

Abstract
Background
DNA-launched infectious system is a useful tool with high rescue efficiency that allows the introduction of mutations in specific positions to investigate the function of an individual viral element. Rescued virus particles could be harvested by directly transfecting the DNA-launched recombinant plasmid to the host cells, which will reduce labor and experimental cost by skipping the in vitro transcription assay.
Methods
A total of four overlapping fragments covering the entire viral genome were amplified and then were assembled into a transformation vector based on pIRES2-EGFP to establish the DNA-launched infectious system of duck hepatitis A virus type 1 (DHAV-1), named pIR-DHAV-1. Reverse transcription polymerase chain reaction (RT-PCR) detection, quantitative real-time polymerase chain reaction (qRT-PCR), western blotting assay and indirect immunofluorescence (IFA) were conducted for rescued virus identification. A total of 4.0 μg of recombinant plasmid of pIR-DHAV-1 and in vitro transcribed product of 4.0 μg of RNA-launched infectious clone named pR-DHAV-1 were transfected into BHK-21 cells to analyze the rescue efficiency. Following that, tissue tropism of rescued virus (rDHAV-1) and parental virus (pDHAV-1) were assayed for virulence testing in 1-day-old ducklings.
Results
Rescued virus particles carry the designed genetic marker which could be harvested by directly transfecting pIR-DHAV-1 to BHK-21 cells. The qRT-PCR and western blotting results indicated that rDHAV-1 shared similar growth characteristics with pDHAV-1. Furthermore, DNA-launched infectious system possessed much higher rescue efficiency assay compared to RNA-launched infectious system. The mutation at position 3042 from T to C has no impact on viral replication and tissue tropism. From 1 h post infection (hpi) to 48 hpi, the viral RNA copies of rDHAV-1 in liver were the highest among the six tested tissues (with an exception of thymus at 6 hpi), while the viral RNA copy numbers in heart and kidney were alternately the lowest.
Conclusion
We have constructed a genetically stable and highly pathogenic DNA-launched infectious clone, from which the rescued virus could be harvested by direct transfection with recombinant plasmids. rDHAV-1 shared similar growth characteristics and tissue tropism with pDHAV-1. The DNA-launched infectious system of DHAV-1 possessed higher rescue efficiency compared to the traditional RNA-launched infectious system.
Keywords: DHAV-1, DNA-launched infectious clone, Ribozyme, Rescue efficiency
Background
Duck virus hepatitis (DVH), which was first described in Long Island in 1949 [1], is commonly recognized as an acute and fatal disease of ducklings. DVH is caused by duck hepatitis virus (DHV) types 1, 2 and 3, and no antigenic relationships have been found among them [2–5]. In the three different serotypes of DHVs, DHV-1 was considered as the most common and most virulent serotype [1, 6]. According to the Virus Taxonomy the Ninth Report of the International Committee on Taxonomy of Viruses (ICTV), DHV-1 was classified as a member of Picornaviridae and renamed as duck hepatitis A virus (DHAV) [7]. As the only member of the genus Avihepatovirus in the family Picornaviridae, DHAV has been further classified into three serotypes: the DHAV serotype 1 (DHAV-1, the classical serotype) [8, 9], the DHAV serotype 2 (DHAV-2, a serotype isolated in Taiwan) [10], and the DHAV serotype 3 (DHAV-3, a serotype isolated in South Korea and China) [11, 12]. There is no cross-neutralization between DHAV-1 and DHAV-2 [10] yet limited cross-neutralization between DHAV-1 and DHAV-3 [13]. Both DHV-2 and DHV-3 were later identified as astroviruses based on sequence analysis of a 391 nt RNA-dependent RNA polymerase [14].
DHAV-1 is a non-enveloped, single-stranded and positive-sense RNA virus, and possesses a short genome of approximately 7.7 kb excluding the poly (A) tail at the 3′ end, with a long open reading frame (ORF) flanked by 5′ and 3′ untranslated regions (UTRs) [8]. The ORF encodes a large polyprotein, which is subsequently cleaved into three capsid proteins (VP0, VP1 and VP3) and nine nonstructural proteins (2A1, 2A2, 2A3, 2B, 2C, 3A, 3B, 3C and 3D) [8]. The 5′ UTR of DHAV-1 contains an internal ribosome entry site (IRES) [15]. The 3′ UTR of DHAV-1, composed of three double-stranded hairpin stems, is 314 nt in length and is the longest among all Picornaviridae members [16].
Reverse genetics systems have been considered as powerful tools for studying all aspects of virus biology, including virus pathogenesis and vaccine development [17]. Up to date, there are at least four types of viral RNA rescued system, including RNA-launched system [18], helper virus-launched system [19] and two DNA-launched systems based on cellular RNA polymerases I and II [20–22]. Although the infectious DHAV genomic RNA could be harvested in vitro from a full-length DHAV cDNA clone by the RNA-launched reverse genetics system [23, 24], the operative strategy still depends on in vitro transcription. Compared to the RNA-launched infectious clone, the RNA polymerases II-based DNA-launched infectious system is able to generate homogenous RNA transcripts from transfected cDNA clone in vivo, allowing for higher rescue efficiency with less cost and labor by skipping in vitro RNA transcription [25–27]. For porcine reproductive and respiratory syndrome virus (PRRSV), by the introduction of ribozyme elements at both termini of the viral genomic cDNA, an improved DNA-launched infectious clone improved the rescue efficacy with approximately 10–50-fold higher than the in vitro-transcribed RNA-based system [28]. The hammerhead ribozyme is a catalytic motif responsible for the self-cleaving activity of RNA virusoids [29], while the hepatitis delta virus ribozyme also contains site-specific self-cleavage with the existence of divalent cations and a cytidine nucleotide [30]. After transcription, the ribozymes at both termini of DNA-launched infectious clones could lead into the self-cutting and releasing of the viral mRNA under the condition of existing Mg2+ or other divalent ions and a cytidine nucleotide. By this way, the viral RNA can be harvested by direct transfection with the recombinant plasmid without in vitro transcription [31].
In the previous study, a highly pneumovirulent strain of DHAV-1, LY0801 (originally named LY01), was isolated from a 7-day-old duckling exhibiting typical duck viral hepatitis (DVH) in 2008 from an outbreak of severe DVH in Shandong province, China [9]. Based on the virulent strain LY0801, we firstly established a DNA-launched infectious clone of DHAV-1, which possessed higher rescue efficiency compared to the traditional RNA-launched infectious system.
Methods
Cell, viruses and antibodies
BHK-21 cells were cultured at 37 °C in 5% CO2 in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml Streptomycin Sulfate. The construction of this infectious cDNA clone of DHAV-1 was based on the 5th passage of LY0801 strain (accession no. FJ436047). LY0801 is a virulent strain of DHAV-1 isolated in 2008 from an outbreak of severe DVH in Shandong province, China [9]. The LY0801 virus produced an acute and fatal disease in one-week-old ducklings. The mortality of duck flock was 80%, and the sick ducklings died quickly with typical hemorrhagic hepatitis. After propagated in the allantonic cavities of 10-day-old duck embryos and adapted to BHK-21 cells for five passages, the LY0801 strain was used for the full-length genomic cDNA construction. Anti-DHAV-1 monoclonal antibody (mAb) 4F8, which could recognize the epitope “75GEIILT80” in VP1 of DHAV-1, was stored in the veterinary molecular etiology laboratory of Shandong Agricultural University [32]. Fluorescein isothiocyanate (FITC)-labeled goat anti-mouse antibody was purchased from KPL (MD, USA), Horseradish Peroxidase (HRP)-labeled goat anti-mouse antibody was purchased from Abcam (MA, USA).
