

Abstract
Leishmania affects millions of people worldwide. Its genome undergoes constitutive mosaic aneuploidy, a type of genomic plasticity that may serve as an adaptive strategy to survive distinct host environments. We previously found high rates of asymmetric chromosome allotments during mitosis that lead to the generation of such ploidy. However, the underlying molecular events remain elusive. Centromeres and kinetochores most likely play a key role in this process, yet their identification has failed using classical methods. Our analysis of the unconventional kinetochore complex recently discovered in Trypanosoma brucei (KKTs) leads to the identification of a Leishmania KKT gene candidate (LmKKT1). The GFP‐tagged LmKKT1 displays “kinetochore‐like” dynamics of intranuclear localization throughout the cell cycle. By ChIP‐Seq assay, one major peak per chromosome is revealed, covering a region of 4 ±2 kb. We find two largely conserved motifs mapping to 14 of 36 chromosomes while a higher density of retroposons are observed in 27 of 36 centromeres. The identification of centromeres and of a kinetochore component of Leishmania chromosomes opens avenues to explore their role in mosaic aneuploidy.
Keywords: centromeres, ChIP‐sequencing, fluorescent in situ hybridization, kinetoplastid kinetochores, Leishmania
Subject Categories: Cell Cycle; Microbiology, Virology & Host Pathogen Interaction
Introduction
Leishmania sp. is the causative agent of leishmaniasis. It is a vector‐transmitted disease of large medical and veterinary importance affecting millions of people and domesticated animals around the world. The species belongs to the trypanosomatids, divergent eukaryotes that present original features, among which is their chromosomal organization. Indeed, genes are organized in large strand‐specific polycistronic transcription units. The genes of each unit are not functionally related and are separated by either convergent or divergent “strand‐switch regions” (SSRs) 1. The genetics of this protozoan parasite is also marked with a high level of homozygosity explained by frequent automixy and high rates of asymmetric chromosome allotments during mitosis, generating a constitutive “mosaic aneuploidy” 2, 3. These allotments are most likely secondary to a defect in the regulation of DNA replication, followed by permissive chromosome segregation 3. Yet very little is known about chromosomal segregation in trypanosomatids.
Centromeres are essential structures that allow proper segregation of the chromosomes during mitosis. They may be defined by specific and often repetitive DNA sequences. But such sequences evolve rapidly and are extremely diverse in eukaryotes making them difficult to identify in most organisms 4. However, the kinetochore proteins connecting the spindle microtubules to the centromeres are more conserved in late metazoans 5. Finally, the molecular architecture of centromeric chromatin, particularly the assembly histone H3 variants, plays an essential role in defining centromere identity and inheritance 6. Conventional components of kinetochores, like the variant histone CenH3 that specifies centromere location in most eukaryotes, were not recognized in the trypanosomatid genomes 7, 8, 9. Additionally, ultrastructural studies of the Leishmania nucleus did not detect canonical kinetochores but only six “electron‐dense plaques” assimilated with them; this number is not consistent with the 34–36 heterologous chromosomes present in Leishmania 10. A few specific studies have identified proteins that are essential to segregation, but we are far from a global view of the process. One of the major players in this process are centromeres, which have long remained elusive in Leishmania. Indeed, Leishmania centromeres could not be identified from genome sequencing, from in silico searches for repetitive motifs, by telomere‐associated chromosome fragmentation, nor even by position mapping of topoisomerase II binding sites via etoposide‐mediated cleavage. Even though these methods have revealed the centromeric sequences in a large spectrum of organisms, ranging from human to several protozoan pathogens (including Trypanosoma brucei, T. cruzi, Toxoplasma gondii, and Plasmodium falciparum) 11, 12, 13, 14. Such studies have located centromere domains on eight of the 11 chromosomes in T. brucei, as well as a limited region required for mitotic stability of chromosomes 1 and 3 in T. cruzi.
A recent description of novel and unconventional kinetochore proteins in T. brucei has hinted toward a method for centromere identification in Leishmania 15, 16. The authors of this work showed that trypanosomatids possess a unique kinetochore machinery comprising at least 20 members called kinetoplastid kinetochore proteins (KKTs). Moreover, KKT orthologues are present in the Leishmania genome. In the present work, we aimed to identify and characterize the centromeres of L. major. For this purpose, we used chromatin immunoprecipitation coupled to high‐throughput sequencing (ChIP‐Seq) of L. major KKT1 (LmKKT1) and were able to define discrete sites of protein binding, or putative centromere regions, for each of the 36 chromosomes constituting the L. major genome. Using immunofluorescence coupled to fluorescent in situ hybridization (FISH), we performed colocalization experiments using centromeric sequences as FISH probes and a GFP‐fused LmKKT1. Our results should allow a better understanding of genome organization, evolution of centromeres in eukaryotes, and cell cycle progression in these parasites.
Results and Discussion
LmKKT1 follows a “kinetochore‐like” dynamic localization throughout the cell cycle
Both the centromeres and kinetochores of Leishmania have long remained elusive. The recent description of 20 kinetochore KKT proteins in the related parasite T. brucei 15, 16 allowed for the in silico identification of 20 syntenic L. major orthologues (http://tritrypdb.org/tritrypdb/; Appendix Table S1) 17. In this study, we used LmjF.36.1900, the putative LmKKT1, which has 36% identity and 53% similarity to TbKKT1 (Tb927.10.6330). To further support the involvement of LmKKT1 in the kinetochore macromolecular complexes, we studied the subcellular localization of this protein in L. major throughout the cell cycle. Leishmania cells combine two features that allow for one to follow the cell cycle by DAPI staining: (i) the persistence of the nuclear envelope, hence of DAPI staining, during the whole cycle; (ii) the intracellular distribution of two DNA‐containing organelles, the nucleus, and the kinetoplast (the dense and complex network of circular DNA molecules at the apex of the single mitochondrion). Indeed, the nuclear and mitochondrial cell cycles coexist, and while independent they are strictly coordinated 18. Thus, the cell cycle sequence consists of (i) interphase cells containing one nucleus and one kinetoplast (1N1K), followed by (ii) the duplication of the kinetoplast leading to 1N2K cells, and then (ii) synchronous but independent processes of mitosis and karyokinesis that lead to 2N2K, and eventually (iii) by cytokinesis.
