

Abstract
Purpose
To assess vitreous levels of inflammatory cytokines and neurotrophins (NTs) in diabetic retinopathy (DR) and elucidate their potential roles.
Methods
A prospective study was performed on 50 vitreous samples obtained from patients with DR (n = 22) and the nondiabetic controls (n = 28). All patients were candidates for vitrectomy. Inflammatory cytokine and NT levels were determined with ELISA. Potential source and role of NTs was determined by using human retinal Müller glia and mouse photoreceptor cells and challenging them with TNF-α or IL-1β, followed by detection of NTs and cell death.
Results
Vitreous NT levels of all DR patients were significantly higher than those of nondiabetic controls (nerve growth factor [NGF, P = 0.0001], brain-derived neurotrophic factor [BDNF, P = 0.009], neurotrophin-3 [NT-3, P < 0.0001], neurotrophin-4 [NT-4, P = 0.0001], ciliary neurotrophic factor [CNTF, P = 0.0001], and glial cell–derived neurotrophic factor [GDNF, P = 0.008]). Similarly, the levels of inflammatory mediators IL-1β (P < 0.0001), IL-6 (P = 0.0005), IL-8 (P < 0.0001), and TNF-α (P < 0.0001) were also higher in eyes with DR. Interestingly, inflammatory cytokine and NT levels, particularly TNF-α (P < 0.05), IL-8 (P < 0.004), NT-3 (P = 0.012), NGF (P = 0.04), GDNF (P = 0.005), and CNTF (P = 0.002), were higher in eyes with nonproliferative diabetic retinopathy (NPDR) than in eyes with active proliferative diabetic retinopathy (PDR). Cytokine stimulation of Müller glia resulted in production of NTs, and GDNF treatment reduced photoreceptor cell death in response to inflammation and oxidative stress.
Conclusions
Together, our study demonstrated that patients with DR have higher levels of both inflammatory cytokines and NTs in their vitreous. Müller glia could be the potential source of NTs under inflammatory conditions to exert neuroprotection.
Keywords: diabetic retinopathy, inflammation, neurotrophins, vitreous humor, human
Diabetic retinopathy (DR) is a disease of progressive vascular neurodegeneration and it remains a significant cause of blindness in middle-aged adults in the United States.1 Although the pathogenesis of DR is likely multifactorial and remains unknown, it appears to be associated with increased inflammation induced by oxidative stress,2 generation of reactive oxygen species,3 advanced glycation end products, and a plethora of inflammatory mediators.4 Several clinical studies have demonstrated that diabetics have increased serum levels of inflammatory markers, including C-reactive protein, interleukin 6 (IL-6), and tumor necrosis factor-α (TNF-α), suggesting that inflammatory processes play a considerable role in the pathogenesis of DR.5 Inflammation is generally considered a beneficial host response toward invading pathogens or tissue injury.6 Prolonged inflammation, as in diabetes, however, can be destructive and maladaptive, leading to irreversible damage to delicate tissues, such as the retina.7 Indeed, DR exhibits features of chronic neuroinflammation, but the precise relationship between inflammatory alterations in DR and the loss of neural function is currently being investigated.8,9
More recently, in addition to being a vascular complication of diabetes, DR has been recognized as a neurodegenerative disease with reported vision deficits at the onset of diabetes.10 The neuronal changes in DR include retinal cell death, loss of ganglion cells,11 reduced inner retinal thickness, neurofilament abnormality, and declined ERG.10,11 These physiological conditions trigger neuronal survival signaling, including the production of neurotrophins (NTs).12 Given the critical role played by NTs in regulating neuronal functions, it is not surprising then that neurodegenerative disorders, including DR, are associated with altered NT levels and/or expression pattern of their receptors.
NT levels have been shown to be elevated in vitreous samples in animal studies with induced proliferative diabetic retinopathy (PDR).13 While glial cell line–derived neurotrophic factor (GDNF)14 has been shown to be increased in vitreous samples of patients with DR, to our knowledge, a comprehensive analysis of other NT levels, including levels of nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), neurotrophin-4 (NT-4), and ciliary neurotrophic factor (CNTF), have not been evaluated in DR. Therefore, the objectives of our study were (1) to analyze and compare NT levels in the vitreous of patients with or without DR, (2) to find correlation between the levels of vitreous NTs and inflammatory mediators in patients with DR, (3) to analyze the effect of panretinal photocoagulation (PRP) ablation of the ischemic peripheral retina in DR on NT levels, and (4) to determine the potential mechanism of NT production and its functional role under inflammatory/oxidative stress conditions.
Materials and Methods
Patient Population and Vitreous Sample Collection
This was a prospective study conducted according to the tenets of the Declaration of Helsinki. All patients were candidates for vitrectomy and had signed a preoperative informed consent with their approval to use the excised vitreous fluid for analysis and clinical research. The study design and protocol were approved by the Wayne State University School of Medicine Institutional Review Board. Exclusion criteria included patients younger than 18 years, a history of previous intravitreal anti–vascular endothelial growth factor (VEGF) injections, a history of or current uveitis, current or recent topical or systemic steroid use, a history of penetrating eye injury, a history of blunt trauma to the eye, or previous intraocular surgery within the past 6 months. Patients were excluded from analysis of intravitreal inflammatory cytokines if they underwent a pars plana lensectomy for retained lens fragments due to complicated cataract surgery.
