

Abstract
The human selenoprotein family contains 25 members that share the common feature of containing the amino acid, selenocysteine (Sec). Seven selenoproteins are localized to the endoplasmic reticulum (ER) and exhibit different structural features contributing to a range of cellular functions. Some of these functions are either directly or indirectly related to calcium (Ca2+) flux or homeostasis. The presence of the unique Sec residue within these proteins allows some to exert oxidoreductase activity, while the function of the Sec in other ER selenoproteins remains unclear. Some functional insight has been achieved by identifying domains within the ER selenoproteins or through the identification of binding partners. For example, selenoproteins K and N (SELENOK AND SELENON) have been characterized through interactions detected with the inositol 1,4,5-triphosphate receptors (IP3Rs) and the SERCA2b pump, respectively. Others have been linked to chaperone functions related to ER stress or Ca2+ homeostasis. This review summarizes the details gathered to date regarding the ER-resident selenoproteins and their effect on Ca2+ regulated pathways and outcomes in cells.
Graphical abstract
Introduction
Selenium is an essential micronutrient that plays an important role in several aspects of human health. Deficient intake of selenium can lead to a wide variety of health disorders ranging from impaired gestational development, poor immunity, cognitive and neurodegenerative problems, and endocrine imbalances [1]. The biological effects of selenium are exerted mainly through the actions of selenoproteins, which contain this element in the form of the 21st amino acid selenocysteine (Sec) [2]. There are 25 selenoproteins in humans [3] that exhibit a wide range of biological functions with nearly half of the members still uncharacterized in terms of their function. Animal models have demonstrated the essential role of selenoproteins, as mice deficient in tRNASec, the selenocysteine-specific tRNA required for selenoprotein synthesis, do not survive early embryogenesis [4]. The most studied selenoproteins include those that function as enzymes. The significance of these enzymes and other members of the selenoprotein family to human health have been demonstrated in patients with errors in the synthesis process or in the selenoproteins themselves [5]. A number of studies in humans have found that defects in expression or function of selenoproteins can impinge upon a variety of physiological processes involving the muscular and skeletal systems, respiratory system, neurological system, and endocrine system. Perhaps the best example of how selenoproteins affect multiple systems is the syndrome in which there is defective production or function of one protein involved in selenoprotein synthesis, the SECIS binding protein 2 (SECISBP2). Mutations in SECISBP2 give rise to a spectrum of health problems depending on the patient and/or type of mutation present [6].
There are 25 genes encoding selenoproteins in humans that were revealed in a 2003 hallmark study by the Gladyshev laboratory following the completion of the human genome project [3]. While several new members of the family were identified in that study, some selenoproteins had already been characterized as redox regulating enzymes containing the Sec residue located within the catalytic site. The defining characteristic of selenoproteins is the presence of the 21st amino acid Sec, which itself has special properties. The Sec residue is isosteric to cysteine (Cys) with selenium in place of sulfur. This results in a highly reactive site in the enzyme that is better at nucleophilic exchange reactions compared to Cys [7]. The lower pKa of the selenol side chain of Sec compared to thiol of Cys means that Sec is mostly deprotonated at physiological pH [8], and this further enhances its oxidoreductase activity.
Subsequent to the complete identification of the selenoprotein family members in 2003, the nomenclature was recently revised [9]. Selenoprotein enzymes with known functions are designated according to these functions. These include the glutathione peroxidases (GPXs), the thioredoxin reductases (TXNRDs), iodothyronine deiodinases (DIOs), methionine-R-sulfoxide reductase 1 (MSRB2) and selenophosphate synthetase 2 (SEPHS2). Those selenoproteins without a clearly demonstrated enzymatic function are named with the root symbol SELENO followed by a letter. A list of selenoproteins with the characteristics of each is shown in Table 1. As indicated in this table, several selenoproteins have been associated with a cellular function involving Ca2+, either directly or indirectly, and the remainder of this review will elaborate on ER-resident selenproteins and what is known regarding their Ca2+ relevant functions.
Table 1.
Summary of Selenoprotein Functions
| Selenoprotein | Abbreviation(s) | Function |
|---|---|---|
| Cytosolic glutathione peroxidase | GPX1 | Reduces cellular H2O2. |
| Gastrointestinal glutathione peroxidase | GPX2 | Reduces peroxide in gut. |
| Plasma glutathione peroxidase | GPX3 | Reduces peroxide in blood. |
| Phosholipid hydroperoxide glutathione peroxidase | GPX4 | Reduces phospholipid peroxide. |
| Olfactory glutathione peroxidase | GPX6 | Importance unknown. |
| Thioredoxin reductase Type I | TRXRD1, TR1 | Localized to cytoplasm and nucleus; regenerates reduced thioredoxin. |
| Thioredoxin reductase Type II | TRXRD2, TR3 | Localized to mitochondria; regenerates reduced thioredoxin. |
| Thioredoxin reductase Type III | TRXRD3, TR2, TGR | Testes-specific expression; regenerates reduced thioredoxin. |
| Iodothyronine Deiodinase Type I | DIO1 | Important for systemic active thyroid hormone levels. |
| Iodothyronine Deiodinase Type II | DIO2 | ER enzyme important for local active thyroid hormone levels. |
| Iodothyronine Deiodinase Type III | DIO3 | Inactivates thyroid hormone. |
| Methionine sulfoxide reductase B1 | MSRB1, SelR | Reduces sulfoxidated methionines on proteins. |
| Selenoprotein F | SELENOF, Sep15 | ER thiol-disulfide oxidoreductase possibly involved in protein folding. |
| Selenoprotein H | SELENOH, SelH | Nuclear localization; involved in redox sensing and transcription. |
| Selenoprotein I | SELENOI, SelI, EPT1 | Possibly involved in phospholipid biosynthesis. |
| Selenoprotein K | SELENOK, SelK | Transmembrane protein localized to ER and involved in calcium flux in immune cells and ER associated degradation in cell lines. |
| Selenoprotein M | SELENOM, SelM | Thioredoxin-like ER-resident protein that may be involved in regulation of body weight and energy metabolism. |
| Selenoprotein N | SELENON, SelN, SEPN1, SepN | Transmembrane protein localized to ER; mutations lead to multiminicore disease and other myopathies. |
| Selenoprotein O | SELENOO, SelO | Mitochondrial protein that contains a C-X-X-U motif suggestive of redox function. |
| Selenoprotein P | SELENOP, SelP, Sepp1 | Secreted into plasma for selenium transport to tissues. |
| Selenoprotein S | SELENOS, SelS, SEPS1, SELENOS, VIMP | Transmembrane protein found in ER involved in ER associated degradation. |
| Selenoprotein T | SELENOT, SelT | ER protein involved in calcium mobilization, unclear exactly how. |
| Selenoprotein V | SELENOV, SelV | Testes-specific expression. |
| Selenoprotein W | SELENOW, SelW, SEPW1 | Putative antioxidant role, perhaps important in muscle growth. |
| Selenophosphate synthetase | SPS2 | Involved in synthesis of all selenoproteins, including itself. |
Calcium Homeostasis and ER Stress
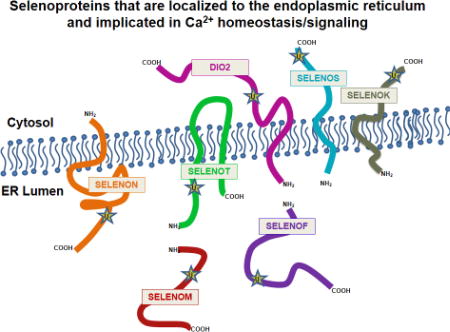
Within the cell, cytosolic levels of free Ca2+ are kept low under baseline conditions by the synchronized action of a variety of channels, pumps, exchangers, and Ca2+-binding proteins. The endoplasmic reticulum (ER) represents the primary storage site for intracellular Ca2+, with concentrations reaching high micromolar levels in this organelle, whereas resting cytosolic concentrations typically range from 50 to 200 nM [10]. Seven of the twenty-five members of the selenoprotein family are localized to the ER, including SELENOF, SELENOK, SELENOM, SELENON, SELENOS, SELENOT, and the type-2 iodothyronine deiodinase [11]. Structural features of these ER selenoproteins are shown in Figure 1. Cytosolic Ca2+ is maintained at nanomolar levels by plasma membrane Ca2+ ATPases and SR/ER Ca2+ ATPase b (SERCAb) that pump Ca2+ into the extracellular space and the ER lumen, respectively. In response to various stimuli, Ca2+ is released from the ER by means of two classes of channels, the inositol 1,4,5-trisphosphate receptors (IP3Rs) and the ryanodine receptors (RyRs). For example, immune cells stimulated through cell surface receptors generate inositol 1,4,5-trisphosphate (IP3) that binds to the IP3Rs in the ER membrane to release Ca2+ stored in the lumen of this organelle. Loss of Ca2+ from ER stores is sensed by stromal interaction molecule 1 (STIM1). This alters the structure of STIM1 and causes its oligomerization and it then engages the Ca2+ channel protein Orai1 or transient receptor potential canonical 1 (TRPC1) in the plasma membrane to open and allow influx of millimolar levels of Ca2+ [12]. This process is referred to as store operated Ca2+ entry (SOCE) and is crucial for cell proliferation, migration and other functions. It is important to note that SOCE is emerging as a potential target for cancer therapeutics [13]. When different cell types transform and progress to metastatic tumor cells, Ca2+ homeostasis and signaling is altered in a process referred to as Ca2+ remodeling [14]. Certain selenoproteins contribute to SOCE and some may also be involved in Ca2+ remodeling in cancer, and these issues that are discussed in greater detail below.
