

Abstract
We report the identification of a novel reassortant clade 2.3.4.4 H5N8 virus from a dead grey heron in Korea in 2017. Outbreaks of clade 2.3.4.4 H5 HPAIVs have been reported worldwide, and they have evolved into multiple genotypes among wild birds. Phylogenetic analysis indicated that this virus likely originated from Qinghai Lake and Western Siberia and further evolved through reassortment with Eurasian LPAI during the 2016 fall migration of wild birds. Enhanced surveillance and comparative genetic analysis will help to monitor the further evolution and dissemination of clade 2.3.4.4 HPAIVs.
Electronic supplementary material
The online version of this article (doi:10.1007/s00705-017-3547-2) contains supplementary material, which is available to authorized users.
Keywords: Avian influenza virus, Wild bird, H5N8, Clade 2.3.4.4, Influenza
Avian influenza virus (AIV) belongs to the genus Influenzavirus A of the family Orthomyxoviridae. Sixteen hemagglutinin (HA) subtypes (H1 to H16) and nine neuraminidase (NA) subtypes (N1 to N9) have been identified in avian species. To date, all identified highly pathogenic AIVs (HPAIVs) that cause high mortality in poultry have been reported to be of the H5 or H7 subtype. Since 1996, the A/goose/Guangdong/1/1996 (Gs/Gd) lineage of H5 HPAIV has spread to more than 60 countries and resulted in outbreaks with considerable economic losses to the poultry industry, infections in wild birds, and clinical, often fatal, cases in humans [1].
The Gs/Gd H5 HPAIVs have evolved into 10 genetically distinct HA clades (0–9) and have undergone reassortment with neuraminidase and internal genes from other viruses to generate novel reassortant viruses with NA gene subtypes N1, N2, N3, N5, N6, N8, and N9 [2–4]. The HA gene of clade 2.3.4.4 H5 HPAIVs has evolved into four distinct genetic groups, A-D [5]. In early 2014, outbreaks of groups A and B of H5N8 clade 2.3.4.4 were reported in South Korea, followed by outbreaks of group A H5N8 HPAI in Europe and North America in late 2014 [6, 7]. Aquatic migratory birds are suspected to play a key role in the global spread of group A H5N8 clade 2.3.4.4 from East Asia to Europe and North America [8]. In contrast, there have been less widespread reports of the group B viruses since their original detection in China in 2013 and South Korea in early 2014 [6].
In May 2016, novel reassortant group B clade 2.3.4.4 H5N8 viruses were detected in wild birds at Qinghai Lake in China and Lake Uvs-Nuur at the Russia-Mongolia border, and they were subsequently detected in various regions of Europe, Africa, and India in the fall of 2016 [9–12]. Meanwhile, novel reassortant group C clade 2.3.4.4 H5N6 viruses were identified in Korea and Japan in the fall of 2016, and they have since caused numerous outbreaks in domestic poultry and wild birds in Korea [13, 14]. In January 2017, during HPAI surveillance of wild birds in Korea, we isolated a group B H5N8 clade 2.3.4.4 virus from the carcass of a grey heron (Ardea cinerea). In this study, we sequenced this H5N8 virus, A/grey heron/Korea/W779/2017(H5N8) (W779), and conducted a comparative phylogenetic analysis to trace its origin and understand its genetic relationship to other group B H5N8 clade 2.3.4.4 viruses identified in Eurasia and Africa in 2016-2017.