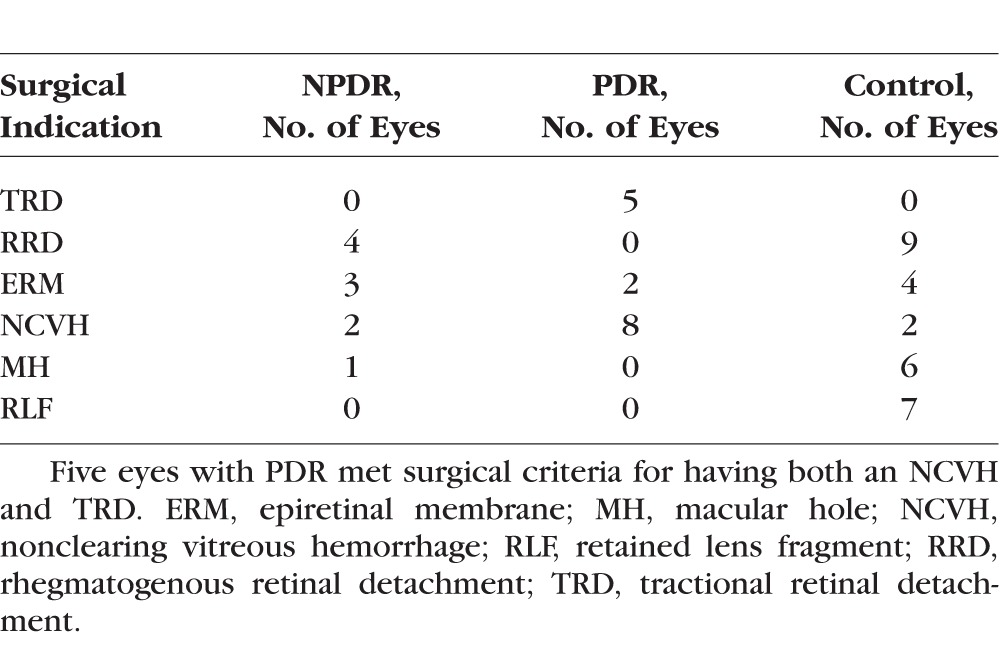
Undiluted vitreous fluid samples were obtained from 50 eyes from 50 patients (DR, N = 22; Nondiabetic, N = 28) from 2013 to 2015 during pars plana vitrectomy. The indications for vitrectomy were rhegmatogenous and tractional retinal detachment, nonclearing vitreous hemorrhage, epiretinal membrane peel, macular hole repair, or retained lens fragments. (Table 1) The preoperative degree of DR was graded from the current preferred practice patterns outlined by the American Academy of Ophthalmology. Undiluted vitreous samples were collected by manual suction into a syringe through the aspiration line of vitrectomy before opening the infusion line. Samples were transferred to sterile polypropylene screw cap conical bottom vials and were immediately snap-frozen in liquid nitrogen until transfer to a −80°C freezer for storage and assays.
Table 1.
Surgical Indications for Vitrectomy
Vitreous Neurotrophins and Cytokine Analysis
Vitreous levels of NGF (Cat. No. DY 256), BDNF (Cat. No. DY248), NT-3 (Cat. No. DY267), NT-4 (Cat. No. DY268), CNTF (Cat. No. DY257), GDNF (Cat. No. DY212), interleukin 1β (IL-1β, Cat. No. DY201), IL-6 (Cat. No. DY206), IL-8 (Cat. No. DY208), and TNF-α (Cat. No. DY210) were determined by ELISA as per manufacturer's instruction (R&D Systems, Minneapolis, MN, USA). For ELISA assay, total protein concentration in the vitreous was determined by the Micro BCA protein assay kit (Thermo Scientific, Rockford, IL, USA) and equal amount (20 μg) of protein was used for ELISA assay.
Cell Culture and Treatment
The immortalized human Müller glial cells (MIO-M1cell line)15 were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 1% penicillin-streptomycin, and L-glutamine (10 μg/mL). Cells were grown in serum-free media before TNF-α challenge. For NT expression, MIO-M1 cells were challenged with recombinant TNF-α (100 ng/mL), IL-1β (100 ng/mL), IL-4 (100 ng/mL), or dexamethasone (100 μM) for indicated time points. The conditioned media was centrifuged at 10,000g for 10 minutes to remove cell debris. The clear supernatant was used for dot blot to measure the expression of various NTs.
For the neuroprotection study, mouse cone photoreceptor cell line 661W, as described in our previous study,16 was used. The 661W cell line was maintained in DMEM supplemented with 10% FBS, 10 μg/mL L-glutamine, 1% penicillin-streptomycin, 40 μg/L hydrocortisone, 40 μg/L progesterone, 32 mg/L putrescine, and 40 μl/L β-mercaptoethanol. Photoreceptor cell death was induced by challenging 661W cells with H2O2 (100 μM) or TNF-α (100 ng/mL) for 24 hours in the presence and absence of GDNF (100 ng/mL). The cell death and protection by GDNF treatment was assessed by TUNEL staining using an ApopTag fluorescein in situ apoptosis detection kit as described previously.17
Dot Blot Analysis
To measure the NT level in MIO-M1, dot blot was performed as described previously.15 Briefly, 50 μL conditioned media was loaded onto a 0.2-μm nitrocellulose membrane by using a BIO-DOT apparatus (Bio-Rad, Hercules, CA, USA) and vacuum suction. The membrane was fixed in 10% formaldehyde in Tris buffer saline (TBS) for 1 hour at room temperature (RT). The membrane was blocked in 5% skim milk made in TBST (TBS containing 0.05% Tween 20) for 1 hour at RT and incubated with primary antibody for various NTs (GDNF, NGF, NT-3, and NT-4) overnight at 4°C. On the following day, the blot was washed three times in TBST and incubated with respective anti-mouse or anti-rabbit horseradish peroxidase (HRP) conjugates (Bio-Rad) for 1 hour at RT. The blot was developed using SuperSignal West Femto maximum Sensitivity Substrate (Thermo Scientific) via chemiluminescence using a Kodak image station, 4000R Pro molecular imaging system (Carestream Health, Inc., Rochester, NY, USA). Dot intensity was quantified using ImageJ analysis software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA).
Statistical Analysis
Values of vitreous concentrations of cytokines and NTs were reported as mean ± standard deviation (SD). All of the analyses were performed by using the Statistical Package for the Social Science software version 16.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad prism V7.02 (GraphPad Software, La Jolla, CA, USA). The unpaired t-test was used for the normally distributed continuous variables, comparing the means from two independent groups, and a P value < 0.05 was considered statistically significant. One-way ANOVA was performed for the dot blot densitometry analysis. The relationship between NTs and cytokines was determined by Spearman rank correlation.
Results
Demographics
A total of 50 vitreous samples from 50 patients (50 eyes) were collected. Among these 22 patients had DR and 28 patients were nondiabetic controls. The mean age of all subjects was 64.7 years (range, 42–90) and 39 patients were male and 11 were female. Among diabetics, the mean age was 67.8 years (range, 46–90), whereas the mean age of nondiabetics was 61.5 years (range, 42–78). Among patients with DR, at the time of vitrectomy 3 had mild NPDR, 4 had moderate NPDR, 3 had severe NPDR, and 12 had PDR (Table 1). All patients were candidates for vitrectomy performed at the Kresge Eye Institute of Wayne State University, Detroit, Michigan, United States.