LmKKT1 recombinant proteins C‐ and N‐terminally fused to the GFP were thus followed both in 1N1K and in 2N2K cells. No phenotype was observed in the mutants as compared to the wild‐type, in terms of cell morphology, motility, cell growth, and nucleus–kinetoplast patterns, suggesting that the episomal expression of LmKKT1‐GFP did not perturb the parasite. The localization pattern of LmKKT1, whether N‐ or C‐terminally tagged, was suggestive of kinetochores. Indeed, fluorescent protein signals were observed in 1N1K cells as 8–12 spots in the nuclei (Fig 1A and B, left panels), a number far lower than that of the 36 heterologous chromosomes that constitute the genome, but consistent with the low number of “electron‐dense plaques” observed in electron microscopy and regarded as kinetochores 19. In 2N2K cells, LmKKT1 relocated as two large foci to the spindle poles (Fig 1A and B, right panels and Fig 1C and D). This pattern is reminiscent in particular of that of the nucleoporin Mlp2, which is found adjacent to the centromeric sequences in T. brucei and is involved in chromosomal distribution during mitosis 20. Considering that KKT proteins localize and enrich at the centromeres of the megabase chromosomes in T. brucei 15, we regarded LmKKT1 as a bona fide orthologue and a good candidate to aid in the search for the centromeres of L. major using ChIP‐Seq.
Figure 1. Subcellular localization of LmKKT1 during the cell cycle.
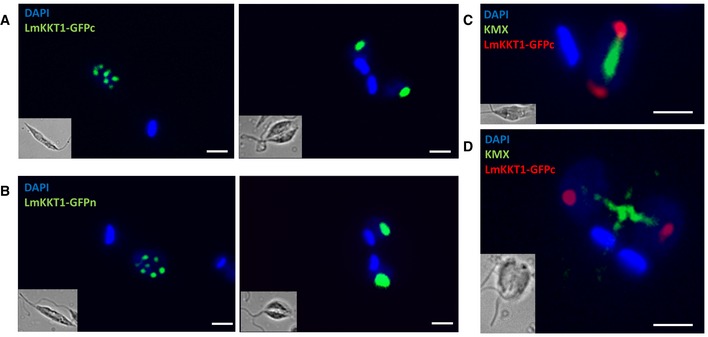
-
A, BInterphase (1N1K) cells: (A) LmKKT1‐GFPn or (B) LmKKT1‐GFPc in green. During interphase (1N1K cells), LmKKT1 was essentially found in a limited number of foci that localized at the periphery of the nucleolus.
-
C, DDividing (2N2K) cells: (C, D) LmKKT1‐GFPc in red and the mitotic spindle was labeled with the anti‐tubulin antibody KMX in green. During mitosis, LmKKT1 clustered in two foci which repositioned to the spindle poles at anaphase.
Identification of Leishmania major KKT1 binding sites
ChIP‐Seq was performed using Leishmania promastigote cells expressing an episomal GFP‐tagged LmKKT1. Trials using either MNase digestion or sonication to process the chromatin of L. major did not yield good results, but a combination of both techniques gave satisfactory sheared DNA fragments. ChIP‐Seq data were obtained for LmKKT1 (ChIP‐LmKKT1) and the control (INPUT‐LmKKT1). A fold‐change analysis between ChIP and INPUT showed a single clear peak per chromosome for LmKKT1 in the genome of L. major, except in Chr. #29 (Fig 2 and Appendix Fig S1).
Figure 2. Schematic representation of the LmKKT1 ChIP‐Seq results.

Mapping of the centromeres in the Leishmania major nuclear genome. Graphs show the distribution of LmKKT1 ChIP‐INPUT fold‐change results in the 36 chromosomes of L. major (numbered 1–36; size scale 0.1 Mb). The y‐axis represents the mean‐smoothed fold‐change profile over a window of 200 nt after filtering ChIP and input data for low mapping (threshold of 0.1 RPM). For each chromosome, the upper panel tracks show the polycistronic transcription units or directional gene clusters; genes transcribed from right to left are represented in red, and those from left to right in blue (obtained from TriTrypDB). The lower tracks show the fold change of the read depth between LmKKT1‐ChIP‐Seq and LmKKT1‐INPUT‐Seq samples, represented across the chromosome (x‐axis) and viewed on “genome browser” using the TriTrypDB version 8.0 database platform.
Centromeres and LmKKT1 colocalize in the nucleus during mitosis
The peaks revealed by ChIP‐Seq were validated by immunolocalization combined to FISH. For this purpose, FISH was performed on paraformaldehyde‐fixed LmKKT1‐GFP parasites, using the following DNA probes: the LmKKT1 binding sequences of Chr. #13 and of Chr. #27, a Chr. #27‐specific BAC (LB00646; 419,066..510,374) distant from the LmKKT1 binding sequence (LmjF.27: 983200.. 987599), and the telomeric repeats. In interphase cells, both LmKKT1‐GFP and the LmKKT1 binding sequences of Chr. #27 were located at the periphery of the nucleolus. We could detect 8–12 dots for LmKKT1‐GFP and two for the centromere of Chr. #27 (Fig 3A, left panel). During the course of mitosis, we observed a comigration of LmKKT1 and the centromeric FISH probes, gradually traveling together from the periphery of the nucleolus toward the spindle poles. Thus, in 2N2K cells, both the centromeric probes and LmKKT1 relocated at the spindle poles at anaphase (Fig 3A). Similar patterns and dynamics of fluorescence were observed for Chr. #13 (Fig 3B). Signals obtained with either the Chr. #27‐specific probe LB646 or the telomeric probe were very different: LB646 showed no dynamics of relocation toward the spindle poles, and the telomeres remained at the nuclear periphery throughout the cell cycle (Fig 3C). Therefore, LmKKT1 binding sites on Chr. #13 and 27 were validated as centromeres using immunofluorescence combined with FISH. For a few other chromosomes, our results correlate well with previously mapped regions required for the stability of Leishmania chromosomes. Indeed, “pre‐genomics” work had shown that the “right” end of Chr. #1 conferred mitotic stability to an artificial chromosome made of construct repeats and of this “right” end, comprising a subtelomeric ~20‐kb cluster of 272‐bp repeats 21: From the mapping data available, it clearly appears that the LmKKT1 binding site is located precisely at the “left” end of the ~272‐bp repeat cluster. Similarly, the LmKKT1 binding site on Chr. #19 correlates well with a 30‐kb region involved in the mitotic stability of an extrachromosome originating from the mirror duplication of one subtelomeric end of this chromosome 22, 23. By crossing the sequence and mapping data from 2,000 to those in TriTrypDB, we could infer that the centromere identified here using ChIP‐Seq is precisely located in a ~10‐kb region devoid of CDSs but rich in poly(dA)n, poly(dT)n, poly(dC)n, and poly(dG)n, which was suspected at the time to be responsible for this mitotic stability 23. For Chr. #29, given the very “noisy” enrichment observed using the fold‐change analysis, we also used a subtraction method that yielded a clearer enrichment pattern with a double peak (Appendix Fig S1), both peaks being separated by 25 kb and sharing sequence similarities. The exploration of these sequences using PCR amplifications leads us to suspect misassembly of the reference sequence in this region of Chr. #29 (Appendix Fig S2 and Appendix Table S2). In addition to these major peaks, rare minor peaks were observed on nine chromosomes (Chr. # 05, 09, 16, 20, 21, 25, 30, 33, and 36) (Fig 2) using the fold‐change method but not the subtraction method. We hypothesize that these peaks might be either secondary centromeres or non‐centromeric peaks (termed by Akiyoshi and Gull: “false‐positive signals” 15). It should be stressed here that ChIP‐Seq gives a global picture of the parasite cell population; hence, one cannot rule out that a minority of cells uses the minor peaks as centromeres, while the majority uses the main peaks. In total, the whole of these data allows us to infer that we have identified the bona fide centromeres of the 36 Leishmania chromosomes.
