

Abstract
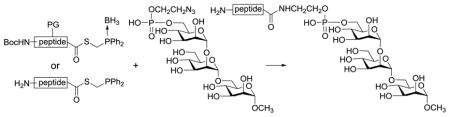
A new strategy has been developed for GPI glycan-peptide conjugate synthesis based upon a traceless Staudinger reaction between a peptide phosphinothioester and a GPI glycan azide. The strategy was first studied and optimized with simple peptides and GPI glycans, which offered excellent yields of the desired conjugates in both organic and aqueous solvents. It was then used to successfully synthesize an analog of the human CD52 antigen containing the whole CD52 peptide sequence and the conserved trimannose motif of all GPI anchors.
Graphical Abstract
Post-translational protein modification by glycosylphosphati-dylinositols (GPIs), a class of complex glycolipid (Figure 1), is ubiquitous in eukaryotes.1–7 It is predicted that ca. 0.5–1.5% of the proteins in the entire eukaryotic genome and up to 20% of surface proteins on some cells are GPI-linked.8–10 GPI lipid chain intercalation into the plasma membrane helps anchor GPI-linked proteins onto the extracellular surface. GPIs and GPI-anchored proteins play a pivotal role in various biological and pathogenic events, such as cell recognition and adhesion, signal transduction, molecule trafficking, membrane fluidity, bacterial infection, Alzheimer disease and cancer development, etc.11–20 To study the structures and functions of GPIs and GPI-anchored proteins, it is essential to have access to these molecules in homogeneous form, which is difficult to achieve by biological means. Furthermore, the study of these molecules is challenging due to the amphiphilic property of GPIs. To address the problem, water soluble GPI glycan-linked peptides or proteins are useful. In this context, chemical synthesis has become a potentially powerful tool to acquire structurally homogeneous and defined GPI-anchored proteins and related conjugates.
Figure 1.

Representative structure of GPI-anchored proteins
As depicted in Figure 1, all GPI-anchored proteins have their polypeptide C-termini linked to the conserved phosphoethano-lamine (EtNP) group at the Man-III 6-O-position. Thus, current synthetic studies on GPI-linked peptides or proteins have been focused on late-stage regioselective coupling of GPI derivatives with peptides or proteins, because this may lead to broadly applicable methods. One of the methods was based on regioselective conjugation of extensively protected, C-terminus free peptides or glycopeptides with GPIs carrying a free EtNP group, which was followed by global deprotection.21,22 However, this is not suitable for large proteins. To address the issue, the Nakahara, Bertozzi, and Seeberger groups23–27 have probed the native chemical ligation (NCL) of peptides or proteins as C-terminal thioesters with cysteine-modified GPIs, and Engelhard28 studied a modular synthetic method that used GPI-linked peptides carrying a cysteine at their N-terminus. Alternatively, our group developed a chemoenzymatic method that employed sortase A (SrtA) to mediate peptide or protein ligation with glycine-modified GPIs.29–32 Although NCL and SrtA-based methods have the potential for broad application, they produce artificial products with a cysteine residue or a signal peptide inserted between the polypeptide sequence and the GPI anchor, respectively. Thus, the synthesis of natural GPI-anchored peptides or proteins and related conjugates remains an essentially unsolved problem.
In this research, we explored a novel strategy for GPI glycan-peptide coupling (Scheme 1) based upon a traceless Staudinger ligation reaction developed by the Raines group33–35 that has been successfully used in protein synthesis.36 We envisioned that a regioselective reaction between phosphinothioester 2 and azide 3 would result in GPI glycan-peptide conjugate 4. An important feature of this strategy is that the ligation reaction has no structural restrictions to the thioester and the azide.
Scheme 1.

Synthesis of GPI glycan-peptide conjugates via traceless Staudinger ligation
Furthermore, the mechanism of this ligation reaction determines that the method should not result in amino acid epimerization. Therefore, it was expected to result in various conjugates having peptides naturally linked to GPI glycans.