Vitreous Levels of Neurotrophins and Inflammatory Cytokines
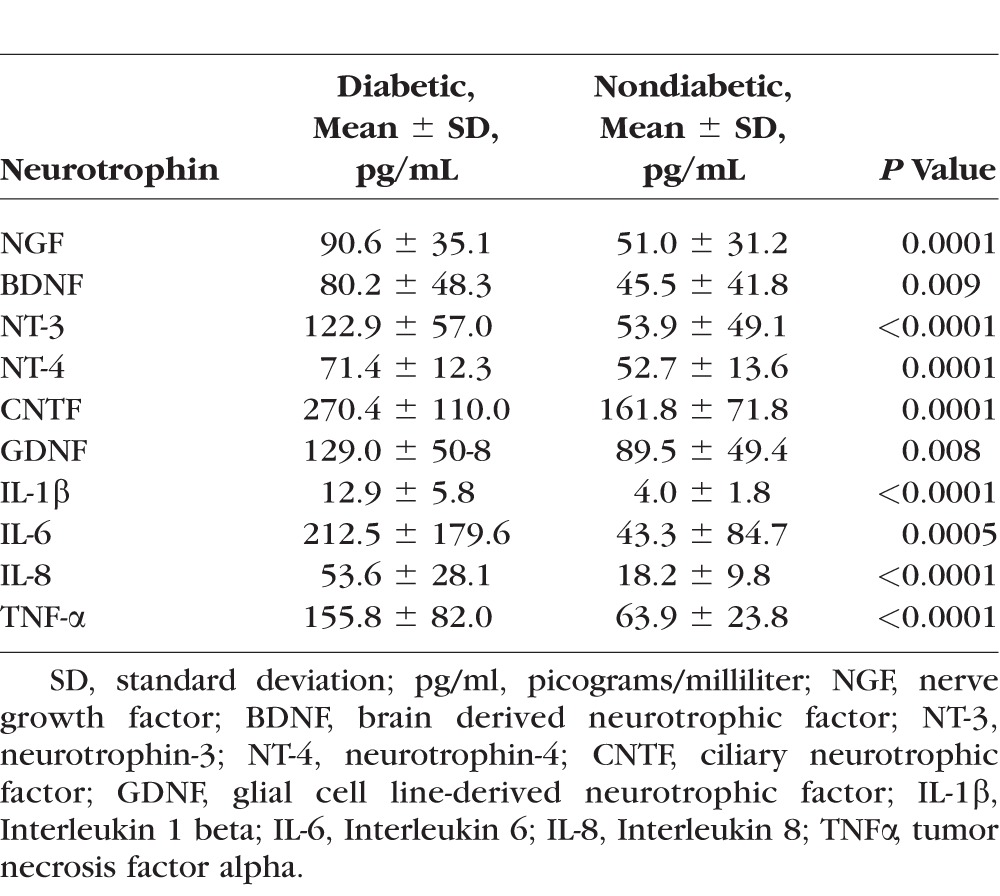
Our analysis showed that NTs, including NGF, BDNF, NT-3, NT-4, CNTF, and GDNF, were present in all vitreous samples of diabetic and nondiabetic eyes, but levels were on average higher in DR eyes than in nondiabetic eyes. There was a significant increase in levels of NGF (P = 0.0001), BDNF (P = 0.0091), NT-3 (P < 0.0001), NT-4 (P = 0.0001), CNTF (P = 0.0001), and GDNF (P = 0.0079) in eyes with DR compared to nondiabetic eyes (Fig. 1A; Table 2). Among eyes with DR, eyes with NPDR compared to eyes with active PDR had higher vitreous concentrations of NGF (P = 0.04), BDNF (P = 0.15), NT-3 (P = 0.01), NT-4 (P = 0.48), CNTF (P = 0.001), and GDNF (P = 0.004) (Fig. 1B). NTs in eyes with NPDR compared to nondiabetic eyes had higher average vitreous concentrations of NGF (P < 0.0001), BDNF (P < 0.0001), NT-3 (P < 0.0001), NT-4 (P < 0.0001), CNTF (P < 0.0001), and GDNF (P < 0.0001). The Spearman correlation analysis revealed a strong correlation (0.513; P < 0.0001) between NT and cytokine levels in patients with DR. The subgroup analysis also revealed a strong correlation between NTs and cytokines in NPDR (0.366; P = 0.02) and PDR (0.535; P = 0.001).
Figure 1.
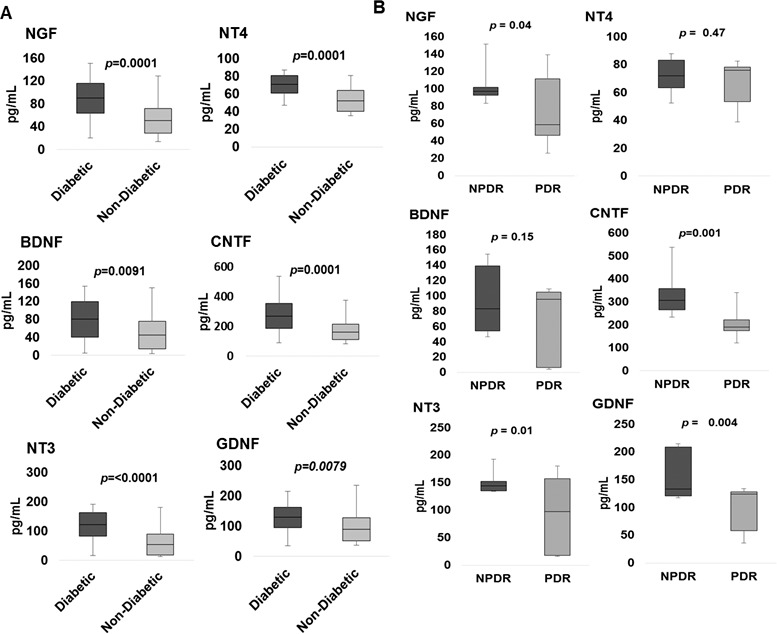
Box-and-whisker plot analysis showing comparison of vitreous NGF, BDNF, NT-3, NT-4, CNTE, and GDNF levels in diabetics (n = 22) compared to nondiabetics (n = 28) (A) and diabetic patients with NPDR compared to active PDR (B).
Table 2.