The dead grey heron from which we isolated A/grey heron/Korea/W779/2017(H5N8) was found in Jeonju, South Korea (35°52’4.15”N, 127°6’28.08”E), on January 27, 2017. Tissue samples were collected from the carcass and homogenized with phosphate-buffered saline (PBS) to a 10% (wt/vol) final concentration. The swabs were collected into 1 ml of antibiotic-containing PBS (gentamicin, 250 μg/mL). After centrifugation, 100 μl of medium from each tissue and swab sample was inoculated into the allantoic cavity of a 10-day-old specific-pathogen-free (SPF) embryonated chicken egg. The allantoic fluid was harvested after 72 h of incubation at 37 °C. AIV was identified by a hemagglutination assay and an H5 subtype-specific reverse transcription polymerase chain reaction (RT-PCR) assay [15]. The isolate was identified as H5N8 AIV by subtyping RT-PCR [16]. The complete genomes were sequenced using influenza A universal primers as described previously [17]. Nucleotide sequences were deposited in the GenBank database (accession nos. MF155629-MF155636). For phylogenetic analysis, by reference sequences were selected based on sequence homology as determined by GISAID EpiFLU BLAST searches (http://platform.gisaid.org), including clade 2.3.4.4 H5 HPAIVs identified in 2016-2017 and related low-pathogenic strains [3]. For Bayesian phylogenetic analysis and estimation of the time to the most recent common ancestor (tMRCA), Bayesian analysis was performed for all eight gene segments using BEAST version 1.8.4. We employed a Hasegawa-Kishino-Yano (HKY) substitution model with four gamma categories and specified an uncorrelated lognormal relaxed clock and GMRF Bayesian skyride tree prior for each segment. The Markov chain Monte Carlo (MCMC) method was employed with 50 million chain lengths to draw inference under this model. BEAST output was analyzed with TRACER v1.4 (https://beast.bio.ed.ac.uk/tracer) with 10% burn-in. Maximum-clade-credibility (MCC) trees with median heights were generated for each dataset using TreeAnnotator version 1.8.4 and visualized using the program FigTree v1.4.2. Estimated tMRCA values were obtained from MCC trees with common ancestor heights.
We considered the W779 virus to be an HPAIV on the basis of the deduced amino acid sequence at the HA proteolytic cleavage site (PLREKRRKR/G). In the HA, the receptor-binding sites maintained Q226 and G228 (H3 numbering), which is suggestive of preferential binding to the sialic acid-2,3-NeuAcGal, as is typical for avian influenza viruses [18]. The established markers of NA inhibitor resistance (E119A, H274Y, and N294S, N2 numbering) and amantadine resistance (V27A and S31N, M2 protein) were not found [19, 20]. In addition, phenotypic markers related to increased potential for transmission and pathogenicity to mammals such as markers in PB2 (E158G, E627K, and K701N), PB1 (Y436H), and PA (T515A) were also not found [21, 22].
Phylogenetic analysis of the HA gene showed that the W779 virus clustered within group B of H5 clade 2.3.4.4 HPAIV (Fig. 1 and Supplemental Figure 1). However, the characterization of the complete genome revealed that this virus had likely undergone reassortment with Eurasian low-pathogenic AIVs (LPAIVs). Particularly, the PB2, PB1, HA, NA, M, and NS genes of the W779 virus clustered with group B H5N8 clade 2.3.4.4 HPAIV identified in Western Siberia (Uvs-Nuur Lake), China (Qinghai Lake), Europe, and India in 2016-2017 (Supplemental Figures 1 and 2). On the other hand, the PA gene of the W779 virus was phylogenetically distinct from those of H5N8 viruses from Qinghai Lake and Uvs-Nuur Lake but clustered with an Indian isolate (A/painted stork/India/10CA03/2016, India 10CA03), an Italian isolate (A/turkey/Italy/17VIR538-1/2017), and a Russian isolate (A/gadwall/Chany/97/2016, Chany 97). The NP gene was phylogenetically distinct from H5N8 viruses identified at Qinghai Lake, Uvs-Nuur Lake, India (India 10CA03), Russia (Chany 97), and Europe.
Fig. 1.
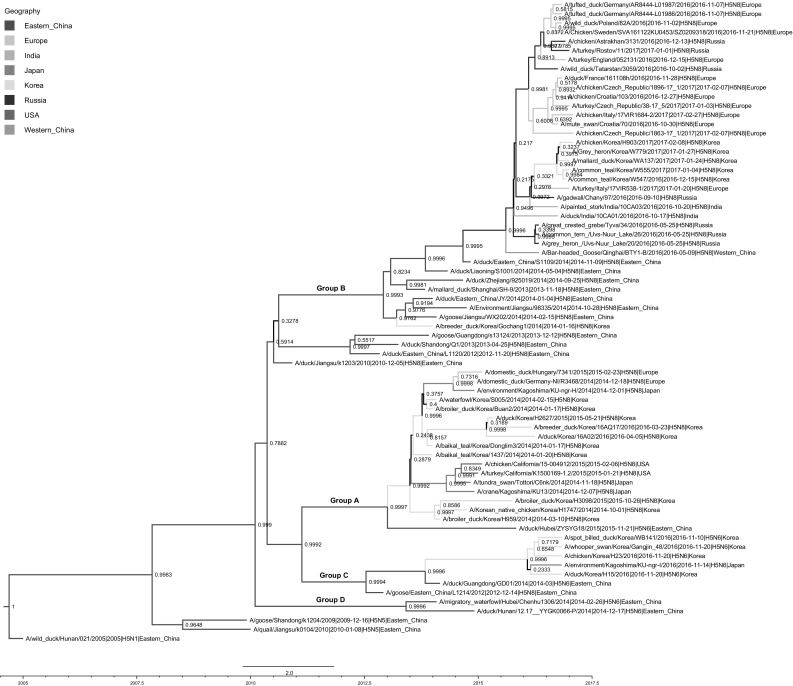