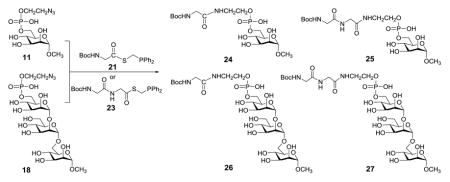
Our study commenced with the preparation of GPI glycan azido derivatives 11 and 18 (Scheme 2) utilized to probe the above strategy. Phosphoramidite 7 was obtained from reacting ethanolazide with purchased 6 in the presence of tetrazolide and employed to phosphorylate mannose derivative 9 by a two-step one-pot protocol.37–39 As the target GPI glycans contained an azide group in their structure, the para-methoxybenzyl (PMB) group, which can be removed by oxidation or under mildly acidic condition,40–42 was employed for permanent protection of the carbohydrate hydroxyl groups. The reaction gave a diastereomeric mixture 9 (R:S 1:1), which was globally deprotected on treatments with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to remove the cyanoethyl group utilized to protect the phosphate and with 10% trifluoroacetic acid (TFA) to remove the PMB groups, to eventually provide 11 readied for the coupling with peptide phosphinothioesters.
Scheme 2.

Synthesis of the azido derivative 11 of a short GPI glycan analog
The azido derivative 18 of the conserved trimannose phosphate of GPI anchors was prepared by a similarly strategy. As shown in Scheme 3, glycosylation of 8 with Schmidt glycosyl donor 12 and 2'-O-deacetylation of the resultant disaccharide afforded 13. This glycosylation reaction was highly α-stereoselective, as it was favored by both anomeric and neighboring group participation effects. Subsequently, 13 was glycosylated with 14, which was followed by selective desilylation to afford trimannoside 16. All the glycosidic linkages in 16 had the expected α-configuration, verified by the JC1,H1 coupling constants (171.6, 171.2, and 168.9 Hz). Finally, phosphorylation of 16 and global deprotection of the product according to above-mentioned procedures gave 18.
Scheme 3.

Synthesis of the azido derivative 18 of the GPI core trimannose
Glycine and diglycine phosphinothioesters 21 and 23 were used as models to explore and optimize the ligation reaction. They were prepared by condensing phosphinothiol 19 with 20 and 22 (Scheme 4) in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI).
Scheme 4.

Synthesis of peptide phosphinothioesters 21 and 23
With both the azides, 11 and 18, and phosphinothioesters, 21 and 23, in hand, we set out to explore their ligation and optimize the reaction conditions. The results are listed in Table 1. The reaction between 11 and 21 in DMF was slow at room temperature, thus after two days of reaction conjugation product 24 was isolated only in a moderate yield (55%, Entry 1). Then, we examined the reaction at elevated temperature (40 °C), which gave an excellent yield (96%) after one day of reaction (Entry 2). Moreover, it was found that switching the solvent from DMF to a mixture of DMF and H2O (1:1) did not significantly affect the ligation yield (Entry 3). Subsequently, we examined the ligations among more complex substrates (Entries 4–9), and all gave excellent yields of desired conjugation products in both DMF and the DMF and H2O mixture.
Table 1.
Results of the ligation reactions between 11/18 and 21/23
| |||||||
|---|---|---|---|---|---|---|---|
| entry | azide (1 equiv) | thioester (1.5 equiv) | temp (°C) | solventa | time (day) | product | yieldb (%) |
| 1 | 11 | 21 | rt | DMF | 2 | 24 | 55 |
| 2 | 11 | 21 | 40 | DMF | 1 | 24 | 96 |
| 3 | 11 | 21 | 40 | DMF/H2O | 1 | 24 | 93 |
| 4 | 11 | 23 | 40 | DMF | 1 | 25 | 93 |
| 5 | 11 | 23 | 40 | DMF/H2O | 1 | 25 | 92 |
| 6 | 18 | 21 | 40 | DMF | 1 | 26 | 93 |
| 7 | 18 | 21 | 40 | DMF/H2O | 1 | 26 | 91 |
| 8 | 18 | 23 | 40 | DMF | 1 | 27 | 92 |
| 9 | 18 | 23 | 40 | DMF/H2O | 1 | 27 | 89 |
DMF:H2O ratio 1:1.
Isolated yields.