Quantitative Analysis and Comparison of Intravitreal Levels of Various Neurotrophins in Eyes With Diabetic Retinopathy (N = 22) and Nondiabetic Eyes (N = 28)
The assessment of inflammatory cytokines revealed overall higher levels in eyes with DR than in nondiabetic controls (Fig. 2). There was a significant increase in levels of IL-1β (P < 0.0001), IL-6 (P = 0.0005), IL-8 (P < 0.0001), and TNF-α (P < 0.0001) in eyes with DR compared to nondiabetic eyes (Fig. 2A). The comparative analysis of cytokine levels in eyes with NPDR (N = 12, excluding eyes with PDR) and in nondiabetic control eyes (N = 21, excluding retained lens fragment eyes) also revealed a significant increase in levels of IL-1β (P < 0.0001), IL-6 (P = 0.0011), IL-8 (P < 0.0001), and TNF-α (P < 0.001). Interestingly, levels of some cytokines were higher in diabetic eyes with NPDR than with active PDR, with an increase in levels of IL-8 (P = 0.004) and TNF-α (P < 0.05), while IL-1β (P = 0.08) and IL-6 (P = 0.41) concentrations were comparable (Fig. 2B).
Figure 2.
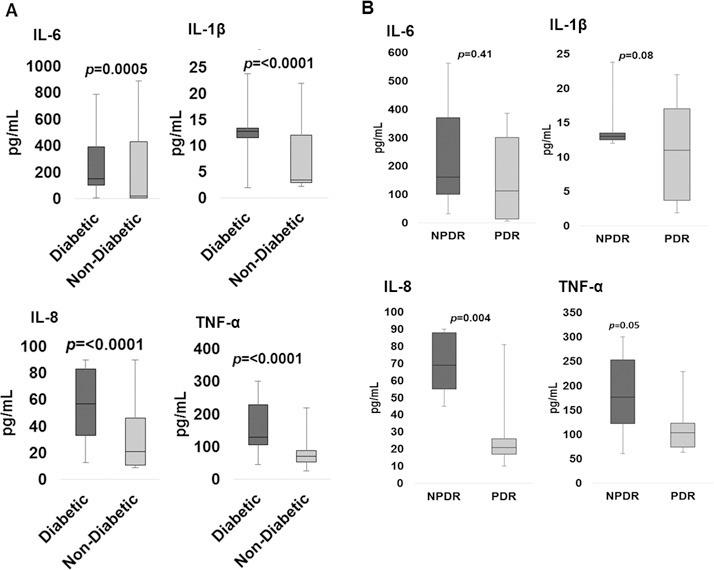
Box-and-whisker plot analysis showing comparison of vitreous IL-6, IL-1β, IL-8, and TNF-α in diabetics (n = 22) compared to nondiabetics (n = 28) (A) and diabetic patients with NPDR compared to PDR (B).
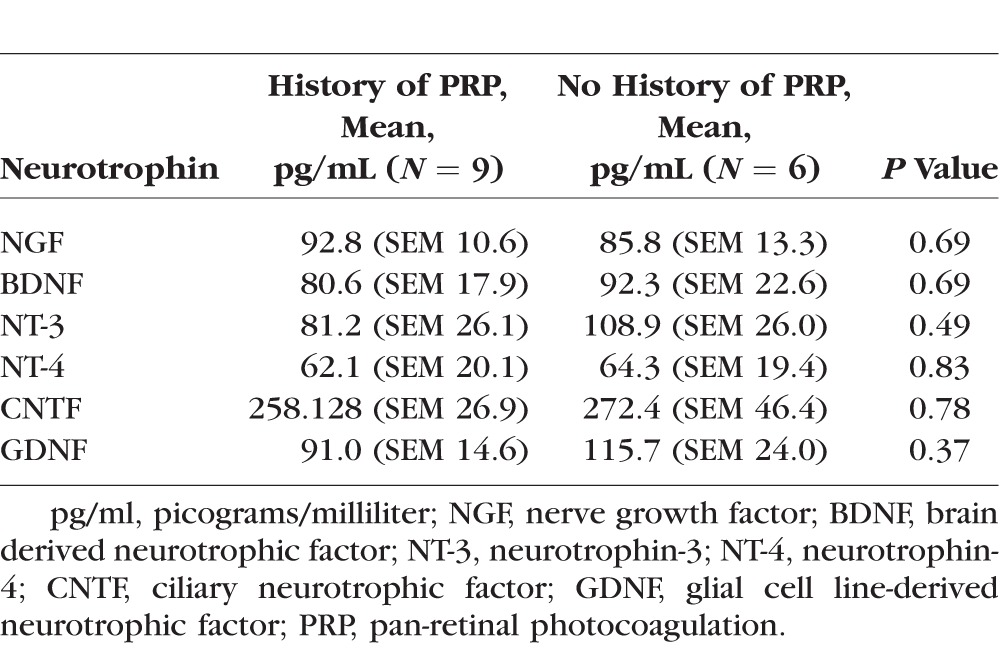
In a subgroup analysis of eyes with severe NPDR and PDR, nine (60%) vitreous samples were collected from eyes with a history of PRP, and six (40%) control samples were collected from eyes without a history of PRP (No PRP). The average patient age was 57.9 years (range, 44–76) in the PRP group and 65.3 years (range, 42–71) in No PRP. The comparative analysis of PRP versus No PRP revealed no significant difference in the vitreous levels of either NTs (Fig. 3A; Table 3) or inflammatory cytokines (Fig. 3B).
Figure 3.
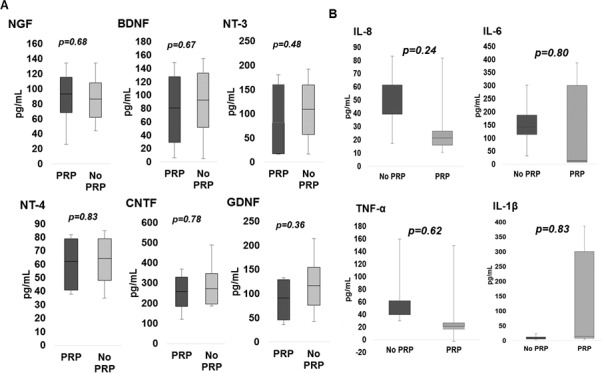
Box-and-whisker plot analysis of vitreous neurotrophin (A) and inflammatory cytokine (B) levels in diabetics who have undergone peripheral retinal ablation with PRP compared to diabetics without a history of PRP.
Table 3.