Figure 1.

Domain organization of ER selenoproteins. This diagram illustrates the relative position of Sec residues (red), signal peptides (green), transmembrane domains (yellow), thioredoxin-like motifs (pink), EF hand motifs (orange), and ER retention signals (white triangles). Established binding/interaction sites are denoted by three-quarter circle symbols.
In addition to its integral role in Ca2+ signaling, the ER is also critically important for the folding, assembly, and post-translational modification of nascent proteins destined for secretion or residence in the cell membrane. Newly synthesized proteins destined for modification in the ER are marked by an N-terminal signal peptide that tags them for insertion into the Sec61 translocon spanning the ER membrane. Within the ER, a host of chaperones and enzymes act to ensure that proteins mature and reach their optimal conformation. These include glucose-regulated protein (GRP) chaperones, glucosidases, transferases, protein disulfide isomerases (PDIs), and the Ca2+-regulated chaperones, calnexin and calreticulin. ER homeostasis can be disrupted by a variety of insults, such as the accumulation of misfolded proteins [15], elevated levels of reactive oxygen species (ROS) [16, 17], and abnormalities in Ca2+ signaling [18, 19], which are all generically referred to as ER stress. These disturbances trigger the unfolded-protein response (UPR), a protective counter-measure that acts to reestablish balance and promote survival by increasing the production of chaperones involved in protein folding, inhibiting global translation, and eliminating chronically misfolded proteins. The UPR is conveyed by three ER stress sensors: PKR-like endoplasmic reticulum kinase (PERK), inositol-requiring kinase 1-alpha (IRE1α), and activation of transcription factor 6 (ATF6) that are negatively regulated by the chaperone GRP78 under baseline conditions [15, 20]. Increasing levels of misfolded proteins causes GRP78 to dissociate from these sensors, resulting in activation of the UPR. Proteins that fail to properly fold are eliminated by means of ER-associated degradation (ERAD), a multistep process in which misfolded proteins are selected, retrotranslocated across the ER membrane, ubiquitinated, and finally degraded by cytosolic proteasomes. In cases of severe and/or sustained ER stress where the UPR fails to restore homeostasis, apoptotic cell signaling pathways are activated [21]. In particular, sustained UPR activation of Jun amino-terminal kinase (JNK), a downstream mediator of the UPR, has been demonstrated to be pro-apoptotic in a wide variety of cell types and contexts [22–24]. Furthermore, severe ER stress promotes apoptosis by increasing ER-to-mitochondria Ca2+ transfer at mitochondria-associated membranes, leading to disturbances in the mitochondrial membrane potential and release of cytochrome c [25]. Of clinical importance, chronic ER stress is implicated in the pathogenesis of many chronic diseases, including type 2 diabetes [26, 27], cardiovascular disease [28] and multiple neurodegenerative disorders [29].
There is substantial evidence indicating that Ca2+ homeostasis, protein folding, and oxidative stress are extensively intertwined. Redox imbalance and ER stress typically increase cytosolic Ca2+ levels and diminish ER Ca2+ stores by a variety of mechanisms. For example, in addition to its interactions with the aforementioned ER stress sensors, GRP78 also binds to the Sec61 translocon and prevents Ca2+ leak from the ER to cytosol under unstressed condtions [30, 31]. Under conditions of ER stress, GRP78 dissociates from Sec61 and increased ER Ca2+ leak ensues. Similarly, oxidation of cysteine residues on RyRs promotes ER Ca2+ leakage [32, 33], whereas ROS-dependent thiol modification of IP3Rs increases ligand affinity and thereby promotes Ca2+ efflux [34, 35]. In particular, studies in cell culture have reported that selenium deprivation augments IP3 triggered ER Ca2+ release [36]. Furthermore, the redox environment of the ER is more oxidizing than the surrounding cytosol [37, 38], and this appears to be influenced, at least in part, by ER Ca2+ levels [39]. When ER Ca2+ stores become depleted, the redox state in this organelle becomes more reducing and modulates redox-sensitive proteins via disruption of intramolecular disulfide bonds. In particular, the SERCA2b pump contains a pair of cysteine residues in the fourth lumen loop that forms a disulfide bond when ER Ca2+ levels are high, which serves to inactivate the pump [40]. When ER Ca2+ levels decline, this disulfide bond is reduced and the pump becomes fully active.
Regulation of Ca2+ signaling by ER-resident selenoproteins
Various ER-resident selenoproteins have been shown to modulate Ca2+ flux into and out of the ER lumen, promote protein folding, and respond to ER luminal redox status. The following section will review what is currently known regarding these individual selenoproteins and how they impact Ca2+-related processes such as proliferation, survival, and apoptosis. The structural and topological features of the seven ER-resident selenoproteins suggest diversity even within this subgroup (Figure 1). How the domains and topologies may be related to potential functions is discussed in detail below.
Selenoprotein F (SELENOF, previously Sep15)
SELENOF, formerly known as SEP15, is a 15 kDa selenoprotein that was first identified in 1998 by Gladyshev and colleagues in a human T-cell line [41]. It shares a common evolutionary origin with SELENOM, and together they comprise a distinct clade within the thioredoxin superfamily. The primary sequence of SELENOF contains a cleavable N-terminal signal peptide, a thioredoxin-like fold, and a C-x-U redox motif. It also possesses a distinct cysteine-rich region in its N-terminus that is required for interaction with its binding partner, UDP-glucose:glycoprotein glucosyltransferase (UGGT) [42, 43]. Due to the fact that its sequence does not contain an ER retention signal, the localization of SELENOF to the ER is thought to be dependent upon UGGT binding. Within the ER lumen, SELENOF is believed to mediate disulfide bond formation on a distinct set of glycoproteins that serve as UGGT substrates. Studies performed on Drosophila SELENOF determined that the equilibrium redox potential was −225 mV, a value that falls between the established redox potentials of protein disulfide isomerase (−175 mV) and cytosolic thioredoxin (−270 mV) [44].
Evidence to date has largely implicated SELENOF in protein folding due to its documented interaction with UGGT. UGGT is an essential component of the calnexin/calreticulin cycle, a quality control pathway that promotes folding of N-linked glycoproteins within the ER. SELENOF associates tightly with UGGT in a 1:1 complex that glucosylates misfolded protein domains, tagging them for another round of protein folding. Calnexin itself is a Ca2+-binding ER-resident chaperone that can function in protein quality control or Ca2+ signaling, depending upon its palmitoylation status [45]. When palmitoylated, calnexin is preferentially distributed to mitochondria-associated membranes (MAMs) and regulates ER Ca2+ levels via interactions with the SERCA2b pump. Moreover, ER stress reduces calnexin palmitolyation, allowing it to migrate away from MAMs into the ER lumen, where it acts as a chaperone.
Whereas SELENOF has been linked to protein folding, it has yet to be directly associated with Ca2+ homeostasis. Interestingly, induction of ER stress with the SERCA pump inhibitor, thapsigargin, was observed to trigger rapid degradation of SELENOF, whereas administration of the ER stress agents, tunicamycin or brefeldin A, upregulated SELENOF expression [46]. This indicates that SELENOF may be preferentially targeted to disturbances related to protein folding rather than Ca2+ dysregulation.
Expression of SELENOF is highly sensitive to levels of dietary Se, and is most prevalent in tissues with secretory functions, such as liver, kidney, thyroid, testes, and prostate [44]. This protein has been of significant interest to cancer researchers, as altered levels have been reported in several cancers and the gene is located on chromosome 1p31, a region often deleted or mutated in cancer. One of the initial reports on SELENOF found that expression levels were reduced in lung cancer, hepatocarcinoma, and a prostate cancer cell line [47]. Conversely, subsequent studies have reported SELENOF upregulation in human liver cancer cell lines, HepG2 and Huh7 [48], and shown that SELENOF can promote colon cancer [49]. In particular, knockdown of SELENOF inhibited growth of cultured colon cancer cells [50] and SELENOF KO mice were less susceptible to chemically-induced colon cancer than control mice [51].
Constitutive KO mice for SELENOF have been developed and initially characterized. They were observed to be viable and fertile with normal brain morphology [52]. Early development of cataracts was the primary defect found to result from SELENOF deletion. The authors of this paper observed enriched SELENOF expression during lens development and suggested that formation of cataracts were a consequence of impaired folding of lens proteins due to SELENOF deficiency. SELENOF KO mice also displayed elevated levels of oxidative stress in liver, yet when ER stress pathways were assessed in this tissue, no significant differences were observed. Earlier studies conducted in NIH3T3 cells, also found that SELENOF knockdown did not increase baseline ER stress, suggesting that SELENOF is either non-essential or its loss can be compensated for by similar proteins [46].
Selenoprotein K (SELENOK)
This small (~10 kDa) selenoprotein is a single spanning transmembrane protein localized to ER membrane [53]. It contains a highly disordered cytosolic region that contains a Sec residue located near the C-terminus and it is expressed in most tissues with highest levels detected in immune cells [53–55]. Two cellular functions have emerged for SELENOK including participation in the ER associated protein degradation (ERAD) pathway and regulation of Ca2+ flux from the ER. The role in ERAD was initially pursued based on evidence that SELENOK was transcriptionally upregulated during ER stress [56], and this led to the identification of an ER stress response element (ERSE) in the promoter of human SELENOK [57]. Subsequent studies showed that SELENOK is bound within complexes in the ER membrane along with SELENOS, valosin-containing protein (VCP; p97), and the Derlins [57, 58]. One may expect that a deletion in SELENOK may lead to increased ER stress and enhanced apoptosis, but perhaps due to redundancy or compensation this has not turned out to be the case. In fact, SELENOK knockout mice appear healthy and fertile, and cells from these mice show no signs of ERAD defects or ER stress [53].