To demonstrate the application potential of this synthetic strategy, it was probed for the synthesis of an analog 34 of the human CD52 antigen (Scheme 5), having the intact CD52 peptide sequence linked to the conserved trimannose moiety of GPI anchors. The CD52 peptide 30 was prepared by solid-phase peptide synthesis on a peptide synthesizer, using standard fluorenylmethyloxycarbonyl (Fmoc) chemistry, serine-modified acid-sensitive 2-chlorotrityl resin 28 as solid-phase support, and (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), hydroxybenzotriazole (HOBt) and N,N-diisopropylethylamine (DIPEA) as condensation reagents. The N-terminal glycine in 30 was protected with the tert-butyloxycarbonyl (Boc) group, which was later removed simultaneously with other protecting groups on peptide side chains under acidic condition. Peptide 30 was converted into thioester 32 upon condensation with phosphinothiol 31 by a reported method.43 In 31, the phosphine was protected as a borane complex to prevent its oxidation by oxygen in the air. Eventually, global deprotection of 32 under acidic condition in the presence of Et3SiH afforded free peptide phosphinothioester 33 (MALDI-TOF MS: calcd for [M - H]−, 1420.5; found 1420.7). However, HPLC purification of 33 was difficult due to the oxidation of its phosphine moiety in the air. Therefore, 33 generated upon deprotection was precipitated with cold ether and directly used for ligation with GPI glycan.
Scheme 5.

Synthesis of an analog of the human CD52 antigen 33
Both the protected and deprotected peptides 32 and 33 were investigated for the ligation with 18 (Scheme 5). Deprotection of the phosphine in 32 with 1,4-diazabicyclo[2.2.2]octane (DABCO) and ligation with 18 were carried out in one pot to afford smoothly the coupling product, verified by MALDI-TOF MS data (calcd for [M+Na]+, 2678.4; found, 2678.4). Thereafter, the peptide was globally deprotected by the above-described protocol, which was followed by HPLC purification, to produce the desired GPI glycan-peptide conjugate 34 in a 78% overall yield. No isomers of 34 resulting from amino acid epimerization were noticed during the HPLC purification and by NMR analysis. Similarly, 33 derived from deprotection of 32 under acidic condition was used to couple with 18 in the presence of DIPEA, and this reaction afforded 34 in a 66% overall yield. Compound 34 was confirmed by MALDI-TOF MS data (calcd for [M - H]−, 1829.7; found, 1829.9). It should be noted, however, that the presence of a base, e.g., DIPEA, to scavenge the trace amount of TFA left from the deprotection step was important in the latter case, as the ligation did not proceed well under acidic condition. In addition, a small amount of oxidized 33 (MALDI-TOF MS: calcd for [M - H]−, 1436.5; found 1436.5), which was unreactive with GPI azide, was also observed as a side product in the latter ligation.
In summary, we have described herein a novel strategy for GPI glycan-peptide conjugate synthesis based on the regiospecific traceless ligation of a GPI azide and a peptide phosphinothioester. The synthetic strategy was first optimized using simple peptides and then successfully applied to the preparation of an analog of the human CD52 antigen having its whole peptide sequence naturally linked to the conserved trimannose motif of all GPI anchors. An important feature of this strategy was that the ligation reaction between phosphinothioester and azide did not require a specific amino acid or sequence at the peptide C-terminus or on the GPI anchor. Thus, it is broadly useful for accessing various GPI glycan-peptide conjugates. These GPI conjugates, as water soluble analogs of natural GPI-anchored peptides and proteins, should be very useful for their detailed structural analysis by techniques such as NMR spectroscopy and for biological studies. Moreover, the ligation proceeded smoothly in a mixture of DMF and H2O. This was a significant discovery as it would render the ligation method applicable to complex structures, including the ligation of proteins with intact GPI anchors, which needs to be carried out in the proper mixture of water and an organic solvent, such as DMF, to dissolve both substrates. Therefore, this study has also provided the proof of concept for a new synthetic strategy for GPI-anchored proteins. In this context, protein phosphinothioesters can be prepared by the intein technology.44,45 Currently, the application of this traceless Staudinger ligation to the synthesis of GPI-anchored proteins and other GPI conjugates is under further investigation.
Supplementary Material
Acknowledgments
The authors thank NIH/NIGMS for the support (R01GM090270) of this research work.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors, and all authors have given approval to the final version of this manuscript.
Supporting Information, including synthetic procedures and NMR and MS spectra and other analytical data of all new compounds, is available free of charge on the ACS Publications website.