Quantitative Analysis and Comparison of Intravitreal Levels of Various Neurotrophins in Severe Nonproliferative Diabetic Retinopathy and Proliferative Diabetic Retinopathy Comparing History of Panretinal Photocoagulation
Müller Glia Secrete Neurotrophins in Response to Inflammatory Stimuli
We previously reported that in response to infectious or inflammatory stimuli, retinal glial cells (Müller glia and microglia) get activated and produce more inflammatory mediators along with protective molecules.15,18,19 We hypothesized that diabetes-induced increase in inflammatory mediators triggers the production of NTs by Müller glia, the major glial cell types in the retina.20 To test this, we challenged human retinal Müller glia (MIO-M1 cell line) with recombinant TNF-α or IL-1β for 8 and 24 hours. Our data showed that both TNF-α and IL-1β–challenged Müller glia secrete NTs in conditioned media (Figs. 4A, 4B). To determine whether TNF-α or IL-1β–stimulated NT production is specific to inflammatory stimuli, MIO-M1 cells were challenged with an anti-inflammatory cytokine, IL4 (Fig. 4C), and an anti-inflammatory drug, dexamethasone (Fig. 4D). To this end our results indicate that Müller glia were unable to secrete NTs in the conditioned media in response to these anti-inflammatory agents.
Figure 4.
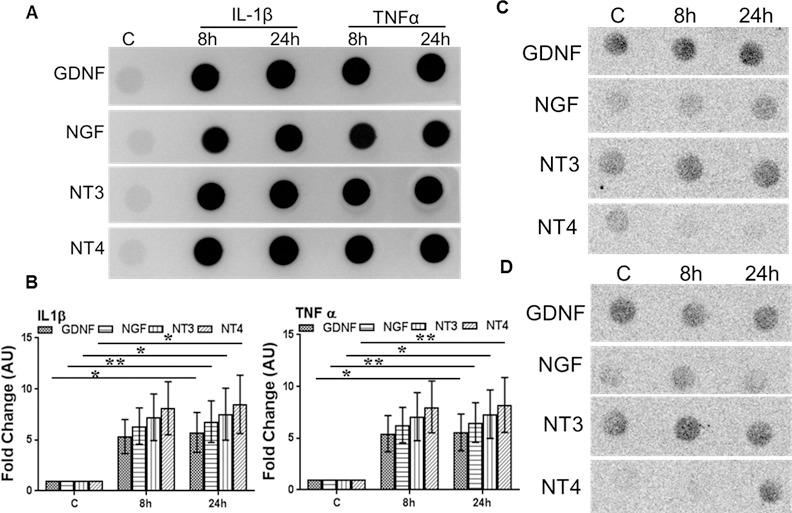
Inflammatory cytokines induce NT production in Müller glia. Human retinal Müller glia (MIO-M1 cell line) were challenged with recombinant TNF-α or IL-1β (100 ng/mL each) (A, B), IL-4 (100 ng/mL) (C), and dexamethasone (100 μM) (D) for the indicated time points. The secretion of NTs into the conditioned media was detected by dot blot, and the intensities of the dots were quantitated by densitometric analysis, using ImageJ and presented as fold-changes (arbitrary units; AU), using a value of 1 for the control samples (B). Statistical analysis was performed by using a 1-way ANOVA (*P < 0.01; **P = 0.001), for comparisons of control versus stimulated cells. Data points and bars represent mean ± SD of triplicates from three independent experiments.
These findings indicate that Müller glia could be one of the potential sources of increased NT levels in the vitreous of patients with DR under chronic inflammatory conditions. Since NTs are known to exert neuroprotection, we then investigated whether NTs can diminish inflammation or oxidative stress–induced neuronal cell death, using mouse cone photoreceptor cell line 661W. Our data showed that GDNF treatment attenuated cell death (TUNEL stain) of 661W cells in response to inflammatory (TNF-α) and oxidative stress (H2O2) conditions, as in the case of DR (Fig. 5).
Figure 5.
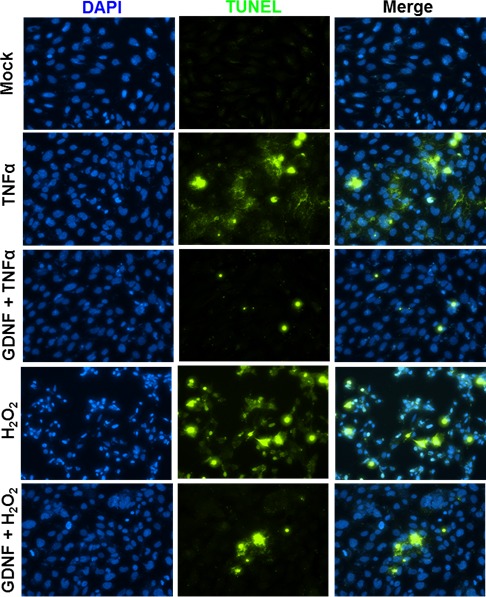
GDNF protects oxidative stress and inflammation-induced photoreceptor cell death. Mouse cone photoreceptor cells (661W cell line) were challenged with H2O2 (100 μM) and TNF-α (100 ng/mL) in the presence and absence of GDNF (100 ng/mL) for 24 hours. The neuroprotective effect of GDNF was measured by measuring the cell death by TUNEL staining (blue, 4′,6-diamidino-2-phenylindole [DAPI] nuclear stain; green, TUNEL+ve cells).
Discussion
DR is a multifactorial disease affecting the retina with an extremely complex pathobiology involving a variety of cell types (both residential and infiltrated), molecules, and factors. From the clinical presentation, DR is broadly classified as NPDR or PDR.21 NPDR represents an early stage of the disease in which symptoms are either mild or nonexistent. This progresses to PDR, a more advanced form of the disease where neovascularization is evident in the retina. Because of the severity of PDR, significant research effort has been devoted in determining the pathogenesis of transition from NPDR to PDR and to identify potential biomarkers to aid in early diagnosis, including ETDRS.22,23 Clearly, a plethora of studies have implicated inflammation in the pathogenesis of DR, with progression to PDR being strongly correlated with increased inflammatory mediators.24 While inflammation is a protective host response to infection or tissue injury, it must be resolved quickly to prevent collateral tissue damage, especially in the retina.25 How inflammation is initiated and persists in DR or whether it is a cause or effect still remains the key questions in the field. Similarly, how increased inflammatory milieu in DR influences other growth factors, including NTs, is not clearly understood. We postulate that increased inflammatory mediators in DR could trigger the production of protective agents to counterregulate the harmful effects of chronic inflammation and induce neuroprotection. In the current study, we showed that patients with DR have high levels of both inflammatory mediators and NTs in their vitreous. Moreover, a comparative analysis revealed higher vitreous inflammatory mediators in NPDR than in PDR. Furthermore, in a proof-of-principle in vitro study, we demonstrated the mechanistic basis of NT production and its potential role in protecting retinal neurons under inflammatory and oxidative stress conditions.
