

Abstract
Thymic regulatory T cells (tTreg) or induced Tregs (iTregs) suppress murine acute graft-versus-host disease (GVHD). Previously we demonstrated that plasmacytoid dendritic cell (pDC) indoleamine 2,3-dioxygenase (IDO) fosters the in vitro development of human iTregs via tryptophan depletion and kynurenine (Kyn) metabolites. We now show that stimulation of naïve CD4+ T cells in low tryptophan (Low Trp) plus Kyn supports human iTreg generation. In vitro, low Trp+Kyn iTreg and tTregs potently suppress Teffector cell proliferation equivalently but are phenotypically distinct. As compared to tTreg or Teffector, bioenergetics profiling reveals that Low Trp+Kyn iTreg have increased basal glycolysis and oxidative phosphorylation and also use glutaminolysis as an energy source. Low Trp+Kyn iTreg viability was reliant on IL-2 in vitro. Although in vivo IL-2 administration increased Low Trp+Kyn iTreg persistence upon adoptive transfer into immune deficient mice given peripheral blood mononuclear cells to induce GVHD, IL-2 supported iTregs did not improve recipient survival. We conclude that Low Trp+Kyn create suppressive iTregs that have high metabolic needs that will need to be addressed prior to clinical translation.
Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) is limited by immune cell-mediated graft-versus-host disease (GVHD) (1), significant in 40–70% of recipients, with skin, liver and gut representing major GVHD target organs (2). Immune regulation is critical for maintaining homeostasis. Regulatory T-cells (Treg) are required to suppress self-reactive lymphocytes, and can develop in the thymus (tTreg) or be induced in vivo (peripheral Tregs) or in vitro (iTreg). Adoptive Treg transfer is effective at preventing autoimmunity, organ rejection and GVHD in preclinical models (1, 3). We completed first-in-human clinical trials of ex vivo expanded umbilical cord blood (UCB) CD4+25hi tTreg in patients receiving HSCT (4, 5). As predicted by allogeneic and xenogeneic murine models (6), high tTreg doses (1×108/kg, or ~1:1 tTreg:UCB T cell) were needed to effectively ameliorate Grade II-IV GVHD (9% vs. 45% for matched controls, p<0.05) (5), thus requiring large-scale expansion with retention of function from a relatively rare UCB tTreg population.
While third-party UCB tTreg fits nicely into a UCB clinical trial, peripheral blood (PB) Treg from histocompatible family members are more amenable for solid organ graft studies and HSCT by enabling closer minor histocompatibility antigen matching. Because expanding human PB tTreg can result in loss of suppression due to the proliferative advantage of contaminating Teffectors after purification, we explored clinically relevant approaches to generate iTreg. iTreg therapeutics are of interest because co-transfer of both tTreg and iTreg in a rodent inflammatory bowel disease model was shown to be necessary for optimal disease suppression (7) suggesting future trials could combine tTregs and iTregs. We found that clinically relevant numbers of iTreg (>200 × 109) could be induced by in vitro expansion for 14 days in the presence of TGFß and rapamycin (8). Because murine Treg induced by food allergens in the gut, a primal GVHD target organ, through TGFß-secreting DC can be re-programmed into Teffectors (9), we sought iTreg approaches that might result in high yields, potency and stability and do not rely on TGFß.
Previously, we observed that human Tregs can be induced by plasmacytoid DC (pDC) through indoleamine 2,3-dioxygenase (IDO)-mediated depletion of tryptophan (Trp) or by exposure to immune suppressive kynurenines (Kyn), produced as catabolites of Trp metabolism (10, 11). IDO is implicated in maternal-fetal acceptance (12), tumor immunity (13), autoimmunity (14, 15) and alloimmunity reactions, such as GVHD (16, 17). Exposure of murine tTregs to IDO in vitro or in vivo markedly enhances their suppressor function, “super-charging” their potency (18–21). Because pDCs are rare and difficult to purify and expand, we sought to determine whether an optimally suppressive iTreg could be generated using conditions that mimicked pDCs support (i.e. Low Trp+Kyn) amenable for clinical translation.
We report that clinically relevant numbers of iTreg can be generated by stimulating naïve human CD4+ T-cells with a GMP-compliant artificial APC (aAPC) in media containing Low Trp+Kyn. These iTreg are as suppressive as tTreg in vitro. However, bioenergetics profiling showed that, unlike tTreg, Low Trp+Kyn iTreg displayed remarkably active oxidative phosphorylation, driven in part through glutamine catabolism. Low Trp+Kyn iTreg also had higher glycolytic metabolism, and were more sensitive to IL-2 deprivation, known to support glycolysis (22). Low Trp+Kyn iTreg had decreased viability in vitro and in vivo. Ultra-low dose IL-2 unable to sustain iTreg persistence in vivo and recipient survival was not significantly increased compared to GVHD mice receiving no iTregs. These results demonstrate that clinically relevant numbers of highly suppressive Low Trp+Kyn iTreg can be generated, but that the in vivo persistence of iTreg after adoptive transfer will need to be improved by overcoming their higher metabolic needs or by optimizing IL-2 supplementation in vivo to achieve GVHD prevention in vivo.
Materials and Methods
Cell purification and culture
CD4+25++ tTreg and CD4+25-45RA+ T-cells were purified from leukapheresis products (Memorial Blood Center, St.Paul,MN) using negative selection (Miltenyi Biotec) on an AutoMACS (Depletion 2.1). Unbound cells were washed away and CD25++ tTregs were purified by positive selection using cGMP-grade anti-CD25 microbeads. Naive CD4+ T-cells (CD4+25-45RA+) were isolated from the flow-through: remaining CD25int cells were depleted using a high-dose, anti-CD25 step, then CD45RA fraction was purified by positive selection using anti-CD45RA microbeads. Bead incubations and washes were carried out as specified by the manufacturer (30′ at room temperature for cGMP-grade beads).
Purified cells were stimulated with a GMP-licensed, K562 cell line (KT) engineered to express CD86 and the high affinity Fc Receptor (CD64) (KT64/86) (2:1 Treg:KT) (23), irradiated with 10,000 cGray and incubated with anti-CD3 mAb (Orthoclone OKT3, Janssen-Cilag). CD25++ tTregs cells were cultured in X-Vivo-15 media (BioWhittaker) while CD4+25-45RA+ were cultured in Trp-free RPMI media (Gibco) supplemented with either normal (25uM) or low levels of L-tryptophan (5μM, Sigma) (Teffectors and Low Trp+Kyn iTreg, respectively). rIL-2 (300 IU/ml, Novartis) was added (day 2 for tTregs and day 0 for all others) and maintained throughout culture. For tTreg, Rapa (Rapammune, Wyeth-Ayerst) was added at 109 nM on day 0 and with media supplementation. Low Trp+Kyn iTreg cultures were treated with 5 kynurenines,(10μM each) added on day 0 and maintained throughout culture.
Suppression assays
Suppression was assessed using a 5-carboxyfluorescein-diacetate-succinimide ester (CFSE) inhibition assay (24). Peripheral blood mononuclear cells (PBMCs) were purified, labeled with CFSE (InVitrogen), and stimulated with anti-CD3 mAb-coated beads (Dynal) in 96-well round-bottom plates with or without cultured tTreg or iTreg (Treg:PBMC at ratios of 1:4–1:16). Assays were harvested on day 4, stained with anti-CD4 and/or -8 mAb and data acquired by FACScalibur or LSRII. Data were analyzed using the proliferation platform in FlowJo (Treestar, Ashland, OR), and suppression determined from the Division Index.
Statistical analysis
Data were analyzed by ANOVA or Student’s t-test. Survival effects were assessed by Mantel-Cox (Prism 5). Probability (P) values ≤0.05 were considered statistically significant.
Additional information on materials, cell purification, media and culture conditions and xenogeneic GVHD studies are provided in Supporting Information.
Results
Expansion of naïve CD4+ non-Tregs in the presence of low Trp+Kyn induces suppressive function
PBMC were isolated from apheresis products, and CD4+25-45RA+ T-cells and CD4+25++127− T-cells (tTreg) were MACS-purified from the same apheresis units. Purified tTreg were 80 ± 4% CD127-Foxp3+ while CD4+25-45RA+ cells contained 5 ± 2 % CD127-Foxp3+ cells (Figure S1A,B). Purified tTreg and CD4+25-45RA+ T-cells were expanded as shown in Figure S2A. CD4+25-45RA+ T-cells cultured in normal media had significantly higher expansion than tTreg or CD4+25-45RA+ T-cells grown in Low Trp+Kyn (Figure 1A). In vitro expanded tTreg were 80 ± 2% CD127-Foxp3+ (Figure 1B). While expression of Foxp3 in CD4+25-45RA+ cells grown in normal Trp without Kyn was low, CD4+25-45RA+ T-cells grown in Low Trp+Kyn had a significantly higher percentage of CD127-Foxp3+ T-cells, although Foxp3 mean fluorescent intensity was significantly lower than tTreg (Figure 1C). A significantly higher number of human CD4+25-45RA+ T-cells expanded in Low Trp+Kyn and tTregs were CD25hiFoxp3+, compared to Teff (Figure 1D) with significantly higher (~10-fold) CD25 levels in Low Trp+Kyn iTreg vs tTreg (Figure 1E).