References
- 1.Ferguson MAJ, Williams AF. Ann Rev Biochem. 1988;57:285. doi: 10.1146/annurev.bi.57.070188.001441. [DOI] [PubMed] [Google Scholar]
- 2.Englund PT. Ann Rev Biochem. 1993;62:121. doi: 10.1146/annurev.bi.62.070193.001005. [DOI] [PubMed] [Google Scholar]
- 3.Ikezawa H. Biol Pharm Bull. 2002;25:409. doi: 10.1248/bpb.25.409. [DOI] [PubMed] [Google Scholar]
- 4.Cole RN, Hart GW. In: New Comprehensive Biochemistry. Montreuil J, Vliegenthart JFG, Schachter H, editors. Vol. 29. Elsevier; Amsterdam: 1997. p. 69. [Google Scholar]
- 5.Udenfriend S, Kodukula K. Ann Rev Biochem. 1995;64:563. doi: 10.1146/annurev.bi.64.070195.003023. [DOI] [PubMed] [Google Scholar]
- 6.McConville MJ, Ferguson MAJ. Biochem J. 1993;294:305. doi: 10.1042/bj2940305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulick MG, Bertozzi CR. Biochemistry. 2008;47:6991. doi: 10.1021/bi8006324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenhaber B, Bork P, Eisenhaber F. J Mol Biol. 1999;292:741. doi: 10.1006/jmbi.1999.3069. [DOI] [PubMed] [Google Scholar]
- 9.Eisenhaber B, Bork P, Eisenhaber F. Protein Eng. 2001;14:17. doi: 10.1093/protein/14.1.17. [DOI] [PubMed] [Google Scholar]
- 10.Nakayasu ES, Yashunsky DV, Nohara LL, Torrecilhas ACT, Nikolaev AV, Almeida IC. Mol Syst Biol. 2009;5:1. doi: 10.1038/msb.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diep DB, Nelson KL, Raja SM, Pleshak EN, Buckley JT. J Biol Chem. 1998;273:2355. doi: 10.1074/jbc.273.4.2355. [DOI] [PubMed] [Google Scholar]
- 12.Horejsi V, Cebecauer M, Cerny J, Brdicka T, Angelisova P, Drbal K. Immunol Lett. 1998;63:63. doi: 10.1016/s0165-2478(98)00054-6. [DOI] [PubMed] [Google Scholar]
- 13.Varma R, Mayor S. Nature. 1998;394:798. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- 14.Sambamurti K, Sevlever D, Koothan T, Refolo LM, Pinnix I, Gandhi S, Onstead L, Younkin L, Prada CM, Yager D, Ohyagi Y, Eckman CB, Rosenberry TL, Younkin SG. J Biol Chem. 1999;274:26810. doi: 10.1074/jbc.274.38.26810. [DOI] [PubMed] [Google Scholar]
- 15.Kasahara K, Sanai Y. Glycoconjugate J. 2000;17:153. doi: 10.1023/a:1026576804247. [DOI] [PubMed] [Google Scholar]
- 16.Marmor MD, Julius M. J Biol Regul Homeostatic Agents. 2000;14:99. [PubMed] [Google Scholar]
- 17.Menon AK, Baumann NA, Rancour DM. In: Carbohydrates in Chemistry and Biology. Hart GW, Hart G, Sinay P, Ernst B, Sinaikin P, editors. Vol. 4. Wiley-VCH; New York: 2000. p. 757. [Google Scholar]
- 18.Trotter J, Klein C, Kramer E. Neuroscientist. 2000;6:271. [Google Scholar]
- 19.Onda H, Ohkubo S, Shintani Y, Ogi K, Kikuchi K, Tanaka H, Yamamoto K, Tsuji I, Ishibashi Y, Yamada T, Kitada C, Suzuki N, Sawada H, Nishimura O, Fujino M. Biochem Biophys Res Commun. 2001;285:235. doi: 10.1006/bbrc.2001.5149. [DOI] [PubMed] [Google Scholar]
- 20.Mayor S, Riezman H. Nat Rev Mol Cell Biol. 2004;5:110. doi: 10.1038/nrm1309. [DOI] [PubMed] [Google Scholar]
- 21.Xue J, Shao N, Guo Z. J Org Chem. 2003;68:4020. doi: 10.1021/jo034213t. [DOI] [PubMed] [Google Scholar]