Cloning and sequencing the full-length genome of DHAV-1
To determine the complete sequence of LY0801 strain, the viral RNA was extracted from allantoic liquids of dead duck embryos with a E.Z.N.A.™ Viral RNA Kit (Omega Bio-Tek, Norcross, GA, USA) followed by RNA transcription using RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Burlington, Canada) according to the manufacturer′s instruction. Nine pairs of specific primers were designed to amplify 9 overlapping PCR fragments (Table 1). PCR products were purified using a gel purification kit (Invitrogen, CA, USA), and then were TA-cloned into the pMD18-T vector (TaKaRa, Dalian, China) following the manufacturer′s protocol. The TA-cloned products were then transfected into E. coli DH5α competent cells (TaKaRa, Dalian, China), and the positive clones were selected by PCR amplification and then were sent to a commercial service for sequencing with dideoxy terminal termination method by 3730xl DNA Analyzer (ThermoFisher, MA, USA) by Sangon Biotech Co., Ltd., Shanghai, China. The 5′ sequences of the viral genome were determined by 5′ rapid amplification of cDNA ends (RACE). The complete genomic sequence of the 5th passage of the DHAV-1 LY0801 strain in BHK-21 cell culture was compared with the FJ436047 sequence by the Clustal W method (DNA Star LaserGene software, DNAStar Inc. Madison, WI) and then used for the construction of DNA-launched infectious clone.
Table 1.
PCR primers used in this study
| Primer | Primer sequences (5′ → 3′) |
|---|---|
| DHAV-Seq-1F | TTT GAA AGC GGG TGC AT |
| DHAV-Seq-1R | TTA TAG TGT GTG GGA CTC GAC C |
| DHAV-Seq-2F | GAC TAG TTC CTG AGG GAC AGA TGT T |
| DHAV-Seq-2R | TCC CTG ATT GTC AAA TGG TCG G |
| DHAV-Seq-3F | ACA ACT GGT GGT GCC ATT TGT GT |
| DHAV-Seq-3R | CTG CCA AAA GTT GCC TCT GAT GTG C |
| DHAV-Seq-4F | TGG ATG ACC TCA CTT CAG AGT ATG C |
| DHAV-Seq-4R | TTG ACT GCA TGT GAT CAC CTG CTG G |
| DHAV-Seq-5F | TAA ATG GTG AAG TCA CAA TCA AG |
| DHAV-Seq-5R | GGC CAA AAT CAT CAA AAG CAT |
| DHAV-Seq-6F | CTC TTG GTA TAT GGA TAT CTG GTG GT |
| DHAV-Seq-6R | GTA GCA ATC AAT TTA GAC ACA TCT |
| DHAV-Seq-7F | AGA TGA GAT TAG GGA CAT |
| DHAV-Seq-7R | TGG GTA TAA CAT CAC TAC TC |
| DHAV-Seq-8F | AGA CAC ATG TTG CTG AAA AAC T |
| DHAV-Seq-8R | AGA ACA CAG TCA TCC CCA TAA CT |
| DHAV-Seq-9F | CTG ATG AGA TAT GGC AGG TA |
| DHAV-Seq-9R | TTT TTT TTT AGG TAG GGT AGG GAA T |
| pIR-BamHI-XhoI-F | GGG ATC CTA CTC TAG ACA TAA TCA GCC ATA CCA CAT TTG TAG AGG |
| pIR-BamHI-AscI-R | GAA GGA TCC ACT GGC GCG CCT CGA GAT CTG AGT CCG GTA G |
| 5HeadRibo-F | ACA TGG CGC GCC ACA TCA TCT GAT GAG TCC GTG AGG ACG AAA CGG TAC CCG CGT GAG GAC GAA ACG GTA CCC GGT ACC GTC ATT TTG AAA GCG GGT GCA TGC ATG GCC AT |
| 5HeadRibo-R | AGG TGG ATC CAC TAT TGT CAC CTT C |
| 3endRibo-F | CTG ATG AGA TAT GGC AGG TA |
| 3endRibo-R | GCT CTA GAG TCC CAT TCG CCA TTA CCG AGG GGA CGG TCC CCT CGG AAT GTT GCC CAG CCG GCG CCA GCG AGG AGG CTG GGA CCA TGC CGG CCT TTT TTT TTT TTT TTT TTT TTA GGT AGG GTA GGG AAT AGT AAA GT |
| Cloning 2-F | TAG TGG ATC CAC CTG AAA CAC C |
| Cloning 2-R | ACC AGAT ATC CAT ATA CCA AGA GGT T |
| BamH I-detect-F | ACA ACT GGT GGT GCC ATT TGT GT |
| BamH I-detect-R | TTG ACT GCA TGT GAT CAC CTG CTG G |
| RT-qPCR-F | AGA CAC ATG TTG CTG AAA AAC T |
| RT-qPCR-R | AGA ACC AGT TGT CGT TTG GTC |
| DHAV-1-Probe | Cy5-ATG CCA TGA CAC TAT CTC ATA TGA GTC AGC-BHQ-2 |
Assembly of the full-length cDNA clone
A total of 9 overlapping fragments covering the entire viral genome based on the cDNA of the 5th passage of LY0801 strain were amplified for construction of DNA-launched infectious clone. The fragment IR was amplified with the primers pIR-BamHI-XhoIF/pIR-BamHI-AscI-R (Table 1) based on the vector pIRES2-EGFP (Clontech, Mountain View, CA) to introduce corresponding enzyme restriction sites at both terminuses. The fragments were then digested with BamH I and ligated with T4 DNA ligase (TaKaRa, Dalian, China), yielding a vector named pIR to delete enhanced green fluorescent protein (EGFP) gene. The fragments clone 1 containing the hammerhead ribozyme and 1 to 3042 of LY0801 sequence was amplified with 5HeadRibo-F/R, while the clone 4 possessing the hepatitis delta virus ribozyme and 6820 to 7710 of viral sequence was amplified with 3HeadRibo-F/R, respectively (Fig. 1). The ribozyme sequences were shown in italic in Table 1. By overlapping PCR, four fragments covering the full-length genome were amplified with the four pairs of primers, namely 5HeadRibo-F/R (clone 1), Cloning 2-F/R (clone 2), DHAV-Seq-6F/9R (clone 3) and 3endRibo-F/R (clone 4) (Table 1). The four fragments were assembled into pIR vector by one multi-step strategy (Fig. 1).
Fig. 1.
