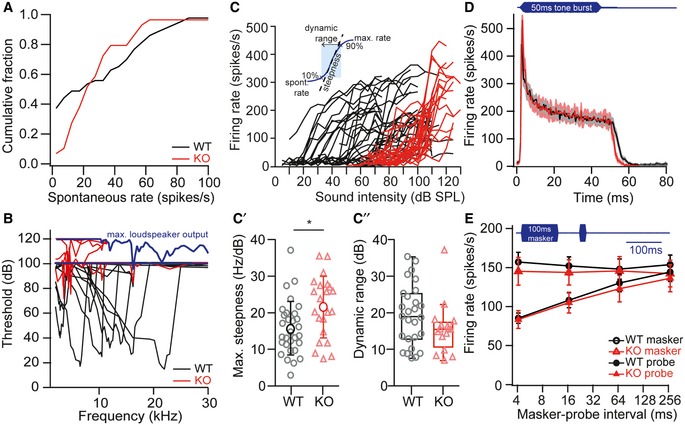
Figure 3. Elevated sound thresholds and lack of low spontaneous rate fibers in Lrba‐KO animals.
-
ADistribution of spontaneous firing rates of SGN from WT and Lrba‐KO mice aged 5–6 weeks. Mutants (red, n = 54) show a significant reduction in the fraction of low spontaneous rate fibers compared to WT (black, n = 49, P < 0.0001, Kolmogorov–Smirnov test).
-
BSound thresholds of Lrba‐KO (red) SGNs are highly elevated and lack the asymmetric V‐shaped frequency tuning curve that is typical of WT SGNs (representative examples). The maximum stimulus intensity (blue line) was initially restricted to 100 dB to avoid noise damage. Since mutant SGNs typically did not always respond at that intensity, it was increased up to 120 dB in later experiments with Lrba‐KO.
-
C–C″SGN firing rates in response to tone bursts of varying intensities: Mutants show elevated thresholds, a steeper increase in firing rate (C′, WT n = 30, KO n = 22, *P = 0.011, unpaired Student's t‐test) and a trend for a reduced dynamic range (C″, WT n = 27, KO n = 16, P = 0.126, unpaired Mann–Whitney U‐test). Data in (C′) are presented as means ± s.d.; Boxplots in (C″) show median, inner/outer quartile + outlier range.
-
DIn response to 50‐ms tone burst stimulation (characteristic/best frequency, 30 dB above threshold in WT, at least 10 dB above threshold in KO), response patterns, peak and steady‐state firing rates as well as first spike latencies were comparable between both genotypes (WT n = 37, KO n = 22). Data are presented as means ± s.e.m. (gray and light red shadings).
-
ETop: illustration of the stimulus paradigm: 100‐ms masker tone and 15‐ms probe tone separated by a silent interval of variable duration. Both stimuli were presented at characteristic/best frequency, 30 dB above threshold in WT, at least 10 dB above threshold in KO. Bottom: Forward masking effect and time course of recovery were comparable between WT (n = 20) and KO (n = 14) SGNs. Data are presented as means ± s.e.m.
Taken together, the in vivo data were mostly consistent with a deficit of active cochlear amplification in Lrba‐KO mice, for example, through corrupted OHC electromotility or mechanotransduction deficits; however, the reduction in spontaneous SGN spike rates and the shallow amplitude growth functions of ABR/CAPs (Fig 2C and Appendix Fig S2B) suggested an additional impairment of IHC synaptic transmission (“synaptopathy”): in case of a pure OHC dysfunction, an increased steepness would have been expected 20. Given the essential role of the highly homologous BEACH protein neurobeachin for neurotransmission at conventional synapses and neuromuscular junctions 21, 22, 23, 24, 25, we also considered a role for LRBA in the development and presynaptic function of IHC ribbon synapses. We therefore studied OHC and IHC maturation and function in vitro using electrophysiological recordings and immunohistochemistry. We found no alterations in voltage‐dependent nonlinear capacitance changes in OHC at postnatal days 15–16 (Fig EV1; Appendix Supplementary Methods), which would have suggested a change in the trafficking and/or membrane insertion of the OHC motor protein prestin or a developmental delay. In addition, we neither found changes in IHC BK channel clustering, nor in ribbon synapse number or in depolarization‐induced calcium currents and exocytosis (Fig EV2; Appendix Supplementary Methods), which would have pointed to deficits in IHC maturation, synapse formation, or presynaptic vesicle release mechanisms, respectively.
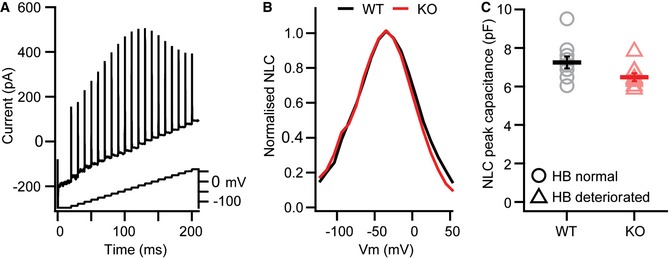
Figure EV1. OHC electromotility remains unaffected in Lrba mutants.
-
AMotor protein levels in the OHC membrane leaflet were determined electrophysiologically by analyzing the nonlinear capacitance (NLC) of WT and Lrba‐KO OHCs in response to a stair‐step protocol.
-
B, CRepresentative current traces to quantify; (B) NLC voltage dependence; and (C) peak capacitance. In these latter experiments, no statistically significant differences between WT and Lrba mutants could be detected (WT n = 10, Lrba‐KO n = 9; P = 0.063; Student's t‐test). Data are presented as means ± s.e.m.
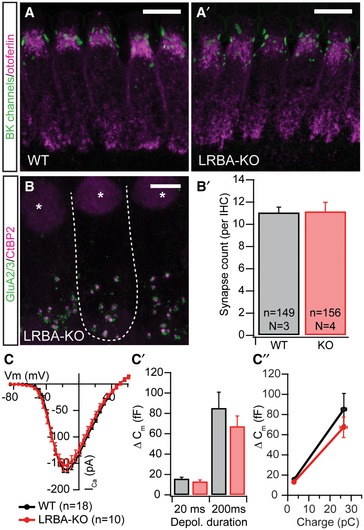
Figure EV2. Normal maturation, synapse count, and synaptic function in Lrba mutant IHCs.
-
A, A′Spotlike BK channel (green) staining at the IHC neck region indicates normal IHC maturation in Lrba mutant mice; IHCs have been counterstained for otoferlin (magenta). Scale bar: 5 μm.
-
B, B′Synapse count in apical turn IHCs from p14–16 WT and Lrba‐KO mice, as assessed by presynaptic CtBP2 (magenta) and postsynaptic GluA2/3 (green) co‐staining remains unaltered, as quantified in (B′). Data are presented as means ± s.e.m. The dashed line in (B) outlines a single IHC, and asterisks indicate nuclei. Scale bar: 10 μm.
-
C–C″Electrophysiologically recorded whole‐cell Ca2+ current–voltage relationship from p14–17 IHCs show similar voltage dependence and current amplitudes in both genotypes. (C′) Exocytic ∆Cm in response to step depolarizations to the respective maximum Ca2+ current potential for either 20 ms (to deplete readily releasable vesicles) or 200 ms (to probe sustained exocytosis) revealed no statistically significant difference in synaptic release between WT (n = 18; N = 16) and Lrba‐KO IHCs (n = 10; N = 7) (P = 0.24 and P = 0.51, respectively; Student's t‐test). (C″) Similarly, release efficiencies for both depolarization durations (i.e., ∆Cm per QCa2+) appear unchanged in Lrba mutants. Data are presented as means ± s.e.m.
LRBA is concentrated near the kinociliar basal body at the apex of mature hair cells
Since we did not find an impairment of OHC electromotility and IHC synaptic function in vitro, we searched for a common function in both cell types. Therefore, we analyzed its subcellular localization in hair cells by immunofluorescence (IF) (Fig 4). From p0, LRBA was diffusely distributed throughout the cytosol of IHCs and OHCs; however, in both hair cell types, LRBA clustered at a plane beneath the cuticular plate and was highly enriched—that is, showed the highest fluorescence intensity signal (i.e., yellow to white pixels in Fig 4A′–D″)—in the areas void of cuticular plate actin, especially where the kinocilium passes through the cuticular plate (the so‐called fonticulus/kinociliar pore; Fig 4A′–D″). Here, LRBA appeared to outline the pore (Fig 4E–E″) and its distribution overlapped with Rab11, a marker for recycling endosomes (Fig EV3). From p7, LRBA progressively concentrated at the pericuticular necklace (Fig 4C–D″) and fully encircled the cuticular plate of IHCs. This subcellular distribution pattern at the fonticulus and pericuticular necklace 26 was maintained after the degeneration of the kinocilium—which commonly occurs by p10 in this mouse strain—and even past the onset of hearing (data not shown).
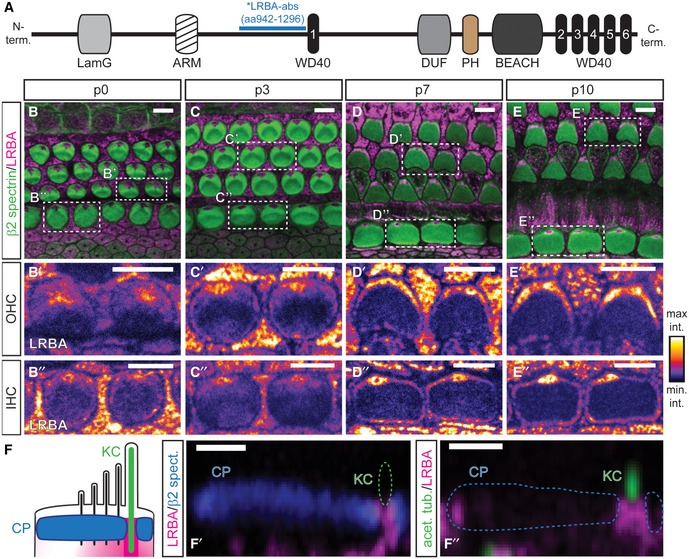
Figure 4. LRBA localizes to the kinocilium basal body and progressively encircles the cuticular plate.