Figure 1. In vitro expansion of naïve CD4 T-cells in the presence of Low Trp+Kyn induces suppressive function.
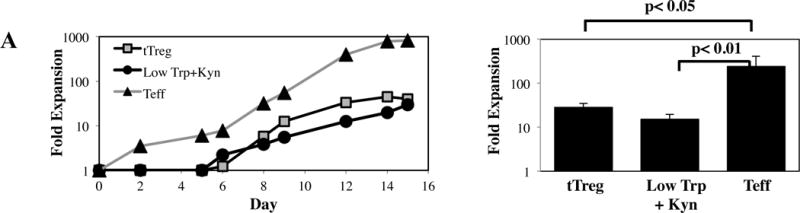
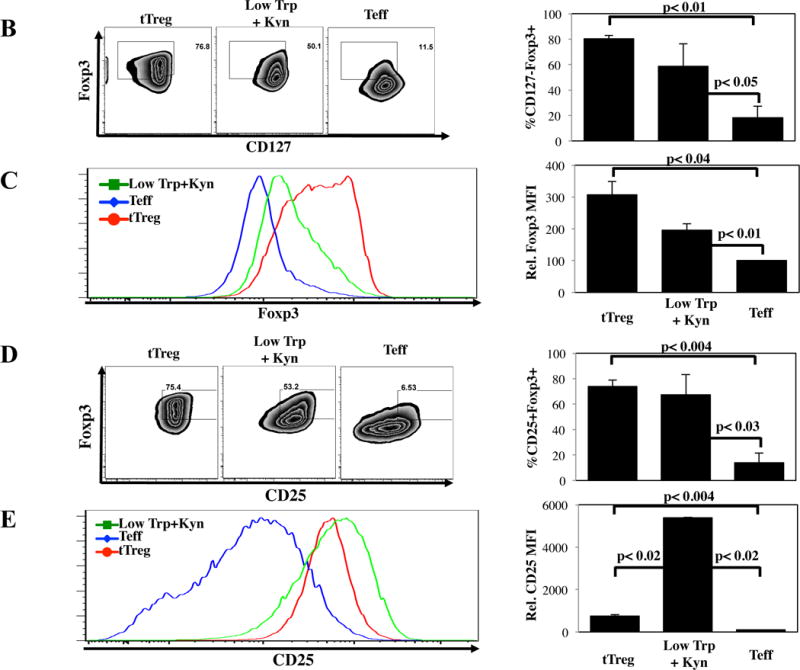
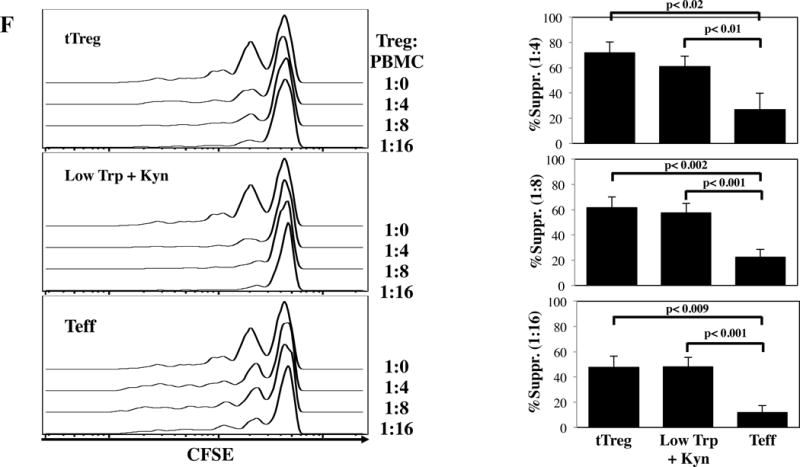
tTreg (CD4+25++127−) and naïve CD4+ T-cells (CD4+ 25–45RA+) were purified from peripheral blood using magnetic beads, stimulated with a cell line expressing CD86 and CD64 (KT64/86) to which anti-CD3 mAb is bound, and cultured for 14 days as described. A) Representative example and average fold expansion for the indicated cell types on day 14 of culture. B) Representative example and summary of CD127 and Foxp3 expression in tTregs and naive T-cells cultured ± Low Trp+Kyn. C) Representative example and summary of relative Foxp3 expression (MFI) in tTregs and naive T-cells cultured ± Low Trp+Kyn. D) Representative example and summary of CD25 and Foxp3 expression in tTreg and naive T-cells cultured ± Low Trp+Kyn. E) Representative example and average CD25 expression (± SEM) on CD4-gated cells. F) Suppressive function of cultured cells was determined using a CFSE-based proliferation assay in which expanded tTreg or naive T-cells were incubated with CFSE-loaded allogeneic PBMC at doses from 1:4 to 1:16 (expanded cell:PBMC) and stimulated with anti-CD3 beads for 4 days.
tTreg potently suppressed CD8+ T-cell division using a 5-carboxyfluorescein diacetate succinimide ester based proliferation assay (Figure 1F). CD4+25-45RA+ T-cells expanded in Low Trp+Kyn were as suppressive as tTreg. Few cells of either Treg type expressed effector cytokines IFNγ, IL-4 or IL-17, though IL-10 secretion was observed (Figure S3A–D). In contrast to tTregs, Low Trp+Kyn iTreg did not show Foxp3 Treg Specific Demethylated Region (TSDR) (25) hypomethylation, nor did they express Helios, an Ikaros family transcription factor associated with tTreg origin (Figures S3E and F) (26). tTreg expression of CD39 and adenosine production has been linked to suppressive function (27–29). While Low Trp+Kyn iTreg expressed CD39, expression was not significantly higher than Teffs, which had minimal suppressive function (Figure S2G). To determine whether suppression was contact-dependent, assays were performed in transwell plates with expanded cells either in the lower chamber with the PBMC or separated by the transwell membrane. tTreg suppression was greatly decreased when contact was prevented. Close contact was required for Low Trp+Kyn iTreg suppression, whether measured by division index or number of CD8+ cells remaining (a measure for cytolysis-mediated suppression) (Figure S2H and I, respectively).
Since the goal is to use Low Trp+Kyn iTreg as adoptive cellular therapy in patients with normal Trp and lacking kynurenines, we assessed the stability and suppressive function of the Low Trp+Kyn iTreg re-cultured for the final 3 days in media with normal Trp (25mM) and no kynurenines. As shown in Figure S4, no significant change was observed in Foxp3 or CD25 expression, and cultures maintained suppressive function, suggesting adoptive transfer of Low Trp+Kyn iTreg would retain suppressor function in vivo.
Low Trp+Kyn iTreg have higher levels of oxidative phosphorylation compared to tTreg or Teff
Metabolism is intimately linked to T cell differentiation and function (22). The metabolic program of Low Trp+Kyn iTreg was of particular interest given their generation under conditions of essential amino acid (Trp) depletion. Both the basal and maximal respiratory capacity (oxygen consumption rate; OCR) of iTreg were significantly higher than tTreg or Teff cultures (Figure 2A). Increased OCR did not result from increased mitochondria, as all three cultures stained similarly with MitoTracker Green, which measures mitochondrial mass (Figure 2B).
Figure 2. Low Trp + Kyn iTreg have increased oxidative function relative to tTreg or Teff cells.