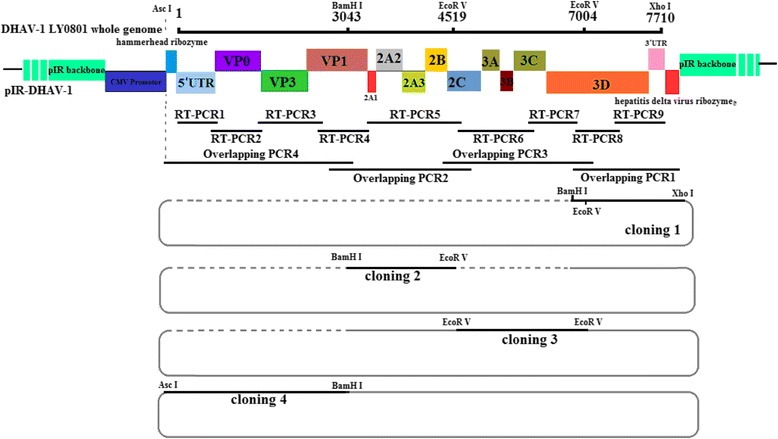
Strategy for the construction of a full-length DNA-launched infectious clone based on a virulent strain LY0801 of DHAV-1. The fragment IR was derived from the plasmid pIRES2-EGFP (Clontech) using primers pIR-BamHI-XhoIF and pIR-AscI-BamHIR, and then was digested with BamH I and ligated with T4 DNA ligase to yield pIR vector. The LY0801 virus genome with two ribozyme sequences introduced at the both ends was placed downstream of the CMV promoter in pIR vector to construct the DNA-launched infectious clone, pIR-DHAV-1. A total of 4 overlapping fragments amplified from total RNAs extracted from the 5th passage of LY0801 virus were assembled successively into the pIR vector by unique restriction enzyme indicated for each fragment. A copy of hammerhead ribozyme sequence and Asc I restrict enzyme site were engineered at the 5′ end while a copy of hepatitis delta virus ribozyme (HDVRs) sequence and Xho I restrict enzyme site were engineered at the 3′ end of the cDNA copy of DHAV full-length genome, respectively. The base T at position of 3042 was mutated into base C to generate a BamH I restrict enzyme site which was treated as a genetic marker to distinguish the rescued virus from the parental virus
The fragment cloning 1 and pIR vector was digested with BamH I and Xho I and then ligated by T4 DNA ligase. The ligation products were then transformed into E. coli DH5α competent cells, yielding a recombinant plasmid named pIR-1. By introducing an EcoR V restriction enzyme site into pIR-1 through ligation of cloning 1, a newly recombinant plasmid named pIR-12 was obtained by transforming the ligation of BamH I/ EcoR V-digested products of pIR-1 and fragment cloning 2 into E. coli DH5α competent cells. The fragment cloning 3 was amplified with primer DHAV-Seq-6F/9R, including two EcoR V restriction enzyme sites on both terminuses (lie in position 4517–4522 and 7002–7007 of viral genome). After digestion of EcoR V, the fragment cloning 3 was inserted into vector pIR-12, yielding the recombinant plasmid pIR-123. The base at the position 3042 was mutated from T to C (the mutation causing no amino acid sequence change) by the designed primers to generate a BamH I restrict enzyme site, which was used as a genetic marker to distinguish the rescued virus from the parental virus. Following that, fragment cloning 4 was inserted into pIR-123 after digestion with AscI and BamH I. The DNA-launched infectious clone of DHAV-1 was named as pIR-DHAV-1.
Transfection of BHK-21 cells
When reaching around 70% confluence in a six-well plate, BHK-21 cells were transfected with 4.0 μg of purified plasmid pIR-DHAV-1 per well using 10 μl Lipofectamine 2000 Transfection Reagent (Invitrogen) according to the manufacturer′s protocols. After incubation for 4 h, the cells were washed by phosphate-buffered saline (PBS; 8.1 mM Na2HPO4, 1.5 mM KH2PO4, 140 mM NaC1, 3.0 mM KC1, pH 7.2) for five times and fresh MEM with 2% FBS was added to each well.
Detection of the rescued virus by IFA
At 60 h post transfection (hpt), the rescued viruses were harvested and propagated in BHK-21 cells. Equal amount of pDHAV-1 was propagated in BHK-21 cells as positive control while the same amount of PBS was conducted as the negative control. At 48 hpi, the cells were fixed by a mixture of acetone and formaldehyde (1:1) for 30 min at room temperature. After washed five times with PBS, the cells were incubated with anti-DHAV-1 mAb 4F8 (dilution of 1:1000 with PBS) for 1 h at 37 °C. FITC-labeled goat anti-mouse antibody (dilution of 1:1000 with PBS) was used as secondary-antibody and the cells were incubated at 37 °C for 1 h in the dark. The stained cells were analyzed by fluorescence microscopy (Leica AF6000).
SDS-PAGE and western blotting
To further confirm whether the infectious virus particles could be harvested from DNA-launched infectious system, the transfected product was used to propagate in BHK-21 cells. The supernatants were collected and centrifuged at 3000 rpm for 5 min at 4 °C to remove the floating cells and debris. The BHK-21 cells were manually harvested by a cell scraper, washed for three times with PBS and resuspended with equal amount of PBS. For Western blot analysis of cell culture media, cells were collected as previously described, washed three times with PBS and then resuspended with supernatant. Cell lysates (200 μl) were then added with 50 μl 5× SDS loading buffer and boiled at 100 °C for 10 min and incubated for 5 min on ice. The samples were run on SDS 12%-polyacrylamide gels and transferred onto a polyvinylidene fluoride (PVDF) membrane (ThermoFisher, MA, USA) by standard procedures. Following that, the PVDF membrane was blocked in 5% non-fat milk in Tris-Buffered Saline saline Tween-20 (TBST) (500 ml NaCl, 0.05% Tween 20, 10 mM TRIS-HCl pH 7.5) for 1 h on a horizontal table. Anti-DHAV-1 monoclonal antibody 4F8 (dilution of 1:500) was incubated at 4 °C for 8 h. The membrane was washed four times with TBST and incubated with HRP-conjugated goat anti-mouse antibody (Abcam, MA, USA, dilution of 1:3000) at 4 °C for 4 h. The PVDF membrane was then visualized with hydrogen peroxide and 3, 3′-diaminobenzidine tetrahydrochloride (DAB) (Sigma, USA). The BHK-21 cells infected with pDHAV-1 or with pIR vector were conducted as positive and negative controls, respectively.
Identification of the genetic marker in rescued virus
During the construction of the recombinant plasmid, the base T at position 3042 was replaced by base C to create a genetic marker to distinguish rDHAV-1 from the parental virus pDHAV-1. The transfected products were harvested at 60 hpt and were immediately used for RNA extraction as previously described. Following that, total RNA was used for RT-PCR amplification with primers BamH I-detect-F/R (Table 1) diluted in RNAase free water (TaKaRa, Dalian, China). RT-PCR/PCR was performed utilizing a one-step RNA PCR kit (TaKaRa, Dalian, China). The RT-PCR/PCR condition was 50 °C for 30 min, 94 °C for 2 min, and then 30 cycles of 94 °C for 30 s, 50 °C for 30 s, 72 °C for 1 min, and with a final step of 72 °C for 5 min. During the RNA extraction assay, the DNaseI Digestion kit (OMEGA, GA, USA) was used to remove the residual recombinant plasmid. The 2069 bp fragment including the genetic marker was then digested by restriction enzyme BamH I and verified by electrophoresis in 2% agarose gel.
Comparison of growth characteristics between the parental virus and the rescued virus
To examine the growth characteristics of the rescued virus, BHK-21 cells were respectively infected with pDHAV-1 and rDHAV-1 at 0.1 MOI (multiplicity of infection, with viral copies of 4.71 × 104 and cell numbers of 4.69 × 105), and incubated at 37 °C in 5% CO2. MEM supplemented with 10% FBS was added to each well 2 hpt, and the cells were cultured at 37 °C in 5% CO2. The supernatant (1.0 ml) was collected every 12 h from 12 hpi to 72 hpi, while the cells were scraped from the plate and resuspended with the same amount of PBS. The cell culture samples were collected by scraping the cells with a cell scraper and resuspended with the supernatant. The samples were treated with three freeze-thaw cycles, and the equal amounts of samples were immediately used for RNA extraction. The extracted RNA was then used for qRT-PCR amplification as previously described [33]. With the primers RT-qPCR-F/R and the DHAV-1-Probe (Table 1), a TaqMan real-time RT-PCR assay for quantitative detection of DHAV-1 was conducted in a total volume of 25 μl, containing 12.5 μl 2 × One step RT-qPCR buffer (with ROX), 0.4 μM of forward primer and reverse primer, 0.2 μM of probe, 0.9 μl EnzyMix, 2 μl template RNA. PCR was initiated by 95 °C denaturation for 5 min followed by 40 cycles at 95 °C for 15 s, annealing at 60 °C for 45 s, and fluorescence was measured at every annealing step. Non-template control (NTC) samples were included in each run. The supernatants and cells samples from pDHAV-1 and rDHAV-1 group at each detection point were also conducted with western blotting assay as previously described. A 10-mm glass coverslip was put into the 6-well plate before the equal number of BHK-21 cells were seeded in the 6-well plate. When reaching around 70% confluence, the glass coverslip was taken out from the plate and was immediately used for cell counting with Countess II FL (ThermoFisher, MA, USA).