Closer examination of the immune cells from SELENOK knockout mice found that receptor mediated Ca2+ flux from ER stores, or SOCE as described above, was impaired in SELENOK deficient immune cells. The reason for this impairment was eventually found to be due to the role SELENOK plays as a cofactor along with the enzyme, DHHC6, in palmitoylating the IP3Rs on particular cysteine residues [59]. This was shown to stabilize the IP3Rs and promote efficient SOCE. Thus, lower SELENOK levels caused by either lower dietary selenium intake or genetic factors leads to less IP3Rs in the ER membrane and lower capacity for SOCE in immune cells. While this does not appear to render cells more sensitive to stress or apoptosis, several immune cell functions that rely on efficient SOCE are compromised in SELENOK deficient immune cells. These include proliferation, migration, cytokine secretion, and protection against pathogens [53].
Exactly how the Sec residue within SELENOK is involved in Ca2+ flux or ERAD functions is not well understood. Unlike the selenoezymes that are well characterized, SELENOK does not have a thioredoxin-fold motif or oxidoreductase catalytic domain itself. For catalyzing palmitoylation of the IP3Rs, it is conceivable that the Sec residue in the SELENOK cofactor is utilized by the catalytic domain of the DHHC6 enzyme to more efficiently transfer the palmitoyl group to the cysteine residues of the IP3Rs. In fact, the redox chemistry of the Sec residue is consistent with promoting acyl transferase reactions [60]. Interestingly, the Sec residue in this selenoprotein exhibits some peroxidase activity in vitro [61], but whether this contributes to in vivo function has not yet been determined. Overall, SELENOK plays at least two important roles in the ER membrane related to cellular Ca2+. Reducing its expression or interfering with its function does not result in altered ER Ca2+ stores, but does hinder Ca2+ flux in a manner that does not induce increased cell stress or death. SELENOK deficiency leads to impaired functions of immune cells and perhaps other cell-types yet to be fully scrutinized (e.g. neurons, myocytes, etc.). It also has been reported that knockdown of SELENOK in a mouse T cell line affects calcium homeostasis in a manner that may involve calcium homeostasis endoplasmic reticulum protein (CHERP) [62]. Although the mechanism linking SELENOK and CHERP was not determined, this study underscores the notion that the IP3Rs are likely not the only calcium related protein related to SELENOK protein levels. Finally, it is worth noting that SELENOK may be important in cancer progression due to its role in promoting SOCE. While there is limited data regarding levels of SELENOK in tumors compared to healthy tissues, our laboratory has found in unpublished studies that SELENOK contributes to growth and migration of melanoma cells consistent with its crucial role in SOCE. Work is on-going to determine the precise role SELENOK plays in primary and secondary tumor formation and whether it may serve as an effective therapeutic target.
Selenoprotein M (SELENOM)
Selenoprotein M (SELENOM) is a thiol-disulfide oxidoreductase with enriched expression in the brain that is ancestrally related to SELENOF [63]. Major structural features include an active site consisting of a Sec-containing thioredoxin-like motif and an ER retention tetrapeptide in the C-terminal domain. The C-terminal region of SELENOM does not assume a defined secondary structure and is speculated to participate in substrate interactions [43]. Immunohistochemical analysis of SELENOM expression in the mouse brain showed high levels in several discrete regions, including the arcuate and paraventricular nuclei of the hypothalamus and the CA2/CA3 region of the hippocampus [64]. Furthermore, SELENOM levels in brain are highly responsive to Se supplementation [65]. Functionally, SELENOM has been documented to be neuroprotective and promote Ca2+ homeostasis. Overexpression of SELENOM in neuronal cell culture was found to decrease superoxide production, reduce cytosolic Ca2+ flux, and prevent apoptosis induced by oxidative challenge with hydrogen peroxide [66]. In contrast, SELENOM knockdown resulted in decreased cell viability in conjunction with increased ROS production and elevated baseline levels of cytosolic Ca2+.
The initial characterization of the phenotypic effects of SELENOM deletion in mice led to the unexpected finding that SELENOM exerts a significant influence on energy homeostasis [64]. Indeed, SELENOM KO mice displayed increased body weight, elevated white adipose tissue levels, and reduced hypothalamic leptin sensitivity relative to wild-type counterparts. Yet, SELENOM KO mice showed no apparent deficits in motor coordination, anxiety-like behavior, or cognition. Nonetheless, it should be noted that these behavioral tests were all conducted on young adult mice fed a Se-adequate diet. Thus, it is unclear whether further perturbations may arise when mice age or are challenged with Se-deficiency.
While the current literature on SELENOM is limited, several studies suggest that it may be protective against Alzheimer’s disease. For example, SELENOM expression levels were found significantly reduced in transgenic mice overexpressing a human mutant presenilin 2 (PS2) gene that causes early onset familial Alzheimer’s disease (FAD) [67]. Importantly, FAD-linked presenilin-2 mutants are known to disrupt Ca2+ homeostasis, as they have been documented to inhibit SERCA2b activity, increase ER Ca2+ leakage, and promote Ca2+ shuttling from the ER to mitochondria [68, 69]. Furthermore, with respect to Alzheimer’s disease, elevated levels of amyloid beta (Aβ) are known to promote ER Ca2+ release and apoptosis [70, 71], and in vitro studies have found that SELENOM protects against Aβ-induced toxicity [72, 73]. One of these studies reported that SELENOM attenuates Aβ-induced toxicity by binding the transition metals Zn2+ and Cd2+ in its thioredoxin-like active site [73]. Yet, this study was performed using a Sec-to-Cys SELENOM mutant and zinc-binding has yet to be verified with endogenous Sec-containing SELENOM. In addition, rats overexpressing SELENOM display increased activation of the ERK pathway and elevated glutathione peroxidase and superoxide dismutase activity in brain [74]. When combined with Se supplementation, SELENOM overexpression decreased γ-secretase activity, which drives production of Aβ42, the most pathogenic form of the Aβ peptide implicated in the development of Alzheimer’s. More recent studies on cortical tissue utilized two-dimensional electrophoresis to identify which proteins were most affected by SELENOM overexpression [75]. These analyses determined that five proteins were significantly elevated: brain-type creatine kinase (CKB), E3 ubiquitin ligase RING1 (RING1), lactate dehydrogenase B (LDHB), synaptotagmin-15 (SYT15), and eukaryotic initiation factor 4H (EIF4H). Both creatine kinase and lactate dehydrogenase are established enzymes in energy metabolism, providing additional corroborating evidence for the influence of SELENOM on this process.
The mechanistic basis of how SELENOM regulates Ca2+ homeostasis has yet to be elucidated. It presumably acts by reducing disulfide bonds on proteins involved in Ca2+ shuttling within the ER lumen, with prospective partners including the IP3Rs, RyRs, SERCA2b, and PS2. Further studies are warranted to: 1) unveil the precise molecular nature of how SELENOM influences Ca2+ homeostasis and 2) determine the signaling pathways affected by SELENOM that contribute to obesity.
Selenoprotein N (SELENON)
This 70 kDa transmembrane protein is localized to the ER and SELENON mRNA is expressed highest during development with lower, ubiquitous expression detected in adult tissues [76, 77]. There are two isoforms of the SELENON gene product, one corresponding to the full-length transcript and the second excluding exon 3 via splicing. Both transcripts are detected in skeletal muscle, brain, lung and placenta, with isoform 2 being the predominant transcript [78]. The full-length transcript has two potential in-frame Sec incorporation sites, whereas isoform 2 codes for only one Sec residue. Experimental evidence indicates that only the single Sec-containing isoform 2 is successfully translated [85]. The SELENON amino acid sequence contains a redox motif S-C-U-G that is similar to the G-C-U-G motif in TXNRDs, which suggests it can exert oxidoreductase activity. However, no enzymatic activity or specific biological function has yet been identified for SELENON. This is quite surprising given the abundance of evidence in humans and rodent models implicating defective SELENON expression in congenital myopathy characterized by muscle weakness, spinal rigidity, and respiratory insufficiency due to impaired diaphragm function [79, 80]. The diseases attributed to mutations in the SELENON gene include rigid spine muscular dystrophy (RSMD1), multi-minicore disease, rare cases of congenital fiber type disproportion, and desmin-related myopathy with Mallory body-like inclusions, all of which are collectively referred to as SEPN1-relative myopathies [81]. These muscle-related disorders that may involve respiratory insufficiency, but SEPN1-relative myopathies exhibit varied outcomes with a wide spectrum of severity and the onset can range from infancy to adulthood.