-
ASchematic illustration of LRBA protein domains and the LRBA‐antibody binding sequence (*LRBA‐abs). LamG, laminin G domain; ARM, Armadillo/β‐catenin‐like repeats; WD40, WD40 repeat; DUF, domain of unknown function; PH, pleckstrin homology domain; BEACH, BEACH domain.
-
B–E″Developmental range (B–B″) p0, (C–C″) p3, (D–D″) p7, (E–E″) p10 showing LRBA clustering at a central position beneath the hair bundle in both, IHCs and OHCs. Samples were stained with specific antibodies against β2‐spectrin (green; cuticular plates) and LRBA (magenta) as well as fluorophore‐conjugated phalloidin (blue; stereocilia). Close‐ups of individual LRBA signals for (B′, C′, D′, E′) OHCs and (B″, C″, D″, E″) IHCs are presented in an intensity‐coded lookup table for clarity and show two adjacent hair cells surrounded by supporting cells as indicated in the dashed boxes in (B, C, D, E) (please also refer to Appendix Fig S1C and D for Lrba‐KO stainings, where all LRBA signal from hair cells and supporting cells is entirely lost). Scale bars: 5 μm.
-
FSchematic yz overview of the LRBA localization as derived from stainings with the indicated (F′) cuticular plate (CP) and (F″) kinocilium (KC) markers. Panels (F′, F″) show representative yz views through single p7 IHC cuticular plates at a central plane through the kinociliar pore. LRBA immunofluorescence appears to line the pore. Scale bars: 1 μm.
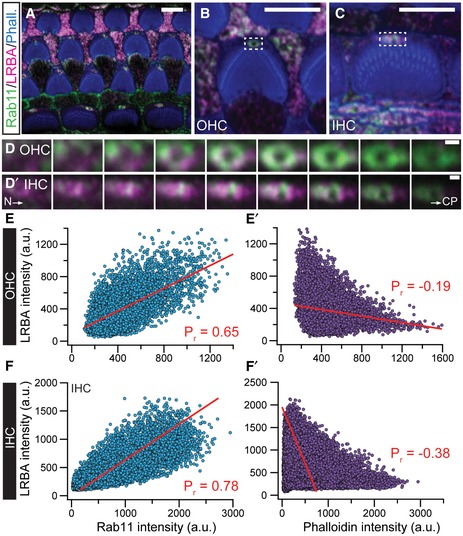
Figure EV3. LRBA partially colocalizes with the intracellular transport vesicle‐associated protein Rab11 at the kinociliar pore of cochlear hair cells.
-
Ap7 wild‐type organs of Corti were co‐stained with specific antibodies against Rab11 (green) and LRBA (magenta). Fluorophore‐conjugated phalloidin (blue) was used to visualize actin‐rich stereocilia. Representative single optical section of an organ of Corti overview at the height of the cuticular plates. Scale bars: 5 µm.
-
B, CRepresentative magnified views of a single (B) OHC and (C) IHC, respectively. Scale bars: 5 µm.
-
D, D′Detailed incremental z‐stacks of the dashed boxes in (B and C), illustrating the overlapping signal between Rab11 and LRBA (as indicated by “white pixels”) in ascending optical planes (step size 200 nm) from the hair cell neck (N) through the cuticular plate (CP) of a representative (D) OHC and (D′) IHC. Scale bars: 1 µm.
-
E–F′Pixel‐based intensity analysis performed on the representative example images shown in panels (B and C) for LRBA and Rab11 or (E′–F′) LRBA and phalloidin as an internal control. Pearson's correlation coefficients (Pr; marked in red in the respective panels) were determined by fitting the data sets with linear regressions and illustrate the positive correlation for LRBA and Rab11. In contrast, LRBA and phalloidin signals show a negative correlation and hence do not colocalize (please also refer to Fig 4F–F″ in this context). Scale bars: 1 µm.
Hair cells of Lrba mutants exhibit severely altered hair bundle morphology
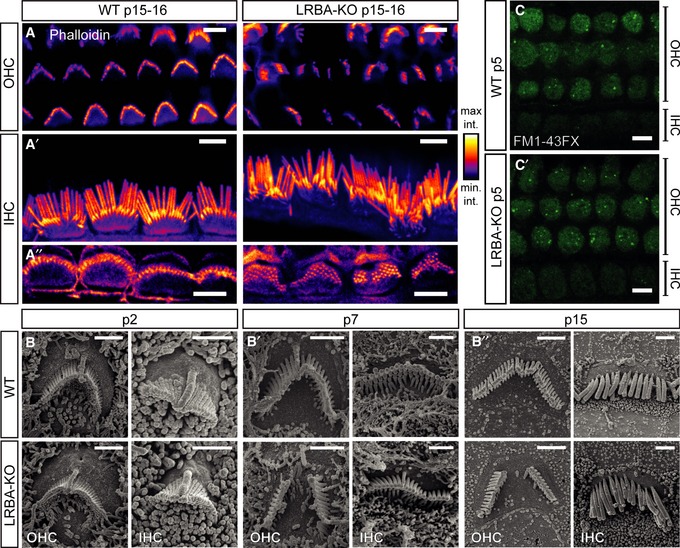
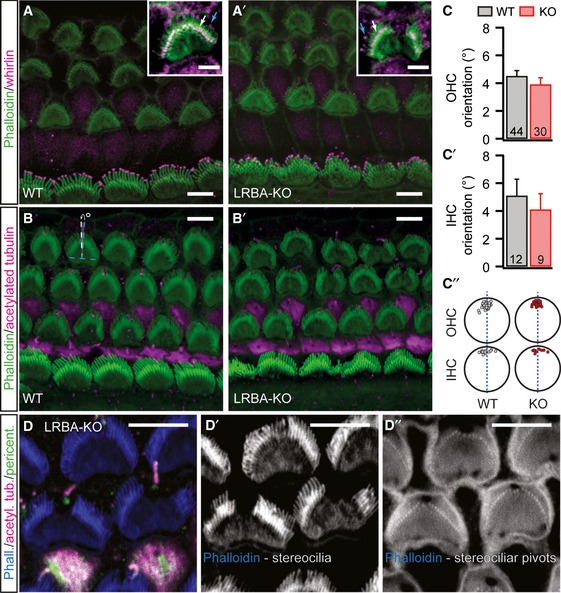
When labeling wild‐type and Lrba mutant organs of Corti with fluorophore‐conjugated phalloidin (Fig 5A), we observed misshaped IHC and OHC hair bundles. While OHCs of p15–16 Lrba‐KO mice showed prominent central gaps in their W‐shaped hair bundles (Fig 5A and B″), IHCs were affected to a lesser extent, but also showed disorganization of their stereociliar arrays (Fig 5A–A″ and B′–B″.) We employed scanning electron microscopy for more detailed analysis and confirmed the abnormal morphology of hair bundles after the onset of hearing (i.e., p15) affecting OHCs and IHCs throughout the entire cochlea (quantified in Appendix Fig S3). In these experiments, the outermost OHC row (OHC3) was best preserved (~30% of affected OHCs), while the middle (OHC2) and innermost OHC rows (OHC1) exhibited the characteristic gap in ~70% of cases after the onset of hearing. Moreover, OHC1 were most severely affected: often more than half of the hair bundle was lost. In contrast, major deformations of IHC hair bundles, such as loss of stereocilia and general hair bundle disorganization, were observed in only 10–15% of cases and remained relatively constant along the tonotopic axis.
Figure 5. Hair bundles of Lrba‐KO hair cells develop a prominent central gap in their stereociliar array.
-
A–A″Representative confocal projections of p15–16 OHC and IHC HBs labeled with a fluorophore‐conjugated phalloidin. While (A) Lrba‐KO OHCs display prominent central gaps, (A′) IHCs display general, but mild, disorganization. (A″) Single confocal planes through stereociliar arrays in close proximity to the respective cuticular plate insertion points in both genotypes. All images are shown with an intensity‐coded lookup table for improved visualization of the fluorescence intensity distribution. Scale bars: 5 μm.
-
B–B″SEM images from p2, p7, and p15 animals illustrate the developmental aspect of hair bundle degeneration in Lrba mutant hair cells. Initially, hair bundles develop normally in Lrba‐KO, but increasingly exhibit central gaps (in case of OHCs) and mildly distorted organization (in case of IHCs) from ˜p7. Scale bars: 1.5 μm.
-
C, C′FM1‐43FX uptake in response to transient exposure is comparable between p5 wild‐type and Lrba mutant cochlear OHCs and IHCs, indicating functional mechanotransduction in both genotypes (shown are single optical sections through a plane below the OHC cuticular plates). Scale bars: 5 μm.
During the course of these experiments, we also assessed earlier postnatal stages in order to determine whether hair bundles are formed normally and stereocilia are lost through subsequent degeneration, or whether morphogenesis itself is compromised. Here, several lines of experimental evidence suggest that hair bundles initially develop normally and then degenerate prior to the onset of hearing: (i) at p2 hair bundle morphology is indistinguishable from wild‐type littermates but deteriorates progressively with age (Fig 5B), (ii) we did not observe any obvious differences in stereociliar lengths between wild‐type and Lrba‐KO hair bundles, nor did we find mistargeting of the developmentally relevant stereociliar protein whirlin 27, which was properly expressed at the tips of the stereocilia—both suggestive of normal stereociliar formation and elongation (Fig EV4A), and finally (iii) planar cell polarity, as well as kinociliar morphology and position, appeared normal (Fig EV4B–D), even in hair cells that showed a prominent central gap.