Comparison of rescue efficiency between RNA- and DNA-launched infectious clones
The RNA-launched infectious clone of DHAV-1 of LY0801 strain (named as pR-DHAV-1) was established previously by introducing a sp6 promoter following with a copy of DHAV-1 genome into pcDNA™3.1/V5-His A vector. The recombinant plasmids of RNA-launched infectious clone were linearized at the 3′ terminus of viral genome with Xho I and then were purified according to the manufacturer′s instruction (OMEGA, Norcross, GA, USA). The purified product was measured with a spectrophotometer (Eppendorf, Germany) to determine the concentration, and then used for in vitro transcription using SP6 RiboMAX™ Express LargeScale RNA Production System (Promega, USA) following the manufacturer′s instruction. RNase-free DNase I (TaKaRa, Dalian, China) was added into in vitro transcribed products and incubated at 37 °C for 15 min to digest residual DNA template, and the in vitro transcribed RNA was then purified with RNeasy kits (QIAGEN). During the construction of the RNA-launched infectious clone, the base A at position 3949 was replaced by C to generate a unique restriction site BamH I to distinguish the rescued virus from parental virus. To identify the rescue efficiency of pR-DHAV-1 and pIR-DHAV-1, 4.0 μg of recombinant plasmid of pIR-DHAV-1 or the in vitro transcribed RNA of 4.0 μg of pR-DHAV-1 were transfected into BHK-21 cells. At 48 hpt, IFA assay was conducted with mAb 4F8 and FITC-labeled goat anti-mouse antibody to measure the difference on rescue efficiency. The experiment was repeated three times. The same amount of pIR empty vector was transfected into BHK-21 cells as the negative control. In order to identify the difference in rescue efficiency between the DNA-launched infectious system and RNA-launched infectious system, cell cultures of the two groups were collected at 48 hpt to conduct the western blotting assay.
To compare the rescue efficiency between the pR-DHAV-1 and pIR-DHAV-1 infectious system on infectious virus level, the cell lysates in the both transfection groups were collected and treated with three freeze-thaw cycles, and then were used to infect BHK-21 cells. Cell lysates (generation 1) were collected at 48 hpi, and 100 μl of cell lysate was used for qRT-PCR measurement while the rest 900 μl was used to infect BHK-21 cells. The rescued virus propagation characteristics of pR-DHAV-1 and pIR-DHAV-1 from generation 1 to 5 were measured by qRT-PCR.
Duckling virulence study of rDHAV-1
Thirty 1-day-old ducklings were divided into three groups and then inoculated intramuscularly with 0.25 ml of 104.0 LD50 of rDHAV-1, 104.0 LD50 of pDHAV-1 of strain LY0801 and aseptic PBS (pH 7.2), respectively. The ducklings were kept under the optimal condition, clinical signs and mortality were observed daily. Ducks received analgesia (carprofen, 5 mg kg−1, i.p.) before the start of surgery. Based on that, ducks were anaesthetized with isoflurane (3.5%) and subjected to execution. Total RNA was extracted from the liver of the dead ducklings for RT-PCR using the primers BamH I-detect-F/R (Table 1). The amplified fragments were digested with restriction enzyme BamH I and then were verified by electrophoresis in 2% agarose gel, and then were sent to a commercial service for sequencing with methods and instrument by Sangon Biotech Co., Ltd., Shanghai, China. The nucleotide sequence and amino acid sequence in both rDHAV-1 group and pDHAV-1 group were analyzed by the Clustal W method (DNA Star LaserGene software, DNAStar Inc. Madison, WI).
The animal experiments were carried out in accordance with the guidelines issued by Shandong Agricultural University Animal Care and Use Committee (SDAUA-2014-014).
Dynamic analysis of viral load
Ducks received analgesia (carprofen, 5 mg kg−1, i.p.) before the start of surgery. Ducks were anaesthetized with isoflurane (3.5%) and maintained under anaesthesia (2–2.5%) throughout the inoculation. Fifty 1-day-old healthy ducklings were injected subcutaneously with 0.25 ml of 104.0 LD50 of rDHAV-1. The heart, liver, kidney, spleen, thymus and bursa of Fabricius (BF) of the 1-day-old ducklings were collected at 1, 6, 12, 18, 24, 48 and 72 h post inoculation. According to the manufacturer′s instruction, virus RNA was isolated from tissue samples using the E.Z.N.A.™ Viral RNA Kit (Omega Bio-Tek, Doraville, USA) and then were measured by qRT-PCR. Non-template control (NTC) samples were included in each run.
The animal experiments were carried out in accordance with the guidelines issued by Shandong Agricultural University Animal Care and Use Committee (SDAUA-2014-014).
Results and discussion
Sequence comparison
A sequence comparison between the complete genomic sequence of the fifth passage of the DHAV-1 LY0801 strain in BHK-21 cell culture and the FJ436047 sequence was performed. Both the 3′ end poly(A) tail of the two viral strains were 21 bases in length. Comparing with FJ436047, there were six base changes after 5 passages: A changed to G at nucleotide position 1289 in VP0 gene, C to G at nucleotide position 2397 in VP1 gene, G to A at nucleotide position 3566 and T to C at nucleotide position 3587 and in 2A3 gene, G to A at nucleotide position 5699 in 3C gene, and A to G at nucleotide position 6839 in 3D gene. These mutations caused no change of the amino acids (Table 2). Comparing with the 5th passage of LY0801 strain, there was no base change in pIR-DHAV-1 or rDHAV-1 except for the mutation (from T to C) at 3042 position.
Table 2.
Summary of sequence differences between LY0801 strain and 5th passage of LY0801 strain
| Base positiona | LY0801 strain | 5th passage of LY0801 | Amino acid change | Location |
|---|---|---|---|---|
| 1289 | A | G | silent | VP0 |
| 2397 | C | G | silent | VP1 |
| 3566 | G | A | silent | 2A3 |
| 3587 | T | C | silent | 2A3 |
| 5699 | G | A | silent | 3C |
| 6839 | A | G | silent | 3D |
aNucleotide position and sequence are based on LY0801 strain (GenBank accession no. FJ436047)
Detection of the rescued virus
The full-length cDNA clone of the 5th passage of LY0801 strain was assembled into the vector pIR by inserting four overlapping fragments into pIR vector using the restriction sites BamH I at nucleotide position 3042, EcoR V at 4519 and 7004, and Asc I and Xho I at both terminuses. The recombinant plasmids were digested with appropriate enzymes and verified by electrophoresis in 2% agarose gel (Fig. 2). The monolayer BHK-21 cells that infected with the transfection product of recombinant plasmid pIR-DHAV-1 was detected by IFA with anti-DHAV-1 monoclonal antibody 4F8. The green fluorescence indicated the viral protein expression in the pIR-DHAV-1 group and positive control group, no specific green fluorescence was observed in the negative control group (Fig. 3). In order to confirm the rescued virus could be harvested from the DNA-launched infectious system, western blot assay was conducted with anti-DHAV-1 monoclonal antibody 4F8. The replication level of viral particles of pDHAV-1 and rDHAV-1 at 48 hpi was determined. Western blot result indicated that rescued virus could be rescued by direct transfection with pIR-DHAV-1 (Fig. 4A).
Fig. 2.