- 22.Shao N, Xue J, Guo Z. Angew Chem Int Ed. 2004;43:1569. doi: 10.1002/anie.200353251. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka Y, Nakahara Y, Hojo H, Nakahara Y. Tetrahedron. 2003;59:4059. [Google Scholar]
- 24.Paulick MG, Forstner MB, Groves JT, Bertozzi CR. PNAS. 2007;104:20332. doi: 10.1073/pnas.0710139104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paulick MG, Wise AR, Forstner MB, Groves JT, Bertozzi CR. J Am Chem Soc. 2007;129:11543. doi: 10.1021/ja073271j. [DOI] [PubMed] [Google Scholar]
- 26.Becker CFW, Liu X, Olschewski D, Castelli R, Seidel R, Seeberger PH. Angew Chem, Int Ed. 2008;47:8215. doi: 10.1002/anie.200802161. [DOI] [PubMed] [Google Scholar]
- 27.Roller R, Vilotijevic I, Michel D, Seeberger PH, Silva DV. J Pept Sci. 2014;20:S47. [Google Scholar]
- 28.Schumacher MC, Resenberger U, Seidel RP, Becker CFW, Winklhofer KF, Oesterhelt D, Tatzelt J, Engelhard M. Biopolymers (Pept Sci) 2010;94:457. doi: 10.1002/bip.21380. [DOI] [PubMed] [Google Scholar]
- 29.Guo X, Wang Q, Swarts BM, Guo Z. J Am Chem Soc. 2009;131:9878. doi: 10.1021/ja903231v. [DOI] [PubMed] [Google Scholar]
- 30.Wu Z, Guo X, Guo Z. Chem Commun. 2010;46:5773. doi: 10.1039/c0cc00828a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z, Guo X, Wang Q, Swarts BM, Guo Z. J Am Chem Soc. 2010;132:1567. doi: 10.1021/ja906611x. [DOI] [PubMed] [Google Scholar]
- 32.Wu Z, Guo X, Gao J, Guo Z. Chem Commun. 2013;49:11689. doi: 10.1039/c3cc47229a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nilsson BL, Kiessling LL, Raines RT. Org Lett. 2001;3:9. doi: 10.1021/ol006739v. [DOI] [PubMed] [Google Scholar]
- 34.Soellner MB, Nilsson BL, Raines RT. J Org Chem. 2002;67:4993. doi: 10.1021/jo025631l. [DOI] [PubMed] [Google Scholar]
- 35.Nilsson BL, Hondal RJ, Soellner MB, Raines RT. J Am Chem Soc. 2003;125:5268. doi: 10.1021/ja029752e. [DOI] [PubMed] [Google Scholar]
- 36.Tam A, Soellner MB, Raines RT. J Am Chem Soc. 2007;129:11421. doi: 10.1021/ja073204p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindh I, Stawinski J. J Org Chem. 1989;54:1338. [Google Scholar]
- 38.Murakata C, Ogawa T. Tetrahedron Lett. 1991;32:101. [Google Scholar]
- 39.Nikolaev AV, Chudek JA, Ferguson MAJ. Carbohydr Res. 1995;272:179. doi: 10.1016/0008-6215(95)00067-4. [DOI] [PubMed] [Google Scholar]
- 40.Swarts BM, Guo Z. J Am Chem Soc. 2010:6648. doi: 10.1021/ja1009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Roy B, Liu X. Chem Commun. 2011;47:8952. doi: 10.1039/c1cc13264d. [DOI] [PubMed] [Google Scholar]
- 42.Xie L, Zhu S, Shen X, He L, Yang J. J Org Chem. 2010;75:5764. doi: 10.1021/jo101231r. [DOI] [PubMed] [Google Scholar]
- 43.Muhlberg M, Jaradat DMM, Kleineweischede R, Papp I, Dechtrirat D, Muth S, Broncel M, Hackenberger CPR. Bioorg Med Chem. 2010;18:3679. doi: 10.1016/j.bmc.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 44.Berrade L, Camarero JA. Cell Mol Life Sci. 2009;66:3909. doi: 10.1007/s00018-009-0122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaha NH, Muir TM. Chem Sci. 2014;5:446. doi: 10.1039/C3SC52951G. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.