Previous studies analyzing intravitreal inflammatory cytokines in DR have shown a significant increase in levels of IL-6, IL-8, IL-1β, and TNF-α.26–28 These studies, however, have mainly focused on PDR. In the present study, the inflammatory cytokines IL-6, IL-8, IL-1β, and TNF-α were shown to be significantly elevated in vitreous samples not only of PDR, but also of patients with mild to severe NPDR. Interestingly, levels of cytokines were higher in diabetic eyes with NPDR than with active PDR. Because most (8 of 15) of our PDR patients had vitreous hemorrhage and because of the known compromised blood–retinal barrier in PDR, there is a possibility that serum/plasma proteins might have leaked into the vitreous and diluted the relative amount of NTs and cytokines. However, we did not find a significant difference in average concentration of total vitreous protein in PDR (4.72 ± 3.1 μg/μL) versus NPDR (4.86 ± 2.4 μg/μL) patients. Our findings also corroborated with those of Loukovaara et al.,29 showing no major differences in vitreous protein levels in a large cohort of PDR and NPDR patients. Hence, we concluded that differential levels of inflammatory mediators or NTs, in our study, are unlikely due to altered concentration of total vitreous proteins in PDR. Overall, these results highlight the increased activity of these inflammatory cytokines in the early stages of DR. One of the potential implications of these findings is to therapeutically target inflammatory milieu in NPDR patients to prevent progression to PDR. Indeed, a recent study by Wykoff and colleagues30 supports this notion whereby intravitreal corticosteroid injections in NPDR eyes have been shown to decrease the rate of progression from NPDR to PDR in patients in FAME A and B trials receiving intravitreal fluocinolone. Additionally, a DRCR.net study called Protocol W is being started in order to compare the reduction in progression from NPDR to PDR when anti-VEGF and PRP laser are applied to severe NPDR patients. Moreover, new Food and Drug Administration (FDA) indications for intravitreal ranibizumab have changed to now incorporate any degree of DR. Hence, an early indication of increased inflammatory mediators in eyes of diabetic patients, as reported in our study, may allow initiating therapeutic strategies to prevent or halt the progression to PDR.
Another unique aspect of our study is the simultaneous assessment of inflammatory mediators and NTs in vitreous of patients with DR. Neurotrophins, released from glial cells such as Müller cells, are a group of functionally and structurally related growth factors that play a critical role in the development, survival, maintenance, and repair of the nervous system, as well as play essential roles in angiogenesis and fibrosis.31 Several studies have shown that inflammation alters the expression of neurotrophins. In the brain, administration of proinflammatory cytokines or lipopolysaccharides (LPS) causes a significant reduction in BDNF expression.32 Similarly, BDNF levels are reported to be reduced in DR,33 whereas GDNF levels are increased.14,33 This study provided evidence that NTs are present in the vitreous in both diabetics and nondiabetic patients. Moreover, this human study showed vitreous levels of NTs, including NGF, BDNF, NT-3, NT-4, CNTF, and GDNF, to be statistically significantly elevated in patients with DR when compared to patients without a history of diabetes mellitus. In previous animal studies of NT levels, both NGF and BDNF are undetected in both diabetic and control samples13; however, our results showed that both NGF and BDNF are not only present in the human vitreous, but also expressed at a higher concentration in diabetics versus nondiabetics.
Ablating the peripheral retina in PDR has been shown to result in regression of undesired neovascularization.34 Our subanalysis of intravitreal levels of NTs in diabetic patients who had a history of PRP, compared to control patients of diabetics with either severe NPDR or PDR who met criteria for PRP but had no history of PRP, did not show a statistically significant difference in local levels of NTs between the two groups. The small sample size limits the interpretation and power of this analysis; however, PRP did not appear to alter NT levels in our study.
NTs signal by binding and activating a dual receptor system consisting of Trks (TrkA, TrkB, and TrkC) and p75NTR. Signaling via Trks mediates the prosurvival effects of NTs on neurons, whereas activation of p75NTR triggers apoptosis.35,36 Moreover, p75NTR has recently been shown to be essential in angiogenesis and is upregulated in hypoxic and dystrophic retina conditions, including DR.36–39 Hence, NTs exert opposite effects mechanistically depending on which type of receptors they interact with and the target cell types involved.40 While we did not aim to estimate NT receptor concentrations in our study, in part because of limited sample availability, previous studies13 have shown increased expression of Trk A, TrkB, and p75NTR in the vitreous of DR patients. The increased availability of ligands and NT receptors suggests the possibility of enhanced ligand–receptor interactions and given the known role of NTs in neuroprotection, one would expect that increased NT levels should result in reduced cell death in DR. However, the functional outcome of NT interaction with its respective receptors is difficult to study in humans. While several clinical trials have been performed to treat neurodegenerative diseases by increasing the supply of neurotrophic factors, their clinical use is somewhat limited owing to difficulties in maintaining the optimum levels and their poor pharmacokinetics profiles.12,36 Currently, the use of neuroprotective agents in DR is an ongoing debate.41,42 As DR is now being increasingly accepted as a neurodegenerative disease,8,43 the better understanding of NT production and its signaling in the retina may accelerate new therapeutic approaches in the management of DR.
To investigate potential mechanisms of increased NT production in DR (Fig. 6), we postulated that increased inflammatory milieu may potentiate retinal glial cells such as Müller glia to secrete neurotrophic factors as reported in several disease conditions.43–45 Indeed, our in vitro data showed that TNF-α and IL-1β stimulation of Müller glia resulted in increased production of NTs, suggesting that Müller glia are one of the potential sources of NTs in the retina. Both increased inflammatory mediators and oxidative stress in DR have been shown to accelerate cell death in a variety of retinal cells. To assess the biological function of NT under these conditions, we challenged cone photoreceptor cells with H2O2 and TNF-α in presence of GDNF. Our data showed that GDNF treatment inhibited photoreceptor cell death induced by oxidative stress and inflammation. We have previously reported that infectious stimuli, such as those evoked in bacterial endophthalmitis, trigger the production of NTs in the retina to counterregulate inflammation and induce protection (Kumar and Singla. IOVS 2012;53:ARVO E-Abstract 2769). Similarly, exogenous administration of BDNF upregulates anti-inflammatory cytokine IL-10 and reduces the expression of inflammatory cytokine TNF-α in experimental stroke.46 Thus, apart from exerting neuroprotective effects, secreted NTs may also counterregulate inflammation in DR (Fig. 6).
Figure 6.
