

Abstract
We report the non-decarbonylative Mizoroki–Heck reactions of amide derivatives. The transformation relies on the use of nickel catalysis and proceeds using sterically hindered tri- and tetrasubstituted olefins to give products containing quaternary centers. The resulting polycyclic or spirocyclic products can be obtained in good yields. Moreover, a diastereoselective variant of this methodology demonstrates its value for accessing adducts bearing vicinal, highly substituted sp3 stereocenters. Our results demonstrate that amide derivatives can be used as building blocks for the assembly of complex scaffolds.
Keywords: nickel, catalysis, Mizoroki–Heck, quaternary centers
Graphical Abstract
We report the first non-decarbonylative Mizoroki–Heck reactions of Boc-activated amide derivatives. Our results demonstrate that amide derivatives can be used as building blocks for the assembly of complex scaffolds.
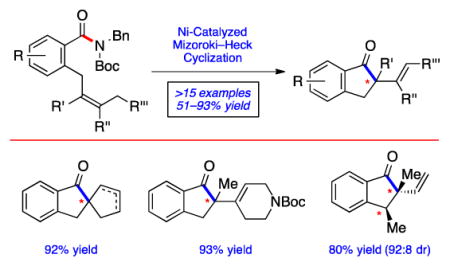
The introduction of quaternary carbon centers remains a popular topic in modern chemical synthesis.[1] Such motifs are often difficult to access due to the steric challenge associated with constructing a fully substituted carbon center. One attractive means to install quaternary centers is via the intramolecular Mizoroki–Heck reaction.[2] Most notably, the Pd-catalyzed Mizoroki–Heck cyclization of aryl halides and triflates has been the subject of intense investigation for decades and has been utilized to assemble many sterically demanding scaffolds. On the other hand, the corresponding Mizoroki–Heck cyclization of acyl electrophiles to furnish ketone products bearing quaternary carbons has not been reported.
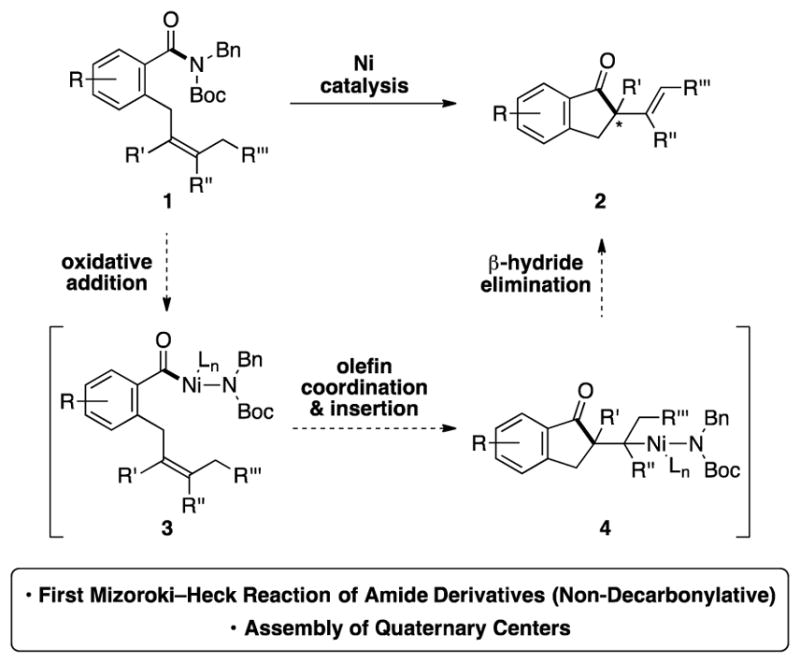
Considering the aforementioned deficiency concerning the Mizoroki–Heck cyclization of acyl electrophiles, we pursued the transformation shown in Figure 1. In the presence of an appropriate nickel catalyst, imide 1, derived from the corresponding secondary amide upon Boc-activation, would be converted to cyclized products 2, bearing the desired quaternary centers. Mechanistically, the conversion would proceed by a sequence akin to classical Mizoroki–Heck chemistry involving oxidative addition (1→3), olefin coordination and insertion (3→4), followed by β-hydride elimination[3] (4→2). It should be noted that amide derivatives have recently been employed in Pd- and Ni-catalyzed couplings for carbon–heteroatom[4] and carbon–carbon[5,6,7] bond formation, although never for the synthesis of quaternary centers.[8] Moreover, precedent for the desired olefin insertion is available from Stambuli’s Pd-catalyzed Mizoroki–Heck cyclization of benzoic anhydrides, albeit without quaternary stereocenter formation,[9,10] and Pd-catalyzed carbonylative Mizoroki–Heck reactions of aryl halides and triflates.[11] Herein, we describe the development and scope of the Ni-catalyzed Mizoroki–Heck cyclization of amide derivatives.[12] The transformation provides a new means to build complex scaffolds using non-precious metal catalysis.[13]
Figure 1.