Temporally structured maximum-clade-credibility phylogenetic tree (years on the horizontal axis) of the hemagglutinin gene of HPAIV H5 clade 2.3.4.4 viruses. The Korean 2017 H5N8 isolate used in this study is colored in red. Branches are colored according to location. The posterior probabilities of Bayesian analysis in which the associated taxa clustered together are shown next to the branches
We estimated that ancestors of the novel H5N8 HPAIV identified in Korea emerged among wild birds during May-September 2016, based on the tMRCA for each gene segment of H5N8 HPAIV identified in Korea during December 2016-February 2017 (Table 1), which corresponds to timing of HPAIV detection in Qinghai Lake, Western Siberia, and Russia. Based on these data, we speculate that the W779 virus is a descendant of group B H5N8 viruses identified in waterfowl in Siberia (or nearby high latitudes) during May-September 2016 that acquired PA and NP segments from a gene pool of Eurasian LPAIV before detection in Korea in the winter of 2016-2017. All genes of A/grey heron/Korea/W779/2017(H5N8) were phylogenetically distinct from group A of clade 2.3.4.4 H5N8 viruses that circulated in South Korea in 2014-2016 and group C of clade 2.3.4.4 H5N6 viruses isolated in South Korea in 2016. Collectively, the phylogenetic analysis indicated that the W779 virus is a novel reassortant virus of group B clade 2.3.4.4 H5N8 that shares genetic ancestry with H5N8 HPAIV from Qinghai Lake and Western Siberia and likely evolved through reassortment with Eurasian LPAIVs.
Table 1.
Time of the most recent common ancestor for each gene segment of H5N8 viruses isolated in Korea, 2016-2017
| Mean | 95% HPD | Posterior probability | |
|---|---|---|---|
| PB2 | August 2016 | June – October 2016 | 0.9992 |
| PB1 | August 2016 | July – October 2016 | 0.9993 |
| PA | July 2016 | May – October 2016 | 0.9995 |
| HA | September 2016 | July – October 2016 | 0.9997 |
| NP | May 2016 | October 2015 – October 2016 | 0.9997 |
| NA | May 2016 | June – October 2016 | 0.9689 |
| M | August 2016 | June – October 2016 | 0.4849 |
| NS | September 2016 | June – October 2016 | 0.9995 |
Previous studies also suggested that group B clade 2.3.4.4 H5N8 viruses identified from stopover and wintering regions of wild birds (Europe, India, and Russia) in fall and winter 2016 were genetically close to H5N8 viruses identified from breeding regions in Western Siberia during spring 2016 [9–12]. The W779 virus had a different genome constellation from other reassortant H5N8 viruses identified in Europe, Russia and India during 2016-2017, suggesting that the W779 virus evolved by an independent reassortment event from other reassortant H5N8 viruses (Supplemental Figure 1).
It is believed that wild birds have played an important role in dissemination of Gs/Gd HPAIV, as seen in the spread of clade 2.2 H5N1 HPAIV from Qinghai Lake and circumpolar breeding areas to Europe and East Asia in 2005-2006 [23, 24] and clade 2.3.4.4 H5N8 HPAIV from Siberia to Europe, Asia, and North America in 2014 [8]. Based on previous reports, possible long-distance dissemination of virus via wild bird migration, and our phylogenetic analysis in this study, we suspected that migratory wild birds introduced the novel reassortant H5N8 HPAIVs into Korea. Since 2014, outbreaks of clade 2.3.4.4 H5 HPAIV have been reported in various geographic regions, and they have evolved into multiple genotypes [5]. Enhanced surveillance and comparative genetic analysis will help to monitor the further evolution and dissemination of clade 2.3.4.4 HPAIVs.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Figure 1 Temporally structured maximum-clade-credibility phylogenetic tree (years on the horizontal axis) of the PB2, PB1, PA, HA, NP, NA, M, and NS genes of HPAIV H5 clade 2.3.4.4 viruses. The Korean 2017 H5N8 isolate used in this study is colored in red. The posterior probabilities of Bayesian analysis in which the associated taxa clustered together are shown next to the branches. (PDF 7 kb)
Acknowledgements
We thank the National Institute of Environmental Research (NIER) and the Ministry of Environment for their efforts in the control of HPAIVs in wild birds. This work was supported by the National Institute of Environmental Research, Grant no. NIER-2017-01-01-008.