The topology of SELENON features its Sec located within the ER lumen [82]. Thus, the potential oxidoreductase activity of SELENON may be directed at the contents of the ER and may involve redox sensing, protein-folding, chaperoning, or some combination of these functions. One potential role for SELENON has been uncovered with evidence that it can reverse H2O2 induced inhibition of SERCA2b. In particular, SELENON may reduce disulfide bonds between a L4 cysteine pair, allowing SERCA2b driven Ca2+ pumping activity to proceed [83]. Consistent with this notion, SELENON-deficient myotubes showed higher basal Ca2+ levels in the cytosol and lower Ca2+ levels in the ER [84]. Furthermore, the absence of SELENON was associated with abnormal susceptibility to H2O2 induced oxidative stress, demonstrated by increased cell death. A healthy cell phenotype was restored by pretreatment with the antioxidant N-acetylcysteine. There likely are other proteins that are targeted by SELENON for reduction of disulfide bonds, perhaps Ca2+ handling proteins or proteins in the process of conformational folding. Also, SELENON is not the only redox regulator of L4 cysteine pair in SERCA2b [85], so other activities by this putative selenoenzyme are likely to be important for preventing SEPN1-relative myopathies. These muscle-related disorders that may involve respiratory insufficiency, likely due to defects in diaphragm function, and muscle dysfunctions are the common feature for the wide variety of disorders associated with mutations in the SELENON gene [86]. The molecular mechanisms by which the different mutations affect SELENON expression or function remains to be determined, but it may be related to SERCA2b function as described in more detail below.
An interesting feature of SELENON is the presence of at least one EF hand motif located in a position predicted to be within the ER lumen [87]. EF hand motifs bind Ca2+, and proteins containing these motifs exhibit structural changes when transitioning between the bound or unbound state [88]. In this manner, SELENON may sense Ca2+ within the ER lumen and switch between Ca2+ bound and unbound states. The presence or absence of Ca2+ bound to SELENON’s EF hand motif are likely to induce conformational changes in the protein, although experimental evidence for this is lacking. This may be related to the finding in zebrafish that SELENON interacts with the ryanodine receptor 1 (RyR1), which is the major component of the RyR intracellular Ca2+ release channel [89]. This is important given that mutations affecting SELENON and RyR1 result in an overlapping spectrum of congenital myopathies. The relevance of this model is evident in the fact that selenoprotein N function causes disruption of muscle architecture in the zebrafish embryo [90]. Interestingly, loss of function of either SELENON or RyR1 in developing zebrafish led to defects in Ca2+ mobilization in the embryo as measured by free Ca2+ levels around the Kupffer's Vesicle, a site used for Ca2+ flux measurement in the zebrafish embryo. Thus, the evidence from this embryonic model strongly supports the notion that SELENON along with RyR1 regulate Ca2+ flux to support muscle tissue development. It is frustrating that a specific molecular or enzymatic activity has not yet been elucidated to explain this effect, but the S-C-U-G and/or EF hand motifs are likely to be involved and further investigation may reveal how sometime in the future.
Selenoprotein S (SELENOS)
SELENOS is another of the many selenoproteins discovered in silico by the Gladyshev laboratory [3]. Prior to its identification as a selenoprotein, it was known in the literature as TANIS, a novel gene found upregulated in response to fasting in the obese/diabetic animal model Pssamomys obesus [91]. In humans, the SELENOS gene encodes two alternative transcripts that differ in their 3’ UTR sequences [92]. Experiments conducted in vitro revealed that read-through of the UGA/Sec codon occurred in one variant, whereas the other transcript encoded a truncated protein that lacked Sec. Full-length SELENOS is predominantly localized to the ER membrane and consists of 189 amino acids. Although not ancestrally related to SELENOK, SELENOS shares several important topological features worth noting. These include a single-pass ER transmembrane domain and proline residues located in a glycine-rich C-terminal region that mediates binding with VCP [93]. Due to its interaction with VCP, SELENOS has also been referred to in the literature as VIMP, an acronym for VCP-interacting membrane protein [94]. VCP is a particularly important ATPase at the ER membrane that uses the energy of ATP hydrolysis to move or segregate proteins within complexes [95]. The VCP chaperone binds to both SELENOS and SELENOK, but how the interactions occur and whether the cytosolic-facing Sec residues located near the C-terminus are involved is presently unclear. For SELENOS, the Sec residue (position 188) is known to bond with an adjacent Cys residue (position 174), to yield a Cys174-Sec188 selenosulfide bond [96, 97]. Furthermore, studies using a recombinant SELENOS where the Sec residue was replaced by Cys, observed a reduction potential of −200 mV and determined it to be a substrate of thioredoxin [98].
SELENOS is widely expressed, with highest levels reported in pancreas, adipose tissue, testis, and stomach. Its promoter contains an ER stress response element and two binding sites for nuclear factor kappa B [99]. Likewise, various studies have observed SELENOS upregulation in response to inflammatory cytokines [100], ER stress, and glucose deprivation [101]. Among humans, genetic variation of SELENOS has been linked to inflammation, as a single nucleotide polymorphism in the promoter region (−105G»A) was identified that significantly associated with inflammatory cytokine levels and exhibited diminished responsiveness to challenge with ER stress [100]. Additional experiments in macrophages demonstrated that SELENOS knockdown increased release of IL-6 and TNF-α, corroborating the role of SELENOS in inflammation. The link to inflammation is further supported by prior studies that identified the acute-phase inflammatory protein, serum amyloid A, as an interaction partner of SELENOS by yeast two-hybrid screening [91].
In terms of function, several studies have established SELENOS as an essential player in ERAD. It is an important component of a multiprotein complex that translocates misfolded proteins across the ER membrane for cytosolic degradation by the ubiquitin-proteasome system. In addition to SELENOS, this complex includes VCP, the Derlin chaperone proteins, SELENOK, and E3 ubiquitin ligases [58, 94]. Recent studies demonstrated that formation of this retrotranslocation complex is contingent upon SELENOS-mediated recruitment of VCP to the ER membrane [58]. Moreover, experiments in cell culture by another group found that SELENOS knockdown impairs cell viability and increases tau phosphorylation in response to agents that cause ER stress [102]. These findings, along with the observation of pronounced SELENOS expression in neurofibrillary tangles in postmortem human brain, suggest that SELENOS may be protective against neurodegeneration. To date, there is no evidence directly implicating SELENOS in Ca2+ regulation. Nonetheless, it is highly likely that SELENOS exerts an indirect influence on Ca2+ signaling by maintaining ER homeostasis through its documented role in ERAD.
Selenoprotein T (SELENOT)
SELENOT is a thioredoxin-like enzyme localized to the ER membrane that was initially identified using an algorithm that searched for mRNAs containing a selenocysteine insertion element (SECIS) in the 3’ untranslated region (UTR) [103]. Structurally, SELENOT contains a signal peptide, a thioredoxin-like fold with a Sec residue in the active site, and two hydrophobic stretches (residues 87–102 and 125–143) indicative of transmembrane domains [104]. It is presumed to modulate ER thiol redox balance and recent in vitro studies have shown that it exhibits thioredoxin reductase-like activity [105].
SELENOT is widely expressed at high levels during embryogenesis and early development, but expression levels are very low postnatally, with the exception of endocrine tissues such as the pancreas, pituitary, testis, and thyroid [106]. In adult tissues, SELENOT expression is most pronounced in secretory cells, such as pancreatic islets and testicular Leydig cells, which produce insulin and testosterone, respectively [106, 107]. Although expression levels are typically low in non-endocrine tissues, exposure to noxious stimuli leads to significant upregulation. In the mouse brain, SELENOT is barely detectable under baseline conditions but is significantly elevated in the nigrostriatal pathway in response to treatment with the neurotoxin MPTP [105]. Moreover, increased SELENOT expression was also found in the striatum of postmortem samples from individuals with Parkinson’s disease. In addition, while SELENOT levels are normally undetectable in liver, expression was reported to be strongly induced during the acute phase of liver regeneration [106]. One of the first papers describing SELENOT identified it as a target gene of the neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) during differentiation of PC12 cells [108]. This paper determined that SELENOT upregulation was dependent upon increased levels of cAMP and intracellular Ca2+ flux in response to PACAP. More recent studies investigating the nature of the PACAP-induced signal activating SELENOT gene transcription unveiled a pathway involving protein kinase A, AMP kinase, and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1-α) that is coupled to mitochondrial biogenesis [104].
Along with having its expression regulated by Ca2+, SELENOT itself contributes to Ca2+ homeostasis. Silencing of SELENOT was observed to impair Ca2+ mobilization induced by PACAP, whereas SELENOT overexpression elevated baseline cytosolic Ca2+ levels [108]. Later studies performed on gastric smooth muscle by a different group, reported that siRNA-mediated SelT knockdown diminished baseline cytosolic Ca2+ content and significantly reduced the enzymatic activity of myosin light chain kinase, a key regulator of muscle contraction [109]. It is currently speculated that SELENOT modulates Ca2+ signaling by means of a redox mechanism involving thiol groups on Ca2+ channels and pumps, but the precise nature of how this occurs has yet to be elucidated.
With respect to function, SELENOT is one of only four selenoproteins known to cause embryonic lethality in mice (GPX4, TXNRD1, and TXNRD2 being the others) and among this group, it is the only one present in the ER [105, 110]. Conditional knockout mice have been generated that ablate SELENOT expression in pancreatic β-cells [107] and nervous tissue [105, 110]. In mice where SELENOT knockout was restricted to pancreatic β-cells, researchers observed alterations in the size and number of pancreatic islets, along with impairments in glucose-stimulated insulin secretion and glucose tolerance. Two recent papers have bred floxed SELENOT mice with another strain expressing Cre under the control of the Nestin promoter (Nestin-Cre) in order to selectively silence SELENOT in neural cells. These mice were reported to exhibit reduced brain volume during the first postnatal week that was associated with increased apoptotic death of immature neurons [110]. Brain size normalized as the mice matured and the predominant behavioral phenotype in adulthood was hyperactivity in the open field apparatus. Subsequent studies showed that these mice were more vulnerable to MPTP-induced neurotoxicity, as they exhibited increased levels of oxidative stress, extensive neurodegeneration in dopaminergic neurons, and severe motor deficits in comparison to controls [105]. This demonstrates that although SELENOT is minimally expressed in the adult brain, it does protect dopaminergic neurons against toxic insults.