Designed nickel-catalyzed Mizoroki–Heck reaction of amide derivatives to forge quaternary centers; Boc=tert-butyloxycarbonyl, Bn=benzyl.
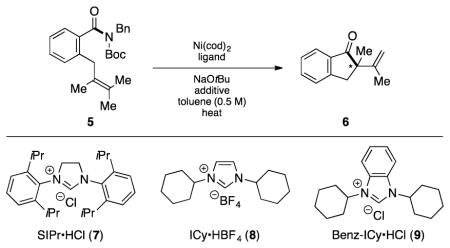
After some initial experimentation, we arrived at 5 as a suitable test substrate (Table 1).[14] This substrate contains the N-Bn,Boc imide-type motif,[15] which we have previously found to be reactive using Ni/SIPr (7) combinations,[4,5] in addition to a sterically encumbered tetrasubstituted olefin. The Mizoroki–Heck cyclization of 5 was attempted under a variety of reaction conditions,[16] with a selection of key results using Ni(cod)2, NHC ligands, and toluene as solvent at 100 °C depicted. Unfortunately, attempts to conduct the desired cyclization using SIPr•HCl (7) in the presence of NaOtBu were unsuccessful (entry 1). However, by switching to NHC precursor 8 the Mizoroki–Heck product 6 was obtained, albeit in modest yield (entry 2). Further improvements were seen when benzimidazolium salt 9 was employed,[17] which gave rise to the desired product 6 in 76% yield (entry 3). We also probed the Ni to ligand ratio and found that employing a 1:1 ratio of Ni(cod)2 to 9 (rather than a 1:2 ratio), led to diminished yields (entry 4). Efforts to optimize the Ni loading were also undertaken. Although using 10 mol% Ni(cod)2 gave the desired product (entry 5), the use of 15 mol% Ni(cod)2 gave excellent yields (entry 6) and was found more generally effective across a range of substrates studied subsequently. During the course of our studies, we also evaluated a series of additives used previously in Ni-catalyzed couplings.[18] These efforts demonstrated that the reaction temperature could be lowered to 60 °C, provided that t-amyl alcohol was employed as the additive, to deliver product 6 in 95% yield (entry 7).[19] It should be noted that: (a) Ni-catalyzed Mizoroki–Heck reactions to form quaternary centers are rare,[20] (b) there are no prior examples of Ni-catalyzed Mizoroki–Heck reactions involving tetrasubstituted olefins in the literature,[21] and (c) decarbonylation products were not observed during reaction development.
Table 1.
Evaluation of ligand effects and reaction conditions for the conversion of 5 to Mizoroki–Heck cyclization product 6, bearing a quaternary center.[a]
| |||||
|---|---|---|---|---|---|
| Entry | Ni(cod)2loading | Ligand (loading) | Additive | Temp. | Yield [b] |
| 1 | 20 mol% | 7 (40 mol%) | none | 100 °C | 0% |
| 2 | 20 mol% | 8 (40 mol%) | none | 100 °C | 24% |
| 3 | 20 mol% | 9 (40 mol%) | none | 100 °C | 76% |
| 4 | 20 mol% | 9 (20 mol%) | none | 100 °C | 67% |
| 5 | 10 mol% | 9 (20 mol%) | none | 100 °C | 51% |
| 6 | 15 mol% | 9 (30 mol%) | none | 100 °C | 91% |
| 7 | 15 mol% | 9 (30 mol%) | t-amyl alcohol[c] | 60 °C | 95% |
Conditions unless otherwise stated: 5 (1.0 equiv, 0.1 mmol), Ni(cod)2 (mol% as shown), 7–9 (mol% as shown), toluene (0.5 M), NaOtBu (1.1x ligand loading) heated at the specified temperature for 24 h in a sealed vial.