Figure 3. Localization and relocation dynamics of LmKKT1 and centromeres during the cell cycle progress in L. major .
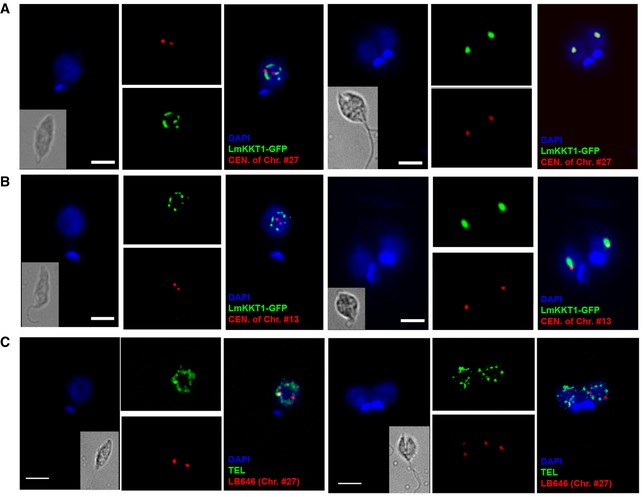
-
A, BImmunofluorescence for LmKKT1 coupled to FISH for centromeric DNA sequences. Interphase (1N1K) cells and dividing (2N2K) cells are shown on the left and right sides, respectively, of each panel. Representative images of the localization of LmKKT1‐GFP (green) and the centromere (CEN) of chromosome #27 (red in A) or of chromosome #13 (red in B) during the cell cycle progress. In mitosis (right), LmKKT1 binding sites colocalize with LmKKT1‐GFP foci. For Chr. #13, the non‐perfect colocalization is explained by the fact that the identified KKT1 binding site (coordinates LmjF.13:143300..145099) and the DNA probe target (LmjF.13:91772..118155) are distant by > 30 kb.
-
CDouble FISH labeling of non‐centromeric regions during the cell cycle progress: chromosome #27‐specific probe LB646 (red) and telomeric (TEL) probe (green).
Analysis of the Leishmania major KKT1 binding sites and comparative analysis of the centromeres in trypanosomatids
The LmKKT1 major peaks determined here by ChIP‐Seq are narrow. They vary in size from 2 to 10 kb (mean size at 4 ±2 kb). In T. brucei, the etoposide mapping method which first allowed identifying the putative centromeric domains and initial estimates from the genome sequencing project suggested that these arrays ranged from 2 to 8 kb 14; however, the use of long‐range restriction endonuclease mapping later indicated that their sizes vary from 20 to 120 kb depending on the chromosome 12. According to the position of the major peak on the chromosome length, from the extremity to the middle of the chromosome core, chromosomes may be categorized as telocentric or acrocentric (N = 2), submetacentric (N = 22), and metacentric (N = 12) (Fig 4A and Table EV1). None of the putative centromeres were found intragenic: One is located in subtelomeric position beyond the last annotated gene on the chromosomal arm, seven are in a convergent SSR, fifteen in a divergent SSR, and thirteen in intergenic regions. Since T. brucei and Leishmania exhibit very different genome structure but a high degree of shared synteny 24, we applied this same analysis to the centromeres of T. brucei. We analyzed the TbKKT3 binding site sequences identified in T. brucei using the ChIP‐sequencing performed by Akyioshi and Gull 15. Chr. #6, 10, and 11 of T. brucei were found to be telocentric, Chr. #5, 7, and 8 acrocentric, Chr. #1 and 2 submetacentric, Chr. #3, 4, and 9 metacentric, and centromere of Chr. #29 remains elusive. No centromere was found intragenic, and all but two are in intergenic regions within a polycistronic transcription unit. These two centromeres belong to Chr. #4, which is positioned in a divergent SSR, and Chr. #10, which stands at the end of its chromosome. The putative centromeric sequences are 57% GC‐rich, which is similar to the overall mean GC‐richness of the Leishmania genome (60%). The centromeric sequences showed no GC content signature based on a sliding window set at 100 nucleotides for the L. major sequences, whereas in T. brucei a decrease in GC content and/or a pattern of repeats could be found in all but two sequences (Fig EV1A). We looked at the 71 genes neighboring these putative centromeric sequences (Table EV2): 63 correspond to hypothetical proteins, three to tRNAs (Chr. #11 and 17), and four to rRNA genes (Chr. #5, 9 and 15 for the 5S unit and Chr. #27 for the 18S unit); finally, we found that the sequence identified for Chr. #2 is adjacent to the spliced leader RNA (SLRNA) gene array. With respect to the genes neighboring the centromere sequences, similarly to what was observed in Leishmania, rRNA and tRNA genes were found for the T. brucei centromeric sequences of Chr. #1, 2, 6, and 7 (Table EV3). This being said, we could not find any synteny between the centromeric loci in L. major and T. brucei. Indeed, we searched all chromosomes for gene orthologues flanking the centromeres in both organisms. In most cases, we found the orthologous genes of L. major in T. brucei, but they were not located close to a centromere with one exception, on LmjF Chr.