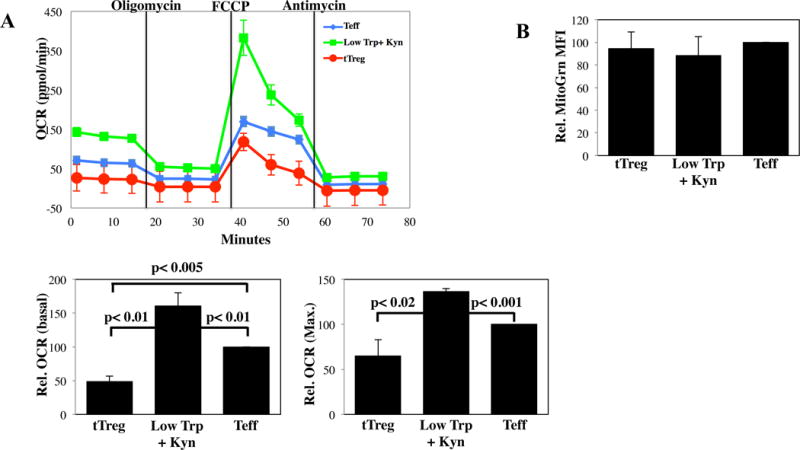
PB tTregs, Low Trp+Kyn iTregs and Teffs were expanded 14 days and metabolic pathway utilization determined. A) Representative example and average basal and maximal Oxygen Consumption Rate for each culture as determined by MitoStress kit (Seahorse) (n=4). B) Relative Mitotracker Green fluorescence of tTreg, Low Trp+Kyn iTreg and Teff cultured cells. Summary data from 3–5 independent experiments.
Low Trp+Kyn iTreg also have increased glucose consumption and production of lactate compared to tTreg or Teff
Extracellular acidification rate (ECAR) is a measure of aerobic glycolysis, and results from lactate and pyruvate export. As expected, Teff had a significantly higher ECAR than tTreg, both under basal conditions and at maximal glycolytic capacity (Figure 3A). Unexpectedly, Low Trp+Kyn iTreg had even higher basal ECAR and a similar maximal glycolytic capacity compared to Teff. Notably, this metabolic profile (increased OXPHOS and glycolysis) is shared with rodent T-cells proliferating in response to alloantigens during GVHD (30). Consistent with increased ECAR, Low Trp+Kyn iTreg expressed higher levels of the glucose transporters GLUT1 and GLUT2 than tTreg or Teff (Figure 3B). Low Trp+Kyn iTreg consumed significantly more glucose and excreted more lactate compared to Teff, while tTreg excreted the lowest amounts of lactate (2.8- and 1.9-fold vs. Low Trp+Kyn iTreg or Teff, respectively) (Figure 3C and D).
Figure 3. Low Trp + Kyn iTreg have increased biosynthetic demands relative to Teff and tTreg.
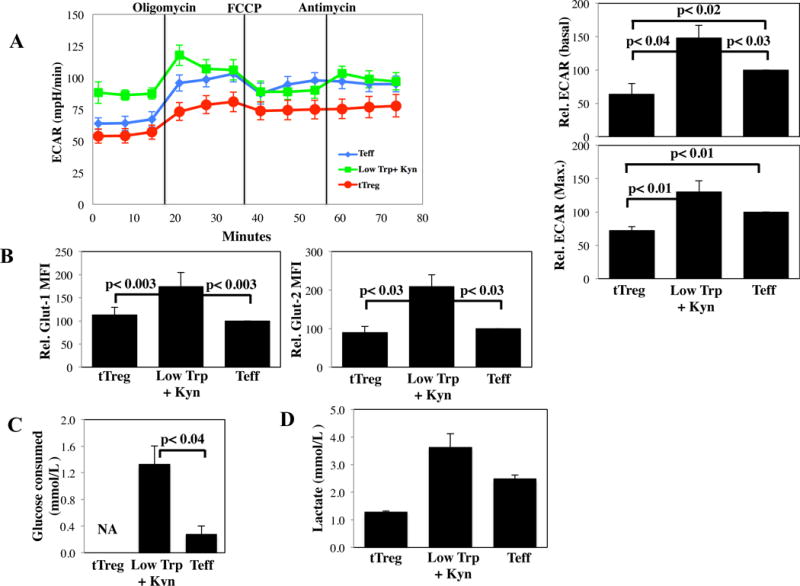
PB tTregs, Low Trp+Kyn iTregs and Teffs were expanded 14 days and metabolic pathway utilization determined. A) Representative example and average relative basal and maximal ECAR values (n=4). B) Summary of Glut-1 (glucose transporter) and Glut-2 expression on cultured cells. The indicated cells were re-stimulated for 24 hours with αCD3/28 beads and, after harvest, the supernatants were assayed for the consumption of glucose (C) and the production of lactate (D). Note that tTreg are expanded in clinically defined media that includes high levels of glucose, and thus these assays are not applicable.
Low Trp+Kyn iTreg display higher rates of glutamine oxidation than tTreg or Teff
CD4 T-cells can produce energy by glutaminolysis (31), a process in which glutamine (Gln) is catabolized to glutamate (Glu) and then α-ketoglutarate (αKG), where it enters the TCA cycle to contribute to OXPHOS (32). To assess whether glutaminolysis contributed to high basal OXPHOS in Low Trp+Kyn iTreg, we measured glutamine consumption at 24 hours from the same anti-CD3/28 mAb-bead stimulated samples used for glucose consumption. While glutamine levels decreased 130 μM in Teff cultures, the decrease was 330 μM in Low Trp+Kyn iTreg cultures, indicating that iTreg cultures are more actively using glutaminolysis for energy (Figure 4A). tTreg are expanded in clinically defined media that includes high glutamine levels and thus these assays are not applicable. In addition to intracelluilar Gln transport, the large neutral amino acid transporter (LAT1, a heterodimer of CD98 and SLC3A2) can also use intracellular Gln as an exchange-partner to drive the import of Trp (33). Low Trp/Kyn iTreg CD98 expression was 7- and 4-fold higher than on tTreg or Teff, respectively (Figure 4B). Low Trp+Kyn iTreg expressed 5-fold higher levels of GLS2, the mitochondria-associated glutaminase responsible for converting glutamine to glutamate, compared to Teff (Figure 4C).
Figure 4. Low Trp+Kyn iTreg display higher rates of glutamine oxidation relative to tTreg or Teff.
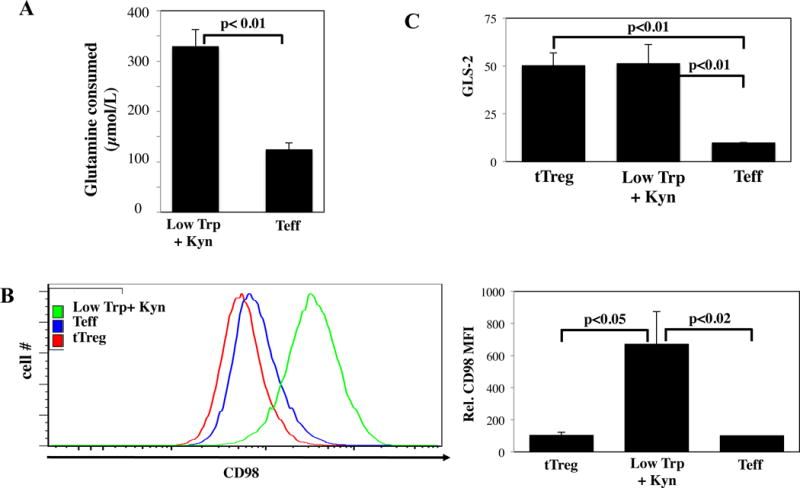
Ex vivo expanded PB tTregs, Low Trp+Kyn iTregs, and Teffs were assayed for characteristics related to glutaminolysis. A) The indicated cells were re-stimulated for 24 hours with αCD3/28 beads and, after harvest, the supernatants were assayed for glutamine consumption. Note that tTreg are expanded in clinically defined media that includes high levels of glutamine and thus these assays are not applicable. B) Representative example and summary of CD98 expression on cultured cells as determined by flow cytometry. C) RNA was isolated from the indicated cells and the relative expression of GLS-2 determined by qRT-PCR.
Fatty Acid esterification and intracellular triglycerides support Low Trp+Kyn iTreg metabolism
tTreg and memory CD8 T cells (Tmem) use fatty acid oxidation (FAO) to drive energy production (34). Compared to Teff, tTreg had increased expression of two key enzymes involved in shuttling FA to the mitochondria (CPT1a and ACSL3) and DGAT1, a triacylglycerol (TAG) synthase, which converts diacylglycerol to TAG, a critical substrate for FAO (35) (Figure S5). Fatty acids (FA) used to fuel CD8 Tmem are not derived from extracellular sources, but through a futile cycle of de novo fatty acid synthesis (FAS) followed by FAO (36); long-chain FA uptake is lower in CD8 Tmem than in CD8 Teff (37). Similar to CD8 Tmem, tTreg had decreased signs of FAS compared to CD4 Teff, including low rate of FA uptake (measured with BoDipyC1–C12, a fluorescent FA analog) and decreased expression of SLC27A2, a long-chain FA transport molecule, (Figure 5). In contrast to tTreg and CD8 Tmem, Low Trp+Kyn iTreg exhibited a reciprocal phenotype, with increased FA uptake and SLC27A2 expression, and decreased expression of CPT1a and ACSL3.