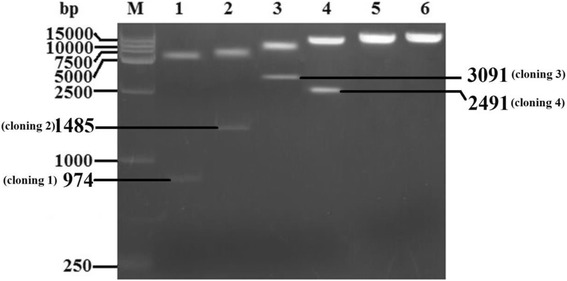
Recombinant plasmids digested with appropriate enzymes during construction. Four fragments that amplified with the four pairs primers 3endRibo-F/R, Cloning 2-F/R, DHAV-Seq-6F/9R and 5HeadRibo-F/R were assembled into pIR vector by one multi-step strategy, respectively. The yield recombinant plasmids were digested with appropriate enzymes and verified by electrophoresis in 2% agarose gel. (M) DNA Marker DL2000; (1) The recombinant plasmid that consisted of pIR vector and cloning 1 was digested with BamH I and Xho I; (2) The recombinant plasmid that consisted of pIR vector and cloning 1 and cloning 2 was digested with BamH I and EcoR V; (3) The recombinant plasmid that consisted of pIR vector and cloning 1 to cloning 3 was digested with EcoR V; (4) The complete DNA-launched infectious clone was digested with Asc I and BamH I; (5) The complete DNA-launched infectious clone digested with BamH I; (6) The complete DNA-launched infectious clone digested with Xho I
Fig. 3.
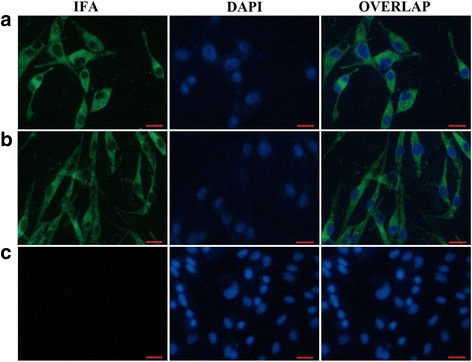
IFA observation of transfected/infected BHK-21 cells. (a) BHK-21 cells were transfected with the recombinant plasmid DNA of pIR-DHAV-1; (b) BHK-21 cells were infected by the parental virus of LY0801 strain; (c) BHK-21 cells were transfected with the plasmid DNA of pIR. After passaging for three times, IFA was conducted by incubation of anti-DHAV-1 mAb 4F8 (dilution of 1:1000 with PBS) for 1 h at 37 °C, followed by incubation in 37 °C for 1 h with FITC-labeled goat anti-mouse antibody. Red bars represent 10 μm
Fig. 4.
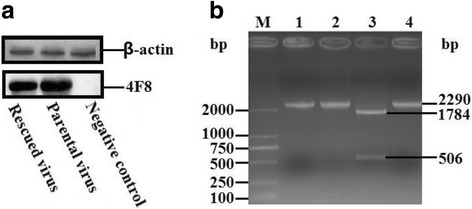
Identification of the rescued virus. (a) Western blotting assay. The BHK-21 cells infected with parental virus and rescued viruses were harvested at 48 hpi. Anti-DHAV-1 monoclonal antibody 4F8 (dilution of 1:500) and HRP-conjugated goat anti-mouse antibody (dilution of 1:3000) were used to conduct the western blot assay. The BHK-21 cells transfected with pIR vector were used as negative control. (b) Identification of the genetic marker in the rescued virus. The BamH I restriction enzyme site was introduced into the recombinant plasmid to create a genetic marker to distinguish the rescued virus from the parental virus (without BamH I enzyme site). Two 2290 bp-fragments derived from parental and rescued virus with primers DHAV-3F and DHAV-4R were digested with BamH I and analyzed on a 2.0% agarose gel. (M) DNA Marker DL2000; 1. Fragment amplified with template of parental RNA; 2. Fragment derived from the parental virus digested with BamH I; 3. Fragment derived from the rescued virus digested with BamH I; 4. Fragment amplified with template of rescued virus RNA
Identification of the genetic marker in rescued virus
In order to distinguish rDHAV-1 from pDHAV-1, nucleotide T at position 3042 was mutated into C to create a restriction enzyme site BamH I as genetic marker (Fig. 1). Fragments including the genetic marker were amplified by RT-PCR from RNA extraction products of the rescued virus and the parental virus with primers BamH I-detect-F and BamH I-detect-R (Table 1). After digested by BamH I, the fragment amplified from the rDHAV-1 was cleaved into a fragment of 1784 bp and a fragment of 506 bp, while the fragment amplified from the pDHAV-1 could not be cleaved into two fragments (Fig. 4B).
rDHAV-1 and pDHAV-1 shared similar growth characteristics
To compare the growth characteristics between rDHAV-1 and pDHAV-1, the growth kinetics of the two viruses were analyzed by infection of BHK-21 cells with the respective virus at 0.1 MOI. From 12 hpi to 72 hpi, the rDHAV-1 showed similar replication and infectivity efficiency with the pDHAV-1 in supernatants (Fig. 5A) and in BHK-21 cells (Fig. 5B). The viral RNA copies peaked at 60 hpt in cells and 48 hpt in supernatants. Western blotting results showed that the viral protein expression level in BHK-21 cells was higher compared to the supernatants group, and the capsid protein levels in pDHAV-1 and rDHAV-1 group were equally similar at each monitoring point (Fig. 5C, D). These data demonstrated that the rDHAV-1 and pDHAV-1 shared similar growth characteristics in both supernatants and BHK-21 cells.
Fig. 5.
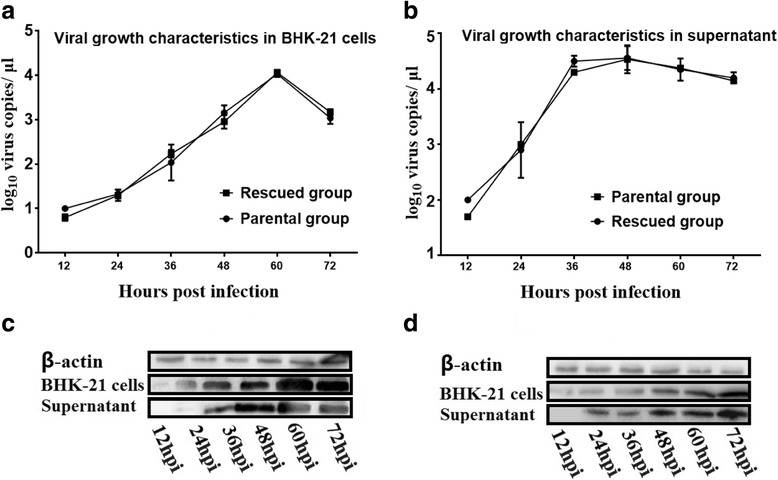
Identification the growth characteristics of rDHAV-1. BHK-21 cells were infected with the parental virus and the rescued virus at 0.1 MOI, respectively. Supernatants and cells were separately collected and used for RNA extraction and qRT-PCR amplification and western blotting. (a) Growth curves of the parental/rescued virus in supernatants; (b) Growth characteristics of the parental/rescued virus in BHK-21 cells. (c) Viral replication levels of the rescued viruses in supernatants and BHK-21 cells. (d) Viral replication levels of the parental viruses in supernatants and BHK-21 cells
Rescue efficiency of RNA- and DNA-launched infectious clone
The rescue efficiency of pR-DHAV-1 and pIR-DHAV-1 was measured by IFA method. The green fluorescence was first observed at 18 hpt in pIR-DHAV-1 group, whereas weak green fluorescence was first observed at 24 hpt in the pR-DHAV-1 group. At 48 hpt, approximately 3–5% of the BHK-21 cells transfected with the pR-DHAV-1 RNA transcripts presented specific green fluorescence, whereas a much higher percentage of FITC-positive cells, approximately 50–60%, were observed when transfected with the pIR-DHAV-1 plasmid DNA (Fig. 6A). Furthermore, we investigated expression levels of viral capsid proteins by western blotting assay. Obviously, the viral capsid protein concentration derived from pIR-DHAV-1 group was much higher than the pR-DHAV-1 group (Fig. 6B), indicating that the DNA-launched infectious system provided more sufficient viral capsid proteins for packaging infectious virions comparing to the RNA-launched system.