#12 and Tb Chr.#27: the flanking genes for the Chr. #12 centromere in L. major are LmjF12.0510 and .0520; their orthologues in T. brucei are Tb927.1.3560 and Tb927.1.3820; the first one is separated from the centromere of Tb Chr. #1 by one gene, whereas the second one is nearby but separated by a rDNA cluster, several putative genes and two SSRs (Fig EV1A). Running a search for repeat sequences using “Tandem Repeats Finder” confirmed that all TbKKT3 binding sites harbor repetitive sequences (Table EV3). Using the same parameters then for the T. brucei search in Leishmania, we found that the sequences in Leishmania are essentially non‐repetitive, which is similar to the case in T. cruzi but not in T. brucei. Indeed, hits were obtained for only six chromosomes (Table EV4). Chr. #27 centromere displayed a minor cluster of 63 bp repeated 10 times. The centromere of Chr. #1 was found at the very end of the chromosome, at the “left” end of a 12‐ to 20‐kb minisatellite cluster located at the “right” end of this chromosome 25, 26, which were not included in the reference sequence of L. major Friedlin 1. However, it is not presently possible to correlate the presence of this centromere with that of this minisatellite. We ran a MEME search to further characterize the binding sites of LmKKT1. The search yielded two different and specific AT‐rich motif patterns. One of the motifs is MAAAAARARVDYNBKYBYWYDSTWWW (IUPAC nomenclature for nucleotides, see legend of Fig 4B), and the second is WWKKTTKSAAARAAAAA (Fig 4B). These were found solely at the centromeres of LmjF Chr. #1, 2, 3, and 4 for the former, and at the centromeres of LmjF Chr. #9, 10, 11, and 12 for the latter. They were also found once on Chr. # 18, 19, 26, 27, 35, and 34, but not at the centromeres. Finally, degraded versions of these two motifs mapped only once to the centromeres of LmjF Chr. #6, 13, 14, 21, 25, and 26. The MEME search yielded a single motif for all the centromeres of T. brucei, which is DHHNVYRYDHHNWNVHNHNHHWBHBCWNW (Appendix Fig S3). The motif “characterizing” all T. brucei centromeres was non‐specific, as it could be found 3,181 times in the T. brucei genome, as well as 1,698 times in the L. major genome.
Figure 4. Centromeres and other genome landmarks in Leishmania major .
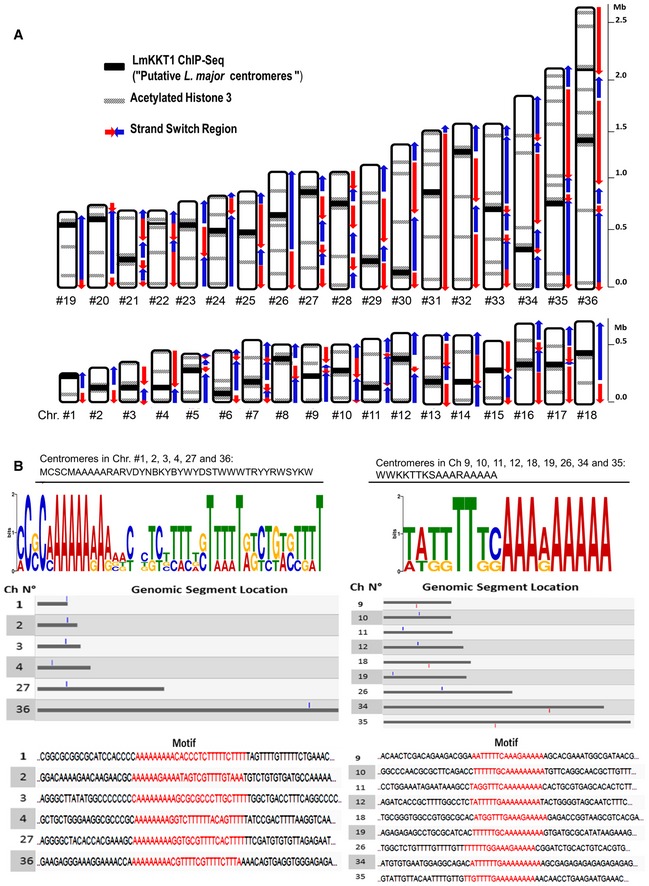
- Schematic representation of LmKKT1 binding sites in the L. major karyotype. A representative mapping of centromere localization, strand‐switch regions of polycistronic units, and acetylated H3 peaks 28 is shown for all L. major chromosomes. The maps were realized from data previously published in different studies and available in TriTrypDB.
- DNA motifs found in some of the LmKKT1 binding sites. All putative centromeric regions were used to perform a MEME search for finding common motif patterns. Two motif patterns for LmKKT1 binding sites are shown in graphical view with the representation of nucleotide distribution components in different colors and sizes (upper panel) using the IUPAC nomenclature for nucleotides (C: cytosine, A; adenine, G: guanine, T: thymidine, M: A or C, W: A or T, S: C or G, B: T, C or G, V: A, T or G, D: A, G or C, R: purines, Y: pyrimidines, N: any nucleotide). The results of a search for these DNA sequences in the L. major genome in TriTrypDB are shown in red (bottom panel). Blue and red boxes indicate sense and anti‐sense.
Figure EV1. Centromeric regions of Leishmania major and Trypanosoma brucei .