Yields reflect an average of two experiments and were determined by 1H NMR analysis using hexamethylbenzene as an internal standard.
3.0 equiv of t-amyl alcohol was used; Bn=benzyl, Boc=tert-butyloxycarbonyl, cod=bis(1,5-cyclooctadiene)nickel(0).
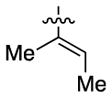
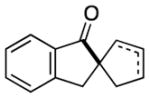
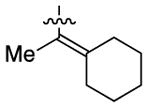
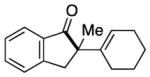
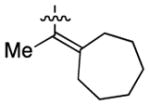
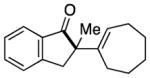
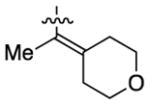
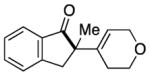
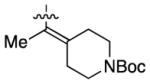
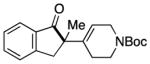
Having identified conditions to achieve the nickel-catalyzed cyclization, we evaluated the scope with respect to the tethered alkene (Table 2).[22,23] It was found that a trisubstituted olefin[24] analog of our parent substrate could be employed to furnish terminal olefin product 10 in 71% yield (entry 1). We also examined substrates in which the trisubstituted olefin was embedded in a ring. Using both 5- and 6-membered ring substrates, the desired Mizoroki–Heck cyclization proceeded smoothly to give the corresponding spirocyclic products, 11 and 12, respectively, as mixtures of olefin isomers (entries 2 and 3).[25] Returning to the more challenging tetrasubstituted olefins, a series of substrates bearing exocyclic olefins were prepared and evaluated. Whereas utilization of a substrate containing a 5-membered ring led to product 13 in 51% yield (entry 4), the use of 6- and 7-membered ring-containing substrates furnished products 14 and 15, respectively, in good yields (entries 5 and 6). Lastly, two heterocyclic substrates were examined. We were delighted to find that our methodology proved tolerant of a tetrahydropyran and a protected piperidine, thus giving rise to tricycles 16 and 17, respectively, in excellent yields (entries 7 and 8).
Table 2.
Mizoroki–Heck cyclization of a variety of tri- and tetrasubstituted olefin substrates.
| |||
|---|---|---|---|
| Entry | Alkene | Product | Yield [a] |
| 1 |
|
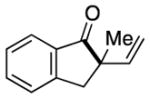 10 |
71%[b] |
| 2 |
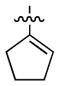
|
 11 |
92%[b] |
| 3 |
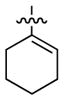
|
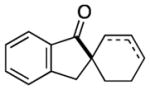 12 |
75%[b] |
| 4 |
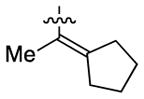
|
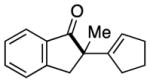 13 |
51% |
| 5 |
|
 14 |
96% |
| 6 |
|
 15 |
80% |
| 7 |
|
 16 |
91% |
| 8 |
|
 17 |
93% |
Yields shown reflect the average of two isolation experiments.
Reaction performed at 100 °C in the absence of t-amyl alcohol; Bn=benzyl, Boc=tert-butyloxycarbonyl, cod=bis(1,5-cyclooctadiene)nickel(0).