Figure 5. Fatty Acid esterification and intracellular triglycerides support Low Trp+Kyn iTreg metabolism.
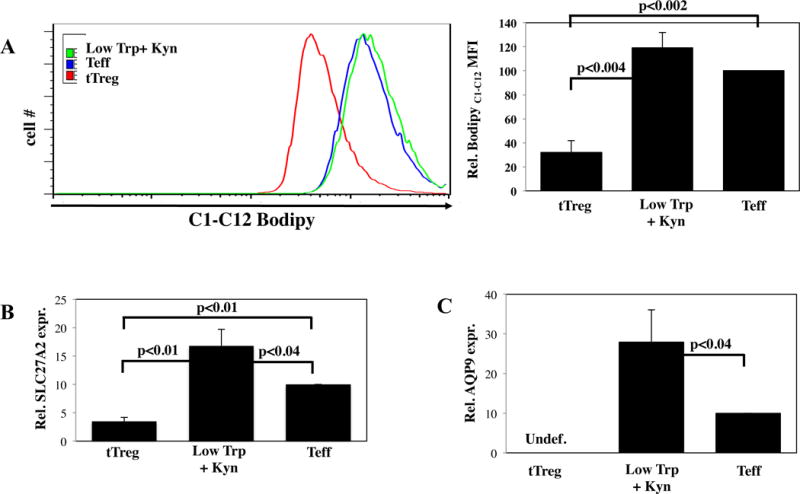
Ex vivo expanded PB tTregs, Low Trp+Kyn iTregs, and Teffs were assayed for characteristics related to FAS/FAO. A) Representative example and summary of fatty acid uptake as measured by flow cytometry with BodipyC1-C12. RNA was isolated from the indicated cells and the relative expression of B) SLC27A2 and C) AQP9 determined by qRT-PCR. Results are from 3–5 independent experiments
Human and murine CD8 Tmem survival and self-renewal was recently shown to be dependent upon molecules involved in TAG synthesis (38). Specifically, CD8 T-cells that lacked the glycerol channel Aquaporin 9 (AQP9), or were treated with a AQP9-specific inhibitor, had impaired memory responses and AQP9−/− Tmem survival was rescued by overexpression of AQP9. AQP9 expression, driven by IL-7R signaling in CD8 Tmem, was very low/absent in tTreg, which have greatly reduced IL-7R expression (Figure 5C). AQP9 expression, observed in Teff, was highest in Low Trp+Kyn iTreg. In summary, while the predominant phenotype of tTreg is FAO, Low Trp+Kyn iTreg are more closely linked with FAS.
Low Trp+Kyn iTreg adoptive transfer transiently suppress clinical manifestations of xenogeneic GVHD but not to a sufficient magnitude to prolong survival
To determine whether Low Trp+Kyn iTreg could suppress T-cell activation in vivo, a xenogeneic model of GVHD was used in which human CD4+25++ and CD4+25-45RA+ T-cells were purified from the same donor, expanded in vitro as tTreg and Low Trp+Kyn iTreg, and transferred into immune deficient NSG mice along with HLA-A2 mismatched PBMC (8). tTreg and Low Trp+Kyn iTreg were ≥75% CD25+Foxp3+ and were highly suppressive in vitro (Figure 6A). In this model, disease severity correlates with the peripheral blood (PB) expansion of human PBMC-derived CD4+ and CD8+ T-cells (24). To assess PBMC-derived T cell expansion, recipients were bled on day 13, PBMCs stained for human CD45, CD4, CD8 and HLA-A2, and PBMC-derived (HLA-A2+) T-cells were enumerated (Figure 6B). Mice receiving only human PBMC had 112±25 HLA-A2+CD4+ T-cells/μl of blood, and 81±10 HLA-A2+CD8+ T-cells/μl of blood. In contrast, HLA-A2+CD4+ or CD8+ T-cells were virtually undetectable in mice that also received tTreg. Mice receiving adoptive transfer of Low Trp+Kyn iTreg also had significantly fewer PBMC-derived CD4+ T-cells but not fewer CD8+ T-cells in PB.
Figure 6. Adoptive transfer of Low Trp + Kyn iTreg does not increase survival in a xenogeneic model of GVHD.
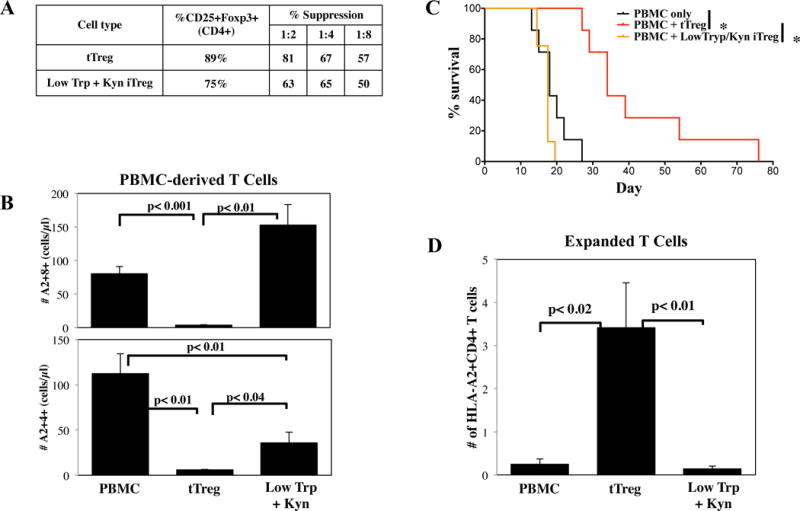
Ex vivo expanded tTreg and Low Trp+Kyn iTregs (15 × 106 cells) were co-transferred with allogeneic, HLA-A2 mismatched PBMC (15 × 106 cells) into non-lethally irradiated NSG mice to assess the ability to ameliorate xenogeneic GVHD. A) Characteristics of the cultures that were used. B) Animals were bled on day 13 to assess disease progression by quantitating the number of PBMC-derived (i.e. human CD45+ A2−) CD4+ and CD8+ T cells. C) Kaplan–Meier survival curves for mice receiving PBMC only or PBMC + tTreg or Low Trp+Kyn iTregs (*=p<0.05). D) Animals were bled on day 13 to determine Treg persistence by quantitating the number of human CD45+HLA-A2+ cells expressing CD4. n= 7 mice per group.
In comparison to animals receiving PBMC only, mice receiving PBMC plus expanded tTregs had significantly decreased weight loss and other clinical manifestations of GVHD (ruffled fur, hunched back, decreased activity) from day 13 onward (Figure S6A and B) along with prolonged survival (Figure 6C). While Low Trp+Kyn iTreg adoptive transfer had positive effects on CD4 T-cell expansion, no significant effects on weight loss, clinical scores or survival were observed.
Low Trp+Kyn iTregs have greatly decreased persistence following adoptive transfer in xenogeneic GVHD recipients
In vivo persistence of adoptively transferred tTreg correlates with efficacy of preventing xenogeneic GVHD in NSG mice (24). To assess the relative persistence of expanded tTreg and Low Trp+Kyn iTreg, expanded Treg (HLA-A2-CD4+) were enumerated from day 13 PB, prior to pathological injury. Results from mice receiving PBMC only, and thus no HLA-A2− cells, defined the assay limit of detection. As shown in Figure 6D, while the number of tTreg in PB was 10-fold over background, Low Trp+Kyn iTregs were not higher than background, suggesting their lack of efficacy may be due to diminished viability.
Low Trp+Kyn iTregs are highly dependent on IL-2 for survival
IL-2 signaling supports glycolysis (22). Because Low Trp+Kyn iTregs express 10-fold higher levels of CD25 than tTreg, Low Trp+Kyn iTregs may be more sensitive to cytokine withdrawal induced apoptosis (39) if IL-2 bioavailability is limiting in vivo. To assess the effects of IL-2 depletion on tTreg, Low Trp+Kyn iTreg and Teff cell viability, cultures were harvested on day 14 and were then cultured for another 7 days in fresh media containing 300U/ml (normal) or 30U/ml IL-2 (low). While similar numbers were recovered from tTreg and Teff cultures grown in 300U/ml and 30U/ml of IL-2, Low Trp+Kyn iTreg recovery was decreased 5-fold (Figure S7), confirming that Low Trp+Kyn iTreg are more dependent on high dose IL-2 than tTreg or Teff. We compared Low Trp+Kyn iTreg cultures expanded with 300- vs. 1000 U/ml of IL-2 to determine whether decreased viability observed in Low Trp+Kyn iTreg cultures resulted from a sub-optimal IL-2 concentration, but observed no concentration-dependent difference in expansion, phenotype or suppressive function (data not shown).