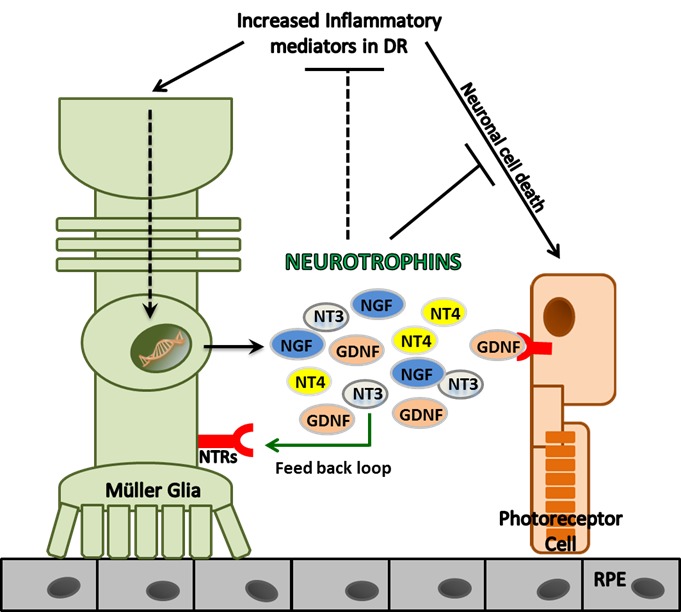
Schematic of the relationship between inflammatory mediators and NTs in diabetic retinopathy. Patients with diabetic retinopathy have increased levels of inflammatory mediators in vitreous. This increased inflammatory milieu triggers the production of NTs in retinal cells, such as Müller glia, which in turn exert neuroprotective and anti-inflammatory effects to prevent neuronal cell death in the retina.
In conclusion, to our knowledge, this study is the first to simultaneously assess vitreous levels of inflammatory cytokines and NTs in DR patients. Our comparative analysis of inflammatory mediators in PDR versus NDPR, demonstrating higher levels in NDPR, suggests that early anti-inflammatory therapy may prevent disease progression to PDR. Moreover, our in vitro data provides mechanistic insight into the production and functional role of NTs under inflammatory conditions, evoked in DR.
Acknowledgments
Supported by an unrestricted grant from Research to Prevent Blindness (RPB) to the Department of Ophthalmology/Kresge Eye Institute at Wayne State University, Detroit, Michigan, United States. The immunology resource core is supported by a National Institutes of Health Center Grant P30EY004068.
Disclosure: J.D. Boss, None; P.K. Singh, None; H.K. Pandya, None; J. Tosi, None; C. Kim, None; A. Tewari, None; M.S. Juzych, None; G.W. Abrams, None; A. Kumar, None
References
- 1. Klein R, Klein BE, Moss SE, Wong TY. . The relationship of retinopathy in persons without diabetes to the 15-year incidence of diabetes and hypertension: Beaver Dam Eye Study. Trans Am Ophthalmol Soc. 2006; 104: 98– 107. [PMC free article] [PubMed] [Google Scholar]
- 2. Kowluru RA, Kowluru A, Mishra M, Kumar B. . Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res. 2015; 48: 40– 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kowluru RA, Chan P-S. . Oxidative stress and diabetic retinopathy. Exp Diabetes Res. 2007; 2007: 43603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu Y, Chen H, Su SB. . Neuroinflammatory responses in diabetic retinopathy. J Neuroinflamm. 2015; 12: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang J, Kern TS. . Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011; 30: 343– 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar A, Kumar A. . Role of Staphylococcus aureus virulence factors in inducing inflammation and vascular permeability in a mouse model of bacterial endophthalmitis. PLoS One. 2015; 10: e0128423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shaw PX, Stiles T, Douglas C,et al. . Oxidative stress, innate immunity, and age-related macular degeneration. AIMS Mol Sci. 2016; 3: 196– 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kadlubowska J, Malaguarnera L, Waz P, Zorena K. . Neurodegeneration and neuroinflammation in diabetic retinopathy: potential approaches to delay neuronal loss. Curr Neuropharmacol. 2016; 14: 831– 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stem MS, Gardner TW. . Neurodegeneration in the pathogenesis of diabetic retinopathy: molecular mechanisms and therapeutic implications. Curr Med Chem. 2013; 20: 3241– 3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barber AJ. . A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry. 2003; 27: 283– 290. [DOI] [PubMed] [Google Scholar]
- 11. Bikbova G, Oshitari T, Baba T, Yamamoto S. . Neurotrophic factors for retinal ganglion cell neuropathy—with a special reference to diabetic neuropathy in the retina. Curr Diabetes Rev. 2014; 10: 166– 176. [DOI] [PubMed] [Google Scholar]
- 12. Semkova I, Krieglstein J. . Neuroprotection mediated via neurotrophic factors and induction of neurotrophic factors. Brain Res Rev. 1999; 30: 176– 188. [DOI] [PubMed] [Google Scholar]
- 13. Abu El-Asrar AM, Mohammad G, De Hertogh G,et al. . Neurotrophins and neurotrophin receptors in proliferative diabetic retinopathy. PLoS One. 2013; 8: e65472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nishikiori N, Mitamura Y, Tashimo A,et al. . Glial cell line-derived neurotrophic factor in the vitreous of patients with proliferative diabetic retinopathy. Diabetes Care. 2005; 28: 2588. [DOI] [PubMed] [Google Scholar]
- 15. Singh PK, Shiha MJ, Kumar A. . Antibacterial responses of retinal Muller glia: production of antimicrobial peptides, oxidative burst and phagocytosis. J Neuroinflamm. 2014; 11: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singh PK, Kumar A. . Retinal photoreceptor expresses Toll-like receptors (TLRs) and elicits innate responses following TLR land bacterial challenge. PLoS One. 2015; 10: e0119541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Singh PK, Kumar A. . Mitochondria mediates caspase-dependent and independent retinal cell death in Staphylococcus aureus endophthalmitis. Cell Death Discov. 2016; 2: 16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kochan T, Singla A, Tosi J, Kumar A. . Toll-like receptor 2 ligand pretreatment attenuates retinal microglial inflammatory response but enhances phagocytic activity toward Staphylococcus aureus. Infect Immun. 2012; 80: 2076– 2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumar A, Shamsuddin N. . Retinal Muller glia initiate innate response to infectious stimuli via toll-like receptor signaling. PLoS One. 2012; 7: e29830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shamsuddin N, Kumar A. . TLR2 mediates the innate response of retinal Muller glia to Staphylococcus aureus. J Immunol. 2011; 186: 7089– 7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilkinson CP, Ferris FL III, Klein RE,et al. . Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology. 2003; 110: 1677– 1682. [DOI] [PubMed] [Google Scholar]