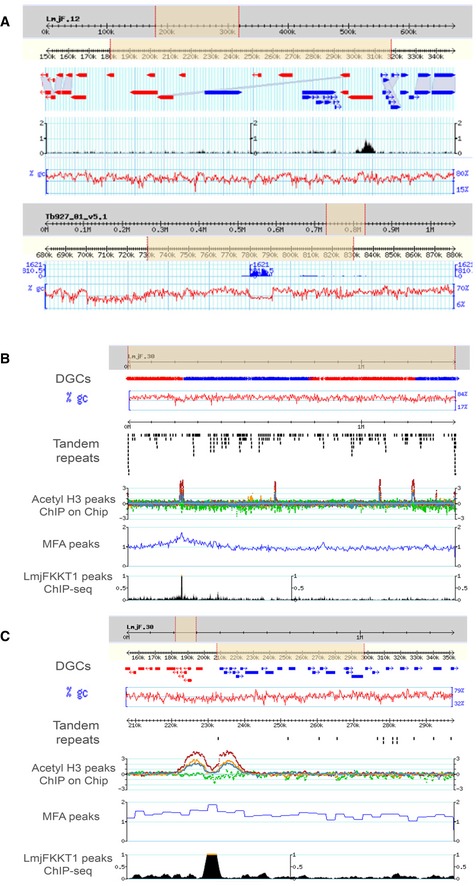
-
AAbsence of synteny between the centromeric regions of L. major and T. brucei: The flanking genes for the chr. #12 centromere in Leishmania are LmjF12.0510 and .0520; their orthologues in T. brucei are Tb927.1.3560 and Tb927.1.3820; the first one is separated from the centromere of Tb Chr. #1 by one gene, whereas the second one is nearby but separated by an rDNA cluster, several putative genes and two SSRs. Screenshots from TryTripDB.
-
B, CL. major chromosome 30: centromere (LmKKT1 binding site) and known genome landmarks. (B) View of the whole chromosome. (C) Zoom‐in view.
Genome landmarks
We further analyzed our data in the frame of known Leishmania genome landmarks. Transcription initiation and termination sites are in great part located at SSRs 27. The first genome‐wide maps of DNA‐binding protein occupancy were established with TBP, SNAP50, histone H3, and acetylated histone H3 28. Only 184 peaks of acetylated H3 were found in the entire genome; and each of the 58 divergent SSRs contained peaks of acetylated H3. It has been proposed that histone H3 acetylation marks the origins of polycistronic transcription in L. major. Here, in all chromosomes but Chr. #1 and 4, LmKKT1 binding sites were found colocalized or adjacent to acetylated H3 peaks (Figs 4A and EV1B and C). This putative association between centromeres and transcription start sites may seem unexpected, as centromeres were generally considered as transcriptionally inactive, yet, in the last decade, several reports have shown that pervasive transcription may occur at centromeres, at least in higher eukaryotes 29, 30. SSRs thus appear to concentrate most of the essential chromosomal functions, and even more so in Leishmania than in T. brucei. As hypothesized by others, this may be due to major constraints on genome dynamics imposed by polycistronic transcription; yet the differences observed between Leishmania and T. brucei are not fully explained. In addition, up to 5% of the trypanosomatid genome sequence is made of retroposons divided into subclades and subfamilies ranging from short extinct truncated transposable elements termed short interspersed degenerated retroposons (SIDERs) 31, 32 element to long active elements termed large degenerate Ingi‐related elements (LmDIRE) 33, 34. Thus, ~1,800 LmSIDER retroposons (categorized in two subgroups comprising ~700 LmSIDER1 and ~1,100 LmSIDER2) are dispersed in the genome of L. major and correspond to ~2% of the genome sequence. LmSIDERs are found almost exclusively within the 3′‐untranslated regions (3′UTRs) and play a role in mRNA instability 31. We found members of the LmSIDER1 subgroup in six centromeres (Chr. #2, 7, 11, 18, 29, and 36) and members of the LmSIDER2 subgroup in 20 centromeres (Chr. #3, 7, 8, 11, 14, 15, 17, 19, 20–22, 24–26, 28, 29, 31–34) and in the close vicinity (< 1 kb) to the centromere in three additional chromosomes (Chr. #12, 13, and 23). We also found LmDIRE in Chr. #7, 12, 18, and 29 (see Appendix Table S2). Thirty‐four LmSIDER and LmDIRE elements representing a sequence length of 16 kb were found in the 149 kb of centromeric sequence identified here by ChIP‐Seq. Therefore, LmSIDER sequences correspond to > 10% of the centromeric sequences, suggesting that they are enriched at centromeres. Trypanosoma brucei and T. cruzi centromeric DNA also contain degenerate retroelements: ingi and DIRE in T. brucei, and vestigial interposed repetitive retroelement/short interspersed repetitive elements (VIPER/SIRE) in T. cruzi. The role of these mobile elements within the centromeres remains elusive. Although speculative, it is tempting to hypothesize that the 5′ 79‐nt conserved sequence of these retroelements, which contains a conserved transcription promoter 35, 36, might be conserved for its capacity to bind specific, here kinetochore, proteins. In total, despite the indication that DNA sequence might be important for centromere definition in L. major, the whole of the data presented here do not allow us inferring whether the centromeres are preferentially determined by epigenetic factors or by DNA sequence 37. As in most eukaryotes, centromere loci in L. major display a great interchromosomal sequence heterogeneity.
In conclusion, we have for the first time identified centromeres on all chromosomes of L. major. It appeared difficult to define any common features among the centromeres of the different chromosomes, which would be related to functional significance; hence, a lot of work remains in order to fully understand the chromosomal segregation process in these divergent eukaryotes. Yet, the finding of these sequences together with the description of a kinetochore marker will now provide a tool to further study the composition of this unusual complex. This may result in a better understanding of the molecular events leading to chromosomal segregation in this parasite and shed light on the hypothesis that “permissive” segregation plays a role in mosaic aneuploidy.
Materials and Methods
Leishmania major parasites culture and epitope‐tagged construction of LmKKT1‐GFP expression vectors
Leishmania major strain Friedlin promastigotes (MHOM/IL/81/Friedlin) were grown at 26°C in RPMI 1640 (Gibco BRL) supplemented with 10% fetal bovine serum (FBS) 38. The gene LmjF.36.1900 encoding LmKKT1 was found in TriTrypDB (http://tritrypdb.org/tritrypdb/) and amplified by polymerase chain reaction (PCR) from genomic L. major strain Friedlin DNA. The PCR products were cloned into pGEM‐T‐Easy (Promega®) and then into the expression vectors pTH6cGFPn and pTH6nGFPc 39.
Transfection of L. major
A total of 5 × 107 mid‐log phase cells were resuspended with 100 μg of plasmid DNA. Electroporation was performed in a Bio‐Rad® Gene pulser 2 electroporator using the following conditions: square wave protocol, 1,500 V, 25 F, two pulses of 0.5 ms and 10 s between the pulses. Selective antibiotic hygromycin (Sigma®) was added at 30 μg/ml 24 h later, and stable LmKKT1 transfectants were obtained between 1 and 2 weeks after transfection.