Ultra-low dose IL-2 improves GVHD survival in mice treated with Low Trp+Kyn iTregs, but does not increase overall recipient survival
Clinical trials have now shown that patients treated with ultra-low dose IL-2 have increased Treg numbers (40–43). To determine whether ultra-low dose IL-2 could enhance Low Trp+Kyn iTreg persistence and efficacy, tTreg and Low Trp+Kyn iTreg were purified from the same donor, expanded in vitro (Figure 7A) and were injected with HLA-A2 mismatched PBMC (15×106 each) into NSG mice. A subset of each cohort was given ultralow dose IL-2 (25,000 U/day for 14 days). To assess persistence, animals were bled on days 7 (Figure S8A) and 14 (Figure 7B) Tregs and Teffectors enumerated (Figure 7B). Low Trp+Kyn iTreg were present in similar numbers to tTreg on day 7. Ultra-low dose IL-2 significantly increased the number of day 7 PB Low Trp+Kyn iTreg, although iTregs were not present in higher numbers than tTregs regardless of IL-2 administration. Importantly, Low Trp+Kyn iTreg were not found on day 14, in sharp contrast to tTreg cohorts. Day 14 PB CD4+ and CD8+ Teffectors each were significantly lower in tTreg groups compared to all other cohorts, whereas low Trp+Kyn iTregs did not favorably impact on either day 14 Teffector subset compared to PBMNC controls. Reflective of day 14 Teffector numbers, in vitro expanded tTregs significantly prolonged survival compared to animals receiving PBMC only (Figure 7C), whereas Low Trp+Kyn iTreg adoptive transfer did not. Ultra-low dose IL-2 significantly decreased survival of mice receiving PBMC only or PBMC + Low Trp+Kyn iTreg (p≤0.001 for both). IL-2 decreased the survival of mice receiving PBMC + tTreg, albeit not significantly (median survivals of 53 vs. 38 days). IL-2 significantly increased weight loss and clinical scores from day 11 until time of death (day 15 for Low Trp+Kyn iTreg and day 18 for PBMC only) (Figure S8A, B). Consistent with increased disease severity, IL-2 increased the number of day 14 PB CD4+ and CD8+ T-cells (Figure 7D) in mice receiving either no iTregs or Low Trp+Kyn iTreg. In contrast, mice receiving tTreg had >20-fold fewer PB CD4+ and CD8+ T-cells, and IL-2 had no effect on PBMC-derived T-cell number. The ratio of PB CD4+ to CD8+ T-cells was skewed towards CD8 T-cells in mice receiving Low Trp+Kyn iTreg, exacerbated further by IL-2 treatment. In aggregate these data indicate that ultra-low dose IL-2 favored CD4+ and CD8+ Teffector expansion without sustaining Low Trp+Kyn iTreg persistence, resulting in a failure of iTregs and a reduction in tTregs to prevent GVHD.
Figure 7. Ultra-low dose IL-2 improves xenoGVHD survival in mice treated with Low Trp+Kyn iTregs, but does not increase overall recipient survival.
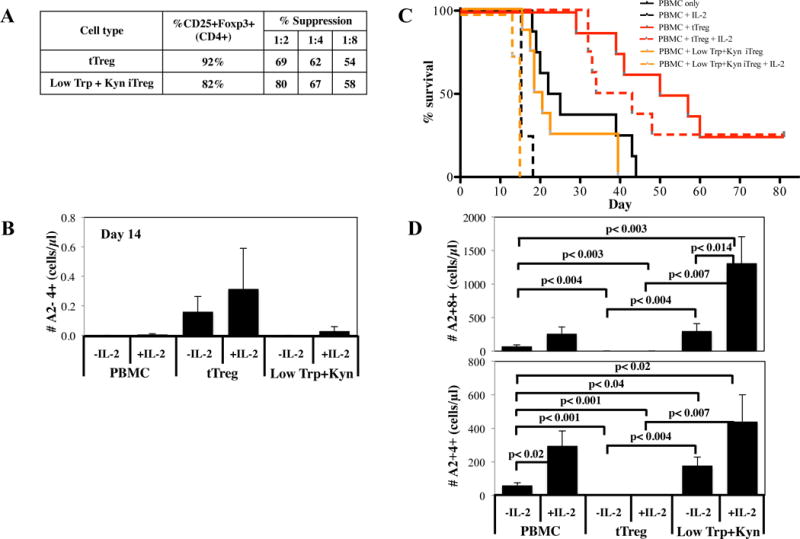
Ex vivo expanded tTreg and Low Trp+Kyn iTregs (15 × 106 cells) were co-transferred with allogeneic, HLA-A2 mismatched PBMC (15 × 106 cells) into non-lethally irradiated NSG mice to assess the ability to ameliorate xenogeneic GVHD. A cohort of mice receiving Low Trp+Kyn iTregs were also given low dose IL-2 (IP; 25000 units/day for 14 days). A) Characteristics of the cultures that were used. B) Animals were bled on day 14 to determine Treg persistence by quantitating the number of human CD45+HLA-A2+ cells expressing CD4. C) Kaplan–Meier survival curves for mice receiving PBMC only or PBMC + tTreg or Low Trp+Kyn iTregs with and without exogenous IL-2 (*=p<0.05). D) Animals were bled on day 14 to assess disease progression by quantitating the number of PBMC-derived (i.e. human CD45+ A2−) CD4+ and CD8+ T cells. n= 8 mice per group.
Low Trp + Kyn iTreg have decreased expression of bcl-2, increased superoxide production, and decreased viability in vitro
Since decreased Low Trp+Kyn iTreg in vivo persistence and efficacy could be due to an overall ‘lack of fitness’ of Low Trp+Kyn iTreg, we assessed the viability of cultures on day 14 by flow cytometry. Low Trp+Kyn iTreg had significantly reduced viability compared to tTreg and Teff cultures (Figure 8A). In keeping with increased basal OXPHOS, Low Trp+Kyn iTreg had increased mitochondrial membrane potential relative to tTreg and Teff, which can lead to increased formation of reactive oxygen species (ROS) (Figure 8B and C). Kyn have been linked to glutathione depletion, increased formation of ROS and apoptosis in activated T-cells (44). To assay ROS generation, we stained cells with dihydroethidine (DHE), a redox-sensitive dye specific for superoxide (45). ROS generation was observed in <8% of tTreg and Teff (Figure 8D), whereas, >25% of Low Trp+Kyn iTregs had demonstrable DHE staining, indicating that decreased viability may be linked to oxidative stress.
Figure 8. Low Trp + Kyn iTreg have decreased expression of bcl-2, increased superoxide production, and decreased viability in vitro.
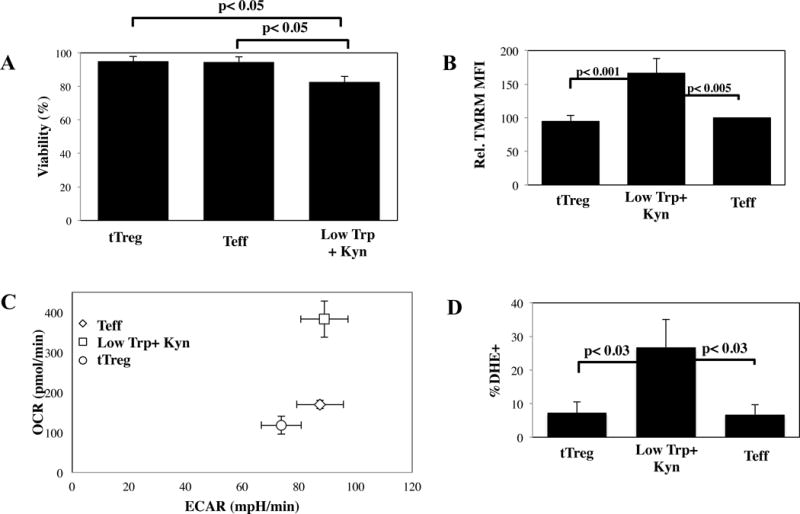
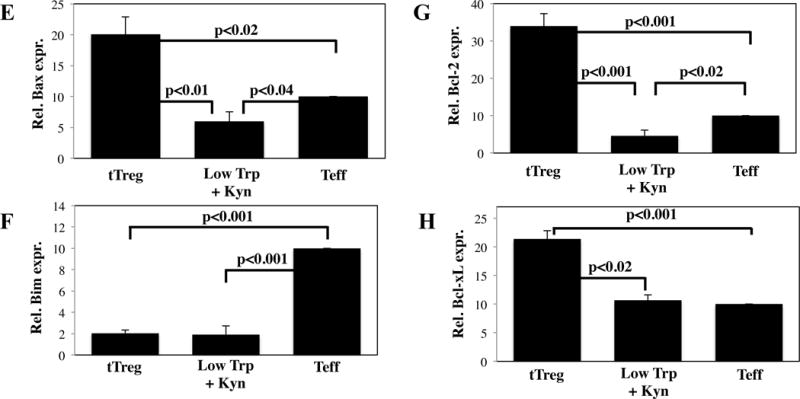
A) Viability in cultures of tTreg, Low Trp+Kyn iTreg and Teff was assessed by flow cytometry using Annexin-V and 7-AAD. B) Average mitochondrial membrane potential (ΔΨm) of the various cultures was measured by flow cytometry using TMRM. C) Representative example of resting OCR and ECR for cultured cells. D) Superoxide production in cultured cells was determined by flow cytometry using DHE. RNA was isolated from cultured cells and the relative expression of the following genes determined: E) Bax, F) Bim, G) Bcl-2 and H) Bcl-xL. Results are from 3–5 independent experiments.