Type 2 Iodothyronine Deiodinase (DIO2)
Thyroid hormone (TH) is regulated by the actions of three selenoproteins: iodothyronine deiodinase 1 and 2 (DIO1 and 2) that catalyze the conversion of the pro-hormone, T4, to the active hormone T3, and iodothyronine deiodinase 3 (DIO3) that catalyzes the deactivation of the hormone [111]. DIO2 is the predominant form in adult tissues that generates active T3 in local tissues [112], and consists of a single transmembrane protein with the Sec near the C-terminus located in the cytosol similar to DIO1 [113]. The activity of DIO2 is regulated at many levels, but degradation through ERAD is particularly interesting. In mammals, DIO2 is ubiquitinated by the E3 ubiquitin ligases, WSB-1 [114] and TEB4 [115]). Ubiquitinated DIO2 interacts with VCP and can be retrotranslocated to the cytosol for proteasomal degradation [116]. However, the degradation of DIO2 is prevented by two ubiquitin-specific peptidases, USP20 and USP33, that promote deubiquitination and counteract the catabolic effects of WSB-1 and TEB4 [117]. In addition, ER stress has been shown to rapidly reduce DIO2 activity [118]. Surprisingly, this observed reduction in activity was not associated with decreased mRNA levels or increased proteasomal degradation, but rather resulted from diminished de novo DIO2 synthesis due to p-eIF2α-mediated inhibition of translation. So, both the synthesis and degradation of DIO2 appears to be dynamically regulated by environmental conditions to control local levels of T3. Also influenced by DIO2 is SERCA2b, which has been found in both slow twitch skeletal muscles and human myocardium. Levels of SERCA2b mRNA are regulated by TH through TH response elements in the promoter of the SERCA2b gene [119]. This has implications for cardiomyocytes, in that active T3 produced by the activity of the DIO2 (and possibly DIO1) can accelerate sarcoplasmic reticulum Ca2+ uptake, thereby improving cardiomyocyte relaxation and overall cardiac diastolic function [120]. A transgenic mouse model was developed in which human DIO2 was highly expressed in the heart and myocardial thyrotoxicity was detected in isolated hearts [121]. Thus, there appears to be an upper limit to the beneficial effects of more active TH available to the cardiomyocytes. Hence, DIO2 may control Ca2+ related functions in cells in a manner that is controlled by ERAD directed turnover of DIO2 levels.
Conclusions
Several selenoproteins are localized to the ER but exhibit diverse structural features and different topologies, with some found in the membrane and others in the lumen. This is consistent with the notion that they play different roles, which has been borne out by reports showing some to be involved in sensing and regulating redox tone, and others regulating protein folding and the chaperoning of misfolded proteins during UPR (Figure 2). In some cases, roles have been demonstrated for ER selenoproteins in regulating Ca2+ flux or homeostasis. Exactly how selenoproteins are involved in these processes is only beginning to be understood. Given that none of these ER selenoproteins are considered ion channels and only one (SELENON) has a Ca2+ binding EF hand motif, it is likely that most play indirect roles in regulating Ca2+ homeostasis and signaling. Examples include SELENON and SELENOK, which indirectly regulate ER Ca2+ levels by modifying the SERCA2b pump and IP3Rs, respectively. In addition to the ER resident selenoproteins discussed above, other well characterized selenoproteins that function as cytosolic or mitochondrial enzymes in redox based reactions are likely to influence Ca2+ homeostasis and Ca2+-dependent signaling. Overall, it remains to be determined exactly how the ER selenoproteins impinge upon Ca2+ homeostasis and Ca2+-dependent signaling, and there is a need for further dissection of molecular mechanisms by which they operate.
Figure 2.

Major topological features of ER selenoproteins. This illustration details the topology of Sec residues and transmembrane domains in ER selenoproteins. Also shown are key proteins involved in Ca2+ homeostasis, protein folding, and ERAD known to interact with individual ER selenoproteins. U represents Sec residues.
Highlights.
Seven of twenty-five selenoproteins are localized to the ER but exhibit diverse structural features and different topologies, with some found in the membrane and others in the lumen.
Some ER selenoproteins have been shown to be involved in sensing and regulating redox tone, and others regulating protein folding and the chaperoning of misfolded proteins during unfolded protein response, and others in regulating Ca2+ flux and homeostasis.
Examples of molecular mechanisms involving these ER selenoproteins include SELENON and SELENOK, which indirectly regulate ER Ca2+ levels by modifying the SERCA2 pump and IP3R, respectively.
The role of the selenocysteine (Sec) residue within the seven ER selenoproteins in some cases involves its oxidoreductase potential, while in others remains unclear.
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) National Institute of Allergy and Infectious Diseases Grant R01AI089999.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rayman MP. Selenium and human health. Lancet. 2012;379:1256–1268. doi: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- 2.Bulteau AL, Chavatte L. Update on selenoprotein biosynthesis. Antioxid Redox Signal. 2015;23:775–794. doi: 10.1089/ars.2015.6391. [DOI] [PubMed] [Google Scholar]
- 3.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 4.Bosl MR, Takaku K, Oshima M, Nishimura S, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp) Proc Natl Acad Sci U S A. 1997;94:5531–5534. doi: 10.1073/pnas.94.11.5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schweizer U, Fradejas-Villar N. Why 21? The significance of selenoproteins for human health revealed by inborn errors of metabolism. FASEB J. 2016;30:3669–3681. doi: 10.1096/fj.201600424. [DOI] [PubMed] [Google Scholar]
- 6.Dumitrescu AM, Di Cosmo C, Liao XH, Weiss RE, Refetoff S. The syndrome of inherited partial SBP2 deficiency in humans. Antioxid Redox Signal. 2010;12:905–920. doi: 10.1089/ars.2009.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hondal RJ, Marino SM, Gladyshev VN. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid Redox Signal. 2013;18:1675–1689. doi: 10.1089/ars.2012.5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dardashti RN, Dery L, Mousa R, Dery S, Reddy PS, Metanis N. The Chemistry of Selenocysteine in Proteins. In: UaTDLS Hatfield PA, Gladyshev VN, editors. Selenium: Its Molecular Biology and Role in Human Health. Springer; New York: 2016. pp. 73–83. [Google Scholar]
- 9.Gladyshev VN, Arner ES, Berry MJ, Brigelius-Flohe R, Bruford EA, Burk RF, Carlson BA, Castellano S, Chavatte L, Conrad M, Copeland PR, Diamond AM, Driscoll DM, Ferreiro A, Flohe L, Green FR, Guigo R, Handy DE, Hatfield DL, Hesketh J, Hoffmann PR, Holmgren A, Hondal RJ, Howard MT, Huang K, Kim HY, Kim IY, Kohrle J, Krol A, Kryukov GV, Lee BJ, Lee BC, Lei XG, Liu Q, Lescure A, Lobanov AV, Loscalzo J, Maiorino M, Mariotti M, Sandeep Prabhu K, Rayman MP, Rozovsky S, Salinas G, Schmidt EE, Schomburg L, Schweizer U, Simonovic M, Sunde RA, Tsuji PA, Tweedie S, Ursini F, Whanger PD, Zhang Y. Selenoprotein Gene Nomenclature. J Biol Chem. 2016;291:24036–24040. doi: 10.1074/jbc.M116.756155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jousset H, Frieden M, Demaurex N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J Biol Chem. 2007;282:11456–11464. doi: 10.1074/jbc.M609551200. [DOI] [PubMed] [Google Scholar]
- 11.Shchedrina VA, Zhang Y, Labunskyy VM, Hatfield DL, Gladyshev VN. Structure-function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxid Redox Signal. 2010;12:839–849. doi: 10.1089/ars.2009.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambudkar IS, de Souza LB, Ong HL. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium. 2016 doi: 10.1016/j.ceca.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villalobos C, Sobradillo D, Hernandez-Morales M, Nunez L. Remodeling of Calcium Entry Pathways in Cancer. Adv Exp Med Biol. 2016;898:449–466. doi: 10.1007/978-3-319-26974-0_19. [DOI] [PubMed] [Google Scholar]