Fig. 6.
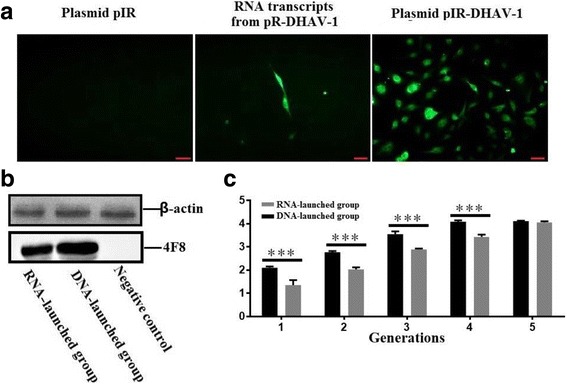
Rescue efficiency of RNA/DNA-launched infectious system. (a) 4.0 μg of the DNA-launched infectious clones or in vitro transcribed product of the same amount of RNA-launched infectious clones were used for BHK-21 transfection. The DNA-launched infectious clone (plasmid pIR-DHAV-1) and the empty vector (plasmid pIR) were transfected directly into BHK-21 cells, while the RNA-launched infectious clone was transcribed in vitro, and then the transcription mixture was transfected into BHK-21 cells. At 48 hpt, IFA observation was conducted after staining with anti-DHAV-1 mAb 4F8 (dilution of 1:1000 with PBS) for 1 h at 37 °C, followed by incubation in 37 °C for 1 h with FITC-labeled goat anti-mouse antibody. Bars represent 10 μm. (b) Western blotting assay. A total of 4.0 μg of recombinant plasmid of DNA-launched infectious clone and in vitro transcribed product from 4.0 μg of RNA-launched infectious clone were transfected into BHK-21 cells to analyze rescue efficiency between DNA-launched system and RNA-launched system. Cell culture was collected at 48 hpt and run on SDS 12%-polyacrylamide gels. Anti-DHAV-1 monoclonal antibody 4F8 (dilution of 1:500) and HRP-conjugated goat anti-mouse antibody (dilution of 1:3000) were used to conduct the western blot assay. BHK-21 cells that transfected with pIR vector were conducted as negative control. (c) BHK-21 cells were infected with transfected product of 4.0 μg of the DNA-launched infectious clones or in vitro transcribed product of the same amount of RNA-launched infectious clones, cell lysates were collected at 48 hpi, and were immediately used for RNA extraction and qRT-PCR measurement. Viral RNA copies were calculated with formula X = 6.7 × 10(40.812-y)/3.285. “X” is a standard of viral copies while “y” is a standard of values derived from one step real-time PCR. Bars represent means and standard of three individual repeats. “***” represents p < 0.001
To compare the rescue efficiency between the RNA-launched infectious system and DNA-launched infectious system, the rescued viral particles from both groups were used to infect BHK-21 cells, and the viral growth characteristics in both groups were measured with qRT-PCR. The results showed that the virus RNA copy number in pIR-DHAV-1 group was constantly higher compared to the pR-DHAV-1 group from generation 1 to generation 4 (Fig. 6C), which demonstrated the higher rescue efficiency of the DNA-launched infectious system than the RNA-launched infectious system.
Virulence of rDHAV-1 and pDHAV-1
The virulence of the rDHAV-1 and pDHAV-1 were tested in 1-day-old ducklings. By identification of the specific genetic marker in rDHAV-1, the ducklings were verified to be infected with the rDHAV-1 or pDHAV-1 and there was no cross-infection with each other (Fig. 7A). A similar pattern occurred in the survival curves of the ducklings inoculated with the same dose of the two viruses (Fig. 7B). At 7 days post inoculation, all the ten ducklings died in the pDHAV-1 group, while nine of the ten ducklings died in the rDHAV-1 group. The nucleotide sequence alignment results showed that all the amplified fragments contain the mutation at the position of 3042 in the rDHAV-1 group, yet not in the pDHAV-1 group (Fig. 7C). The amino acid sequence alignment results indicated that the silent mutation has no impact on viral protein synthesis (Fig. 7D).
Fig. 7.
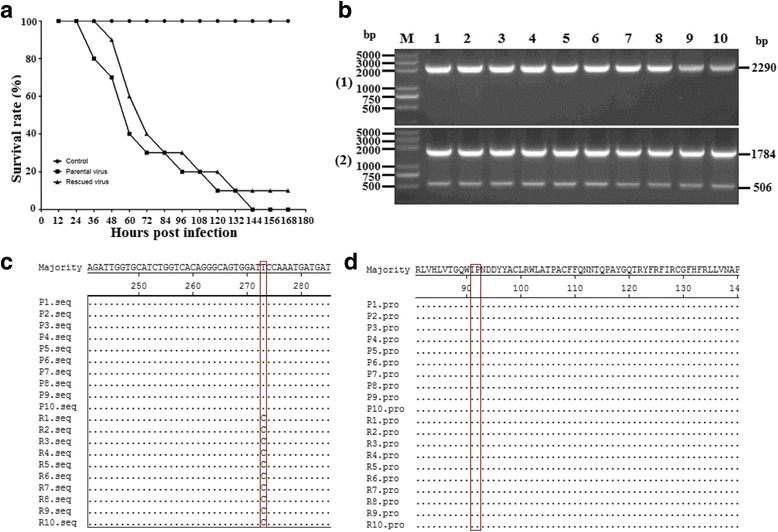
(a) Survival curves of 1-day ducklings after inoculation of 0.25 ml of the parental virus (104.0 TCID50) or the rescued virus (104.0 TCID50). All ten ducklings in control group live in healthy condition. (b) Genetic marker detection of the parental/rescued virus group. Total RNA was extracted from liver tissue of dead ducklings of the rescued virus group and the parental virus group, and then immediately used for transcription using the primers BamH I-detect-F/R. The amplified fragments were then digested with restriction enzyme BamH I and verified by electrophoresis in 2% agarose gel. (M) DNA Marker DL2000; (1) Fragments were amplified with the template RNAs of the parental virus group and then digested with BamH I; (2) Fragments were amplified with the template RNAs of the rescued virus group and then digested with BamH I. Nucleotide sequence alignment (c) and amino acid sequence alignment (d) were also conducted by the Clustal W method (DNA Star LaserGene software, DNAStar Inc. Madison, WI) to investigate genetic marker in both groups
Dynamic analysis of viral load
At 1 hpi, the viral RNA of rDHAV-1 was exclusively detected in the liver, reaching the level of 103.0 ± 0.0 copies/g (Fig. 8A). The rDHAV-1 was first detected in the heart, spleen and thymus at 2 hpi, while the viral RNA was detectable from 6 hpi to 96 hpi in all the collected tissues (Fig. 8). The viral loads increased sharply and reached the peak at 36 hpi in liver, kidney, BF and thymus, whereas the viral loads in spleen and heart reached the peak at 24 hpi and 48 hpi, respectively. From 1 hpi to 48 hpi, the viral RNA copies of rDHAV-1 in liver were the highest among the six tissues (with an exception of thymus at 6 hpi), while the viral RNA copy numbers in heart and kidney were alternately the lowest. Interestingly, the detection result at 96 hpi showed that the viral RNA copies of rDHAV-1 in thymus still remain a high viral load level of 106.06 ± 0.1 copies/g (Fig. 8). The similar curve pattern in the pDHAV-1 group indicated us that the genetic marker causes no impact on viral replication and tissue tropism.
Fig. 8.