Figure EV4. Unaltered targeting of whirlin to stereociliar tips and normal planar cell polarity in Lrba mutant hair cells.
-
A, A′Whirlin (magenta) is found at stereociliar tips of p8 OHCs and IHCs of both, (A) wild‐type and (A′) Lrba mutant animals (blue and white arrows in the insets indicate first‐ and second‐row stereocilia, respectively, for representative OHCs at adjusted brightness for clarity). Stereocilia have been counterstained using fluorophore‐coupled phalloidin (green). Scale bars: 5 μm.
-
B, B′Lrba mutant hair cells exhibit normal planar cell polarity, as assessed by calculating the degrees of deviation (°) from an axis perpendicular to the apico‐basal axis of the organ of Corti that runs through the center of each individual hair bundle (illustrated in B for an OHC; dashed blue lines indicate the perpendicular axes, dashed white line and white arrow illustrate the degrees of deviation). Scale bars: 5 μm.
-
C–C″Quantification of the data from (B) (in C: P = 0.33, in C′: P = 0.56; Student's t‐test). Numbers indicate the number of hair cells analyzed. (C″) Simplified graphical representation of the approximate kinocilia insertion points for OHCs and IHCs in wild‐type and Lrba‐KOs. Data are presented as means ± s.e.m.
-
D–D″Basal body and kinocilium localization are unaffected by stereociliar loss in Lrba‐KO hair cells. Regardless of the extent of stereociliar loss in Lrba mutants (D′), stereociliar pivot points can still be observed (D″). Samples were stained with specific antibodies against pericentrin (green; basal body) and acetylated tubulin (magenta; kinocilia) as well as fluorophore‐conjugated phalloidin (blue, actin‐rich stereocilia). Scale bars: 5 μm.
Next, we set out to probe hair cell mechanotransduction channel function of early postnatal IHCs and OHCs of both genotypes by transient exposure to the lipophilic styryl dye FM1‐43FX 28. In these experiments, we chose p5 animals (i.e., a developmental age group unaffected by hair bundle degeneration), as hair cells from these preparations would reveal functional mechanotransduction deficits occurring alongside—but independent from—stereociliar loss. Here, we would expect to observe mosaic FM1‐43FX uptake in case that functional mechanotransduction is compromised. However, the homogenous styryl dye uptake pattern found in these experiments suggests seemingly normal mechanotransduction channel function in Lrba‐KO hair cells (Fig 5C). Thus, our findings are consistent with the hypothesis that the reduction in the receptor potentials (Fig 2D–F) is due to the loss of central stereocilia from OHCs, and disorganization of IHC hair bundles. In this scenario, the combination of impaired OHC‐mediated cochlear amplification and synaptic release from IHCs would then produce the observed auditory phenotype.
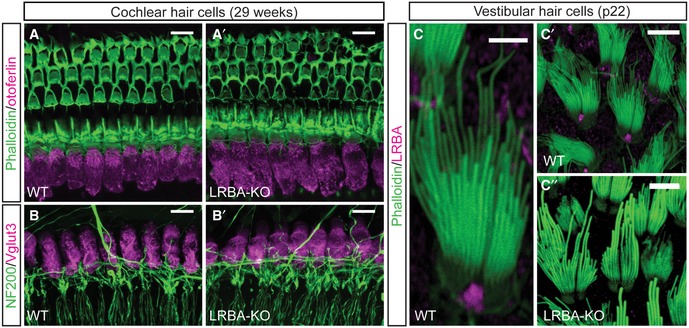
In contrast, vestibular hair cells, which showed a similar LRBA localization at the fonticulus, did not develop morphological abnormalities by p22 (Fig EV5C), an age where cochlear hair bundles were already severely affected. Consistent with seemingly unaltered motor behavior (i.e., normal rotarod performance, unaltered ability to balance on a narrow beam and to swim; not shown), this suggests a cochlear hair cell‐specific phenotype.
Figure EV5. No major loss of cochlear hair cells can be observed in 29‐week‐old Lrba mutants and the afferent innervation pattern is maintained; moreover, vestibular hair cells do not show a prominent hair bundle phenotype.
-
A, A′Representative confocal maximum projections of 29‐week‐old (A) wild‐type and (A′) Lrba‐KO organs of Corti stained with a specific antibody against the exocytic protein otoferlin (magenta). Actin‐rich structures were counterstained with fluorophore‐conjugated phalloidin. Scale bars: 10 μm.
-
B, B′IHC afferent innervation patterns in (B) wild‐type and (B′) Lrba mutants are maintained after 29 weeks. Specimen were stained with specific antibodies against NF200 (green; SGN afferent fibers) and the IHC‐specific marker Vglut3 (magenta). Scale bars: 10 μm.
-
C–C″Utricular hair cells also express LRBA at the fonticulus, but do not show any obvious hair bundle deficits at an age where cochlear hair bundles are degenerated. (C) Detail of a single utricular hair bundle illustrating LRBA localization at the fonticulus. Overviews of (C′) wild‐type and (C″) Lrba‐KO hair bundles from p22 utricles. Scale bars: 5 μm in (C); 10 μm in (C′ and C″).
Reduced levels of RDX and Nherf2 in Lrba mutant OHC hair bundles
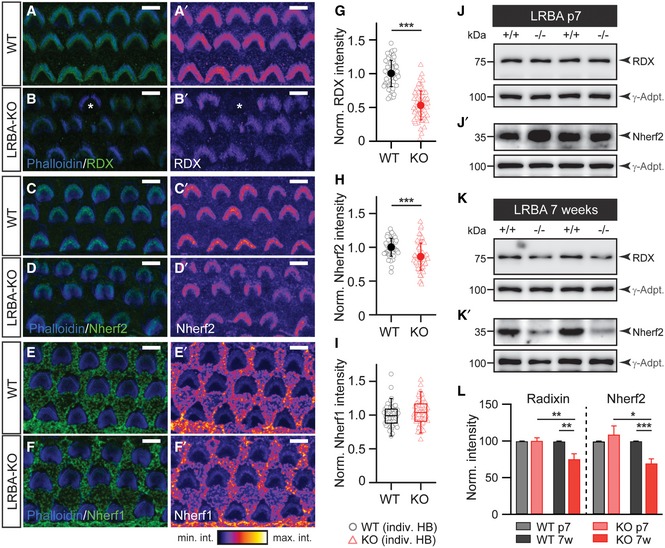
Interestingly, the appearance and position of the basal body and kinocilium as well as the number of actin‐rich stereociliar anchor points in the cuticular plate of cochlear hair cells appeared normal, regardless whether the hair bundle was intact or showed a central gap (Fig EV4D). We thus considered a defect that localized near the stereociliary pivot points. Indeed, the hair bundle phenotype of Lrba mutants closely resembles the one observed in RDX‐KO animals, in which the hair bundle of cochlear hair cells also develops normally until at least p6 but loses its central aspect by p14, while vestibular hair cells are unaffected 12. RDX is a member of the ERM protein family and thought to act as a molecular linker between cytoskeletal actin, the plasma membrane, and intrinsic membrane proteins 29. Prompted by these phenotypic similarities, we assessed stereociliar RDX levels in Lrba mutant OHC hair bundles using semi‐quantitative immunofluorescence analysis with specific antibodies against RDX (for RDX antibody validation, please refer to Appendix Fig S4) and the two RDX‐binding proteins Nherf1 and Nherf2 30 (Fig 6). In these measurements, we found a reduction in stereociliar RDX levels to ~50% of wild‐type levels (Fig 6A, B, and G) alongside a smaller, but statistically significant reduction in the RDX‐interacting protein Nherf2 to ~85% of control (Fig 6C, D, and H). In contrast, Nherf1 levels remained unchanged in Lrba‐KO hair bundles (Fig 6E, F, and I). Since RDX and Nherf2 levels were most strongly affected in these experiments, we next employed protein biochemistry to independently verify our findings with a distinct experimental approach. Here, Western analysis of cochlear extracts revealed a progressive reduction in RDX and Nherf2 expression in Lrba‐KO samples, thereby further implicating an intimate—although, due to the distinct subcellular localizations seemingly indirect—link between LRBA and RDX/Nherf2 (Fig 6J–L). Intriguingly, in these latter experiments, we failed to detect a reduction in whole cochlear RDX or Nherf2 levels from early postnatal samples (p7; Fig 6J–L). Given the strongly reduced stereociliar IF signal at this developmental stage, these findings may appear rather counterintuitive. To explain this conundrum, we propose a scenario where compartmental RDX and Nherf2 targeting to the hair bundle may be specifically compromised without affecting overall cochlear protein levels during early postnatal development; however, with advancing adolescence, the disruption of this pathway may lead to increased RDX and Nherf2 mistargeting and degradation, thereby contributing to the progressive nature of the observed phenotype. In this context, it also has to be taken into consideration that both RDX and Nherf2 are expressed in a range of other cochlear supporting cell types 18, 19, where they might exhibit distinct temporal expression patterns and initially mask the specific reduction observed in hair bundle signals. Finally, our findings that LRBA expression and subcellular distribution appear to be unaffected by loss of RDX again implicate RDX as a downstream target from LRBA (Appendix Fig S5).
Figure 6. Downregulation of RDX and Nherf2 expression in hair bundles of Lrba‐KO mice.