- 14.Villalobos C, Sobradillo D, Hernandez-Morales M, Nunez L. Calcium remodeling in colorectal cancer. Biochim Biophys Acta. 2017 doi: 10.1016/j.bbamcr.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 15.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 16.Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, Marie JC, Pouzet C, Samadi M, Elbim C, O’Dowd Y, Bens M, Vandewalle A, Gougerot-Pocidalo MA, Lizard G, Ogier-Denis E. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol. 2004;24:10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hourihan JM, Moronetti Mazzeo LE, Fernandez-Cardenas LP, Blackwell TK. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol Cell. 2016;63:553–566. doi: 10.1016/j.molcel.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, Lee AS. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol Cell Biol. 2000;20:5096–5106. doi: 10.1128/mcb.20.14.5096-5106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L, Singh BB. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest. 2012;122:1354–1367. doi: 10.1172/JCI61332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 23.Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:701–710. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 24.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 26.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 27.Ozcan L, Tabas I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med. 2012;63:317–328. doi: 10.1146/annurev-med-043010-144749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- 29.Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD. Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000;23:222–229. doi: 10.1016/s0166-2236(00)01548-4. [DOI] [PubMed] [Google Scholar]
- 30.Schauble N, Lang S, Jung M, Cappel S, Schorr S, Ulucan O, Linxweiler J, Dudek J, Blum R, Helms V, Paton AW, Paton JC, Cavalie A, Zimmermann R. BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 2012;31:3282–3296. doi: 10.1038/emboj.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hammadi M, Oulidi A, Gackiere F, Katsogiannou M, Slomianny C, Roudbaraki M, Dewailly E, Delcourt P, Lepage G, Lotteau S, Ducreux S, Prevarskaya N, Van Coppenolle F. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: involvement of GRP78. FASEB J. 2013;27:1600–1609. doi: 10.1096/fj.12-218875. [DOI] [PubMed] [Google Scholar]
- 32.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14:196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan SA, Rossi AM, Riley AM, Potter BV, Taylor CW. Subtype-selective regulation of IP(3) receptors by thimerosal via cysteine residues within the IP(3)-binding core and suppressor domain. Biochem J. 2013;451:177–184. doi: 10.1042/BJ20121600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, Sharma K, Yule DI, Joseph SK, Hajnoczky G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem. 2014;289:8170–8181. doi: 10.1074/jbc.M113.504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng Y, Zhong L, Shen X. Effect of selenium-supplement on the calcium signaling in human endothelial cells. J Cell Physiol. 2005;205:97–106. doi: 10.1002/jcp.20378. [DOI] [PubMed] [Google Scholar]
- 37.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 38.Birk J, Meyer M, Aller I, Hansen HG, Odermatt A, Dick TP, Meyer AJ, Appenzeller-Herzog C. Endoplasmic reticulum: reduced and oxidized glutathione revisited. J Cell Sci. 2013;126:1604–1617. doi: 10.1242/jcs.117218. [DOI] [PubMed] [Google Scholar]
- 39.Enyedi B, Varnai P, Geiszt M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid Redox Signal. 2010;13:721–729. doi: 10.1089/ars.2009.2880. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Camacho P. Ca2+-dependent redox modulation of SERCA 2b by ERp57. J Cell Biol. 2004;164:35–46. doi: 10.1083/jcb.200307010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gladyshev VN, Jeang KT, Wootton JC, Hatfield DL. A new human selenium-containing protein. Purification, characterization, and cDNA sequence. J Biol Chem. 1998;273:8910–8915. doi: 10.1074/jbc.273.15.8910. [DOI] [PubMed] [Google Scholar]
- 42.Korotkov KV, Kumaraswamy E, Zhou Y, Hatfield DL, Gladyshev VN. Association between the 15-kDa selenoprotein and UDP-glucose:glycoprotein glucosyltransferase in the endoplasmic reticulum of mammalian cells. J Biol Chem. 2001;276:15330–15336. doi: 10.1074/jbc.M009861200. [DOI] [PubMed] [Google Scholar]
- 43.Labunskyy VM, Ferguson AD, Fomenko DE, Chelliah Y, Hatfield DL, Gladyshev VN. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J Biol Chem. 2005;280:37839–37845. doi: 10.1074/jbc.M508685200. [DOI] [PubMed] [Google Scholar]
- 44.Ferguson AD, Labunskyy VM, Fomenko DE, Arac D, Chelliah Y, Amezcua CA, Rizo J, Gladyshev VN, Deisenhofer J. NMR structures of the selenoproteins Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J Biol Chem. 2006;281:3536–3543. doi: 10.1074/jbc.M511386200. [DOI] [PubMed] [Google Scholar]
- 45.Lynes EM, Raturi A, Shenkman M, Ortiz Sandoval C, Yap MC, Wu J, Janowicz A, Myhill N, Benson MD, Campbell RE, Berthiaume LG, Lederkremer GZ, Simmen T. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J Cell Sci. 2013;126:3893–3903. doi: 10.1242/jcs.125856. [DOI] [PubMed] [Google Scholar]
- 46.Labunskyy VM, Yoo MH, Hatfield DL, Gladyshev VN. Sep15, a thioredoxin-like selenoprotein, is involved in the unfolded protein response and differentially regulated by adaptive and acute ER stresses. Biochemistry. 2009;48:8458–8465. doi: 10.1021/bi900717p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumaraswamy E, Malykh A, Korotkov KV, Kozyavkin S, Hu Y, Kwon SY, Moustafa ME, Carlson BA, Berry MJ, Lee BJ, Hatfield DL, Diamond AM, Gladyshev VN. Structure-expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J Biol Chem. 2000;275:35540–35547. doi: 10.1074/jbc.M004014200. [DOI] [PubMed] [Google Scholar]
- 48.Guariniello S, Di Bernardo G, Colonna G, Cammarota M, Castello G, Costantini S. Evaluation of the selenotranscriptome expression in two hepatocellular carcinoma cell lines. Anal Cell Pathol (Amst) 2015;2015:419561. doi: 10.1155/2015/419561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsuji PA, Carlson BA, Yoo MH, Naranjo-Suarez S, Xu XM, He Y, Asaki E, Seifried HE, Reinhold WC, Davis CD, Gladyshev VN, Hatfield DL. The 15kDa selenoprotein and thioredoxin reductase 1 promote colon cancer by different pathways. PLoS One. 2015;10:e0124487. doi: 10.1371/journal.pone.0124487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuji PA, Naranjo-Suarez S, Carlson BA, Tobe R, Yoo MH, Davis CD. Deficiency in the 15 kDa selenoprotein inhibits human colon cancer cell growth. Nutrients. 2011;3:805–817. doi: 10.3390/nu3090805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsuji PA, Carlson BA, Naranjo-Suarez S, Yoo MH, Xu XM, Fomenko DE, Gladyshev VN, Hatfield DL, Davis CD. Knockout of the 15 kDa selenoprotein protects against chemically-induced aberrant crypt formation in mice. PLoS One. 2012;7:e50574. doi: 10.1371/journal.pone.0050574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kasaikina MV, Fomenko DE, Labunskyy VM, Lachke SA, Qiu W, Moncaster JA, Zhang J, Wojnarowicz MW, Jr, Natarajan SK, Malinouski M, Schweizer U, Tsuji PA, Carlson BA, Maas RL, Lou MF, Goldstein LE, Hatfield DL, Gladyshev VN. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J Biol Chem. 2011;286:33203–33212. doi: 10.1074/jbc.M111.259218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verma S, Hoffmann FW, Kumar M, Huang Z, Roe K, Nguyen-Wu E, Hashimoto AS, Hoffmann PR. Selenoprotein K knockout mice exhibit deficient calcium flux in immune cells and impaired immune responses. J Immunol. 2011;186:2127–2137. doi: 10.4049/jimmunol.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu J, Rozovsky S. Membrane-bound selenoproteins. Antioxid Redox Signal. 2015;23:795–813. doi: 10.1089/ars.2015.6388. [DOI] [PubMed] [Google Scholar]
- 55.Polo A, Colonna G, Guariniello S, Ciliberto G, Costantini S. Deducing the functional characteristics of the human selenoprotein SELK from the structural properties of its intrinsically disordered C-terminal domain. Mol Biosyst. 2016;12:758–772. doi: 10.1039/c5mb00679a. [DOI] [PubMed] [Google Scholar]
- 56.Du S, Zhou J, Jia Y, Huang K. SelK is a novel ER stress-regulated protein and protects HepG2 cells from ER stress agent-induced apoptosis. Arch Biochem Biophys. 2010;502:137–143. doi: 10.1016/j.abb.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 57.Shchedrina VA, Everley RA, Zhang Y, Gygi SP, Hatfield DL, Gladyshev VN. Selenoprotein K binds multiprotein complexes and is involved in the regulation of endoplasmic reticulum homeostasis. J Biol Chem. 2011;286:42937–42948. doi: 10.1074/jbc.M111.310920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee JH, Park KJ, Jang JK, Jeon YH, Ko KY, Kwon JH, Lee SR, Kim IY. Selenoprotein S-dependent Selenoprotein K Binding to p97(VCP) Protein Is Essential for Endoplasmic Reticulum-associated Degradation. J Biol Chem. 2015;290:29941–29952. doi: 10.1074/jbc.M115.680215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fredericks GJ, Hoffmann FW, Rose AH, Osterheld HJ, Hess FM, Mercier F, Hoffmann PR. Stable expression and function of the inositol 1,4,5-triphosphate receptor requires palmitoylation by a DHHC6/selenoprotein K complex. Proc Natl Acad Sci U S A. 2014;111:16478–16483. doi: 10.1073/pnas.1417176111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reich HJ, Hondal RJ. Why Nature Chose Selenium. ACS Chem Biol. 2016;11:821–841. doi: 10.1021/acschembio.6b00031. [DOI] [PubMed] [Google Scholar]