Dynamic analysis of viral load. Same weight of heart (a), liver (b), kidney (c), spleen (d), thymus (e) and bursa of Fabricius (f) of the 1-day-old ducklings were collected at 1, 6, 12, 18, 24, 48 and 72 hpi, and were immediately used for RNA extraction and qRT-PCR measurement as previously described. Viral RNA copies were calculated with formula X = 6.7 × 10(40.812-y)/3.285. “X” is a standard of viral copies while “y” is a standard of values derived from one step real-time PCR. Bars represent means and standard of three individual repeats
Discussion
Reverse genetics system is particularly useful for RNA viruses since RNA genomes are difficult to manipulate directly. Although the previous research has reported about the establishment of the RNA-launched infectious clone of DHAV-1 [23] and DHAV-3 [24], the in vitro transcription will raise the operation difficulty and experiment cost. The DNA-launched infectious system possessed self-cleaving ribozyme elements at both termini of the viral genomic cDNA, which was expected to retain the authentic terminal nucleotide sequences of the viral genome [28]. The 3′ product of the HDV ribozyme was shown to serve as the substrate for polyadenylation [34]. Here, we established an improved DNA-launched infectious system based on the DHAV-1 LY0801 strain. The viral genome of DHAV-1 was placed under the control of a CMV promoter, allowing the conventional plasmid DNA transfection and generation of homogenous RNA transcripts in vivo by the cellular RNA polymerase II, which could reduce labor and experimental cost. Compared to the RNA-launched infectious system, the DNA-launched infectious system of DHAV-1 improved the viral rescue efficiency approximately 10 to 20 folds (Fig. 6).
The rDHAV-1 and pDHAV-1 shared similar biological characteristics in BHK-21 cells in terms of replication efficiency, the generation of CPE (cytopathic effect), as well as infectivity (Fig. 5). Meanwhile, a similar pattern also occurred in the survival curves of the ducklings inoculated with the rDHAV-1 or pDHAV-1 (Fig. 7A). In order to distinguish the rescued virus from parental virus, the genetic marker was engineered into pIR-DHAV-1. The virus copy number peaked at 48 and 60 hpi in supernatants and BHK-21 cells, respectively. The virus copy number was subsequently followed by a decline, which was consistent with the observation of parental group. Western blot results also confirmed the similar growth characteristics between the rescued virus and parental virus (Fig. 5). The survival curve comparison between the rDHAV-1 and pDHAV-1 also showed almost identical infectivity characteristics. To sum up, the rescued recombinant virus was capable of infecting susceptible host cells and exhibited almost identical replication and infectivity efficiency as 5th passage of LY0801 strain. Based on that, we then investigated viral replication features of rDHAV-1 in young ducklings. In the previous study, the viral RNA could be detected in all the different tissues (heart, liver, kidney, spleen, thymus and BF) of the duckling inoculated with the rDHAV-1, which was consistent with the naturally infected ducks by DHAV-1 [33]. In our experiment, the viral RNA of rDHAV-1 was firstly detected in liver with viral load of 103.0 ± 0.0 copies/g at 1 hpi, whereas the rDHAV-1 was detected in the other five tissues at 2 hpi or 6 hpi (Table 2), which showed that liver might be the most sensitive tissue during the early infection of DHAV-1 via subcutaneously. With an exception of thymus at 6 hpi, the liver tissue exhibited the highest viral load in the six selected tissues from 1hpi to 48 hpi, which could partly explain the severe liver damage induced by the infection of DHAV-1.
Conclusions
We have described an improved DNA-launched reverse genetics system for DHAV-1 with high rescue efficiency, which can be used to save human and financial resources. The rescued virus and parental virus shared similar biological properties and pathogenicity. The DNA-launched reverse genetics system allows the rapid introduction of mutations into the viral genome, which will greatly facilitate the study of the structural and functional relationship of DHAV-1 genes, as well as the development of DHAV-1 vaccine based on the genetically engineered and attenuated mutant clones.
Acknowledgements
Not applicable.
Funding
This study was funded by grants from National Key R&D Program (2016YFD0500800), Funds of Shandong “Double Tops” Program, China (SYL2017YSTD11), Shandong Modern Agricultural Technology & Industry System, China (SDAIT-11-15) and A Project of Shandong Province Agriculture Major Applied Technique Innovation, China.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors′ contributions
JC and SJ designed the experiment and drafted the manuscript. JC, RZ, SL, PL and JL performed the molecular-related experiments. RZ, SL and YW participated in the animal experiments. SL, YW and ZX participated in the design of the experiment, the discussion and helped to draft the manuscript. All authors read and approved the final manuscript.
Abbreviation
- BHK
Baby Hamster Syrian Kidney
- CPE
Cytopathic effect
- DAB
Diaminobenzidine tetrahydrochloride
- DHAV
Duck hepatitis A virus
- DVH
Duck virus hepatitis
- EGFP
Enhanced Green Fluorescent Protein
- FBS
Fetal Bovine Serum
- FITC
fluorescein isothiocyanate
- hpi
hours post infection
- hpt
hours post transfection
- HRP
Horseradish Peroxidase
- IFA
Indirect Immunofluorescence
- LD50
Lethal dose 50%
- MEM
Minimum essential medium
- MOI
Multiplicity of infection
- ORF
Open reading frame
- PRRSV
Porcine reproductive and respiratory syndrome virus
- PVDF
Polyvinylidene fluoride
- qRT-PCR
Quantitative real time polymerase chain reaction
- RACE
rapid amplification of cDNA ends
- RT-PCR
reverse transcription polymerase Chain Reaction
- SDS
Sodium dodecyl sulfate
- TBST
Tris-Buffered Saline saline Tween-20
- UTR
Untranslated regions
Ethics approval
This research was conducted in strict accordance with the recommendations in the Guide for the Institutional Animal Care and Use Commission (IACUC). The animal experiments were carried out in accordance with the guidelines issued by Shandong Agricultural University Animal Care and Use Committee (SDAUA-2014-014). All animal experiments were performed under anesthesia, and every effort was made to minimize suffering.
Consent for publication
Not applicable.
Competing interests
The author(s) declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Junhao Chen, Email: junhao-chenyy@163.com.
Ruihua Zhang, Email: ruirui041127@126.com.
Shaoli Lin, Email: nightingal19903@sina.com.
Pengfei Li, Email: 605118124@qq.com.
Jingjing Lan, Email: 1297563208@qq.com.
Zhijing Xie, Email: xiezhj@sdau.edu.cn.
Yu Wang, Email: wangyu1974-1@163.com.
Shijin Jiang, Phone: +86-538-8245799, Email: sjjiang@sdau.edu.cn.
References
- 1.Levine PP, Fabricant JA. Hitherto-undescribed virus disease of ducks in North America. Cornell Vet. 1950;40:71–86. [Google Scholar]
- 2.Toth TE. Studies of an agent causing mortality among ducklings immune to duck virus hepatitis. Avian Dis. 1969;13:834–846. doi: 10.2307/1588590. [DOI] [PubMed] [Google Scholar]
- 3.Haider SA, Calnek BW. Vitro isolation, propagation, and characterization of duck hepatitis virus type III. Avian Dis. 1979;23:715–729. doi: 10.2307/1589748. [DOI] [PubMed] [Google Scholar]
- 4.Woolcock PR. Duck hepatitis. In: Salf YM, Barnes HJ, Glisson JR, Fadly AM, McDougald LR, Swayne DE (Eds) Diseases of poultry, 11th ed, Iowa state press, Ames, IA, 2003;pp. 343–354.