As shown in Figure 2, the methodology is also tolerant of substituents on the arene. For example, use of substrates containing the fluoride or trifluoromethyl group, both of which are critical in medicinal chemistry,[26] gave rise to products 18 and 19, respectively. The methoxy group was also well tolerated, as shown by the formation of 20 and 21. As demonstrated by the synthesis of 22 and 23, substrates bearing a methyl group could also be utilized. In the latter case, it is notable that the presence of a methyl group ortho to the tethered alkene did not hinder reactivity.
Figure 2.
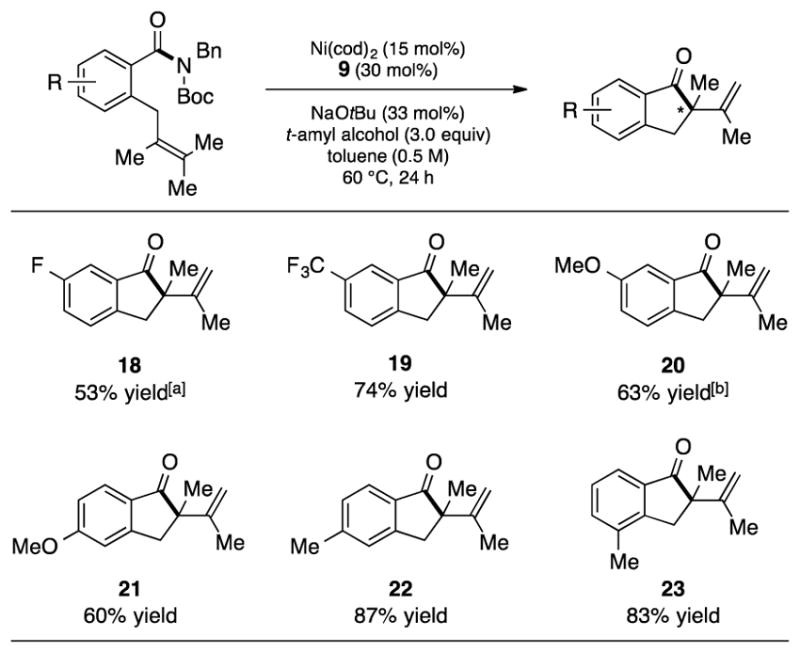
Substituents on the arene motif. Yields shown reflect the average of two isolation experiments. [a] Yield determined by 1H NMR analysis using hexamethylbenzene as an external standard. [b] Reaction performed at 100 °C in the absence of t-amyl alcohol; Bn=benzyl, Boc=tert-butyloxycarbonyl, cod=bis(1,5-cyclooctadiene)nickel(0).
As a further test, we questioned if this methodology could be performed in a diastereoselective sense (Figure 3). Trisubstituted olefin 24,[27] which bears an allylic methyl group, was treated under our optimal reaction conditions. This reaction delivered ketone 25 in 80% yield, as a 92:8 ratio of diastereomers. Of note, 25 contains vicinal sp3 stereocenters, both of which are highly substituted. Prior transition metal-catalyzed methods for the synthesis of 2-vinylindanones[22] have not been demonstrated for the construction of such complexity. The diastereoselectivity seen in the conversion of 24 to 25 can be rationalized by considering the two competing olefin insertion transition states, TS1 and TS2. In both cases, the olefin insertion event is thought to occur via a standard 4-centered transition state, which, in turn, prompts allylic strain arguments.[28] In TS1, A(1,3) strain between the two highlighted hydrogens is minimal and the methyl group rests in a pseudo-equatorial disposition. As such, TS1 is favorable and leads to the major diastereomer of 25 shown, with the methyl groups residing in a cis fashion. On the other hand, the minor diastereomer of 25 (not depicted) is thought to arise from TS2, which displays a less favorable A(1,3) interaction between the highlighted hydrogen and methyl substituents.
Figure 3.