-
A–B′Representative images of radixin immunofluorescence in hair bundles from p7 (A, A′) WT and (B, B′) Lrba‐KO. Few hair bundles completely lacking radixin fluorescence (e.g., marked with * in B and only observed in Lrba‐KOs) have been excluded from analysis to provide a rather conservative estimate. Panels (A′ and B′) show RDX immunofluorescence signal illustrated with an intensity‐coded lookup table for improved visualization of the fluorescence intensity distribution. Scale bars: 5 μm.
-
C–D′Representative images of Nherf2 immunofluorescence in hair bundles from (D) WT and (D′) Lrba‐KO. Panels (C′ and D′) show Nherf2 immunofluorescence signal illustrated with an intensity‐coded lookup table for improved visualization of the fluorescence intensity distribution. Scale bars: 5 μm.
-
E–F′Representative images of Nherf1 immunofluorescence in hair bundles from (E) WT and (F) Lrba‐KO. Panels (E′ and F′) show Nherf1 immunofluorescence signal illustrated with an intensity‐coded lookup table for improved visualization of the fluorescence intensity distribution. Scale bars: 5 μm.
-
GNormalized average fluorescence intensity of RDX expression in WT and Lrba‐KO samples indicates dramatically reduced expression in the mutants (n = 71/65 hair cells; N = 5 cochleae). Quantification of data presented in (A–B′) (mean ± s.d.). ***P < 0.001 (Student's t‐test).
-
HNormalized average fluorescence intensity of Nherf2 expression from WT and Lrba‐KO samples illustrates decreased Nherf2 expression (n = 61/62 hair cells; N = 3 cochleae) in Lrba‐KO. Quantification of data presented in (C–D′) (mean ± s.d.). ***P < 0.001 (Student's t‐test).
-
INormalized average fluorescence intensity of Nherf1 expression from WT and Lrba‐KO samples illustrate comparable Nherf1 expression (n = 59/59 hair cells; N = 3 cochleae). Quantification of data presented in (E–F′). Data are presented as boxplots (median, inner/outer quartile + outlier range, since data were not normally distributed).
-
J, J′Representative Western blots for (J) RDX and (J′) Nherf2 protein expression levels detected in cochlea extracts derived from p7 WT and Lrba‐KO mice. γ‐Adaptin (γ‐Adpt.) was used for normalization.
-
K, K′Representative Western blots for (K) RDX and (K′) Nherf2 protein expression levels detected in cochlea extracts derived from 7‐week‐old WT and Lrba‐KO mice. γ‐Adaptin (γ‐Adpt.) was used for normalization.
-
LNormalized average signal intensities of RDX and Nherf2 expression are significantly decreased in cochlea extracts derived from 7‐week‐old Lrba‐KO mice (p7 WT: N = 5; p7 KO: N = 7; 7‐week‐old WT and KO: N = 5; two‐way ANOVA (genotype × age) followed by one‐sample t‐test against 100%). γ‐Adaptin was used for normalization. Data are presented as means ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001.
Mild hearing impairment in LRBA‐deficient humans
Given the strong hearing phenotype in mice, we were interested whether LRBA is also involved in human hearing loss. Lopez‐Herrera and colleagues showed in 2012 that LRBA deficiency in humans causes a severe immunodeficiency syndrome 31, and only in the past months, larger patient cohorts have been described 16, 32, 33. To assess whether hearing loss also occurs in human patients, we obtained audiological data from two patients with LRBA mutations (Fig 7A). Patient 1, a 31‐year‐old male, described as patient P3 in 31, carries a homozygous nonsense mutation in the LRBA gene (c.5047C>T, p.Arg1683*, reference sequence NM_006726.4), which truncates the protein after 59% of its length and leads to loss of detectable LRBA expression in EBV‐transformed B cells from the patient 31. He developed progressive sensorineural hearing loss, which affected the high frequencies more than the lower frequency range. Consistent with the audiogram, his speech perception thresholds were elevated to 33 dB (right ear) and 50 dB (left ear), respectively. His speech discrimination was moderately impaired and significantly degraded when background noise was present (Appendix Fig S6A). Transitory evoked otoacoustic emissions were detectable only in the left ear between 0.5 and 1.5 kHz, and DPOAE bilaterally at 3 and 4 kHz. Click‐evoked auditory‐evoked potentials had highly elevated thresholds (70 dB right, 60 dB left), but normal latencies.
Figure 7. Hearing impairment in LRBA‐deficient patients.
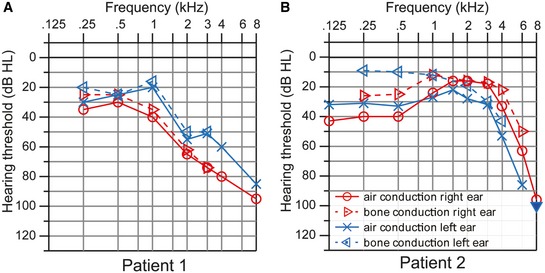
- Hearing thresholds of patient 1 (31 years old) show a sloping configuration with mild‐to‐moderate sensorineural hearing loss at low frequencies and severe impairment at high frequencies.
- Bone conduction hearing thresholds of patient 2 (11 years old) show mild sensorineural hearing loss in the low‐ and mid‐frequency range but a drastic decline at high frequencies. Air conduction thresholds show an additional conductive hearing loss component at low frequencies, which is due to middle ear effusion.
Patient 2, an 11‐year‐old girl, described as patient #1, code 105‐1 in 32, is compound heterozygous for two nonsense/frameshift mutations (c.1420G>A/p.Q473* and c.2834_2837delTCTT/pE945Efs, reference sequences ENST00000510413 and NP_001186211.2) which truncate the protein after 17% and 33%, respectively, of its length and abolish LRBA expression 32. Because of lung disease, she was tracheotomized and had slight bilateral conductive hearing loss explained by middle ear effusion. However, an additional significant high‐frequency hearing loss was noted in the audiogram (Fig 7B). Her discrimination of monosyllabic words was moderately impaired, only partly due to the conductive component of hearing loss. (Appendix Fig S6B).
In both patients, the clinical picture was dominated by immunodeficiency syndrome with hypogammaglobulinemia and severe autoimmune disease. However, the accompanying sensorineural hearing loss could not be explained by ear infections, ototoxic medication, or noise exposure. We thus conclude that human LRBA mutations may cause a new form of syndromic hearing loss; however, the phenotypic severity is much less pronounced in humans than in the Lrba‐KO mouse.
Discussion
Impaired hearing in Lrba‐KO mice likely results from a moderate transduction defect due to instability of stereocilia
Our data set provides a detailed and multidisciplinary description of a mouse model in which a hair bundle defect leads to significant hearing loss, but neither complete deafness nor major hair cell loss. Instead, the hearing deficit in Lrba‐KO mice can be attributed to the loss of a fraction of stereocilia in Lrba‐KO OHCs, strongly impacting the electromechanical feedback that is the basis for active cochlear amplification. An additional transduction deficit in IHCs may contribute to the sound encoding deficit.
In comparison with most other mouse mutants with hair bundle deficits, the Lrba‐KO phenotype is unusual. Stereociliar length and shape, overall hair bundle configuration as well as planar cell polarity appear normal. While initial hair bundle development is normal, the central aspect progressively degenerates with age. The coincidence in timing between the first morphological damage and the developmentally regulated retraction of the kinocilium is intriguing, but not necessarily indicative of a causal relationship. For example, at the time when the inner sulcus opens, sound‐induced shear motion between the basilar and tectorial membranes begins to occur, which could mechanically damage stereocilia at their pivot points. Indeed, the basal insertion points at the cuticular plate were preserved even when stereocilia atop were missing (Fig EV4D). Similar mechanical damage to the pivots preferentially affecting the innermost row of OHCs has been described to occur after noise trauma 34, 35. However, in these experiments, noise trauma appeared to mainly affect the entire tallest row of OHC stereocilia, which are in direct contact with the tectorial membrane. In contrast, LRBA deficiency appears to equally ablate tall and short stereocilia and mostly leaves the lateral parts of the hair bundles intact.
RDX deficiency may be the prevailing mechanism of the hair bundle defect in Lrba‐KO mice
To date, the most similar hair bundle phenotypes reported in literature are from (i) mice with genetic ablation of RDX 12, which is the predominant ERM protein in cochlear hair cells, and (ii) from hair cells that were manipulated with pharmacological compounds that either reduce RDX phosphorylation—such as staurosporine, a non‐selective protein kinase inhibitor 36—or Rho‐associated kinase inhibitors 37. Previous work suggests that RDX is likely involved in mechanical stabilization of the hair bundle by linking the central actin filament core to the cell membrane 38. Moreover, its active, phosphorylated form is abundant in a circumscript membrane domain distal to the stereociliar taper‐shaft boundary in isolated bullfrog vestibular hair cells 39, where it coincides with the adaptor protein Nherf2 (=Slc9a3rl2 or E3KARP, first described in Ref. 40), but not with the closely homologous Nherf1 (=Slc9a3rl1, Ebp50; fig S3 in 30). Both RDX and its direct interaction partner Nherf2 have been shown to be core components of a large protein interaction network in chicken vestibular hair bundles 41. Hence, a decrease in RDX levels would be expected to negatively impact Nherf2 expression. In contrast, it seems unlikely that Nherf2 or Nherf1 are direct effectors of LRBA, since the three KO mouse mutations have distinct morphological and functional consequences (30 and this paper).
Even though additional mechanisms may contribute, the similarity between the phenotypes of Rdx and Lrba‐KO mice suggests that the observed deficit in RDX expression in Lrba mutant IHCs may account for much of the observed auditory phenotype. The remaining RDX expression (~50%) in Lrba‐KO is probably sufficient to preserve some hearing function and prevent the progression of hair bundle degeneration seen in the RDX‐KO at p40, where stereocilia from OHC and IHCs are either entirely lost or fused 12.