Compliance with ethical standards
Conflict of interest
The authors of this study have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Chanjin Woo and Jung-Hoon Kwon contributed equally to this work.
Electronic supplementary material
The online version of this article (doi:10.1007/s00705-017-3547-2) contains supplementary material, which is available to authorized users.
References
- 1.Sonnberg S, Webby RJ, Webster RG. Natural history of highly pathogenic avian influenza H5N1. Virus Res. 2013;178(1):63–77. doi: 10.1016/j.virusres.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qi X, Cui L, Yu H, Ge Y, Tang F. Whole-genome sequence of a reassortant H5N6 avian influenza virus isolated from a live poultry market in China, 2013. Genome Announc. 2014 doi: 10.1128/genomeA.00706-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao K, Gu M, Zhong L, Duan Z, Zhang Y, Zhu Y, Zhao G, Zhao M, Chen Z, Hu S, Liu W, Liu X, Peng D, Liu X. Characterization of three H5N5 and one H5N8 highly pathogenic avian influenza viruses in China. Vet Microbiol. 2013;163(3–4):351–357. doi: 10.1016/j.vetmic.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 4.Lee MS, Chen LH, Chen YP, Liu YP, Li WC, Lin YL, Lee F. Highly pathogenic avian influenza viruses H5N2, H5N3, and H5N8 in Taiwan in 2015. Vet Microbiol. 2016;187:50–57. doi: 10.1016/j.vetmic.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Lee DH, Bahl J, Torchetti MK, Killian ML, Ip HS, DeLiberto TJ, Swayne DE. Highly pathogenic avian influenza viruses and generation of novel reassortants, United States, 2014–2015. Emerg Infect Dis. 2016;22(7):1283–1285. doi: 10.3201/eid2207.160048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee DH, Torchetti MK, Winker K, Ip HS, Song CS, Swayne DE. Intercontinental spread of Asian-origin H5N8 to North America through Beringia by migratory birds. J Virol. 2015;89(12):6521–6524. doi: 10.1128/JVI.00728-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee YJ, Kang HM, Lee EK, Song BM, Jeong J, Kwon YK, Kim HR, Lee KJ, Hong MS, Jang I, Choi KS, Kim JY, Lee HJ, Kang MS, Jeong OM, Baek JH, Joo YS, Park YH, Lee HS. Novel reassortant influenza A (H5N8) viruses, South Korea, 2014. Emerg Infect Dis. 2014;20(6):1087–1089. doi: 10.3201/eid2006.140233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Global Consortium for HN. Related Influenza V Role for migratory wild birds in the global spread of avian influenza H5N8. Science. 2016;354(6309):213–217. doi: 10.1126/science.aaf8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DH, Sharshov K, Swayne DE, Kurskaya O, Sobolev I, Kabilov M, Alekseev A, Irza V, Shestopalov A. Novel reassortant clade 2.3.4.4 avian influenza A (H5N8) virus in wild aquatic birds, Russia, 2016. Emerg Infect Dis. 2017;23(2):359–360. doi: 10.3201/eid2302.161252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, Liu H, Bi Y, Sun J, Wong G, Liu D, Li L, Liu J, Chen Q, Wang H, He Y, Shi W, Gao GF, Chen J. Highly pathogenic avian influenza A (H5N8) virus in wild migratory birds, Qinghai Lake, China. Emerg Infect Dis. 2017;23(4):637–641. doi: 10.3201/eid2304.161866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagarajan S, Kumar M, Murugkar HV, Tripathi S, Shukla S, Agarwal S, Dubey G, Nagi RS, Singh VP, Tosh C. Novel reassortant highly pathogenic avian influenza (H5N8) virus in Zoos, India. Emerg Infect Dis. 2017;23(4):717–719. doi: 10.3201/eid2304.161886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pohlmann A, Starick E, Harder T, Grund C, Hoper D, Globig A, Staubach C, Dietze K, Strebelow G, Ulrich RG, Schinkothe J, Teifke JP, Conraths FJ, Mettenleiter TC, Beer M. Outbreaks among wild birds and domestic poultry caused by reassorted influenza A (H5N8) clade 2.3.4.4 viruses, Germany, 2016. Emerg Infect Dis. 2017;23(4):633–636. doi: 