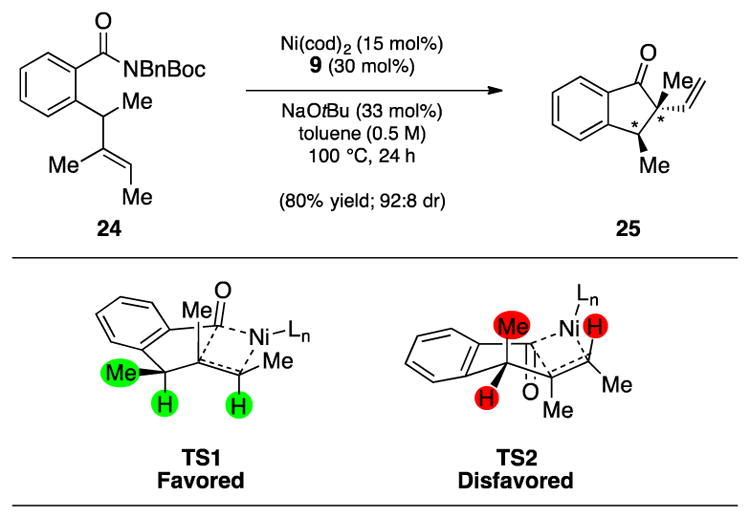
Diastereoselective Mizoroki–Heck cyclization for the introduction of vicinal sp3 stereocenters. Yield and diastereomeric ratio shown reflect the average of two isolation experiments; Bn=benzyl, Boc=tert-butyloxycarbonyl, cod=bis(1,5-cyclooctadiene)nickel(0).
We have developed the Mizoroki–Heck cyclization of amide derivatives to access ketones containing quaternary centers. The transformation is tolerant of variation on both the alkene and aryl moieties, and most notably, proceeds using sterically hindered tetrasubstituted olefins. As a result, polycyclic, spirocyclic, and heteroatom-containing products can be synthesized using this methodology. Moreover, we have demonstrated that a diastereoselective Mizoroki–Heck cyclization proceeds for the controlled formation of an adduct bearing vicinal, highly substituted sp3 stereocenters. In addition to providing a rare Ni-catalyzed Mizoroki–Heck cyclization methodology for accessing quaternary centers and the first Mizoroki–Heck cyclizations of amide derivatives, our results demonstrate that amides, despite once being viewed as unreactive, can be used as building blocks for the preparation of complex scaffolds.
Supplementary Material
Acknowledgments
The authors thank the NIH-NIGMS (R01-GM117016), the Guggenheim Foundation, and the University of California, Los Angeles, for financial support. We are grateful to the NIH-NIGMS (F31-GM113642 to J. M.), the UCLA Graduate Division (Dissertation Year Fellowship and Cota Robles Fellowship (J. M. M.), the Foote Family (J. M.), the Swiss National Science Foundation for an Early Mobility Postdoctoral Fellowship (S. R.), and the UCLA CSST Program (S. D.). Professor Daniel Weix (University of Rochester) is thanked for providing ligands and helpful discussions. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the NIH NCRR (S10RR025631).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.For reviews on quaternary stereocenters, see: Christoffers J, Baro A. Adv Synth Catal. 2005;347:1473–1482.Trost BM, Jiang C. Synthesis. 2006;3:369–396.Quasdorf KW, Overman LE. Nature. 2014;516:181–191. doi: 10.1038/nature14007.Marek I, Minko Y, Pasco M, Mejuch T, Gilboa N, Chechik H, Das JP. J Am Chem Soc. 2014;136:2682–2694. doi: 10.1021/ja410424g.Liu Y, Han SJ, Liu WB, Stoltz BM. Acc Chem Res. 2015;48:740–751. doi: 10.1021/ar5004658.Long R, Huang J, Gong J, Yang Z. Nat Prod Rep. 2015;32:1584–1601. doi: 10.1039/c5np00046g.Zeng XP, Cao ZY, Wang YH, Zhou F, Zhou J. Chem Rev. 2016;116:7330–7396. doi: 10.1021/acs.chemrev.6b00094.Christoffers J, Baro A John Wiley & Sons. Quaternary stereocenters: challenges and solutions for organic synthesis. Wiley-VCH; Weinheim: 2005.