What is the role of LRBA in hair bundle development?
A unifying functional and mechanistic common denominator of BEACH proteins, corresponding to their shared molecular architecture, is still elusive. Here, we find an effect of the Lrba‐KO on the concentration of the actin‐binding ERM protein RDX in the hair bundle, which is reminiscent of the perturbation of actin organization in neuronal dendritic spines and in plants, in mutants for the BEACH proteins neurobeachin and spirrig, respectively 24, 42. In addition to actin binding via the C‐ERMAD domains, ERM proteins bind to plasma membranes via their FERM (band 4.1, ezrin, RDX, moesin) domains, through two cooperative mechanisms: (i) to lipids (phosphatidyl‐inositol 4,5‐bisphosphate) and (ii) to the cytoplasmic domains of multiple transmembrane proteins (directly and/or indirectly via adaptor proteins like Nherf1/2) (reviewed by Ref. 29). These interactions are in turn affected by Rho GTPases and phosphorylation. Future studies will be required to investigate how LRBA deficiency impacts one or several of these molecular factors. The role of LRBA may also be related to intracellular transport processes, as suggested by its subcellular enrichment at the fonticulus and the pericuticular necklace. Here, like in activated T cells 16, LRBA partially colocalized with the recycling endosome marker Rab11, which has also been suspected to act as a functional antagonist of the Drosophila BEACH protein bchs 43. The subcellular localization of LRBA and Rab11 is well compatible with complementary roles in intracellular transport processes that bypass the actin‐rich cuticular plate and deliver proteins as well as lipids to the stereociliar compartments 26, 44, 45. For example, LRBA may be involved in the targeting of membrane proteins to the stereociliar base, and if the intracellular domains of these proteins were anchored by RDX and Nherf2, mislocalization of the former would also reduce the concentrations of the latter.
The elaborate ultrastructural and molecular architecture of the hair bundle and our multidisciplinary approach allowed us to characterize morphological and molecular consequences of a very specific defect resulting from a BEACH protein deficiency. Generally, our findings are consistent with an assumed superordinate role for BEACH proteins in the coordination of the assembly of macromolecular complexes, often involving membrane protein targeting, but sometimes also impacting the actin cytoskeleton.
LRBA loss—impact on sound encoding
Beyond the abnormality of the hair bundles, we did not find any indications for further abnormalities of hair cell morphology or function in Lrba mutants. Specifically, we found that (i) overall hair cell counts, as well as numbers and innervation patterns of IHC afferent synapses, were normal, (ii) presynaptic Ca2+ currents and exocytosis from sensory hair cells were unaltered, and (iii) prestin‐mediated OHC electromotility remained intact. Most of the observed alterations in SGN spike responses (high threshold, poor frequency tuning, and steep spike rate growth with increasing intensity) are well consistent with a primary defect of OHC function. However, it seemed as if we failed to record from a fraction of fibers, which lacked spontaneous spiking and did not respond to our search stimuli. Indeed, our experimental yield was very low in the mutants, despite unchanged numbers of IHC‐SGN synapses. Therefore, we suspect that we were unable to record from a fraction of SGNs from Lrba‐KOs, whose thresholds exceeded the intensity of our search stimulus. This experimental bias likely contributes to the lack of low‐SR fibers in our population, since low‐SR fibers typically have high sound thresholds 46, 47. Another possibility is that, by being forced to stimulate at such high intensities, we actually noise‐damaged low‐SR fibers, which have been shown to be highly susceptible to noise‐induced glutamate excitotoxicity 48. Finally, we argue that diminution of stereociliar numbers, in combination with general stereociliar disorganization, impairs transduction and leads to a concomitant reduction in IHC receptor potentials. Therefore, the amount of depolarization may not suffice to reach the activation threshold of synaptic voltage‐dependent calcium channels at low‐SR synapses and hence fails to initiate synaptic release 49, 50.
In summary, we propose that LRBA contributes to the subcellular targeting of selected hair bundle proteins, including RDX and Nherf2. The resulting degeneration of a fraction of stereocilia then leads to a reduction in the number of available MET channels and impairs hair cell depolarization upon hair bundle deflection. The resulting phenotype is dominated by the defect of active cochlear amplification due to OHC dysfunction, but likely involves an additional IHC transduction deficit.
Clinical implications
Human non‐syndromic autosomal recessive deafness DFNB26 has been described to be associated with a gene locus (4q31) in the chromosomal region where the BEACH protein LRBA is encoded [4q31.3, 51]. However, the genetics of DFNB26 is complex (involving a genetic modifier), and it remains questionable whether LRBA mutations are involved. The clinical phenotype is clearly different, as DFNB26 patients suffer from severe deafness, which has not been described in LRBA‐deficient humans so far. Instead, the latter clinical phenotype is dominated by a severe immunodeficiency syndrome 31. The variable symptoms include autoimmunity, chronic diarrhea, B‐cell deficiency, and hypogammaglobulinemia. At the cellular level, aberrant autophagy 33 and impaired intracellular trafficking of the CTLA4 immune receptor in T cells 16 have been described. No marked immunological abnormalities were detected in our Lrba‐KO mice, likely reflecting species differences with regard to the immune system 52. The published patient data summarize a large variety of possible symptoms, but so far, there has been no mention of hearing impairment. The latter may have remained undetected as a consequence of the (i) relative mildness and hence limited clinical relevance, (ii) slow progression—most of the currently confirmed patients are quite young (the median age of the 41 published patients is 14 years); (iii) large inter‐patient variability, due to genetic or phenotypic variations, or (iv) misdiagnosis due to other potential causes of hearing loss, including otitis, ototoxic medication or noise trauma. In the patients we present in this study, neither of the above causes explained the hearing loss, suggesting that it might be yet another symptom of LRBA deficiency syndrome. The hearing disorder could become more clinically relevant in future, when newly established treatment methods allow for longer survival and improved quality of life in these patients. However, more audiometric and genetic tests will be needed to validate our results and confirm the causal relationship between the genetic and audiological data. From our mutant mouse data, we would predict a phenotype with progressive sensorineural hearing loss, decreased ABR amplitudes with normal latencies and poor speech perception in background noise.
Materials and Methods
Animals
Lrba‐KO mice (laboratory code for the line, Lrba2‐A‐C9) were constructed by constitutive deletion of coding exon 3, generating a frameshift mutation. For a detailed description of the mutation please refer to 17. The mutation was generated in C57BL/6N ES cells, and subsequently propagated in the same background (five or more backcrosses at the time of analysis). Male and female Lrba‐KO mice and wild‐type littermates were used at the ages mentioned in the figure legends. Radixin‐KO animals have previously been described 12, 53.
In vivo recordings
Auditory brainstem response (ABR), distortion product otoacoustic emission (DPOAE), and single SGN recordings were performed as previously described 54. For SGN recordings, an opening was made in the left occipital bone and the cerebellum was partially removed. Subsequently, SGNs were stereotactically approached through the cochlear nucleus with a glass electrode filled with 3 M NaCl and 2% methylene blue. 50 ms noise burst search stimuli of 80 dB for wild‐type and 90–100 dB for Lrba‐KO were presented at 5 Hz through a loudspeaker (JBL2402) to elicit spiking of auditory neurons. Upon detection of a sound‐responsive unit, spontaneous firing rates were determined in silence for 10–30 s. Tuning curves were obtained by varying stimulus intensities and frequencies of 15‐ms tone bursts presented at 15 Hz, to estimate the best threshold of hearing and the characteristic frequency with a 1/32‐octave and 2‐dB precision. In mutants, the automatic algorithm often failed because thresholds were too close to the maximum speaker output. In these cases, the best frequency was determined manually and the threshold was defined as the lowest intensity in which rates clearly increased over spontaneous rate in the rate‐intensity function which employed 25 repetitions of 50‐ms tone bursts at 5 Hz for each 5‐dB step. Poststimulus time histograms to 50‐ms tone burst stimulation or paired stimuli (forward masking) were obtained at the characteristic/best frequency at sound intensities at which spike rates were mostly saturated: 30 dB above threshold in WT and at least 10 dB above threshold in KO.
Spiral ganglion neurons were differentiated from cochlear nucleus neurons by (i) the electrode position (> 1,200 μm below the surface of cochlear nucleus), (ii) their primary‐like PSTHs to suprathreshold 50‐ms tone bursts presented at the characteristic/best frequency, and (iii) their irregular firing pattern, as confirmed by a coefficient of variance of inter‐spike interval of steady‐state firing response, > 0.5. Abnormalities in the responses of cochlear nucleus neurons in Lrba‐KO animals were comparable to those observed in SGNs (not shown).
For electrocochleography, we used Tucker Davis System III hardware, custom‐written MATLAB software and a JBL2402 loudspeaker. Mice were anesthetized (urethane 1.32 mg/kg, xylazine 5 mg/kg, and buprenorphine 0.1 mg/kg i.p.), tracheotomized, and placed in a custom‐made headholder. The left bulla was opened to place a silver ball electrode on the round window membrane and responses to 100 clicks or 12‐ms tone bursts were amplified (50 times, custom‐built amplifier) and sampled at a rate of 50 kHz for 20 ms. Summating potential (notch filtered for stimulus frequency) and cochlear microphonic amplitudes (low‐pass filter: stimulus frequency/4) was determined as the mean or peak to peak amplitude in a window 7–11 ms following stimulus onset, respectively.
Immunohistochemistry and confocal microscopy
Immunohistochemistry was performed on acutely dissected, 4% formaldehyde‐fixed apical turn organ of Corti whole‐mount preparations as described previously 55. Specimens were imaged on a Leica SP2 laser‐scanning confocal microscope with a 1.4 NA 63× oil‐immersion objective. Images were processed using NIH ImageJ software 56 and spectrally deconvolved with a calculated point‐spread‐function for illustrative purposes only. Final figures were assembled for display using Adobe Illustrator software.