- 61.Liu J, Zhang Z, Rozovsky S. Selenoprotein K form an intermolecular diselenide bond with unusually high redox potential. FEBS Lett. 2014;588:3311–3321. doi: 10.1016/j.febslet.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang C, Li R, Huang Y, Wang M, Yang F, Huang D, Wu C, Li Y, Tang Y, Zhang R, Cheng J. Selenoprotein K modulate intracellular free Ca2+ by regulating expression of calcium homoeostasis endoplasmic reticulum protein. Biochem Biophys Res Commun. 2017;484:734–739. doi: 10.1016/j.bbrc.2017.01.117. [DOI] [PubMed] [Google Scholar]
- 63.Korotkov KV, Novoselov SV, Hatfield DL, Gladyshev VN. Mammalian selenoprotein in which selenocysteine (Sec) incorporation is supported by a new form of Sec insertion sequence element. Mol Cell Biol. 2002;22:1402–1411. doi: 10.1128/mcb.22.5.1402-1411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pitts MW, Reeves MA, Hashimoto AC, Ogawa A, Kremer P, Seale LA, Berry MJ. Deletion of selenoprotein M leads to obesity without cognitive deficits. J Biol Chem. 2013;288:26121–26134. doi: 10.1074/jbc.M113.471235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang JQ, Ren FZ, Jiang YY, Lei X. Characterization of Selenoprotein M and Its Response to Selenium Deficiency in Chicken Brain. Biol Trace Elem Res. 2016;170:449–458. doi: 10.1007/s12011-015-0486-1. [DOI] [PubMed] [Google Scholar]
- 66.Reeves MA, Bellinger FP, Berry MJ. The neuroprotective functions of selenoprotein M and its role in cytosolic calcium regulation. Antioxid Redox Signal. 2010;12:809–818. doi: 10.1089/ars.2009.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hwang DY, Cho JS, Oh JH, Shim SB, Jee SW, Lee SH, Seo SJ, Lee SK, Lee SH, Kim YK. Differentially expressed genes in transgenic mice carrying human mutant presenilin-2 (N141I): correlation of selenoprotein M with Alzheimer’s disease. Neurochem Res. 2005;30:1009–1019. doi: 10.1007/s11064-005-6787-6. [DOI] [PubMed] [Google Scholar]
- 68.Brunello L, Zampese E, Florean C, Pozzan T, Pizzo P, Fasolato C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J Cell Mol Med. 2009;13:3358–3369. doi: 10.1111/j.1582-4934.2009.00755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci U S A. 2011;108:2777–2782. doi: 10.1073/pnas.1100735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferreiro E, Oliveira CR, Pereira CM. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis. 2008;30:331–342. doi: 10.1016/j.nbd.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 71.Resende R, Ferreiro E, Pereira C, Resende de Oliveira C. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience. 2008;155:725–737. doi: 10.1016/j.neuroscience.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 72.Chen P, Wang RR, Ma XJ, Liu Q, Ni JZ. Different Forms of Selenoprotein M Differentially Affect Abeta Aggregation and ROS Generation. Int J Mol Sci. 2013;14:4385–4399. doi: 10.3390/ijms14034385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Du X, Li H, Wang Z, Qiu S, Liu Q, Ni J. Selenoprotein P and selenoprotein M block Zn2+ -mediated Abeta42 aggregation and toxicity. Metallomics. 2013;5:861–870. doi: 10.1039/c3mt20282h. [DOI] [PubMed] [Google Scholar]
- 74.Yim SY, Chae KR, Shim SB, Hong JT, Park JY, Lee CY, Son HJ, Sheen YY, Hwang DY. ERK activation induced by selenium treatment significantly downregulates beta/gamma-secretase activity and Tau phosphorylation in the transgenic rat overexpressing human selenoprotein M. Int J Mol Med. 2009;24:91–96. doi: 10.3892/ijmm_00000211. [DOI] [PubMed] [Google Scholar]
- 75.Kim Y, Goo JS, Kim IY, Kim JE, Kwak MH, Go J, Shim S, Hong JT, Hwang DY, Seong JK. Identification of the responsible proteins for increased selenium bioavailability in the brain of transgenic rats overexpressing selenoprotein M. Int J Mol Med. 2014;34:1688–1698. doi: 10.3892/ijmm.2014.1945. [DOI] [PubMed] [Google Scholar]
- 76.Lescure A, Gautheret D, Carbon P, Krol A. Novel selenoproteins identified in silico and in vivo by using a conserved RNA structural motif. J Biol Chem. 1999;274:38147–38154. doi: 10.1074/jbc.274.53.38147. [DOI] [PubMed] [Google Scholar]
- 77.Castets P, Lescure A, Guicheney P, Allamand V. Selenoprotein N in skeletal muscle: from diseases to function. J Mol Med (Berl) 2012;90:1095–1107. doi: 10.1007/s00109-012-0896-x. [DOI] [PubMed] [Google Scholar]
- 78.Moghadaszadeh B, Petit N, Jaillard C, Brockington M, Quijano Roy S, Merlini L, Romero N, Estournet B, Desguerre I, Chaigne D, Muntoni F, Topaloglu H, Guicheney P. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet. 2001;29:17–18. doi: 10.1038/ng713. [DOI] [PubMed] [Google Scholar]
- 79.Rederstorff M, Castets P, Arbogast S, Laine J, Vassilopoulos S, Beuvin M, Dubourg O, Vignaud A, Ferry A, Krol A, Allamand V, Guicheney P, Ferreiro A, Lescure A. Increased muscle stress-sensitivity induced by selenoprotein N inactivation in mouse: a mammalian model for SEPN1-related myopathy. PLoS One. 2011;6:e23094. doi: 10.1371/journal.pone.0023094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maiti B, Arbogast S, Allamand V, Moyle MW, Anderson CB, Richard P, Guicheney P, Ferreiro A, Flanigan KM, Howard MT. A mutation in the SEPN1 selenocysteine redefinition element (SRE) reduces selenocysteine incorporation and leads to SEPN1-related myopathy. Hum Mutat. 2009;30:411–416. doi: 10.1002/humu.20879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arbogast S, Ferreiro A. Selenoproteins and protection against oxidative stress: selenoprotein N as a novel player at the crossroads of redox signaling and calcium homeostasis. Antioxid Redox Signal. 2010;12:893–904. doi: 10.1089/ars.2009.2890. [DOI] [PubMed] [Google Scholar]
- 82.Lescure A, Rederstorff M, Krol A, Guicheney P, Allamand V. Selenoprotein function and muscle disease. Biochim Biophys Acta. 2009;1790:1569–1574. doi: 10.1016/j.bbagen.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 83.Marino M, Stoilova T, Giorgi C, Bachi A, Cattaneo A, Auricchio A, Pinton P, Zito E. SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Hum Mol Genet. 2015;24:1843–1855. doi: 10.1093/hmg/ddu602. [DOI] [PubMed] [Google Scholar]
- 84.Arbogast S, Beuvin M, Fraysse B, Zhou H, Muntoni F, Ferreiro A. Oxidative stress in SEPN1-related myopathy: from pathophysiology to treatment. Ann Neurol. 2009;65:677–686. doi: 10.1002/ana.21644. [DOI] [PubMed] [Google Scholar]
- 85.Appenzeller-Herzog C, Simmen T. ER-luminal thiol/selenol-mediated regulation of Ca2+ signalling. Biochem Soc Trans. 2016;44:452–459. doi: 10.1042/BST20150233. [DOI] [PubMed] [Google Scholar]
- 86.Dai Y, Liang S, Huang Y, Chen L, Banerjee S. Targeted next generation sequencing identifies two novel mutations in SEPN1 in rigid spine muscular dystrophy 1. Oncotarget. 2016;7:83843–83849. doi: 10.18632/oncotarget.13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Petit N, Lescure A, Rederstorff M, Krol A, Moghadaszadeh B, Wewer UM, Guicheney P. Selenoprotein N: an endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum Mol Genet. 2003;12:1045–1053. doi: 10.1093/hmg/ddg115. [DOI] [PubMed] [Google Scholar]
- 88.Dominguez DC, Guragain M, Patrauchan M. Calcium binding proteins and calcium signaling in prokaryotes. Cell Calcium. 2015;57:151–165. doi: 10.1016/j.ceca.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 89.Jurynec MJ, Xia R, Mackrill JJ, Gunther D, Crawford T, Flanigan KM, Abramson JJ, Howard MT, Grunwald DJ. Selenoprotein N is required for ryanodine receptor calcium release channel activity in human and zebrafish muscle. Proc Natl Acad Sci U S A. 2008;105:12485–12490. doi: 10.1073/pnas.0806015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deniziak M, Thisse C, Rederstorff M, Hindelang C, Thisse B, Lescure A. Loss of selenoprotein N function causes disruption of muscle architecture in the zebrafish embryo. Exp Cell Res. 2007;313:156–167. doi: 10.1016/j.yexcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 91.Walder K, Kantham L, McMillan JS, Trevaskis J, Kerr L, De Silva A, Sunderland T, Godde N, Gao Y, Bishara N, Windmill K, Tenne-Brown J, Augert G, Zimmet PZ, Collier GR. Tanis: a link between type 2 diabetes and inflammation? Diabetes. 2002;51:1859–1866. doi: 10.2337/diabetes.51.6.1859. [DOI] [PubMed] [Google Scholar]