- 2.a) Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]; b) Whitcombe NJ, Hii KK, Gibson SE. Tetrahedron. 2001;57:7449–7476. [Google Scholar]; c) Dounay AB, Overman LE. Chem Rev. 2003;103:2945–2964. doi: 10.1021/cr020039h. [DOI] [PubMed] [Google Scholar]; d) Shibasaki M, Vogl EM, Ohshima T. Adv Synth Catal. 2004;346:1533–1552. [Google Scholar]; e) Oestreich M. Top Organomet Chem. 2007;24:169–192. [Google Scholar]; f) Shibasaki M, Ohshima T, Itano W. Sci Synth. 2011;3:483–512. [Google Scholar]; g) Mc Cartney D, Guiry PJ. Chem Soc Rev. 2011;40:5122–5150. doi: 10.1039/c1cs15101k. [DOI] [PubMed] [Google Scholar]; h) Le Bras J, Muzart J. Chem Rev. 2011;111:1170–1214. doi: 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]; i) Jones AC, May JA, Sarpong R, Stoltz BM. Angew Chem, Int Ed. 2014;53:2556–2591. doi: 10.1002/anie.201302572. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:2590–2628. [Google Scholar]
- 3.β-hydride elimination of Ni(II) intermediates is known to be challenging; see: Lin B-L, Liu L, Fu Y, Luo S-W, Chen Q, Guo Q-X. Organometallics. 2004;23:2114–2123.
- 4.For Ni-catalyzed reactions of amide derivatives to build C–O or C–N bonds, see: Hie L, Fine Nathel NF, Shah T, Baker EL, Hong X, Yang Y-F, Liu P, Houk KN, Garg NK. Nature. 2015;524:79–83. doi: 10.1038/nature14615.Baker EL, Yamano MM, Zhou Y, Anthony SM, Garg NK. Nat Commun. 2016;7:11554. doi: 10.1038/ncomms11554.Hie L, Baker EL, Anthony SM, Desrosiers JN, Senanayake C, Garg NK. Angew Chem, Int Ed. 2016;55:15129–15132. doi: 10.1002/anie.201607856.Angew Chem. 2016;128:15353–15356.
- 5.For Ni-catalyzed reactions of amide derivatives to build C–C bonds, see: Weires NA, Baker EL, Garg NK. Nat Chem. 2016;8:75–79. doi: 10.1038/nchem.2388.Simmons BJ, Weires NA, Dander JE, Garg NK. ACS Catal. 2016;6:3176–3179. doi: 10.1021/acscatal.6b00793.Dander JE, Weires NA, Garg NK. Org Lett. 2016;18:3934–3936. doi: 10.1021/acs.orglett.6b01758.Shi S, Szostak M. Chem Eur J. 2016;22:10420–10424. doi: 10.1002/chem.201602202.Liu L, Chen P, Sun Y, Wu Y, Chen S, Zhu J, Zhao Y. J Org Chem. 2016;81:11686–11696. doi: 10.1021/acs.joc.6b02093.
- 6.For Pd-catalyzed reactions of amide derivatives to build C–C bonds, see: Li X, Zou G. Chem Commun. 2015;51:5089–5092. doi: 10.1039/c5cc00430f.Li X, Zou G. J Organomet Chem. 2015;794:136–145.Yada A, Okajima S, Murakami M. J Am Chem Soc. 2015;137:8708–8711. doi: 10.1021/jacs.5b05308.Meng G, Szostak M. Org Lett. 2015;17:4364–4367. doi: 10.1021/acs.orglett.5b02209.Meng G, Szostak M. Org Biomol Chem. 2016;14:5690–5705. doi: 10.1039/c6ob00084c.Liu C, Meng G, Liu Y, Liu R, Lalancette R, Szostak R, Szostak M. Org Lett. 2016;18:4194–4197. doi: 10.1021/acs.orglett.6b01836.Meng G, Shi S, Szostak M. ACS Catal. 2016;6:7335–7339.Cui M, Wu H, Jian J, Wang H, Liu C, Daniel S, Zeng Z. Chem Commun. 2016;52:12076–12079. doi: 10.1039/c6cc06428k.