Antibodies
A rabbit antiserum was raised against recombinant mouse LRBA aa 942–1,296 (Fig 4A; a sequence with no similarity to Neurobeachin, targeting coding exon 22 and the initial 24 codons of exon 23 of mouse LRBA 17), and affinity‐purified on the same fusion protein. The following commercial antibodies were used in this study: mouse anti‐CtBP2 (BD Bioscience, 612044), mouse monoclonal anti‐acetylated tubulin (Sigma‐Aldrich, T7451), mouse anti‐otoferlin (Abcam, ab53233), mouse anti‐beta2 spectrin (BD Bioscience, 612562), mouse anti‐pericentrin (Abcam, ab28144), rabbit anti‐GluA2/3 (Merck Millipore, AB1506), rabbit anti‐radixin (Sigma‐Aldrich, R3653, batch 044M4752), rabbit anti‐Nherf1/EBP50 (Abcam, ab3452), rabbit anti‐Nherf2/SLC9A3R2 (Sigma‐Aldrich, HPA001672), as well as Atto‐488‐ (Sigma‐Aldrich, 49409) or Alexa‐633‐coupled phalloidin (Thermo Scientific, A22284) to visualize actin‐containing stereocilia. AlexaFluor‐488, AlexaFluor‐568 or AlexaFluor‐647 IgG‐coupled, species‐specific secondary antibodies (Thermo Scientific) were used for detection. Radixin antibody specificity was validated in RDX‐KO mice (Appendix Fig S4; 12; the Nherf1 and Nherf2 antibodies have been validated in KO controls 30).
Fluorescence intensity quantification
Maximum projections of confocal z‐stacks of WT and Lrba‐KO acquired at comparable tonotopic positions were used for analysis. Wild‐type and mutant organs of Corti were fixed, immunostained, mounted, and imaged in parallel with identical software and hardware settings (N = 5 sample pairs for RDX; N = 3 for Nherf1 and Nherf2). For background subtraction, fluorescence intensity from four randomly chosen areas per preparation (∅1 μm each), lacking specific signal (e.g., within the center of the cuticular plate), was averaged and subtracted from the respective images. Hair bundles were outlined manually in ImageJ, and the average fluorescence intensity was calculated for each individual hair bundle. In cases where parts of the hair bundle were missing, the separate parts of one hair bundle were analyzed individually and averaged subsequently to obtain a respective grand average per hair bundle. Individual fluorescence intensity values of a given experiment were then normalized to the global average of the corresponding wild‐type preparation.
FM1‐43FX dye uptake
p5 organs of Corti from wild‐type and Lrba‐KO littermates were dissected in parallel as described above, incubated with FM1‐43FX (3 μM in HBSS supplemented with 1.3 mM Ca2+; 10 s) (Thermo Scientific, F35355), washed repeatedly with HBSS, and then fixed with ice‐cold 4% formaldehyde according to the manufacturer's instructions. After washing, organs were mounted with Mowiol and subsequently imaged on a Leica TCS SP2 confocal microscope with a 1.4 NA 63× oil‐immersion objective.
Scanning electron microscopy
For cochlear whole‐mount preparations, freshly dissected cochleae were immediately perfused with PBS (37°C) through a small hole punched in the apex of each cochlea and the round and oval windows followed by initial perfusion fixation with pre‐warmed fixative (2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4). Cochleae were then immersion‐fixed in the same fixative for 2 h at RT and then overnight at 4°C. Subsequently, the cochleae were washed for 30 min in 0.1 M sodium cacodylate buffer (pH 7.4) and then dehydrated in an ascending ethanol series (30%, 50%, 70%, 80%, 90%, and 100%). After critical point drying with liquid CO2 (critical point dryer CPD300, Leica, Wetzlar, Germany), cochleae were mounted on sample holders and postdissected by removing the bony shell as well as Reissner's and tectorial membranes to expose the organ of Corti. Finally, the specimens were sputtered with gold (SCD005, BAL‐TEC, Liechtenstein) and examined with a field emission scanning electron microscope (LEO‐1530, Zeiss, Oberkochen, Germany) operated at 10 keV.
Preparation of total cochlea protein extracts and protein biochemistry
Animals were anesthetized by CO2/O2 exposure and sacrificed by subsequent cervical dislocation. Cochleae were rapidly removed from Lrba‐KO and wild‐type littermate mice at postnatal day 7 (p7) or postnatal day 49 (p49) and dissected in ice‐cold PBS at pH 7.4. The tissue was snap‐frozen in liquid nitrogen and stored at −80°C. For total protein extraction, one cochlea per mouse per sample was transferred into a pre‐cooled 2‐ml Eppendorf tube containing 500 μl ice‐cold RIPA‐lysis buffer [50 mM Tris–HCl pH 7.5, 1% (v/v) IGEPAL, 0.25 % (w/v) Na‐deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM NaF (Sigma‐Aldrich, St. Louis, USA), PhosSTOP phosphatase inhibitors, and complete proteases inhibitors (Roche Holding AG, Basel, Switzerland)] and was incubated for 1 h on ice. To homogenize the tissue, a Teflon‐plunger (Sartorius AG, Göttingen, Germany) was used to crack up cochleae manually. After an additional incubation of 30 min on ice, the tissue was further homogenized using a hand disperser (Polytron PT 1200 CL, Kinematic, Luzern, Switzerland) with a 5‐mm aggregate and a tip speed of 4 m/s for 10 s. Afterward, 500 μl RIPA‐lysis buffer was added and the samples were kept on a rotating wheel at 4°C for additional 60 min. Subsequently, cell debris was removed by centrifugation at 1,000 × g for 10 min at 4°C. Supernatants were retained as total cochlea protein extracts and boiled for 6 min at 95°C in SDS‐sample buffer (62.5 mM Tris–HCL pH 6.8, 10 % (v/v) glycerin, 2% SDS (w/v), 5 % (v/v) β‐mercaptoethanol, 0.002 % (w/v) bromophenol blue) after adjustment of protein concentrations using a BCA assay (Pierce Biotechnology, Waltham, USA).
To determine radixin and Nherf2 protein expression levels, 10 and 20 μg protein, respectively, were subjected to 10% SDS–PAGE and were analyzed by Western blotting (WB) using the following antibodies: rabbit anti‐radixin (radixin, Cat.‐No.: R3653, 1:1,500, overnight incubation at 4°C; Sigma‐Aldrich, St. Louis, USA); rabbit anti‐Nherf2 (SLC9A3R2, Cat.‐No.: HPA001672, 1:500, overnight incubation at 4°C; Sigma‐Aldrich, St. Louis, USA); peroxidase‐conjugated donkey anti‐rabbit (Cat.‐No.: 711‐036‐152, 1:10,000, 1‐h incubation at room temperature; Dianova, Hamburg, Germany). All antibodies were incubated in TBS‐T containing 5% (w/v) dry milk (Roth, Karlsruhe, Germany).
WB signal intensities were analyzed using the ImageJ analysis software (version 1.51f; 56). Signal intensities of radixin and Nherf2 were normalized against loading control signal intensities. Signal intensities from control groups were set to 100% and were compared to experimental groups. Statistical analysis was performed using SPSS (PASW Statistics 18.0, IBM, New York, NY, USA).
Outer hair cell patch clamp
Expression levels and voltage‐dependent characteristics of prestin can be assessed by electrophysiological recordings of charge movement or—equivalently—nonlinear capacitance (NLC), both resulting from prestin's voltage sensor 57. Since NLC amplitude and voltage dependence systematically change with postnatal development 58, 59, NLC can also be used as a measure of OHC functional maturation 60. Normally, prestin expression reaches a mature level around p15 58, 61. The voltage‐dependent NLC was recorded from OHCs of Lrba‐KO mice and WT littermates (p15–16) using a stair‐step stimulus protocol from −130 mV to +60 mV (10‐mV increments, 5 ms step duration) 58, 59. OHCs were perfused with an extracellular solution containing (in mM): KCl (5.8), NaCl (34), lactobionic acid (110), MgCl2 (0.9), CaCl2 (1.3), NaH2PO4 (0.7), d‐glucose (5.6), and HEPES (10); 305–310 mosm/kg, pH 7.3 (NaOH). Patch pipettes were filled with an intracellular solution containing (in mM): CsCl (110), TEA‐Cl (20), MgCl2 (3.5), CaCl2 (2.41), Na2‐ATP (2.5), HEPES (5), EGTA (5); 290–295 mosm/kg, pH 7.3 (KOH). Currents were low‐pass filtered at 10 kHz and sampled at 100 kHz with an EPC‐10 patch‐clamp amplifier and PatchMaster software (HEKA electronics). In these experiments, the cell capacitance (Cm) was not compensated in the whole‐cell configuration. The time constant (τ) of current decay was calculated from mono‐exponential fits to the current transients in response to each voltage step. The input resistance (Rin) was calculated from voltage‐dependent steady‐state currents. Integration of the current transients yielded the charge (Q). Cm(i) at each voltage step was derived after correction of all voltages for series resistance (Rs) errors from the formula: Cm(i) = (Rin/Rm)2 × (Q/Vc) where Rm is the membrane resistance and Vc is the holding potential. Rin and Rs were calculated as reported previously 58. The capacitance was fitted with a derivative of the Boltzmann function: C(V) = Clin + (Qmax/(α × exp((V−V1/2)/α) × (1 + exp((V−V1/2)/(–α)))2)), where Clin is the residual uncompensated linear capacitance, V is the membrane potential, Qmax is the maximum voltage sensor charge moved through the membrane electrical field, V1/2 is the voltage at half‐maximal charge transfer, and α is the slope factor of the voltage‐dependence. Clin was derived from the Boltzmann equation and reflects a measure for the surface membrane area of OHCs. As measure for prestin density in the OHC membrane Qmax is presented as charge density (Qmax/Clin). The NLC was normalized to Cpeak that denotes maximal voltage‐dependent capacitance at V1/2.