Chromatin Immunoprecipitation (ChIP‐Seq)
ChIP was performed as previously described 28 with some modifications. For each assay, 3 × 109 L. major cells were incubated with 1% formaldehyde at 4°C for 5 min and stopped with 2.5 M glycine incubation for an additional 5 min. Chromatin purification and preparation of sheared DNA: spun down cells were resuspended in 100 μl of permeabilization buffer (100 mM KCl, 10 mM Tris pH 8.0, 25 mM EDTA, protease inhibitors), followed by incubation with Digitonin 200 μM final for 15 min RT. Next, the pellet was spun down and resuspended in 100 μl of cold NP‐S Buffer (0.5 mM spermidine, 0.075% NP‐40 (IGEPAL CA‐630), 50 mM NaCl, 10 mM Tris–HCl pH 7.5, 5 mM MgCl2, 1 mM CaCl2). Pellet was washed 3 times with NP‐S buffer, resuspended in 40 μl of NP‐S buffer, and subjected to MNase digestion for 5 min at 37°C. The reaction was stopped by adding 4 μl of EDTA 0.5 M pH 8 (final 10 mM) and centrifuged at 10,000 g for 10 min. Supernatants containing soluble mononucleosomes were collected in a new tube. Solubilized pellets with 100 μl of NP‐S, 0.2% SDS buffer were sonicated for 16 cycles of 30 s on/30 s off using a Bioruptor Pico sonication device (Diagenode®). Sheared chromatin was centrifuged at 10,000 g for 10 min, and the supernatant was added to saved soluble mononucleosomes. After checking chromatin shearing size, an immunoprecipitation assay was performed following the protocol from Lopez‐Rubio et al, with minor modifications 40. Antibody anti‐GFP ChIP grade from AbCam (ab2090) was added to the chromatin sample at a 1/500 dilution and incubated overnight at 4°C with rotation. To collect immune complexes, we incubated the sample with 50 μl per sample of protein G Dynabeads for 2 h at 4°C with rotation. Dynabeads with chromatin complexes were washed for 5 min on a rotating platform with 1 ml of each of the buffers listed below: low salt immune complex wash buffer (0.1% SDS, 1% Triton X‐100, 2 mM EDTA (pH 8.0), 20 mM Tris–HCl (pH 8.1), and 150 mM NaCl) at 4°C; high salt immune complex wash buffer (0.1% SDS, 1% Triton X‐100, 2 mM EDTA (pH 8.0), 20 mM Tris–HCl (pH 8.1), and 500 mM NaCl) at 4°C; LiCl immune complex wash buffer (0.25 M LiCl, 1% NP‐40, 1% deoxycholate, 1 mM EDTA (pH 8.0), and 10 mM Tris–HCl (pH 8.1)) at 4°C; TE buffer (10 mM Tris–HCl (pH 8.0) and 1 mM EDTA (pH 8.0)); two washes at room temperature (RT). Immune complexes were eluted by adding 100 μl of fresh elution buffer (1% SDS and 0.1 M NaHCO) at RT for 15 min with rotation two times. ChIP and input samples were reverse cross‐linked at 65°C overnight, and the DNA was purified on a Qiagen QiaQuick® spin column and analyzed by agarose gel electrophoresis to determine the average genomic fragment size achieved by sonication (300 bp). Single‐end sequencing was carried out on a HiSeq2000 Illumina platform at Institut Pasteur, Paris. Both input DNA and ChIP DNA were sequenced in each case. Sequence tags were mapped using BWA MEM, allowing up to two mismatches to the L. major Friedlin strain genome. Peaks were detected using MACS2, and signal was measured as the fold‐change between ChIP and input using custom Python scripts. In addition to the fold‐change method, we have also used a subtraction method in which peaks were detected using MACS2 and input noise was subtracted after normalization using custom Python scripts. Results were visualized in the TriTrypDB genome browser.
Association of ChIP‐Seq peaks to sequence features
The search for tandem repeats in the L. major centromeres using Tandem Repeats Finder v 4.09 (https://tandem.bu.edu/trf/trf.html) with the following parameters: Alignment Parameters (match: 2, mismatch: 7, indels: 7); Minimum Alignment Score To Report Repeat: 80; Maximum Period Size: 500; and Maximum TR array Size: 10 million bp 41. The search for common motifs on the binding sites of LmKKT1 and TbKKT3 was done using MEME (http://meme-suite.org/ 42) and validated by searching for the DNA Motif in TriTrypDB.
FISH, immunofluorescence coupled to FISH and microscopy imaging
For immunofluorescence assays, transfected L. major cells were grown to mid‐log phase. FISH was performed as described previously 2. Briefly, cells were fixed in 4% paraformaldehyde and 4% acetic acid, air‐dried on microscope immunofluorescence slides and dehydrated in serial ethanol baths (50–100%). Different DNA probes were used. The chromosome 27‐specific cosmid LB646 was kindly provided by Peter Myler (Seattle Biomedical Research Institute) and Christiane Hertz‐Fowler (Sanger Centre) and has been used previously 2. The telomeric probe was obtained from our DNA clone As2666, which consisted in 1,200 bp of the TTAGGG repetition. Centromere‐specific probes were obtained from PCR amplicons. For chromosome 13, we used the following primers: CentroLm13dir, 5′CGATCAAGACGAAGCGCAC3′; and CentroLm13rev, 5′CGAGGCGCGCGAGGATC3′. For chromosome 27, three PCR products were mixed, and the following primers were used: centroLm27dir1 5′AGTGGACTGCCATGGCG3′ and centroLm27rev1 5′CGCGCACTACAATGTCAGT3′; centroLm27dir2 5′GCAGACCGTACTCATCACC3′ and centroLm27rev2 5′GGTAGTACTTGGTACGCTC3′; and centroLm27dir3 5′TCAAGCGCAAAGCAAACCC3′ and centroLm27rev3 5′GGAAAGTGAAGGTGTGTCG3′. PCR products were purified using the gel extraction purification kit (Qiagen®) according to the manufacturer specifications. Probes were labeled with tetramethyl‐rhodamine‐5‐dUTP (Roche®) by using the Nick Translation Mix R (Roche®). Slides were then hybridized with a heat‐denatured DNA probe under a sealed rubber frame at 94°C for 2 min and then overnight at 37°C. The hybridization solution contained 50% formamide, 10% dextran sulfate, 2× SSPE, 250 mg/ml salmon sperm DNA, and 100 ng of labeled double‐strand DNA probe. After hybridization, parasites were sequentially washed in 50% formamide‐2 × SSC at 37°C for 30 min, 2× SSC at 50°C for 10 min, 2× SSC at 60°C for 10 min, and 4× SSC at room temperature. Slides were finally mounted in Vectashield®.