- 92.Bubenik JL, Miniard AC, Driscoll DM. Alternative transcripts and 3’UTR elements govern the incorporation of selenocysteine into selenoprotein S. PLoS One. 2013;8:e62102. doi: 10.1371/journal.pone.0062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee JH, Kwon JH, Jeon YH, Ko KY, Lee SR, Kim IY. Pro178 and Pro183 of selenoprotein S are essential residues for interaction with p97(VCP) during endoplasmic reticulum-associated degradation. J Biol Chem. 2014;289:13758–13768. doi: 10.1074/jbc.M113.534529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 95.Xia D, Tang WK, Ye Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene. 2016;583:64–77. doi: 10.1016/j.gene.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu J, Li F, Rozovsky S. The intrinsically disordered membrane protein selenoprotein S is a reductase in vitro. Biochemistry. 2013;52:3051–3061. doi: 10.1021/bi4001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu J, Rozovsky S. Contribution of selenocysteine to the peroxidase activity of selenoprotein S. Biochemistry. 2013;52:5514–5516. doi: 10.1021/bi400741c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Christensen LC, Jensen NW, Vala A, Kamarauskaite J, Johansson L, Winther JR, Hofmann K, Teilum K, Ellgaard L. The human selenoprotein VCP-interacting membrane protein (VIMP) is non-globular and harbors a reductase function in an intrinsically disordered region. J Biol Chem. 2012;287:26388–26399. doi: 10.1074/jbc.M112.346775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gao Y, Hannan NR, Wanyonyi S, Konstantopolous N, Pagnon J, Feng HC, Jowett JB, Kim KH, Walder K, Collier GR. Activation of the selenoprotein SEPS1 gene expression by pro-inflammatory cytokines in HepG2 cells. Cytokine. 2006;33:246–251. doi: 10.1016/j.cyto.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 100.Curran JE, Jowett JB, Elliott KS, Gao Y, Gluschenko K, Wang J, Abel Azim DM, Cai G, Mahaney MC, Comuzzie AG, Dyer TD, Walder KR, Zimmet P, MacCluer JW, Collier GR, Kissebah AH, Blangero J. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37:1234–1241. doi: 10.1038/ng1655. [DOI] [PubMed] [Google Scholar]
- 101.Gao Y, Feng HC, Walder K, Bolton K, Sunderland T, Bishara N, Quick M, Kantham L, Collier GR. Regulation of the selenoprotein SelS by glucose deprivation and endoplasmic reticulum stress - SelS is a novel glucose-regulated protein. FEBS Lett. 2004;563:185–190. doi: 10.1016/S0014-5793(04)00296-0. [DOI] [PubMed] [Google Scholar]
- 102.Rueli RH, Torres DJ, Dewing AS, Kiyohara AC, Barayuga SM, Bellinger MT, Uyehara-Lock JH, White LR, Moreira PI, Berry MJ, Perry G, Bellinger FP. Selenoprotein S Reduces Endoplasmic Reticulum Stress-Induced Phosphorylation of Tau: Potential Role in Selenate Mitigation of Tau Pathology. J Alzheimers Dis. 2017;55:749–762. doi: 10.3233/JAD-151208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kryukov GV, Kryukov VM, Gladyshev VN. New mammalian selenocysteine-containing proteins identified with an algorithm that searches for selenocysteine insertion sequence elements. J Biol Chem. 1999;274:33888–33897. doi: 10.1074/jbc.274.48.33888. [DOI] [PubMed] [Google Scholar]
- 104.Boukhzar L, Tanguy Y, Abid H, Castex M, Hamieh A, Alsharif I, Cartier D, Prevost G, Falluel-Morel A, Librmann I, Chagraoui A, Anouar Y. Selenoprotein T: From Discovery to Functional Studies Using Conditional Knockout Mice. In: Hatfield DL, Schweizer U, Tsuji PA, Gladyshev VN, editors. Selenium: Its Moleculr Biology and Role in Human Health. Springer; New York: 2016. pp. 275–286. [Google Scholar]
- 105.Boukhzar L, Hamieh A, Cartier D, Tanguy Y, Alsharif I, Castex M, Arabo A, El Hajji S, Bonnet JJ, Errami M, Falluel-Morel A, Chagraoui A, Lihrmann I, Anouar Y. Selenoprotein T Exerts an Essential Oxidoreductase Activity That Protects Dopaminergic Neurons in Mouse Models of Parkinson’s Disease. Antioxid Redox Signal. 2016;24:557–574. doi: 10.1089/ars.2015.6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tanguy Y, Falluel-Morel A, Arthaud S, Boukhzar L, Manecka DL, Chagraoui A, Prevost G, Elias S, Dorval-Coiffec I, Lesage J, Vieau D, Lihrmann I, Jegou B, Anouar Y. The PACAP-regulated gene selenoprotein T is highly induced in nervous, endocrine, and metabolic tissues during ontogenetic and regenerative processes. Endocrinology. 2011;152:4322–4335. doi: 10.1210/en.2011-1246. [DOI] [PubMed] [Google Scholar]
- 107.Prevost G, Arabo A, Jian L, Quelennec E, Cartier D, Hassan S, Falluel-Morel A, Tanguy Y, Gargani S, Lihrmann I, Kerr-Conte J, Lefebvre H, Pattou F, Anouar Y. The PACAP-regulated gene selenoprotein T is abundantly expressed in mouse and human beta-cells and its targeted inactivation impairs glucose tolerance. Endocrinology. 2013;154:3796–3806. doi: 10.1210/en.2013-1167. [DOI] [PubMed] [Google Scholar]
- 108.Grumolato L, Ghzili H, Montero-Hadjadje M, Gasman S, Lesage J, Tanguy Y, Galas L, Ait-Ali D, Leprince J, Guerineau NC, Elkahloun AG, Fournier A, Vieau D, Vaudry H, Anouar Y. Selenoprotein T is a PACAP-regulated gene involved in intracellular Ca2+ mobilization and neuroendocrine secretion. FASEB J. 2008;22:1756–1768. doi: 10.1096/fj.06-075820. [DOI] [PubMed] [Google Scholar]
- 109.Li JP, Zhou JX, Wang Q, Gu GQ, Yang SJ, Li CY, Qiu CW, Deng GZ, Guo MY. Se Enhances MLCK Activation by Regulating Selenoprotein T (SelT) in the Gastric Smooth Muscle of Rats. Biol Trace Elem Res. 2016;173:116–125. doi: 10.1007/s12011-016-0620-8. [DOI] [PubMed] [Google Scholar]
- 110.Castex MT, Arabo A, Benard M, Roy V, Le Joncour V, Prevost G, Bonnet JJ, Anouar Y, Falluel-Morel A. Selenoprotein T Deficiency Leads to Neurodevelopmental Abnormalities and Hyperactive Behavior in Mice. Mol Neurobiol. 2016;53:5818–5832. doi: 10.1007/s12035-015-9505-7. [DOI] [PubMed] [Google Scholar]
- 111.Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23:38–89. doi: 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- 112.Arrojo EDR, Bianco AC. Type 2 deiodinase at the crossroads of thyroid hormone action. Int J Biochem Cell Biol. 2011;43:1432–1441. doi: 10.1016/j.biocel.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baqui MM, Gereben B, Harney JW, Larsen PR, Bianco AC. Distinct subcellular localization of transiently expressed types 1 and 2 iodothyronine deiodinases as determined by immunofluorescence confocal microscopy. Endocrinology. 2000;141:4309–4312. doi: 10.1210/endo.141.11.7872. [DOI] [PubMed] [Google Scholar]
- 114.Dentice M, Bandyopadhyay A, Gereben B, Callebaut I, Christoffolete MA, Kim BW, Nissim S, Mornon JP, Zavacki AM, Zeold A, Capelo LP, Curcio-Morelli C, Ribeiro R, Harney JW, Tabin CJ, Bianco AC. The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol. 2005;7:698–705. doi: 10.1038/ncb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zavacki AM, Arrojo EDR, Freitas BC, Chung M, Harney JW, Egri P, Wittmann G, Fekete C, Gereben B, Bianco AC. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol Cell Biol. 2009;29:5339–5347. doi: 10.1128/MCB.01498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Arrojo EDR, Egri P, Jo S, Gereben B, Bianco AC. The type II deiodinase is retrotranslocated to the cytoplasm and proteasomes via p97/Atx3 complex. Mol Endocrinol. 2013;27:2105–2115. doi: 10.1210/me.2013-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Curcio-Morelli C, Zavacki AM, Christofollete M, Gereben B, de Freitas BC, Harney JW, Li Z, Wu G, Bianco AC. Deubiquitination of type 2 iodothyronine deiodinase by von Hippel-Lindau protein-interacting deubiquitinating enzymes regulates thyroid hormone activation. J Clin Invest. 2003;112:189–196. doi: 10.1172/JCI18348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Arrojo EDR, Fonseca TL, Castillo M, Salathe M, Simovic G, Mohacsik P, Gereben B, Bianco AC. Endoplasmic reticulum stress decreases intracellular thyroid hormone activation via an eIF2a–mediated decrease in type 2 deiodinase synthesis. Mol Endocrinol. 2011;25:2065–2075. doi: 10.1210/me.2011-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Accorroni A, Saponaro F, Zucchi R. Tissue thyroid hormones and thyronamines. Heart Fail Rev. 2016;21:373–390. doi: 10.1007/s10741-016-9553-8. [DOI] [PubMed] [Google Scholar]
- 120.Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. 2005;26:704–728. doi: 10.1210/er.2003-0033. [DOI] [PubMed] [Google Scholar]
- 121.Pachucki J, Hopkins J, Peeters R, Tu H, Carvalho SD, Kaulbach H, Abel ED, Wondisford FE, Ingwall JS, Larsen PR. Type 2 iodothyronin deiodinase transgene expression in the mouse heart causes cardiac-specific thyrotoxicosis. Endocrinology. 2001;142:13–20. doi: 10.1210/endo.142.1.7907. [DOI] [PubMed] [Google Scholar]