Inner hair cell patch clamp
Inner hair cell patch‐clamp electrophysiology experiments were performed as described in 62. In brief, apical coils from murine organs of Corti (p14–17) were acutely dissected from WT or Lrba‐KO mice and Ca2+ currents as well as exocytic membrane capacitance changes (ΔCm) recorded in the perforated‐patch configuration 63. The pipette solution contained (in mM) 130 Cs‐gluconate, 10 tetraethylammonium‐Cl (TEA‐Cl), 10 4‐aminopyridine, 1 MgCl2 · 6H2O, 10 Cs‐HEPES, and 300 μg/ml amphotericin B (Calbiochem), pH 7.2. The extracellular solution was comprised of (in mM): 110 NaCl, 2.8 KCl, 1 MgCl2 · 6H2O, 35 TEA‐Cl, 10 HEPES, 1 CsCl, 2 CaCl2, and 2 mg/ml d‐glucose, pH 7.3. An EPC‐9 amplifier in combination with PatchMaster software (HEKA Elektronik) was used for all recordings. Voltages were corrected for liquid‐junction potentials.
Statistics
Data were processed and analyzed with Igor Pro (Wavemetrics) and OriginPro 8.5 (OriginLabs) and presented as means ± s.e.m. for normally distributed data and as box‐and‐whisker plots (median, inner/outer quartile range, outlier range) for non‐normally distributed data. Statistically significant differences between WT and Lrba‐KO data were assessed using unpaired two‐sample Student's t‐test unless stated otherwise. *P < 0.05, **P < 0.01, ***P < 0.001.
Study approval
Animal handling and experiments complied with national animal care guidelines and were approved by the University of Göttingen Board for animal welfare and the animal welfare office of the state of Lower Saxony. Patient 1 gave informed consent to publish his audiological data specifically acquired for the purpose of this study, which was obtained after approval from the ethics committee of the University of Liège (Nr. B707201420571, 2014/74). The family of patient 2 gave written consent to share clinical and laboratory data for scientific publications (University Freiburg Ethic Committee protocol no. 282/11).
Author contributions
MWK initiated the study and provided the Lrba‐KO mice and antibody. LG, LB, and HF and MHdA performed or supervised behavioral, neurology, and dysmorphology screens at the German Mouse Clinic, respectively, in which reduced acoustic startle responses were detected. The subsequent study was designed by NS and CV. The experimental work was performed by CV (immunohistochemistry, confocal microscopy, IHC, and OHC patch clamp), TB (single‐unit recordings, supervised by NS), NH [scanning electron microscopy (SEM), supervised by MMK and SN], MGL. (OHC patch clamp, supervised by DO), TJH (Western blot, supervised by MK), and NS (electrocochleography, ABR, DPOAE). CS, PPL, and MM provided clinical data. CV, NS and MWK wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
The authors would like to express their gratitude to N. Herrmann, S. Thom, S. Gerke, and C. Senger‐Freitag for expert technical assistance and Dr. T. Pangrsic‐Vilfan for dissecting the utricles used in Fig EV5. Special thanks go to Tobias Moser for critical reading of the manuscript and for general support. This study was financed by the German Research Foundation (grant STR 1027/2 in Priority Program 1608 and project A6 in the Collaborative Research Center 889 to N.S.), the Swedish Research Council (grants 2004‐6221 and 521‐2011‐4573 to M.W.K.), and the German Federal Ministry of Education and Research (Infrafrontier grant 01KX1012 to M.H.A). C.V. is a Creutzfeldt Fellow of the Elisabeth und Helmut Uhl Foundation.
EMBO Reports (2017) 18: 2015–2029
Contributor Information
Christian Vogl, Email: christian.vogl@med.uni-goettingen.de.
Nicola Strenzke, Email: nstrenzke@med.uni-goettingen.de.
References
- 1. Frank T, Rutherford MA, Strenzke N, Neef A, Pangršič T, Khimich D, Fejtova A, Gundelfinger ED, Liberman MC, Harke B, et al (2010) Bassoon and the synaptic ribbon organize Ca2+ channels and vesicles to add release sites and promote refilling. Neuron 68: 724–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Strenzke N, Chakrabarti R, Al‐Moyed H, Müller A, Hoch G, Pangrsic T, Yamanbaeva G, Lenz C, Pan K‐T, Auge E, et al (2016) Hair cell synaptic dysfunction, auditory fatigue and thermal sensitivity in otoferlin Ile515Thr mutants. EMBO J 35: 2519–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nayak GD, Ratnayaka HSK, Goodyear RJ, Richardson GP (2007) Development of the hair bundle and mechanotransduction. Int J Dev Biol 51: 597–608 [DOI] [PubMed] [Google Scholar]
- 4. Ezan J, Montcouquiol M (2013) Revisiting planar cell polarity in the inner ear. Semin Cell Dev Biol 24: 499–506 [DOI] [PubMed] [Google Scholar]
- 5. Fettiplace R, Kim KX (2014) The physiology of mechanoelectrical transduction channels in hearing. Physiol Rev 94: 951–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barr‐Gillespie P‐G (2015) Assembly of hair bundles, an amazing problem for cell biology. Mol Biol Cell 26: 2727–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cosgrove D, Zallocchi M (2014) Usher protein functions in hair cells and photoreceptors. Int J Biochem Cell Biol 46: 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Belyantseva IA, Labay V, Boger ET, Griffith AJ, Friedman TB (2003) Stereocilia: the long and the short of it. Trends Mol Med 9: 458–461 [DOI] [PubMed] [Google Scholar]
- 9. Khan SY, Ahmed ZM, Shabbir MI, Kitajiri S, Kalsoom S, Tasneem S, Shayiq S, Ramesh A, Srisailpathy S, Khan SN, et al (2007) Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum Mutat 28: 417–423 [DOI] [PubMed] [Google Scholar]
- 10. Lee K, Amin Ud Din M, Ansar M, Santos‐Cortez RLP, Ahmad W, Leal SM (2011) Autosomal recessive nonsyndromic hearing impairment due to a novel deletion in the RDX gene. Genet Res Int 2011: 294675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shearer AE, Hildebrand MS, Bromhead CJ, Kahrizi K, Webster JA, Azadeh B, Kimberling WJ, Anousheh A, Nazeri A, Stephan D, et al (2009) A novel splice site mutation in the RDX gene causes DFNB24 hearing loss in an Iranian family. Am J Med Genet A 149A: 555–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitajiri S, Fukumoto K, Hata M, Sasaki H, Katsuno T, Nakagawa T, Ito J, Tsukita S, Tsukita S (2004) Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. J Cell Biol 166: 559–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jogl G, Shen Y, Gebauer D, Li J, Wiegmann K, Kashkar H, Krönke M, Tong L (2002) Crystal structure of the BEACH domain reveals an unusual fold and extensive association with a novel PH domain. EMBO J 21: 4785–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cullinane AR, Schäffer AA, Huizing M (2013) The BEACH is hot: a LYST of emerging roles for BEACH‐domain containing proteins in human disease. Traffic 14: 749–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang JW, Howson J, Haller E, Kerr WG (2001) Identification of a novel lipopolysaccharide‐inducible gene with key features of both A kinase anchor proteins and chs1/beige proteins. J Immunol 166: 4586–4595 [DOI] [PubMed] [Google Scholar]
- 16. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, Zhang Y, Liu Z, Fritz JM, Marsh R, et al (2015) AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349: 436–440 [DOI] [PubMed] [Google Scholar]
- 17. Kurtenbach S, Gießl A, Strömberg S, Kremers J, Atorf J, Rasche S, Neuhaus E, Herve D, Brandstätter JH, Asan E, et al (2017) The BEACH protein LRBA promotes the localization of the heterotrimeric G‐protein golf to olfactory cilia. Sci Rep 7: 8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scheffer DI, Shen J, Corey DP, Chen Z‐Y (2015) Gene expression by mouse inner ear hair cells during development. J Neurosci 35: 6366–6380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen J, Scheffer DI, Kwan KY, Corey DP (2015) SHIELD: an integrative gene expression database for inner ear research. Database (Oxford) 2015: bav071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pauli‐Magnus D, Hoch G, Strenzke N, Anderson S, Jentsch TJ, Moser T (2007) Detection and differentiation of sensorineural hearing loss in mice using auditory steady‐state responses and transient auditory brainstem responses. Neuroscience 149: 673–684 [DOI] [PubMed] [Google Scholar]
- 21. Wang X, Herberg FW, Laue MM, Wüllner C, Hu B, Petrasch‐Parwez E, Kilimann MW (2000) Neurobeachin: a protein kinase A‐anchoring, beige/Chediak‐higashi protein homolog implicated in neuronal membrane traffic. J Neurosci 20: 8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Su Y, Balice‐Gordon RJ, Hess DM, Landsman DS, Minarcik J, Golden J, Hurwitz I, Liebhaber SA, Cooke NE (2004) Neurobeachin is essential for neuromuscular synaptic transmission. J Neurosci 24: 3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Medrihan L, Rohlmann A, Fairless R, Andrae J, Döring M, Missler M, Zhang W, Kilimann MW (2009) Neurobeachin, a protein implicated in membrane protein traffic and autism, is required for the formation and functioning of central synapses. J Physiol 587: 5095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niesmann K, Breuer D, Brockhaus J, Born G, Wolff I, Reissner C, Kilimann MW, Rohlmann A, Missler M (2011) Dendritic spine formation and synaptic function require neurobeachin. Nat Commun 2: 557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nair R, Lauks J, Jung S, Cooke NE, de Wit H, Brose N, Kilimann MW, Verhage M, Rhee J (2013) Neurobeachin regulates neurotransmitter receptor trafficking to synapses. J Cell Biol 200: 61–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasson T, Gillespie PG, Garcia JA, MacDonald RB, Zhao Y, Yee AG, Mooseker MS, Corey DP (1997) Unconventional myosins in inner‐ear sensory epithelia. J Cell Biol 137: 1287–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kikkawa Y, Mburu P, Morse S, Kominami R, Townsend S, Brown SDM (2005) Mutant analysis reveals whirlin as a dynamic organizer in the growing hair cell stereocilium. Hum Mol Genet 14: 391–400 [DOI] [PubMed] [Google Scholar]