For immunofluorescence coupled to FISH, cells were fixed and air‐dried on slides as described above, then treated with 0.2% Triton in 1× PBS and saturated with 2% FBS. The anti‐beta‐tubulin monoclonal antibody KMX (kindly provided by Keith Gull, University of Oxford, UK) diluted to 1/100 was then added during 1 h. Anti‐mouse Alexa488 secondary antibodies (Molecular Probes®) diluted to 1/200 were then added. Slides were fixed again with 4% paraformaldehyde to proceed with the FISH assay. The centromeric and control DNA probes for chromosomes #12 and 27 were obtained by PCR‐amplification of genomic DNA. Slides were finally mounted with Mowiol R (Calbiochem®) and 4′,6‐diamino‐2‐phenylindole (DAPI).
Cells were viewed by phase contrast, and fluorescence was visualized using appropriate filters on a Zeiss® Axioplan2 microscope with an 100× objective. Digital images were captured using a Photometrics CoolSnap CCD camera (Roper Scientifics®) and processed with MetaView R (Universal Imaging®).
Data availability
NGS sequence data are deposited in the European Nucleotide Archive (ENA) database under accession number PRJEB21722 and accessible through the following link: http://www.ebi.ac.uk/ena/data/view/PRJEB21722.
Author contributions
M‐RG‐S, LS, SS, NK, and LC conducted the experiments; CRM performed the bioinformatics analysis; FB made the analysis concerning the retroposons, MP, AS, and YS designed the experiments; and M‐RG‐S, MP, YS, and PB wrote the paper with feedback from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Acknowledgements
We gratefully thank Odile Sismeiro, Caroline Proux, and Jean‐Yves Coppée from Institut Pasteur's Plateforme Transcriptome et Epigenome (PF2) for their essential help in the DNA sequencing. This work was supported by the Agence Nationale de la Recherche (ANR) within the frame of the “Investissements d'avenir” program (ANR‐11‐LABX‐0024‐01 “PARAFRAP”), the Centre National de la Recherche Scientifique (CNRS), the University of Montpellier, and the Centre Hospitalier Universitaire of Montpellier. LS and MRGS were recipients of grants from the French Parasitology Consortium “ParaFrap” (ANR‐11‐LABX‐0024‐01). SS was recipient of a grant from the Fondation pour la Recherche Médicale (ING20160435527).
EMBO Reports (2017) 18: 1968–1977
References
- 1. Myler PJ, Audleman L, deVos T, Hixson G, Kiser P, Lemley C, Magness C, Rickel E, Sisk E, Sunkin S et al (1999) Leishmania major Friedlin chromosome 1 has an unusual distribution of protein‐coding genes. Proc Natl Acad Sci USA 96: 2902–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sterkers Y, Lachaud L, Crobu L, Bastien P, Pagès M (2011) FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major . Cell Microbiol 13: 274–283 [DOI] [PubMed] [Google Scholar]
- 3. Sterkers Y, Crobu L, Lachaud L, Pages M, Bastien P (2014) Parasexuality and mosaic aneuploidy in Leishmania: alternative genetics. Trends Parasitol 30: 429–435 [DOI] [PubMed] [Google Scholar]
- 4. Muller S, Almouzni G (2017) Chromatin dynamics during the cell cycle at centromeres. Nat Rev Genet 18: 192–208 [DOI] [PubMed] [Google Scholar]
- 5. Cheeseman IM, Desai A (2008) Molecular architecture of the kinetochore‐microtubule interface. Nat Rev Mol Cell Biol 9: 33–46 [DOI] [PubMed] [Google Scholar]
- 6. Silva MC, Bodor DL, Stellfox ME, Martins NM, Hochegger H, Foltz DR, Jansen LE (2012) Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev Cell 22: 52–63 [DOI] [PubMed] [Google Scholar]
- 7. Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R et al (2005) The genome of the kinetoplastid parasite, Leishmania major . Science 309: 436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. El‐Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz‐Fowler C et al (2005) Comparative genomics of trypanosomatid parasitic protozoa. Science 309: 404–409 [DOI] [PubMed] [Google Scholar]
- 9. Berriman M, Ghedin E, Hertz‐Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B et al (2005) The genome of the African trypanosome Trypanosoma brucei . Science 309: 416–422 [DOI] [PubMed] [Google Scholar]
- 10. Urena F (1985) Ultrastructure of the mitotic nucleus in Leishmania mexicana sp. tridimensional reconstruction of the mitotic spindle and dense plaques. Microsc Electron Biol Celular 9: 51–65 [PubMed] [Google Scholar]
- 11. Kelly JM, McRobert L, Baker DA (2006) Evidence on the chromosomal location of centromeric DNA in Plasmodium falciparum from etoposide‐mediated topoisomerase‐II cleavage. Proc Natl Acad Sci USA 103: 6706–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Echeverry MC, Bot C, Obado SO, Taylor MC, Kelly JM (2012) Centromere‐associated repeat arrays on Trypanosoma brucei chromosomes are much more extensive than predicted. BMC Genom 13: 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Obado SO, Taylor MC, Wilkinson SR, Bromley EV, Kelly JM (2005) Functional mapping of a trypanosome centromere by chromosome fragmentation identifies a 16‐kb GC‐rich transcriptional “strand‐switch” domain as a major feature. Genome Res 15: 36–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Obado SO, Bot C, Nilsson D, Andersson B, Kelly JM (2007) Repetitive DNA is associated with centromeric domains in Trypanosoma brucei but not Trypanosoma cruzi . Genome Biol 8: R37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akiyoshi B, Gull K (2014) Discovery of unconventional kinetochores in kinetoplastids. Cell 156: 1247–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nerusheva OO, Akiyoshi B (2016) Divergent polo box domains underpin the unique kinetoplastid kinetochore. Open Biol 6: 150206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X et al (2010) TriTrypDB: a functional genomic resource for the Trypanosomatidae . Nucleic Acids Res 38: D457–D462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ploubidou A, Robinson DR, Docherty RC, Ogbadoyi EO, Gull K (1999) Evidence for novel cell cycle checkpoints in trypanosomes: kinetoplast segregation and cytokinesis in the absence of mitosis. J Cell Sci 112(Pt 24): 4641–4650 [DOI] [PubMed] [Google Scholar]