- 22. Relhan N, Flynn HW Jr.. The Early Treatment Diabetic Retinopathy Study historical review and relevance to today's management of diabetic macular edema. Curr Opin Ophthalmol. 2017; 28: 205– 212. [DOI] [PubMed] [Google Scholar]
- 23. Srinivasan S, Raman R, Kulothungan V, Swaminathan G, Sharma T. . Influence of serum lipids on the incidence and progression of diabetic retinopathy and macular oedema: Sankara Nethralaya Diabetic Retinopathy Epidemiology And Molecular genetics Study-II [published online ahead of print May 17, 2017]. Clin Exp Ophthalmol. doi:http://dx.doi.org/10.1111/ceo.12990. [DOI] [PubMed]
- 24. Takeuchi M, Sato T, Tanaka A,et al. . Elevated Levels of Cytokines Associated with Th2 and Th17 Cells in Vitreous Fluid of Proliferative Diabetic Retinopathy Patients. PLoS One. 2015; 10: e0137358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K. . Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci. 2016; 73: 1765– 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murugeswari P, Shukla D, Rajendran A, Kim R, Namperumalsamy P, Muthukkaruppan V. . Proinflammatory cytokines and angiogenic and anti-angiogenic factors in vitreous of patients with proliferative diabetic retinopathy and Eales' disease. Retina. 2008; 28: 817– 824. [DOI] [PubMed] [Google Scholar]
- 27. Adamiec-Mroczek J, Oficjalska-Mlynczak J, Misiuk-Hojlo M. . Roles of endothelin-1 and selected proinflammatory cytokines in the pathogenesis of proliferative diabetic retinopathy: analysis of vitreous samples. Cytokine. 2010; 49: 269– 274. [DOI] [PubMed] [Google Scholar]
- 28. Zhou J, Wang S, Xia X. . Role of intravitreal inflammatory cytokines and angiogenic factors in proliferative diabetic retinopathy. Curr Eye Res. 2012; 37: 416– 420. [DOI] [PubMed] [Google Scholar]
- 29. Loukovaara S, Nurkkala H, Tamene F,et al. . Quantitative proteomics analysis of vitreous humor from diabetic retinopathy patients. J Proteome Res. 2015; 14: 5131– 5143. [DOI] [PubMed] [Google Scholar]
- 30. Wykoff CC, Chakravarthy U, Campochiaro PA, Bailey C, Green K, Cunha-Vaz J. . Long-term effects of intravitreal 0.19 mg fluocinolone acetonide implant on progression and regression of diabetic retinopathy. Ophthalmology. 2017; 124: 440– 449. [DOI] [PubMed] [Google Scholar]
- 31. Taylor S, Srinivasan B, Wordinger RJ, Roque RS. . Glutamate stimulates neurotrophin expression in cultured Muller cells. Brain Res Mol Brain Res. 2003; 111: 189– 197. [DOI] [PubMed] [Google Scholar]
- 32. Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R. . Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front Cell Neurosci. 2014; 8: 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Behl T, Kotwani A. . Downregulated brain-derived neurotrophic factor-induced oxidative stress in the pathophysiology of diabetic retinopathy. Can J Diabetes. 2017; 41: 241– 246. [DOI] [PubMed] [Google Scholar]
- 34. The Diabetic Retinopathy Study Research Group. Photocoagulation treatment of proliferative diabetic retinopathy: clinical application of Diabetic Retinopathy Study (DRS) findings, DRS Report Number 8. Ophthalmology. 1981; 88: 583– 600. [PubMed] [Google Scholar]
- 35. Caporali A, Pani E, Horrevoets AJG,et al. . Neurotrophin p75 receptor (p75NTR) promotes endothelial cell apoptosis and inhibits angiogenesis: implications for diabetes-induced impaired neovascularization in ischemic limb muscles. Circ Res. 2008; 103: e15– e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamashita N, Kuruvilla R. . Neurotrophin signaling endosomes: biogenesis, regulation, and functions. Curr Opin Neurobiol. 2016; 39: 139– 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang J, Zhao J, Bai Y, Huang L, Yu W, Li X. . Effects of p75 neurotrophin receptor on regulating hypoxia-induced angiogenic factors in retinal pigment epithelial cells. Mol Cell Biochem. 2015; 398: 123– 134. [DOI] [PubMed] [Google Scholar]
- 38. Sheedlo HJ, Srinivasan B, Brun-Zinkernagel AM,et al. . Expression of p75(NTR) in photoreceptor cells of dystrophic rat retinas. Brain Res Mol Brain Res. 2002; 103: 71– 79. [DOI] [PubMed] [Google Scholar]
- 39. Mysona BA, Al-Gayyar MM, Matragoon S,et al. . Modulation of p75(NTR) prevents diabetes- and proNGF-induced retinal inflammation and blood-retina barrier breakdown in mice and rats. Diabetologia. 2013; 56: 2329– 2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Casaccia-Bonnefil P, Gu C, Chao MV. . Neurotrophins in cell survival/death decisions. Adv Exp Med Biol. 1999; 468: 275– 282. [DOI] [PubMed] [Google Scholar]
- 41. Hernández C, Dal Monte M, Simó R, Casini G. . Neuroprotection as a therapeutic target for diabetic retinopathy. J Diabetes Res. 2016; 2016: 9508541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hernandez C, Simo R. . Neuroprotection in diabetic retinopathy. Curr Diab Rep. 2012; 12: 329– 337. [DOI] [PubMed] [Google Scholar]
- 43. Honjo M, Tanihara H, Kido N, Inatani M, Okazaki K, Honda Y. . Expression of ciliary neurotrophic factor activated by retinal Muller cells in eyes with NMDA- and kainic acid-induced neuronal death. Invest Ophthalmol Vis Sci. 2000; 41: 552– 560. [PubMed] [Google Scholar]
- 44. Harada T, Harada C, Kohsaka S,et al. . Microglia-Muller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci. 2002; 22: 9228– 9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar A, Pandey RK, Miller LJ, Singh PK, Kanwar M. . Muller glia in retinal innate immunity: a perspective on their roles in endophthalmitis. Crit Rev Immunol. 2013; 33: 119– 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang Y, Wei N, Zhu J,et al. . Effects of brain-derived neurotrophic factor on local inflammation in experimental stroke of rat. Mediators Inflamm. 2010; 2010: 372423. [DOI] [PMC free article] [PubMed] [Google Scholar]