Pro- and anti-apoptotic members of the bcl-2 family are key mediators of the mitochondrial apoptotic pathway, and can have a significant impact on viability. Bax induces mitochondrial outer membrane permeabilization (MOMP) and is allosterically regulated by Bim (activation) and Bcl-2/Bcl-xL (inhibition), which compete for a common binding site (46). To determine whether this pathway contributes to decreased Low Trp+Kyn iTreg viability, the expression of these bcl family members was determined (Figure 8E–H, respectively). tTreg express significantly higher levels of Bax than Low Trp+Kyn iTreg and Teff but, as we reported previously, they maintain a favorable ratio of pro- to anti-apoptotic proteins, expressing low levels of Bim and high levels of Bcl-2 Bcl-xL (24). In contrast, Teff had 5-fold higher Bim expression and lower expression of Bcl-2 and Bcl-x (3.4 and 2.1-fold, respectively). Like tTreg, Low Trp+Kyn iTreg expressed low levels of Bim, suggesting that decreased viability may not be due to MOMP. However, they also expressed significantly less Bcl-2, Bcl-xL and Bax than tTreg. In conclusion, determining the pathways that cause decreased Low Trp+Kyn iTreg viability, and identifying ways to promote iTreg survival via appropriately regulating Bcl-2 pathway members, will be critical steps for clinical translation.
Discussion
Because we previously showed that IDO expressed by pDCs induces Treg function in human T-cells in vitro (10, 16, 17), we sought to determine whether suppressive iTreg could be generated from naïve human CD4+ T-cells using conditions that mimicked pDCs (i.e. Low Trp+Kyn) that would be amenable for clinical translation. Here we show that naïve T-cells expanded in conditions of Low Trp+Kyn are as suppressive as tTregs. Low Trp+Kyn iTregs have advantages over tTregs, including a more simple purification and the initial yield after purification is ~7-fold higher than PBMC tTreg and 400-fold higher than UCB tTregs. Consequently, therapeutic yield would be ~100-fold higher than reported in initial studies for the direct infusion of PBMC tTregs (5, 47, 48). Biochemically, we found that naïve CD4+ T-cells expanded in the presence of low Trp+Kyn displayed a metabolic profile unlike CD4+ or CD8+ T-cells in the literature, in that they were very active in glycolysis, OXPHOS, glutaminolysis and FAS.
One of the most striking features of Low Trp+Kyn iTreg metabolism is greatly increased glutaminolysis. While it was unclear the precise mechanisms responsible driving this pathway in Low Trp+Kyn iTreg, which had very low expression of the transcription factors most closely linked to glutaminolysis (p53 and c-myc) (32), engagement of this pathway would have several benefits. First, glutaminolysis produces a variety of amino acids, which prevents GCN2 activation and suppresses the integrated stress response. Glutaminolysis is uniquely suited for high levels of OXPHOS in that glutamine metabolic pathways lead to products that directly control ROS levels. Specifically, glutamine input is the rate-limiting step for the synthesis of glutathione (GSH) and glutaminolysis generates NADPH, which reduces oxidized GSSG to GSH. NADPH is also necessary for FAS (32), and fits with the FAS-profile displayed by Low Trp+Kyn iTreg (increased expression of molecules involved in uptake or transport of FA and glycerol), distinct from the FAO-profile associated with tTreg (increased expression of molecules involved in shuttling FA to the mitochondria or synthesizing triacylglycerides).
Low Trp+Kyn iTreg metabolism appears to be skewed to help them survive in conditions of low Trp. Specifically, arylhydrocarbon ligands have previously been shown to up-regulate CD98, a subunit of the LAT1 antiporter (49). Since the LAT1 antiporter is capable of exchanging glutamine for Trp (33), it is likely that increased glutaminolysis and CD98 expression maximizes Trp and Leucine uptake (50) to maintain their viability and serve as an energy source.
Decreased Low Trp+Kyn iTreg survival even with ultra-low dose IL-2 may result from ROS-driven mitochondrial permeability transition (51) or apoptosis controlled by Bcl-2 family members (46). This process may be exacerbated by activated, CD25+ Teffectors that receive exogenous IL-2 and produce pro-inflammatory cytokines. While in vitro and in vivo survival is the primary limitation for the clinical translation of Low Trp+Kyn iTreg, it is important to note that there are many drugs available that inhibit these processes, including: Cyclophilin-D/GSK3ß inhibitors, anti-oxidants (e.g. n-acetylcysteine) or z-VAD-fmk to inhibit caspase-3 activation. In conclusion, this work defines the clinical-scale production of iTregs induced in vitro through expansion of Teff under conditions of low Trp+Kyn that are as suppressive in vitro as tTreg. A better understanding of Low Trp+Kyn iTreg metabolic demands and survival requirements are needed prior to clinical translation.
Supplementary Material
Figure S1: Purification phenotype of cells used in this study. tTreg (CD4+25++) and naïve T-cells (CD4+ 25–45RA+) were purified from peripheral blood using magnetic beads and purity assessed by flow cytometry. Representative example and summary for T cell (CD4 and CD25) (A) and tTreg markers (CD127, and Foxp3) (B).
Figure S2: Schema for in vitro expansion of nTreg, Rapa/TGFß iTreg, naïve CD4 Teff and naïve CD4+ LT/Kyn iTreg. tTreg (CD4+25++127−) and naïve CD4+ T cells (CD4+ 25–45RA+) were purified from PBMNCs using magnetic beads. A) Purified cells were stimulated on day 0 with a cell line expressing CD86 and CD64 (KT64/86) to which anti-CD3 mAb is bound. For tTreg, Rapa (109nM) was added on day 0 and high dose IL-2 on day 2. For the indicated cultures of naïve T-cells, IL-2 and Kyn were added on day 0 and were incubated in media containing normal (25 mM) or low (5 mM) concentrations of tryptophan. Low Trp+Kyn iTreg were incubated with low Trp media and fresh Kyn throughout the culture period. To increase expansion, Low Trp+Kyn iTregs and Teff cultures were re-stimulated on day 7. % yield at purification (B) and total cell yield after expansion (C) are calculated based upon a standard-sized apheresis product of 12×109 cells.
Figure S3: Low Trp+Kyn iTreg, like tTreg, secrete IL-10 and not effector cytokines but do not have Foxp3 gene hypomethylation or Helios expression. To assess the potential for secretion of effector cytokines, tTreg, Low Trp+Kyn and Teff cultures were re-stimulated with PMA/Ionomycin for 4 hours in the presence of Brefeldin, and the % of total cells secreting IFNγ (A), IL-17 (B), and IL-4 (C) were assessed by intracellular cytokine staining. To assess the ability of cultured cells to produce IL-10, cultures were re-stimulated overnight with CD3/28 beads and, after harvest, the supernatants were tested for IL-10 (L.D=limit of detection for the assay). E) Methylation status of 10 CpG sites within the TSDR was determined for cultures of tTreg and naive T cells cultured ± Low Trp+Kyn using bisulfite sequencing (n=4). F) Helios expression in cultures of tTreg and naive T cells cultured ± Low Trp+Kyn. To determine whether suppression was contact-dependent, cultured cells were incubated with CFSE-labeled PBMC in transwell plates with cultured cells either in the lower well with the PBMC (cell contact), or in the upper chamber (transwell). Suppression of CD8+ T cell proliferation was assessed by CFSE-dilution (G) and by directly enumerating CD8+ T cells (H). n = 3–5 experiments.