10.3201/eid2304.161949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeong J, Woo C, Ip HS, An I, Kim Y, Lee K, Jo SD, Son K, Lee S, Oem JK, Wang SJ, Kim Y, Shin J, Sleeman J, Jheong W. Identification of two novel reassortant avian influenza a (H5N6) viruses in whooper swans in Korea, 2016. Virol J. 2017;14(1):60. doi: 10.1186/s12985-017-0731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwon JH, Lee DH, Swayne DE, Noh JK, Yuk SS, Erdene-Ochir TO, Hong WT, Jeong JH, Jeong S, Gwon GB, Lee S, Song CS. Reassortant clade 2.3.4.4 avian influenza a (H5N6) virus in a wild Mandarin Duck, South Korea, 2016. Emerg Infect Dis. 2017;23(5):822–826. doi: 10.3201/eid2305.161905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Organisation for Animal Health (2009) OIE Terrestrial Manual
- 16.Fereidouni SR, Starick E, Grund C, Globig A, Mettenleiter TC, Beer M, Harder T. Rapid molecular subtyping by reverse transcription polymerase chain reaction of the neuraminidase gene of avian influenza A viruses. Vet Microbiol. 2009;135(3–4):253–260. doi: 10.1016/j.vetmic.2008.09.077. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146(12):2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 18.Stevens J, Blixt O, Tumpey TM, Taubenberger JK, Paulson JC, Wilson IA. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312(5772):404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 19.Chander Y, Jindal N, Sreevatsan S, Stallknecht DE, Goyal SM. Molecular and phylogenetic analysis of matrix gene of avian influenza viruses isolated from wild birds and live bird markets in the USA. Influenza Other Respir Viruses. 2013;7(4):513–520. doi: 10.1111/irv.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orozovic G, Orozovic K, Lennerstrand J, Olsen B. Detection of resistance mutations to antivirals oseltamivir and zanamivir in avian influenza A viruses isolated from wild birds. PloS One. 2011;6(1):e16028. doi: 10.1371/journal.pone.0016028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hulse-Post DJ, Franks J, Boyd K, Salomon R, Hoffmann E, Yen HL, Webby RJ, Walker D, Nguyen TD, Webster RG. Molecular changes in the polymerase genes (PA and PB1) associated with high pathogenicity of H5N1 influenza virus in mallard ducks. J Virol. 2007;81(16):8515–8524. doi: 10.1128/JVI.00435-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Si YJ, Lee IW, Kim EH, Kim YI, Kwon HI, Park SJ, Nguyen HD, Kim SM, Kwon JJ, Choi WS, Beak YH, Song MS, Kim CJ, Webby RJ, Choi YK. Genetic characterisation of novel, highly pathogenic avian influenza (HPAI) H5N6 viruses isolated in birds, South Korea, November 2016. Euro Surveill. 2017 doi: 10.2807/1560-7917.ES.2017.22.1.30434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Xiao H, Lei F, Zhu Q, Qin K, Zhang XW, Zhang XL, Zhao D, Wang G, Feng Y, Ma J, Liu W, Wang J, Gao GF. Highly pathogenic H5N1 influenza virus infection in migratory birds. Science. 2005;309(5738):1206. doi: 10.1126/science.1115273. [DOI] [PubMed] [Google Scholar]
- 24.Lee YJ, Choi YK, Kim YJ, Song MS, Jeong OM, Lee EK, Jeon WJ, Jeong W, Joh SJ, Choi KS, Her M, Kim MC, Kim A, Kim MJ, Ho Lee E, Oh TG, Moon HJ, Yoo DW, Kim JH, Sung MH, Poo H, Kwon JH, Kim CJ. Highly pathogenic avian influenza virus (H5N1) in domestic poultry and relationship with migratory birds, South Korea. Emerg Infect Dis. 2008;14(3):487–490. doi: 10.3201/eid1403.070767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 Temporally structured maximum-clade-credibility phylogenetic tree (years on the horizontal axis) of the PB2, PB1, PA, HA, NP, NA, M, and NS genes of HPAIV H5 clade 2.3.4.4 viruses. The Korean 2017 H5N8 isolate used in this study is colored in red. The posterior probabilities of Bayesian analysis in which the associated taxa clustered together are shown next to the branches. (PDF 7 kb)