- 19. Urena F (1986) Three‐dimensional reconstructions of the mitotic spindle and dense plaques in three species of Leishmania . Z Parasitenkd 72: 299–306 [DOI] [PubMed] [Google Scholar]
- 20. Morelle C, Sterkers Y, Crobu L, MBang‐Benet DE, Kuk N, Portales P, Bastien P, Pages M, Lachaud L (2015) The nucleoporin Mlp2 is involved in chromosomal distribution during mitosis in trypanosomatids. Nucleic Acids Res 43: 4013–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dubessay P, Ravel C, Bastien P, Stuart K, Dedet JP, Blaineau C, Pages M (2002) Mitotic stability of a coding DNA sequence‐free version of Leishmania major chromosome 1 generated by targeted chromosome fragmentation. Gene 289: 151–159 [DOI] [PubMed] [Google Scholar]
- 22. Wilson K, Beverley SM, Ullman B (1992) Stable amplification of a linear extrachromosomal DNA in mycophenolic acid‐resistant Leishmania donovani . Mol Biochem Parasitol 55: 197–206 [DOI] [PubMed] [Google Scholar]
- 23. Dubessay P, Ravel C, Bastien P, Lignon MF, Ullman B, Pages M, Blaineau C (2001) Effect of large targeted deletions on the mitotic stability of an extra chromosome mediating drug resistance in Leishmania . Nucleic Acids Res 29: 3231–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghedin E, Bringaud F, Peterson J, Myler P, Berriman M, Ivens A, Andersson B, Bontempi E, Eisen J, Angiuoli S et al (2004) Gene synteny and evolution of genome architecture in trypanosomatids. Mol Biochem Parasitol 134: 183–191 [DOI] [PubMed] [Google Scholar]
- 25. Fu G, Melville SE (2002) Polymorphism in the subtelomeric regions of chromosomes of Kinetoplastida. Trans R Soc Trop Med Hyg 96(Suppl. 1): S31–S40 [DOI] [PubMed] [Google Scholar]
- 26. Sunkin SM, Kiser P, Myler PJ, Stuart K (2000) The size difference between Leishmania major friedlin chromosome one homologues is localized to sub‐telomeric repeats at one chromosomal end. Mol Biochem Parasitol 109: 1–15 [DOI] [PubMed] [Google Scholar]
- 27. Martinez‐Calvillo S, Nguyen D, Stuart K, Myler PJ (2004) Transcription initiation and termination on Leishmania major chromosome 3. Eukaryot Cell 3: 506–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thomas S, Green A, Sturm NR, Campbell DA, Myler PJ (2009) Histone acetylations mark origins of polycistronic transcription in Leishmania major . BMC Genom 10: 152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Molina O, Vargiu G, Abad MA, Zhiteneva A, Jeyaprakash AA, Masumoto H, Kouprina N, Larionov V, Earnshaw WC (2016) Epigenetic engineering reveals a balance between histone modifications and transcription in kinetochore maintenance. Nat Commun 7: 13334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blower MD (2016) Centromeric transcription regulates aurora‐B localization and activation. Cell Rep 15: 1624–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bringaud F, Muller M, Cerqueira GC, Smith M, Rochette A, El‐Sayed NM, Papadopoulou B, Ghedin E (2007) Members of a large retroposon family are determinants of post‐transcriptional gene expression in Leishmania . PLoS Pathog 3: 1291–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith M, Bringaud F, Papadopoulou B (2009) Organization and evolution of two SIDER retroposon subfamilies and their impact on the Leishmania genome. BMC Genom 10: 240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bringaud F, Ghedin E, Blandin G, Bartholomeu DC, Caler E, Levin MJ, Baltz T, El‐Sayed NM (2006) Evolution of non‐LTR retrotransposons in the trypanosomatid genomes: Leishmania major has lost the active elements. Mol Biochem Parasitol 145: 158–170 [DOI] [PubMed] [Google Scholar]
- 34. Bringaud F, Rogers M, Ghedin E (2015) Identification and analysis of ingi‐related retroposons in the trypanosomatid genomes. Methods Mol Biol 1201: 109–122 [DOI] [PubMed] [Google Scholar]
- 35. Heras SR, Lopez MC, Olivares M, Thomas MC (2007) The L1Tc non‐LTR retrotransposon of Trypanosoma cruzi contains an internal RNA‐pol II‐dependent promoter that strongly activates gene transcription and generates unspliced transcripts. Nucleic Acids Res 35: 2199–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bringaud F, Ghedin E, El‐Sayed NM, Papadopoulou B (2008) Role of transposable elements in trypanosomatids. Microbes Infect 10: 575–581 [DOI] [PubMed] [Google Scholar]
- 37. Sullivan BA, Blower MD, Karpen GH (2001) Determining centromere identity: cyclical stories and forking paths. Nat Rev Genet 2: 584–596 [DOI] [PubMed] [Google Scholar]
- 38. Dubessay P, Blaineau C, Bastien P, Pages M (2004) Chromosome fragmentation in Leishmania . Methods Mol Biol 270: 353–378 [DOI] [PubMed] [Google Scholar]
- 39. Michaeli S (2011) Trans‐splicing in trypanosomes: machinery and its impact on the parasite transcriptome. Future Microbiol 6: 459–474 [DOI] [PubMed] [Google Scholar]
- 40. Lopez‐Rubio JJ, Siegel TN, Scherf A (2013) Genome‐wide chromatin immunoprecipitation‐sequencing in Plasmodium . Methods Mol Biol 923: 321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Data Availability Statement
NGS sequence data are deposited in the European Nucleotide Archive (ENA) database under accession number PRJEB21722 and accessible through the following link: http://www.ebi.ac.uk/ena/data/view/PRJEB21722.