Figure S4: Low Trp + Kyn iTreg phenotype and suppressive function are stable in vitro. Ex vivo expanded Low Trp+Kyn iTregs and Teffs were harvested on day 12 and were re-suspended in normal media (i.e. 25mM Trp and now Kyn) or re-cultured in Low Trp+Kyn for the final two days of culture. On day 14, cultures were assayed for Treg phenotype by flow cytometry (Foxp3+CD25bright) and for suppressive function. Data are a summary of 3 independent experiments.
Figure S5: tTreg express significantly higher levels of genes involved in FAO compared to Low Trp+Kyn iTreg and Teff. RNA was isolated from in vitro expanded tTreg, Low Trp+Kyn iTreg and Teff cells and the relative expression of the following genes involved in FAO determined: A) CPT1a, B) ACSL3 and C) DGAT1. Results are from 3 independent experiments.
Figure S6: Adoptive transfer of Low Trp + Kyn iTreg does not increase survival in a xenogeneic model of GVHD. Ex vivo expanded tTreg and Low Trp+Kyn iTregs (15 × 106 cells) were co-transferred with allogeneic, HLA-A2 mismatched PBMC (15 × 106 cells) into non-lethally irradiated NSG mice to assess the ability to ameliorate xenogeneic GVHD. Percent initial weight curves A) and clinical scores B) for mice receiving PBMC plus adoptive transfer of tTreg or Low Trp+Kyn iTregs. n= 7 mice per group.
Figure S7: Low Trp+Kyn iTregs are highly IL-2-dependent for survival. To assess cytokine dependence, PB tTregs, Low Trp+Kyn iTregs and Teffs cultures were split on day 14 into their respective media with either normal (300U/ml) or limiting (30U/ml) amounts of IL-2 and re-cultured for another 7 days. Dependence was based upon the number of cells remaining at the end of culture, with 300U/ml designated as 100%. Data are from 3 independent experiments.
Figure S8: Ultra-low dose IL-2 improves xenoGVHD survival in mice treated with Low Trp+Kyn iTregs, but does not increase overall recipient survival. Ex vivo expanded tTreg and Low Trp+Kyn iTregs (15 × 106 cells) were co-transferred with allogeneic, HLA-A2 mismatched PBMC (15 × 106 cells) into non-lethally irradiated NSG mice to assess the ability to ameliorate xenogeneic GVHD. A) Animals were bled on day 7 to determine Treg persistence by quantitating the number of human CD45+HLA−A2+ cells expressing CD4. A cohort of mice receiving Low Trp+Kyn iTregs were also given low dose IL-2 (IP; 25000 units/day for 14 days). Percent initial weight curves B) and clinical scores C) for mice receiving PBMC plus adoptive transfer of tTreg or Low Trp+Kyn iTregs. n= 8 mice per group.
Acknowledgments
The authors thank Dr. David A. Bernlohr and Rocio Foncea (Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota) and Minnesota Obesity Center (NIH P30 DK050456) for assistance in Seahorse experiments, and Charles Lehnen (Department of Pediatrics, University of Minnesota) for technical assistance and animal handling.
This work was supported in part by research grants from the Children’s Cancer Research Fund and Blood and Marrow Transplant Research Fund (K.L.H.), Leukemia and Lymphoma Translational Research Grant R6029–07 (B.R.B.), National Institutes of Health (NIH) grants R01 HL114512-01 (M.L.M, K.L.H.), P01 AI056299 (B.R.B.), NCI P01 CA067493 (B.R.B., J.E.W., J.S.M.) and NHLBI N01HB037164 (J.E.W., J.S.M.), R01 CA157971, (to A.K.) and F31 award, CA177119 (to E.A.H.). This work was also funded in part by an Infrastructure grant (for the XFe96 Seahorse) from the University of Minnesota Academic Health Center (to AK) from the JDRF Collaborative Centers for Cell Therapy and the JDRF Center on Cord Blood Therapies for Type 1 Diabetes (J.L.R., C.H.J.). This work was supported in part by an NIH Clinical and Translational Science Award to the University of Minnesota (8UL1TR000114) and an NIH P30 CA77598 utilizing the shared resource Flow Cytometry Core from the Masonic Cancer Center, University of Minnesota.
Abbreviations
- 3-HAA
3-hydroxyanthranilic acid
- AhR
aryl hydrocarbon receptor
- CFSE
carboxyfluorescein diacetate succinimide ester
- cGMP
current Good Manufacturing Practices
- DHE
dihydroethidine
- ECAR
extracellular acidification rate
- FCCP
fluorocarbonyl cyanide phenylhydrazone
- GrB
Granzyme B
- GVHD
graft versus host disease
- HSCT
hematopoietic stem cell transplantation
- IDO
indoleamine 2,3-dioxygenase
- iTreg
induced Treg
- Kyn
kynurenine
- LAP
Latency Associated Peptide
- mTOR
mammalian Target of Rapamycin
- NSG
NOD-SCID-IL2Rgc
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- PBMC
peripheral blood mononuclear cells
- pDCs
plasmacytoid dendritic cells
- rapa
rapamycin
- ROS
reactive oxygen species
- Teff
effector T cells
- Tmem
memory T cells
- TMRM
tetramethylrhodamine
- Tr1
Type 1 regulatory T cells
- Trp
tryptophan
- tTreg
Thymic Treg
- UCB
umbilical cord blood
Footnotes
Disclosure
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. BRB, JLR, BLL, CHJ are founders of and scientific advisors for Tmunity Therapeutics and, with KLH hold patents for the production and use of iTregs for clinical trials. The other authors have no conflicts of interest to disclose.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- 1.Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity. 2009;30(5):656–665. doi: 10.1016/j.immuni.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012;12(6):443–458. doi: 10.1038/nri3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bluestone JA, Tang Q, Sedwick CE. T regulatory cells in autoimmune diabetes: past challenges, future prospects. J Clin Immunol. 2008;28(6):677–684. doi: 10.1007/s10875-008-9242-z. [DOI] [PubMed] [Google Scholar]
- 4.Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061–1070. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. 2016;127(8):1044–1051. doi: 10.1182/blood-2015-06-653667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hippen KL, Riley JL, June CH, Blazar BR. Clinical perspectives for regulatory T cells in transplantation tolerance. Semin Immunol. 2011;23(6):462–468. doi: 10.1016/j.smim.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35(1):109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, Riley JL, et al. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11(6):1148–1157. doi: 10.1111/j.1600-6143.2011.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noval Rivas M, Burton OT, Wise P, Charbonnier LM, Georgiev P, Oettgen HC, et al. Regulatory T cell reprogramming toward a Th2-cell-like lineage impairs oral tolerance and promotes food allergy. Immunity. 2015;42(3):512–523. doi: 10.1016/j.immuni.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The Indoleamine 2,3-Dioxygenase Pathway Is Essential for Human Plasmacytoid Dendritic Cell-Induced Adaptive T Regulatory Cell Generation. J Immunol. 2008;181(8):5396–5404. doi: 10.4049/jimmunol.181.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moseman EA, Liang X, Dawson AJ, Panoskaltsis-Mortari A, Krieg AM, Liu YJ, et al. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. Journal of immunology. 2004;173(7):4433–4442. doi: 10.4049/jimmunol.173.7.4433. [DOI] [PubMed] [Google Scholar]
- 12.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 13.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 14.Gurtner GJ, Newberry RD, Schloemann SR, McDonald KG, Stenson WF. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology. 2003;125(6):1762–1773. doi: 10.1053/j.gastro.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 15.Szanto S, Koreny T, Mikecz K, Glant TT, Szekanecz Z, Varga J. Inhibition of indoleamine 2,3-dioxygenase-mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis research & therapy. 2007;9(3):R50. doi: 10.1186/ar2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood. 2009;114(24):5062–5070. doi: 10.1182/blood-2009-06-227587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Taylor PA, Mellor AL, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood. 2008;111(6):3257–3265. doi: 10.1182/blood-2007-06-096081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. The Journal of clinical investigation. 2007;117(9):2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma MD, Hou DY, Baban B, Koni PA, He Y, Chandler PR, et al. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity. 2010;33(6):942–954. doi: 10.1016/j.immuni.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009;113(24):6102–6111. doi: 10.1182/blood-2008-12-195354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, et al. An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos. Immunity. 2013;38(5):998–1012. doi: 10.1016/j.immuni.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15(5):981–988. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hippen KL, Harker-Murray P, Porter SB, Merkel SC, Londer A, Taylor DK, et al. Umbilical cord blood regulatory T-cell expansion and functional effects of tumor necrosis factor receptor family members OX40 and 4-1BB expressed on artificial antigen-presenting cells. Blood. 2008;112(7):2847–2857. doi: 10.1182/blood-2008-01-132951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. 2009;9(2):83–89. doi: 10.1038/nri2474. [DOI] [PubMed] [Google Scholar]