- 5.Bosch A, Guix S, Krishna NK, Méndez E, Monroe SS, Pantin-Jackwood M, Schultz-Cherry S. Astroviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus taxonomy. Classification and nomenclature of viruses: ninth report of the international committee on the taxonomy of viruses. Elsevier. London, UK: Academic Press; 2011. pp. 953–959. [Google Scholar]
- 6.Asplin FD. Duck hepatitis: vaccination against two serological types. Vet Rec. 1965;77:1529–1530. [PubMed] [Google Scholar]
- 7.Knowles NJ, Hovi T, Hyypiä T, King AMQ, Lindberg AM, Pallansch MA, Palmenberg AC, Simmonds P, Skern T, Stanway G, Yamashita T, Zell R. Picornaviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus taxonomy. Classification and nomenclature of viruses: ninth report of the international committee on taxonomy of viruses. San Diego, CA, USA: Elsevier Academic Press; 2012. pp. 855–880. [Google Scholar]
- 8.Kim MC, Kwon YK, Joh SJ, Lindberg AM, Kwon JH, Kim SJ. Molecular analysis of duck hepatitis virus type I reveals a novel lineage close to the genus Parechovirus in the family Picornaviridae. J Gen Virol. 2006;87:3307–3316. doi: 10.1099/vir.0.81804-0. [DOI] [PubMed] [Google Scholar]
- 9.Gao J, Chen J, Si X, Xie Z, Zhu Y, Zhang X, Wang S, Jiang S. Genetic variation of the VP1 gene of the virulent duck hepatitis a virus type 1 (DHAV-1) isolates in Shandong province of China. Virol Sin. 2012;27:248–253. doi: 10.1007/s12250-012-3255-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tseng CH, Tsai HJ. Molecular characterization of a new serotype of duck hepatitis virus. Virus Res. 2007;126:19–31. doi: 10.1016/j.virusres.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Kim MC, Kwon YK, Joh SJ, Kim SJ, Tolf C, Kim JH, Lindberg AM, Kwon JH. Recent Korean isolates of duck hepatitis virus reveal the presence of a new geno-and serotype when compared to duck hepatitis virus type 1 type strains. Arch Virol. 2007;152:2059–2072. doi: 10.1007/s00705-007-1023-0. [DOI] [PubMed] [Google Scholar]
- 12.Xu Q, Zhang R, Chen L, Yang L, Li J, Dou P, Wang H, Xie Z, Wang Y, Jiang S. Complete genome sequence of a duck hepatitis a virus type 3 identified in eastern China. J Virol. 2012;86:13848. doi: 10.1128/JVI.02651-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim MC, Kwon YK, Joh SJ, Kim SJ, Tolf C, Kim JH, Sung HW, Lindberg AM, Kwon JH. Recent Korean isolates of duck hepatitis virus reveal the presence of a new genoand serotype when compared to duck hepatitis virus type 1 type strains. Arch Virol. 2007;152:2059–2072. doi: 10.1007/s00705-007-1023-0. [DOI] [PubMed] [Google Scholar]
- 14.Todd D, Smyth VJ, Ball NW, Donnelly BM, Wylie M, Knowles NJ, Adair BM. Identification of chicken enterovirus-like viruses duck hepatitis virus type 2 and duck hepatitis virus type 3 as astroviruses. Avian Pathol. 2009;38:21–30. doi: 10.1080/03079450802632056. [DOI] [PubMed] [Google Scholar]
- 15.Pan M, Yang X, Zhou L, Ge X, Guo X, Liu J, Zhang D, Yang H, Duck Hepatitis A. Virus possesses a distinct type IV internal ribosome entry site element of picornavirus. J Virol. 2012;86:1129–1144. doi: 10.1128/JVI.00306-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding C, Zhang D. Molecular analysis of duck hepatitis virus type 1. Virology. 2007;361:9–17. doi: 10.1016/j.virol.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Usme-Ciro JA, Lopera JA, Enjuanes L, Almazán F, Gallego-Gomez JC. Development of a novel DNA-launched dengue virus type 2 infectious clone assembled in a bacterial artificial chromosome. Virus Res. 2014;180:12–22. doi: 10.1016/j.virusres.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahlquist P, Janda M. cDNA cloning and in vitro transcription of the complete brome mosaic virus genome. Mol Cell Biol. 1984;4:2876–2882. doi: 10.1128/MCB.4.12.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bridgen A, Elliott RM. Rescue of a segmented negative-strand RNA virus entirely from cloned complementary DNAs. Proc Natl Acad Sci U S A. 1996;93:15400–15404. doi: 10.1073/pnas.93.26.15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyer JC, Haenni AL. Infectious transcripts and cDNA clones of RNA viruses. Virology. 1994;198:415–426. doi: 10.1006/viro.1994.1053. [DOI] [PubMed] [Google Scholar]
- 21.Huang YW, Li L, Yu L. The reverse genetics systems for human and animal RNA viruses. Sheng Wu Gong Cheng Xue Bao. 2004;20:311–318. [PubMed] [Google Scholar]
- 22.Neumann G, Whitt MA, Kawaoka YA. Decade after the generation of a negative-sense RNA virus from cloned cDNA-what have we learned? J Gen Virol. 2002;83:2635–2662. doi: 10.1099/0022-1317-83-11-2635. [DOI] [PubMed] [Google Scholar]
- 23.Yun T, Ni Z, Liu GQ, Yu B, Chen L, Huang JG, Zhang YM, Chen JP. Generation of infectious and pathogenic duck hepatitis virus type 1 from cloned full-length cDNA. Virus Res. 2010;147:159–165. doi: 10.1016/j.virusres.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 24.Pan M, Yang X, Du J, Zhou L, Ge X, Guo X, Liu J, Zhang D, Yang H. Recovery of duck hepatitis a virus 3 from a stable full-length infectious cDNA clone. Virus Res. 2011;160:439–443. doi: 10.1016/j.virusres.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Almazán F, González JM, Pénzes Z, Izeta A, Calvo E, Plana-Durán J, Enjuanes L. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci U S A. 2000;97:5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez JM, Penzes Z, Almazán F, Calvo E, Enjuanes L. Stabilization of a full-length infectious cDNA clone of transmissible gastroenteritis coronavirus by insertion of an intron. J Virol. 2002;76:4655–4661. doi: 10.1128/JVI.76.9.4655-4661.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishin VP, Cominelli F, Yamshchikov VFA. minimal' approach in design of flavivirus infectious DNA. Virus Res. 2001;81:113–123. doi: 10.1016/S0168-1702(01)00371-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang YW, Fang Y, Meng XJ. Identification and characterization of a porcine monocytic cell line supporting porcine reproductive and respiratory syndrome virus (PRRSV) replication and progeny virion production by using an improved DNA-launched PRRSV reverse genetics system. Virus Res. 2009;145:1–8. doi: 10.1016/j.virusres.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Pley HW, Lindes DS, DeLuca-Flaherty C, McKay DB. Crystals of a hammerhead ribozyme. J Biol Chem. 1993;268:19656–19658. [PubMed] [Google Scholar]
- 30.Ke A, Zhou K, Ding F, Cate JH, Doudna JAA. Conformational switch controls hepatitis delta virus ribozyme catalysis. Nature. 2004;429:201–205. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- 31.Lehman N, Joyce GF. Evolution in vitro of an RNA enzyme with altered metal dependence. Nature. 1993;361:182–185. doi: 10.1038/361182a0. [DOI] [PubMed] [Google Scholar]
- 32.Zhang R, Zhou G, Xin Y, Chen J, Lin S, Tian Y, Xie Z, Jiang S. Identification of a conserved neutralizing linear B-cell epitope in the VP1 proteins of duck hepatitis a virus type 1 and 3. Vet Microbiol. 2015;180:196–204. doi: 10.1016/j.vetmic.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Lin SL, Cong RC, Zhang RH, Chen JH, Xia LL, Xie ZJ, Wang Y, Zhu YL, Jiang SJ. Circulation and in vivo distribution of duck hepatitis a virus types 1 and 3 in infected ducklings. Arch Virol. 2015;161(2):405–416. doi: 10.1007/s00705-015-2648-z. [DOI] [PubMed] [Google Scholar]
- 34.Düvel K, Pries R, Braus GH. Polyadenylation of rRNA and tRNA-based yeast transcripts cleaved by internal ribozyme activity. Curr Genet. 2003;43(4):255–262. doi: 10.1007/s00294-003-0401-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.