- 28. Gale JE, Marcotti W, Kennedy HJ, Kros CJ, Richardson GP (2001) FM1‐43 dye behaves as a permeant blocker of the hair‐cell mechanotransducer channel. J Neurosci 21: 7013–7025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiang L, Phang JM, Yu J, Harrop SJ, Sokolova AV, Duff AP, Wilk KE, Alkhamici H, Breit SN, Valenzuela SM, et al (2014) CLIC proteins, ezrin, radixin, moesin and the coupling of membranes to the actin cytoskeleton: a smoking gun? Biochim Biophys Acta 1838: 643–657 [DOI] [PubMed] [Google Scholar]
- 30. Kamiya K, Michel V, Giraudet F, Riederer B, Foucher I, Papal S, Perfettini I, Le Gal S, Verpy E, Xia W, et al (2014) An unusually powerful mode of low‐frequency sound interference due to defective hair bundles of the auditory outer hair cells. Proc Natl Acad Sci USA 111: 9307–9312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lopez‐Herrera G, Tampella G, Pan‐Hammarström Q, Herholz P, Trujillo‐Vargas CM, Phadwal K, Simon AK, Moutschen M, Etzioni A, Mory A, et al (2012) Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 90: 986–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gámez‐Díaz L, August D, Stepensky P, Revel‐Vilk S, Seidel MG, Noriko M, Morio T, Worth AJJ, Blessing J, Van de Veerdonk F, et al (2016) The extended phenotype of LPS‐responsive beige‐like anchor protein (LRBA) deficiency. J Allergy Clin Immunol 137: 223–230 [DOI] [PubMed] [Google Scholar]
- 33. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, Mohammadzadeh I, El‐Rajab MA, Massaad M, Chou J, et al (2016) Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol 36: 33–45 [DOI] [PubMed] [Google Scholar]
- 34. Liberman MC (1987) Chronic ultrastructural changes in acoustic trauma: serial‐section reconstruction of stereocilia and cuticular plates. Hear Res 26: 65–88 [DOI] [PubMed] [Google Scholar]
- 35. Gao W, Ding D, Zheng X, Ruan F, Liu Y (1992) A comparison of changes in the stereocilia between temporary and permanent hearing losses in acoustic trauma. Hear Res 62: 27–41 [DOI] [PubMed] [Google Scholar]
- 36. Goodyear RJ, Ratnayaka HSK, Warchol ME, Richardson GP (2014) Staurosporine‐induced collapse of cochlear hair bundles. J Comp Neurol 522: 3281–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Han Y, Wang X, Chen J, Sha S‐H (2015) Noise‐induced cochlear F‐actin depolymerization is mediated via ROCK2/p‐ERM signaling. J Neurochem 133: 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sauvanet C, Wayt J, Pelaseyed T, Bretscher A (2015) Structure, regulation, and functional diversity of microvilli on the apical domain of epithelial cells. Annu Rev Cell Dev Biol 31: 593–621 [DOI] [PubMed] [Google Scholar]
- 39. Zhao H, Williams DE, Shin J‐B, Brügger B, Gillespie PG (2012) Large membrane domains in hair bundles specify spatially constricted radixin activation. J Neurosci 32: 4600–4609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yun CH, Oh S, Zizak M, Steplock D, Tsao S, Tse CM, Weinman EJ, Donowitz M (1997) cAMP‐mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA 94: 3010–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shin J‐B, Krey JF, Hassan A, Metlagel Z, Tauscher AN, Pagana JM, Sherman NE, Jeffery ED, Spinelli KJ, Zhao H, et al (2013) Molecular architecture of the chick vestibular hair bundle. Nat Neurosci 16: 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saedler R, Jakoby M, Marin B, Galiana‐Jaime E, Hülskamp M (2009) The cell morphogenesis gene SPIRRIG in Arabidopsis encodes a WD/BEACH domain protein. Plant J 59: 612–621 [DOI] [PubMed] [Google Scholar]
- 43. Khodosh R, Augsburger A, Schwarz TL, Garrity PA (2006) Bchs, a BEACH domain protein, antagonizes Rab11 in synapse morphogenesis and other developmental events. Development 133: 4655–4665 [DOI] [PubMed] [Google Scholar]
- 44. Kachar B, Battaglia A, Fex J (1997) Compartmentalized vesicular traffic around the hair cell cuticular plate. Hear Res 107: 102–112 [DOI] [PubMed] [Google Scholar]
- 45. Grati M, Schneider ME, Lipkow K, Strehler EE, Wenthold RJ, Kachar B (2006) Rapid turnover of stereocilia membrane proteins: evidence from the trafficking and mobility of plasma membrane Ca(2+)‐ATPase 2. J Neurosci 26: 6386–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liberman MC (1978) Auditory‐nerve response from cats raised in a low‐noise chamber. J Acoust Soc Am 63: 442–455 [DOI] [PubMed] [Google Scholar]
- 47. Taberner AM, Liberman MC (2005) Response properties of single auditory nerve fibers in the mouse. J Neurophysiol 93: 557–569 [DOI] [PubMed] [Google Scholar]
- 48. Furman AC, Kujawa SG, Liberman MC (2013) Noise‐induced cochlear neuropathy is selective for fibers with low spontaneous rates. J Neurophysiol 110: 577–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Frank T, Khimich D, Neef A, Moser T (2009) Mechanisms contributing to synaptic Ca2+ signals and their heterogeneity in hair cells. Proc Natl Acad Sci USA 106: 4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohn T‐L, Rutherford MA, Jing Z, Jung S, Duque‐Afonso CJ, Hoch G, Picher MM, Scharinger A, Strenzke N, Moser T (2016) Hair cells use active zones with different voltage dependence of Ca2+ influx to decompose sounds into complementary neural codes. Proc Natl Acad Sci USA 113: 201605737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Riazuddin S, Castelein CM, Ahmed ZM, Lalwani AK, Mastroianni MA, Naz S, Smith TN, Liburd NA, Friedman TB, Griffith AJ, et al (2000) Dominant modifier DFNM1 suppresses recessive deafness DFNB26. Nat Genet 26: 431–434 [DOI] [PubMed] [Google Scholar]
- 52. Mestas J, Hughes CCW (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172: 2731–2738 [DOI] [PubMed] [Google Scholar]
- 53. Kikuchi S, Hata M, Fukumoto K, Yamane Y, Matsui T, Tamura A, Yonemura S, Yamagishi H, Keppler D, Tsukita S, et al (2002) Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nat Genet 31: 320–325 [DOI] [PubMed] [Google Scholar]
- 54. Jing Z, Rutherford MA, Takago H, Frank T, Fejtova A, Khimich D, Moser T, Strenzke N (2013) Disruption of the presynaptic cytomatrix protein bassoon degrades ribbon anchorage, multiquantal release, and sound encoding at the hair cell afferent synapse. J Neurosci 33: 4456–4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Neef J, Gehrt A, Bulankina AV, Meyer AC, Riedel D, Gregg RG, Strenzke N, Moser T (2009) The Ca2+ channel subunit beta2 regulates Ca2+ channel abundance and function in inner hair cells and is required for hearing. J Neurosci 29: 10730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oliver D, Klöcker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B (2000) Gating of Ca2+‐activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron 26: 595–601 [DOI] [PubMed] [Google Scholar]
- 58. Oliver D, Fakler B (1999) Expression density and functional characteristics of the outer hair cell motor protein are regulated during postnatal development in rat. J Physiol 519(Pt 3): 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang G, Santos‐Sacchi J (1993) Mapping the distribution of the outer hair cell motility voltage sensor by electrical amputation. Biophys J 65: 2228–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cimerman J, Waldhaus J, Harasztosi C, Duncker SV, Dettling J, Heidrych P, Bress A, Gampe‐Braig C, Frank G, Gummer AW, et al (2013) Generation of somatic electromechanical force by outer hair cells may be influenced by prestin‐CASK interaction at the basal junction with the Deiter's cell. Histochem Cell Biol 140: 119–135 [DOI] [PubMed] [Google Scholar]
- 61. Abe T, Kakehata S, Kitani R, Maruya S, Navaratnam D, Santos‐Sacchi J, Shinkawa H (2007) Developmental expression of the outer hair cell motor prestin in the mouse. J Membr Biol 215: 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vogl C, Panou I, Yamanbaeva G, Wichmann C, Mangosing SJ, Vilardi F, Indzhykulian AA, Pangršič T, Santarelli R, Rodriguez‐Ballesteros M, et al (2016) Tryptophan‐rich basic protein (WRB) mediates insertion of the tail‐anchored protein otoferlin and is required for hair cell exocytosis and hearing. EMBO J 35: 2536–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moser T, Beutner D (2000) Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci USA 97: 883–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File