- 26.Gottschalk RA, Corse E, Allison JP. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol. 2012;188(3):976–980. doi: 10.4049/jimmunol.1102964. [DOI] [PubMed] [Google Scholar]
- 27.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6):1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu J, Ni X, Pan X, Lu H, Lu Y, Zhao J, et al. Human CD39hi regulatory T cells present stronger stability and function under inflammatory conditions. Cell Mol Immunol. 2016 doi: 10.1038/cmi.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Younas M, Hue S, Lacabaratz C, Guguin A, Wiedemann A, Surenaud M, et al. IL-7 modulates in vitro and in vivo human memory T regulatory cell functions through the CD39/ATP axis. J Immunol. 2013;191(6):3161–3168. doi: 10.4049/jimmunol.1203547. [DOI] [PubMed] [Google Scholar]
- 30.Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med. 2011;3(67):67ra68. doi: 10.1126/scitranslmed.3001975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105(48):18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016 doi: 10.1038/nrc.2016.131. [DOI] [PubMed] [Google Scholar]
- 33.Cibrian D, Saiz ML, de la Fuente H, Sanchez-Diaz R, Moreno-Gonzalo O, Jorge I, et al. CD69 controls the uptake of L-tryptophan through LAT1-CD98 and AhR-dependent secretion of IL-22 in psoriasis. Nat Immunol. 2016;17(8):985–996. doi: 10.1038/ni.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Man K, Kallies A. Synchronizing transcriptional control of T cell metabolism and function. Nat Rev Immunol. 2015;15(9):574–584. doi: 10.1038/nri3874. [DOI] [PubMed] [Google Scholar]
- 36.Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015;36(2):81–91. doi: 10.1016/j.it.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 37.O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41(1):75–88. doi: 10.1016/j.immuni.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, et al. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell. 2015;161(4):750–761. doi: 10.1016/j.cell.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, et al. Mature T lymphocyte apoptosis–immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- 40.Bell CJ, Sun Y, Nowak UM, Clark J, Howlett S, Pekalski ML, et al. Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J Autoimmun. 2015;56:66–80. doi: 10.1016/j.jaut.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, 3rd, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365(22):2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365(22):2067–2077. doi: 10.1056/NEJMoa1105143. [DOI] [PubMed] [Google Scholar]
- 43.Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H, et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res. 2014;20(8):2215–2225. doi: 10.1158/1078-0432.CCR-13-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1(9):1460–1468. doi: 10.4161/onci.21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free radical biology & medicine. 1998;25(7):826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- 46.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. The EMBO journal. 2011;30(18):3667–3683. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117(14):3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 48.Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood. 2014;124(4):638–644. doi: 10.1182/blood-2014-03-564401. [DOI] [PubMed] [Google Scholar]
- 49.Tomblin JK, Arthur S, Primerano DA, Chaudhry AR, Fan J, Denvir J, et al. Aryl hydrocarbon receptor (AHR) regulation of L-Type Amino Acid Transporter 1 (LAT-1) expression in MCF-7 and MDA-MB-231 breast cancer cells. Biochem Pharmacol. 2016;106:94–103. doi: 10.1016/j.bcp.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14(5):500–508. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Green DR. The end and after: how dying cells impact the living organism. Immunity. 2011;35(4):441–444. doi: 10.1016/j.immuni.2011.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Purification phenotype of cells used in this study. tTreg (CD4+25++) and naïve T-cells (CD4+ 25–45RA+) were purified from peripheral blood using magnetic beads and purity assessed by flow cytometry. Representative example and summary for T cell (CD4 and CD25) (A) and tTreg markers (CD127, and Foxp3) (B).
Figure S2: Schema for in vitro expansion of nTreg, Rapa/TGFß iTreg, naïve CD4 Teff and naïve CD4+ LT/Kyn iTreg. tTreg (CD4+25++127−) and naïve CD4+ T cells (CD4+ 25–45RA+) were purified from PBMNCs using magnetic beads. A) Purified cells were stimulated on day 0 with a cell line expressing CD86 and CD64 (KT64/86) to which anti-CD3 mAb is bound. For tTreg, Rapa (109nM) was added on day 0 and high dose IL-2 on day 2. For the indicated cultures of naïve T-cells, IL-2 and Kyn were added on day 0 and were incubated in media containing normal (25 mM) or low (5 mM) concentrations of tryptophan. Low Trp+Kyn iTreg were incubated with low Trp media and fresh Kyn throughout the culture period. To increase expansion, Low Trp+Kyn iTregs and Teff cultures were re-stimulated on day 7. % yield at purification (B) and total cell yield after expansion (C) are calculated based upon a standard-sized apheresis product of 12×109 cells.
Figure S3: Low Trp+Kyn iTreg, like tTreg, secrete IL-10 and not effector cytokines but do not have Foxp3 gene hypomethylation or Helios expression. To assess the potential for secretion of effector cytokines, tTreg, Low Trp+Kyn and Teff cultures were re-stimulated with PMA/Ionomycin for 4 hours in the presence of Brefeldin, and the % of total cells secreting IFNγ (A), IL-17 (B), and IL-4 (C) were assessed by intracellular cytokine staining. To assess the ability of cultured cells to produce IL-10, cultures were re-stimulated overnight with CD3/28 beads and, after harvest, the supernatants were tested for IL-10 (L.D=limit of detection for the assay). E) Methylation status of 10 CpG sites within the TSDR was determined for cultures of tTreg and naive T cells cultured ± Low Trp+Kyn using bisulfite sequencing (n=4). F) Helios expression in cultures of tTreg and naive T cells cultured ± Low Trp+Kyn. To determine whether suppression was contact-dependent, cultured cells were incubated with CFSE-labeled PBMC in transwell plates with cultured cells either in the lower well with the PBMC (cell contact), or in the upper chamber (transwell). Suppression of CD8+ T cell proliferation was assessed by CFSE-dilution (G) and by directly enumerating CD8+ T cells (H). n = 3–5 experiments.
Figure S4: Low Trp + Kyn iTreg phenotype and suppressive function are stable in vitro. Ex vivo expanded Low Trp+Kyn iTregs and Teffs were harvested on day 12 and were re-suspended in normal media (i.e. 25mM Trp and now Kyn) or re-cultured in Low Trp+Kyn for the final two days of culture. On day 14, cultures were assayed for Treg phenotype by flow cytometry (Foxp3+CD25bright) and for suppressive function. Data are a summary of 3 independent experiments.
Figure S5: tTreg express significantly higher levels of genes involved in FAO compared to Low Trp+Kyn iTreg and Teff. RNA was isolated from in vitro expanded tTreg, Low Trp+Kyn iTreg and Teff cells and the relative expression of the following genes involved in FAO determined: A) CPT1a, B) ACSL3 and C) DGAT1. Results are from 3 independent experiments.
Figure S6: Adoptive transfer of Low Trp + Kyn iTreg does not increase survival in a xenogeneic model of GVHD. Ex vivo expanded tTreg and Low Trp+Kyn iTregs (15 × 106 cells) were co-transferred with allogeneic, HLA-A2 mismatched PBMC (15 × 106 cells) into non-lethally irradiated NSG mice to assess the ability to ameliorate xenogeneic GVHD. Percent initial weight curves A) and clinical scores B) for mice receiving PBMC plus adoptive transfer of tTreg or Low Trp+Kyn iTregs. n= 7 mice per group.
Figure S7: Low Trp+Kyn iTregs are highly IL-2-dependent for survival. To assess cytokine dependence, PB tTregs, Low Trp+Kyn iTregs and Teffs cultures were split on day 14 into their respective media with either normal (300U/ml) or limiting (30U/ml) amounts of IL-2 and re-cultured for another 7 days. Dependence was based upon the number of cells remaining at the end of culture, with 300U/ml designated as 100%. Data are from 3 independent experiments.
Figure S8: Ultra-low dose IL-2 improves xenoGVHD survival in mice treated with Low Trp+Kyn iTregs, but does not increase overall recipient survival. Ex vivo expanded tTreg and Low Trp+Kyn iTregs (15 × 106 cells) were co-transferred with allogeneic, HLA-A2 mismatched PBMC (15 × 106 cells) into non-lethally irradiated NSG mice to assess the ability to ameliorate xenogeneic GVHD. A) Animals were bled on day 7 to determine Treg persistence by quantitating the number of human CD45+HLA−A2+ cells expressing CD4. A cohort of mice receiving Low Trp+Kyn iTregs were also given low dose IL-2 (IP; 25000 units/day for 14 days). Percent initial weight curves B) and clinical scores C) for mice receiving PBMC plus adoptive transfer of tTreg or Low Trp+Kyn iTregs. n= 8 mice per group.