- 7.For decarbonylative couplings of amide derivatives, see: Meng G, Szostak M. Angew Chem, Int Ed. 2015;54:14518–14522. doi: 10.1002/anie.201507776.Angew Chem. 2015;127:14726–14730.Hu J, Zhao Y, Zhang Y, Shi Z. Angew Chem, Int Ed. 2016;55:8718–8722. doi: 10.1002/anie.201603068.Angew Chem. 2016;128:8860–8864.Shi S, Meng G, Szostak M. Angew Chem, Int Ed. 2016;55:6959–6963. doi: 10.1002/anie.201601914.Angew Chem. 2016;128:7073–7077.Shi S, Szostak M. Org Lett. 2016;18:5872–5875. doi: 10.1021/acs.orglett.6b02952.Liu C, Meng G, Szostak M. J Org Chem. 2016;81:12023–12030. doi: 10.1021/acs.joc.6b02294.Yue H, Guo L, Liao HH, Cai Y, Zhu C, Rueping M. Angew Chem, Int Ed. 2017;56:4282–4285. doi: 10.1002/anie.201611819.Angew Chem. 2017;129:4346–4349.
- 8.For pertinent reviews, see: Meng G, Shi S, Szostak M. Synlett. 2016;27:2530–2540.Dander JE, Garg NK. ACS Catal. 2017;7:1413–1423. doi: 10.1021/acscatal.6b03277.Liu C, Szostak M. Chem Eur J. 2017 doi: 10.1002/chem.201605012.
- 9.Miles KC, Le CC, Stambuli JP. Chem Eur J. 2014;20:11336–11339. doi: 10.1002/chem.201403561. [DOI] [PubMed] [Google Scholar]
- 10.For the Pd-catalyzed Mizoroki–Heck cyclization of thioesters to give ortho-quinone methide products, see: Thottumkara AP, Kurokawa T, Du Bois J. Chem Sci. 2013;4:2686–2689.
- 11.a) Negishi EI, Copéret C, Ma S, Mita T, Sugihara T, Tour JM. J Am Chem Soc. 1996;118:5904–5918. [Google Scholar]; b) Hayashi T, Tang J, Kato K. Org Lett. 1999;1:1487–1489. [Google Scholar]; c) Wu XF, Neumann H, Spannenberg A, Schulz T, Jiao H, Beller M. J Am Chem Soc. 2010;132:14596–14602. doi: 10.1021/ja1059922. [DOI] [PubMed] [Google Scholar]; d) Goegsig TM, Nielsen DU, Lindhardt AT, Skrydstrup T. Org Lett. 2012;14:2536–2539. doi: 10.1021/ol300837d. [DOI] [PubMed] [Google Scholar]
- 12.Amide derivatives are considered to be ideally suited for use as synthons in chemical synthesis because of their directing group ability and stability to a variety of reaction conditions.
- 13.For pertinent reviews concerning nickel catalysis, see: Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t.Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299–309. doi: 10.1038/nature13274.Mesganaw T, Garg NK. Org Process Res Dev. 2013;17:29–39.Ananikov VP. ACS Catal. 2015;5:1964–1971.
- 14.Substrates described in this manuscript were derived from commercially available 2-halobenzoic acid derivatives and readily available allylic electrophiles, either via lithiation/displacement or reductive coupling methodologies (see the SI). For reductive couplings, see: Weix DJ. Acc Chem Res. 2015;48:1767–1775. doi: 10.1021/acs.accounts.5b00057.Anka-Lufford LL, Prinsell MR, Weix DJ. J Org Chem. 2012;77:9989–10000. doi: 10.1021/jo302086g.Cui X, Wang S, Zhang Y, Deng W, Qian Q, Gong H. Org Biomol Chem. 2013;11:3094–3097. doi: 10.1039/c3ob40232k.Hansen EC, Pedro DJ, Wotal AC, Gower NJ, Nelson JD, Caron S, Weix DJ. Nat Chem. 2016;8:1226–1230. doi: 10.1038/nchem.2587.
- 15.Other classes of amide derivatives were also evaluated, such as N-Me,Ph, N-Me,Ts, N-Ph,Boc, N-Ph,Ts. However, these efforts either led to no product or the Mizoroki–Heck cyclization product in diminished yields. Similarly, efforts to generate 6- or 7-membered ring products were unsuccessful.
- 16.Extensive variations in bases (e.g. Cs2CO3, K3PO4, NaH), ligands (e.g. terpyridine, IMes, Benz-ItBu), solvents (DME, DMA, PhCF3), Ni and ligand loadings, and temperature were explored.
- 17.NHC precursors 7–9 are commercially available.
- 18.Additives such as morpholine, aniline, Et2NH, and 1-hexanol were also tested. Of all additives examined, t-amyl alcohol proved most successful. For the use of alcohols as additives in Ni-catalyzed cross couplings, see: Harris MR, Hanna LE, Greene MA, Moore CE, Jarvo ER. J Am Chem Soc. 2013;135:3303–3306. doi: 10.1021/ja311783k.
- 19.a) Using entry 7 conditions, cyclized product 6 was obtained in 74% isolated yield, respectively. Product 6 is volatile as described in the SI. b) Product 6 could also be obtained in comparable yield by Mizoroki–Heck cyclization of the N-nBu,Boc derivative of 5, thus demonstrating that this methodology is not restricted to N-benzyl amide derivatives.
- 20.For the Ni-catalyzed Mizoroki–Heck cyclization of aryl halides, albeit without tetrasubstituted olefins, see: Desrosiers J-N, Hie L, Biswas S, Zatalochnaya OV, Rodriguez S, Lee H, Grinberg N, Haddad N, Yee NK, Garg NK, Senanayake C. Angew Chem, Int Ed. 2016;55:11921–11924. doi: 10.1002/anie.201606955.Angew Chem. 2016;128:12100–12103.see also references therein.
- 21.For examples of Mizoroki–Heck reactions involving tetrasubstituted olefins, see: Shuler SA, Yin G, Krause SB, Vesper CM, Watson DA. J Am Chem Soc. 2016;138:13830–13833. doi: 10.1021/jacs.6b08932.Seo JH, Liu P, Weinreb SM. J Org Chem. 2010;75:2667–2680. doi: 10.1021/jo100339k.Liu P, Seo JH, Weinreb SM. Angew Chem, Int Ed. 2010;49:2000–2003. doi: 10.1002/anie.200906818.Angew Chem. 2010;122:2044–2047.Abelman MM, Oh T, Overman LE. J Org Chem. 1987;52:4130–4133.Madin A, O’Donnell CJ, Oh T, Old DW, Overman LE, Sharp MJ. J Am Chem Soc. 2005;127:18054–18065. doi: 10.1021/ja055711h.
- 22.2-Vinylindanones bearing quaternary stereocenters can also be synthesized by the Pd- or Ni-catalyzed α-vinylation of lithium enolates; see Huang J, Bunel E, Faul MM. Org Lett. 2007;9:4343–4346. doi: 10.1021/ol7019839.Grigalunas M, Ankner T, Norrby PO, Wiest O, Helquist P. Org Lett. 2014;16:3970–3973. doi: 10.1021/ol5017965.Grigalunas M, Ankner T, Norrby PO, Wiest O, Helquist P. J Am Chem Soc. 2015;137:7019–7022. doi: 10.1021/jacs.5b02945.Chieffi A, Kamikawa K, Ahman J, Fox JM, Buchwald SL. Org Lett. 2001;3:1897–1900. doi: 10.1021/ol0159470.
- 23.A substrate containing a phenyl (Ph) group at the R′ position was also tested, but was deemed unreactive.
- 24.This substrate was employed as a 7:1 mixture of E to Z olefins.
- 25.See the Supporting Information for details.
- 26.a) Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]; b) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]; c) Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. J Med Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- 27.This substrate was employed as a 4:1 mixture of E to Z olefins.
- 28.Based on calculated rotational energies for conformers of 3-methyl-1-butene and allylic strain arguments, TS2 is estimated to be roughly 0.7 kcal/mol higher in energy compared to TS1. For a review, see: Hoffmann RW. Chem Rev. 1989;89:1841–1860.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.